

Abstract
Despite decades of effort, pancreatic adenocarcinoma (PDAC) remains an intractable clinical challenge. An insufficient understanding of mechanisms underlying tumor cell responses to chemotherapy contributes significantly to the lack of effective treatment regimens. Here, paclitaxel, a first-line chemotherapeutic agent, was observed to interact synergistically with birinapant, a second mitochondrial-derived activator of caspases mimetic. Therefore, we investigated molecular-level drug interaction mechanisms using comprehensive, reproducible, and well-controlled ion-current-based MS1 quantification (IonStar). By analyzing 40 biological samples in a single batch, we compared temporal proteomic responses of PDAC cells treated with birinapant and paclitaxel, alone and combined. Using stringent criteria (e.g. strict false-discovery-rate (FDR) control, two peptides/protein), we quantified 4069 unique proteins confidently (99.8% without any missing data), and 541 proteins were significantly altered in the three treatment groups, with an FDR of <1%. Interestingly, most of these proteins were altered only by combined birinapant/paclitaxel, and these predominantly represented three biological processes: mitochondrial function, cell growth and apoptosis, and cell cycle arrest. Proteins responsible for activation of oxidative phosphorylation, fatty acid β-oxidation, and inactivation of aerobic glycolysis were altered largely by combined birinapant/paclitaxel compared with single drugs, suggesting the Warburg effect, which is critical for survival and proliferation of cancer cells, was alleviated by the combination treatment. Metabolic profiling was performed to confirm substantially greater suppression of the Warburg effect by the combined agents compared with either drug alone. Immunoassays confirmed proteomic data revealing changes in apoptosis/survival signaling pathways, such as inhibition of PI3K/AKT, JAK/STAT, and MAPK/ERK signal transduction, as well as induction of G2/M arrest, and showed the drug combination induced much more apoptosis than did single agents. Overall, this in-depth, large-scale proteomics study provided novel insights into molecular mechanisms underlying synergy of combined birinapant/paclitaxel and describes a proteomics/informatics pipeline that can be applied broadly to the development of cancer drug combination regimens.
Pancreatic adenocarcinoma (PDAC)1 is the fourth leading cause of cancer-related death in the United States and is expected to become the second most common by 2030(1, 2). The median survival of PDAC patients is only 4–6 months, and five-year survival is less than 5%. More than 50% of patients have locally advanced or metastatic cancer at the time of diagnosis (3–5). For these patients, chemotherapy and radiation are the primary options (6). However, only modest improvements in outcome have been achieved owing to a lack of effective drugs, an inability to predict which drugs will be effective in a given patient (7), and a poor understanding of the molecular interactions of chemotherapy drugs.
Combination chemotherapy is employed in most clinical settings because of the potential for additive or synergistic effects of appropriately selected agents and the delay of drug resistance onset (8). Paclitaxel is currently used with gemcitabine as first-line chemotherapy for advanced metastatic pancreatic cancer (9). It inhibits microtubule depolymerization, which is necessary for cell division, and activates tyrosine kinase pathways as well as tumor-suppressor genes, thus promoting mitotic arrest and apoptosis of cancer cells (10, 11). Paclitaxel failed in pancreatic cancer as a single agent, but the nab-paclitaxel (Abraxane®) formulation was approved in combination with gemcitabine (12, 13). However, combined gemcitabine/nab-paclitaxel increased survival by only 2.1 months compared with gemcitabine alone in the MPACT Phase III trial (ClinicalTrials.gov, trial number NCT00844649) (14). Therefore, new drug combinations are urgently needed to improve standard-of-care PDAC therapy.
Birinapant (TL32711) is a bivalent second mitochondrial-derived activator of caspases (SMAC) mimetic (15). SMAC targets inhibitor of apoptosis proteins (IAP), such as cIAP1 and cIAP2, which play a significant role in cell survival and apoptosis resistance, and are overexpressed in PDAC (16, 17). Similar to the function of SMAC, birinapant binds the baculovirus IAP repeat (BIR) domain of IAPs, causing the degradation of TNF receptor associated factor (TRAF)-bound IAPs and activation of caspase8-mediated apoptosis (18). Thus, SMAC mimetics that target IAPs can sensitize various cancer cells, including pancreatic cancer cells, to chemotherapy drugs (19, 20).
This study tested the hypothesis that paclitaxel, a pro-apoptotic drug, would synergize with IAP inhibitors such as birinapant in pancreatic cancer cells. Inhibition of cell proliferation and induction of cell death were employed as efficacy endpoints in a cell culture model, and in parallel, a comprehensive, precise, and large-scale proteomics investigation was conducted to enable global, unbiased interrogation of protein alterations that could shed light upon the molecular mechanisms underlying drug interaction. Liquid chromatography–mass spectrometry (LC-MS)-based proteomics strategies hold significant advantages over conventional transcriptomic analysis. They circumvent the frequent lack of concordance between mRNA and protein expression (21, 22) and could reveal biomarkers of PDAC tumor cell drug sensitivity, which genomic and transcriptomic profiling has been unable to provide (7). A unique ion-current-based proteomics strategy was an important enabling factor in this study, in that it permits reproducible and reliable quantification of the large number of samples required for comprehensive temporal analysis of cellular drug responses (23, 24), provides extremely low levels of missing data (15) and the ability to quantify subtle changes in protein abundance (15, 25), and has well-controlled false-positive discovery rates (26, 27). Using this strategy, we performed quantitative proteomic analysis of PDAC cells treated with birinapant and paclitaxel, alone and in combination. A nano-flow LC system with a long column and an Orbitrap Fusion Lumos MS were employed to achieve the in-depth analysis. The experimental and data analysis processes were stringently controlled to ensure high accuracy, precision, and low missing values, and false-positive alteration of protein abundance was quantified using an experimental null (EN) method (27). Proteomics analysis identified three dysregulated biological processes, including mitochondrial dysfunction, apoptosis induction, and cell cycle arrest, that contributed to the synergistic interactions of birinapant and paclitaxel. These results were validated and extended with additional biological endpoints (e.g. Cell Energy Phenotype Test) and immunoassays.
EXPERIMENTAL PROCEDURES
Cell Culture
The human pancreatic cancer cell line Panc-1 (ATCC, Gaithersburg, MD) was cultured in DMEM (Corning, Corning, NY) with 10% fetal bovine serum. For cell proliferation assays, cells (3.0 × 103 cells/well) were seeded into 96-well plates and treated with a range of concentrations of birinapant and paclitaxel, alone and in combination. After incubation for 72 h, cell proliferation was quantified using the sulforhodamine B assay (28).
Experimental Design and Statistical Rationale
For proteomics analysis, Panc-1 cells were seeded in 100-mm dishes at a density of 3.5 × 105 cells/dish, and replicate dishes were exposed the following day to paclitaxel and birinapant, alone and in combination. The four treatment groups were: 1) vehicle-treated controls (n = 4), 2) birinapant-treated (100 nm; n = 12), 3) paclitaxel-treated (10 nm; n = 12), and 4) birinapant/paclitaxel combined (100 nm/10 nm) (n = 12). Samples were harvested at 6, 24, 48, and 72 h, a time frame that would capture the time course of temporally delayed events that involve signal transduction cascades, such as apoptosis. The cell monolayers were washed with phosphate buffered saline to remove dead cells and debris and then harvested using ACCUTASE (EMD Millipore, Temecula, CA). Because cells detach from the substrate early in the process of apoptosis (29), detached cells were also collected from the culture supernatant. Dead cells or cell debris were removed by a centrifugation-based approach. The cells were then subjected to an exhaustive, reproducible extraction in a high-concentration detergent mixture. Details are in Supplemental Experimental Procedures. The one-way analysis of variance was used to determine the statistical differences between the average protein expressions of each treatment group.
Surfactant-Aided-Precipitation/On-Pellet Digestion
The extracts were processed using a surfactant-aided-precipitation/on-pellet digestion procedure, which provides extensive cleanup to remove detergents and nonprotein matrix components, deep protein denaturation (by both surfactants and precipitation) for rapid, efficient, and reproducible digestion, and thereby achieves reliable quantification of the 40 samples. Details are in the Supplemental Experimental Procedures.
Nano LC/High-Field Orbitrap MS
The nano-flow reverse phase LC included a Spark Endurance autosampler (Emmen, Holland) and an ultrahigh pressure Eksigent (Dublin, CA) Nano-2D Ultra capillary/nano-LC system. Peptide separation employed a long nano-LC column (75-μm inner diameter × 100 cm) with Pepmap 3-μm C18 particles. A large-inner diameter trap (300 μm inner diameter × 1 cm) was packed with Zorbax 5-μm C18 materials to allow large-capacity loading and removal of hydrophobic and hydrophilic matrix components. Mobile phase A was 0.1% formic acid in 2% acetonitrile, and mobile phase B was 0.1% formic acid in 88% acetonitrile. A 4-μg peptide sample was loaded onto the trap with 1% B at 10 μl/min. After the trap was washed for 3 min, a 250-nl/min flow rate was used to back-flush the samples onto the nano-LC column for further separation. The column was enclosed in a heating sheath filled with heat-conductive silicone and warmed homogeneously at 52 °C, which helps improve the chromatographic resolution and reproducibility. The following was the 2.5 h separation gradient used on the column: 4% B for 15 min; 13–28% B for 110 min; 28–44% B for 5 min; 44–60% B for 5 min; 60–97% B for 1 min; 97% B for 17 min. The trap was turned offline at 45 min to flush hydrophobic components.
An Orbitrap Fusion Lumos Mass Spectrometer (Thermo Fisher Scientific, San Jose, CA) was employed for peptide identification and quantification. Data collection was operated in a 3-s cycle using the data-dependent top-speed mode. The MS1 survey scan (m/z 400–1,500) was at a resolution of 120,000, with automated gain control target of 500,000 and a maximum injection time of 50 ms. Precursors were fragmented in HCD activation mode at a normalized collision energy of 35%, and the dynamic exclusion was set with 45 s. Precursors were filtered by quadrupole using an isolation window of 1 Th. The MS2 spectra were collected at a resolution of 15,000 in the Orbitrap, with an automated gain control target of 50,000 and a maximum injection time of 50 ms.
Protein Identification and Quantification
The MS-GF+ searching engine (released on May 17, 2013) was employed to identify peptides by scoring MS/MS spectra against peptides derived from the Uniprot-Swissprot protein database (Homo sapiens, 20,212 entries, released on July 2015). A total of 4,061 proteins was identified. The search parameters were set to 20-ppm tolerance for precursor ion mass and 0.02 Da for fragment ion mass. Two missed cleavages were permitted for fully tryptic peptides. Carbamidomethylation of cysteine was set as a static modification, and a dynamic modification was defined as oxidation at methionine and acetylation at the N-terminal. The FDR of identification was estimated using a target–decoy search strategy that was dependent on a concatenated database of forward and reversed sequences. At least two distinct peptides were required for each identified protein. The FDR for peptide and protein identification was set to 0.1 and 1%, respectively.
Quantitative analysis was performed using the locally developed IonStar pipeline (30). It incorporates SIEVE (v2.2, Thermo Scientific) for chromatographic alignment using the ChromAlign algorithm (31), extraction and procurement of peptide peak areas, and a locally developed R package (available at https://github.com/shxm725/IonStarstat) for data quality control, aggregation, normalization, removal of outliers, and summarization. The ion-current area under the curve of each peptide was integrated into the same frame according to the following criteria: m/z width = 10 ppm; frame time width = 2.5 min. The merged feature intensity file was further processed using the IonStar data processing pipeline for proper data normalization (i.e. total ion-current normalization) and summarization (i.e. sum of intensities). Peptides shared by different proteins were excluded in order to quantify only the unique peptides of each protein. The relative expression ratios of proteins in two groups were calculated by comparing average ion current intensities of replicates in each group, and the statistical significance was evaluated with one-way analysis of variance. The cutoff for altered proteins and false-positive altered protein discovery rate were established using an EN method (27) (below). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (32) partner repository with the dataset identifier PXD007890 and 10.6019/PXD007890.
Functional Analysis
Gene Ontology (GO) annotation was performed with DAVID (Database for Annotation, Visualization and Integrated Discovery) Bioinformatics Resources v6.7 (https://david.ncifcrf.gov/). Functional annotations such as biological process, cellular component, and molecular function were evaluated with the tools integrated in DAVID. Protein–protein interactions were analyzed with STRING (Version 10.0). Heatmaps and hierarchical clustering analyses were obtained using the gplots and reshape2 packages, respectively. In order to analyze the enriched signaling pathways, ingenuity pathway analysis (IPA; http://www.ingenuity.com/products/ipa) was also employed to predict the key upstream and downstream regulators of altered proteins.
Western Blot Analysis
Samples in lysis buffer were electrophoresed on 4–12% polyacrylamide Bis-Tris SDS-PAGE gels (Life Technologies, Grand Island, NY), and the separated proteins were transferred to nitrocellulose membranes using an iBlot system (Life Technologies). After blocking for 1 h, the membrane was incubated with the primary antibody overnight at 4 °C and then with the secondary HRP conjugated antibody (Santa Cruz Biotechnology, Inc., Dallas, TX) for another 1 h. The immunoreactions were activated with the ECL Western substrate (Pierce Biotechnology, Inc., Rockford, IL), and the chemiluminescence was quantified using a Bio-Rad ChemiDoc MP Imaging System (Bio-Rad Laboratories, Hercules, CA). All results were repeated independently three times.
Cell Energy Phenotype Analysis
The oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using a XF96 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA). Briefly, 4,000, 7,000, 12,000, and 17,000 PANC-1 cells in DMEM were seeded in wells of XF96 polystyrene cell culture plates, treated with 1) vehicle, 2) 100 nm birinapant, 3) 10 nm paclitaxel, and 4) birinapant/paclitaxel 100/10 nm and incubated for 6 h, 24 h, 48 h, and 72 h in a humidified, 37 °C incubator with 5% CO2. Each group consisted of six replicates. Because drug-treated cells proliferated at different rates, protein quantification using BCA assays were performed to normalize cell numbers for the OCR and ECAR assays. One day prior to assay, the Seahorse XFp Sensor Cartridge was hydrated at 37 °C in a non-CO2 incubator overnight. Seahorse XF base medium (20 ml) was supplemented with 1 mm pyruvate, 2 mm glutamine, and 10 mm glucose and then warmed to 37 °C, followed by washing with 180 μl prepared medium (pH 7.4) per well. Then 20 μl of stressor mixture (oligomycin and FCCP) were loaded into each injection port of the calibrated sensor cartridge. Each measurement cycle consisted of a mixing time of 2 min and a data acquisition period of 4 min, producing 13 data points for each sample. A baseline OCR or ECAR, recorded before the addition of stressors, was used to normalize and compare samples following addition of the stressor reagents. In this assay, the third and fourth measurement cycles, respectively, were used to generate the absolute OCR (pmol/min) and ECAR (mpH/min) before and after addition of stressor reagents.
RESULTS
Birinapant/Paclitaxel Interaction
To test the hypothesis that paclitaxel, a pro-apoptotic agent approved in first-line PDAC therapy, would synergize with IAP inhibitors such as birinapant, Panc-1 cells were exposed to these drugs, alone and in combination, for various durations (6, 24, 48, 72 h), and cell proliferation was quantified. The IC50 (concentration mediating half-maximal inhibition of proliferation) of paclitaxel was 24.2 nm, and the Imax (maximum inhibition achieved) was 70.2% (Fig. 1A). The IC50 for birinapant was 145 nm, but the Imax was only 35.9%, demonstrating that birinapant alone was unable to inhibit proliferation of most Panc-1 cells. Cell proliferation was quantified for 36 pairs of birinapant/paclitaxel concentrations surrounding the IC50 concentration of each drug for exposure times up to 72 h (Fig. 1B). The experimental data for 72 h of drug exposure were overlaid with a drug interaction response surface that was estimated using a noncompetitive drug interaction model (33) that was modified to include a quantitative drug interaction term Ψ (Fig. 1B). The Ψ term signifies synergistic (Ψ<1), additive (Ψ = 1), or antagonistic (Ψ>1) drug interactions. The surface plot shows the expected response for simple additivity (Ψ = 1) of drug interaction. For most drug concentration pairs, the data points lie below the additivity surface, indicating synergistic drug interaction. The overall score for all concentrations tested was Ψ = 0.61, suggesting synergy between birinapant and paclitaxel.
Fig. 1.

Synergistic interaction of birinapant/paclitaxel combination. (A) Concentration-dependence of cell viability with exposure to single-agent paclitaxel or birinapant for 72 h. The Imax for birinapant was 35.9% whereas the Imax for paclitaxel was 70.2%, demonstrating the limited capability of birinapant alone to inhibit PDAC cell proliferation. (B) Concentration-dependence of combined birinapant/paclitaxel on PDAC cell proliferation. PDAC cells were incubated with 36 combinations of birinapant and paclitaxel concentrations surrounding the IC50 of each drug, represented as data points (x and y axes), and cell numbers were quantified after 72 h of exposure (z axis). The drug interaction surface (mesh) represents the expected effect of the combined drugs if the drug interaction were additive (Ψ = 1); red dots above the surface are observations suggesting antagonism (Ψ > 1); blue dots below the surface suggest synergy (Ψ < 1). The majority of observations were below the surface, indicating overall synergistic interactions between birinapant and paclitaxel.
In-Depth, Reproducible, Well-Controlled Ion Current-Based Proteomic Quantification
To obtain molecular-level insights into these synergistic drug interactions, we investigated cellular drug effects as a function of time for paclitaxel and birinapant, alone and combined. The concentrations to which cells were exposed were selected based upon the concentration/response data of Fig. 1; 10 nm paclitaxel or 100 nm birinapant as single agents inhibited cell growth ∼10–15% and, when combined, inhibited cell growth ∼50%. Thus, the cells were exposed to drug concentrations that are active in inhibiting cell proliferation and initiating apoptosis but do not cause rapid cell death and lysis. A workflow was developed for large-scale, quantitative proteomic profiling of drug interactions, over four time points, with control and three drug treatment groups consisting of three biological replicates per treatment (Fig. 2). The four vehicle controls were picked from each of the four time points and combined into a single group. A total of 40 samples was compared in a single batch using IonStar (30).
Fig. 2.

Experimental design and workflow of this study. Cells were treated with four regimens: 1) vehicle; 2) 10 nm paclitaxel; 3) 100 nm birinapant; 4) combined birinapant/paclitaxel 100/10 mm, and collected at four time points (6 h, 24 h, 48 h, 72 h). All cell lysates were prepared with the surfactant-aided-precipitation/on-pellet digestion procedure for nano LC-MS/MS. Data analysis included ion-current-based MS1 quantification, comprehensive protein identification, and function/signaling pathway annotation. Biological validations were performed with mitochondrial energy phenotype assays and Western blots.
The IonStar experimental/informatics pipeline (30) was optimized to ensure exhaustive and consistent extraction of the 40 samples. The workflow employed extraction in strong detergent cocktails along with high-energy homogenization (34, 35). After reduction/alkylation, the detergents, lipids, fatty acids, salts, and other matrix components were efficiently removed by organic solvent precipitation, and the pellets were digested, reproducibly and robustly, using a surfactant-aided-precipitation/on-pellet digestion procedure (36, 37). High, reproducible protein and peptide recoveries were obtained for all samples (CV%<15% for peptide recovery), and this efficient and reproducible sample preparation laid the foundation for reliable quantification. For LC/MS analysis, a unique nano-LC design was employed that enabled the reproducible analysis of many biological samples (23). This enables reliable quantification of large cohorts with minimal missing data. The 100-cm-long column afforded excellent separation, which enhanced the quantification of low-abundance proteins and permitted better ion current peak-matching among the 40 samples that comprised a single batch of analyzed samples. The chromatogram alignment scores ranged from 0.82–0.94 across the 40 samples (>0.6 being acceptable), indicating excellent consistency in chromatography and detection for this sample set. An Ultra-High-Field Orbitrap Fusion Lumos MS was used for sensitive detection of MS1 signals, as well as in-depth identification. It was observed that higher-energy collisional dissociation-orbitrap significantly outperforms higher-energy collisional dissociation-ion trap for peptide identification in large sample numbers (e.g. ∼23% more peptide identifications over 20 runs; detailed data not shown), and thus, higher-energy collisional dissociation-orbitrap was employed here to assign peptide identifications (IDs) to quantified ion current peaks. IonStar data analysis included accurate chromatogram alignment (ChromAlign), sensitive and comprehensive detection of quantitative features, exact mapping of peptide ID to peptide spectrum matches and effective postgeneration feature quality control, with removal of low-quality quantitative data and aggregation to the protein level (38).
In total, 4,069 proteins were quantified with stringent criteria: <1% protein FDR, <0.1% peptide FDR, and at least two quantified peptides per protein. Of the 4,069 quantified proteins, 4,061 (99.8%) were quantified without any missing data in any sample. This extremely low level of missing data is typical of this ion-current-based workflow (38) and enabled comprehensive investigation of drug mechanisms of interaction across the multiple conditions and time points that comprise the sample set.
The reproducibility of protein quantifications between technical replicates was evaluated, and representative data are shown in Fig. 3A. Excellent reproducibility was observed for replicate analyses of the same pooled sample using ion current (IC)-based quantitation (R2 >0.99), which was far superior to results with spectral counting method SpC (Fig. S1A), especially for low-abundance proteins. The median intragroup coefficient of variation (CV) was <5% for protein quantification among technical replicates (n = 3). By comparison, the median intragroup CVs for biological replicates were <15%, indicating moderate biological variability within the treatment groups (Fig. 3B) and demonstrating that biological variability was significantly greater than technical variability. We also correlated the protein quantitative values by IonStar between two randomly selected biological replicates in each biological group. Good correlations between biological replicates (R2 >0.94) were observed across all groups (Fig. S1B), again indicating low biological variability of this experimental system. Furthermore, low intragroup biological variation (∼10%) was observed for vehicle controls obtained from one of the four time points, indicating the culture time did not have a perceivable effect on the protein expressions when the cell density was maintained well below confluence at all time points.
Fig. 3.
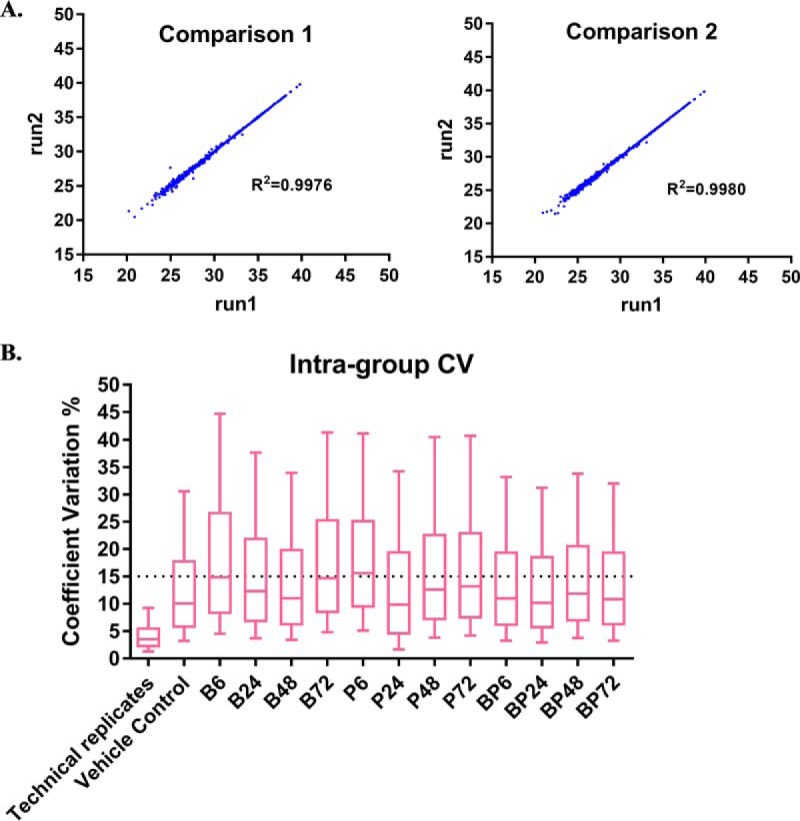
Reproducibility of ion-current-based quantitation. (A) Evaluation of the analytical reproducibility for the ion-current-based quantitation method. The log2 intensity of proteins from two replicate runs of the same sample was compared and the reproducibility of ion-current-based quantitative data was evaluated by linear regression. (B) Box plot (10–90 percentile) of coefficients of variation of protein intensities in each treatment group and technical replicate. Median CV in technical replicates (n = 4) was 3% and CV of biological replicates (n = 3) was measured to be <20%.
For the discovery of altered proteins, an EN method (27) was used to determine the false-positive rate for detection of altered proteins (26). The EN method provides an experimental approach to measure the null distribution and quantifies false positives arising from factors such as technical variability, study-specific features, and biological variability. Here, the EN method determined the optimal fold change cutoff as ≥ 40% (log2 ratio in the range of -0.4854 to 0.4854) and a p value < 0.05. According to these criteria, a total of 541 proteins were altered significantly among the treatment groups. For each treatment group and time point, the measured false altered protein discovery rates were all <1%, indicating confident altered protein discovery. Volcano plots of p values versus ratios for the EN data set and all experimental groups are shown in Fig. S2.
GO Annotation of Altered Proteins
With the cutoff optimized, the significantly altered proteins in each group were identified (Fig. S2B). The total number of altered proteins in the three treatment groups (single-drug and combined drugs) at four time points was 541 (Fig. 4A). Among them, 58 were changed by single-agent birinapant, and 65 were changed by single-agent paclitaxel. When combined, the drugs altered 413 proteins significantly, reflecting more complex molecular-level effects and interactions. Of these, 289 changed proteins were unique to the drug combination group; 34 were shared between the birinapant- and combination groups, whereas 79 were shared between the paclitaxel- and combination groups (Fig. 4A).
Fig. 4.
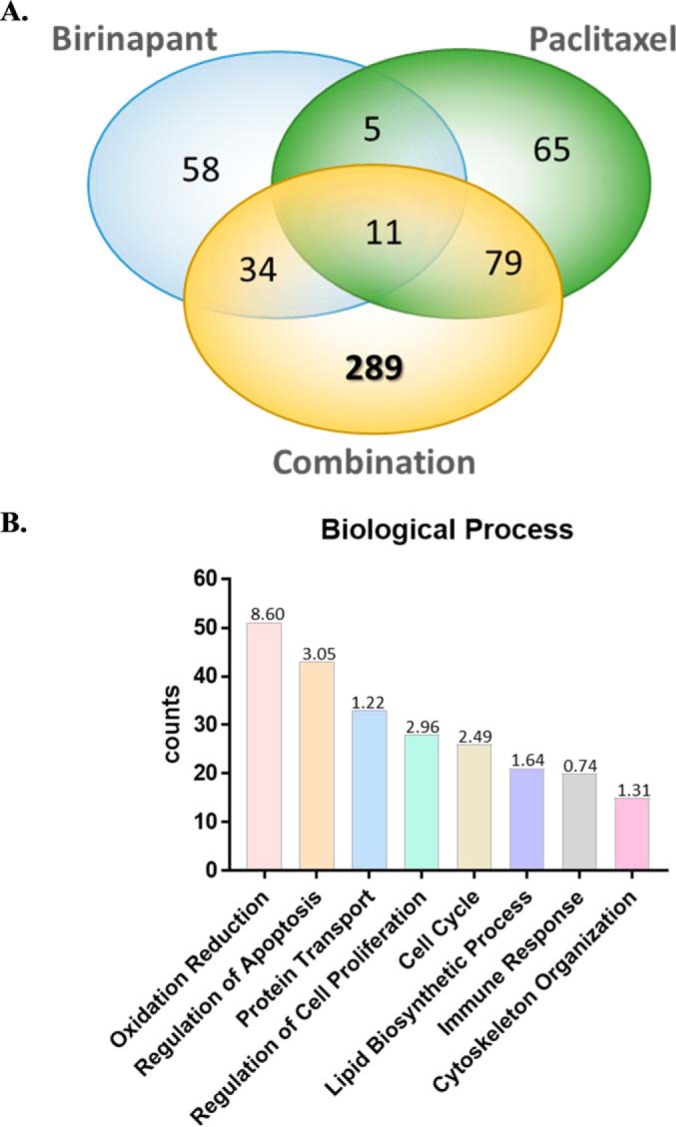
Functional annotation of significantly altered proteins. (A) Venn diagram of total 541 proteins significantly altered in three treatment groups (single-agent and combined drugs) at four time points. Among them, 289 proteins were uniquely altered by combined birinapant/paclitaxel. (B) Bar chart of the top eight mostly enriched biological processes represented by 541 altered proteins in response to drug treatment. These processes included oxidation-reduction, regulation of apoptosis, protein transport, regulation of proliferation, cell cycle, lipid biosynthetic processes, immune response, and cytoskeleton organization. The p value (-log10) of each biological process is shown.
GO analysis indicated the largest number of treatment-altered proteins (51 of 541) were associated with oxidation-reduction (redox), which is involved in critical biological processes such as mitochondrial oxidative phosphorylation (OXPHOS). The second-most enriched biological process (43 proteins) was regulation of apoptosis, which is consistent with the mechanisms of action of these two drugs. In addition, based on the altered proteins, several other functional categories were also identified as perturbed, including protein transport (33 proteins), regulation of cell proliferation (28 proteins), cell cycle (26 proteins), lipid biosynthetic processes (21 proteins), immune response (20 proteins), and cytoskeleton organization (15 proteins) (Fig. 4B). Interestingly, the 289 altered proteins that were unique to the drug combination group were enriched predominantly in three functional categories: apoptosis, cell cycle, and mitochondrial dysfunction (Fig. S3), and these functions therefore became the focus of further investigation as to their contributions to the observed synergistic interactions of combined birinapant/paclitaxel.
Signaling Networks of Altered Proteins
The KEGG database and IPA were employed to analyze the functional roles of altered proteins in the regulation of apoptosis, cell cycle, and mitochondrial function networks. Table I shows the altered protein responses and enriched signaling pathways induced only by the combined drugs.
Table I. Signaling pathways enriched in proteins uniquely altered by combined birinapant/paclitaxel exposure.
| Term | Gene Name | p value (-log10) | Altered proteins # |
|---|---|---|---|
| Mitochondrial Function and Metabolism | |||
| Mitochondrial dysfunction | AIFM1, APP, ATP5A1, ATP5B, ATP5H, ATP5O, COX7A2, COX7A2L, CYB5A, FIS1, NDUFA11, NDUFB4, NDUFB6, PRDX3, SOD2, TXN2, TXNRD2, UQCRC1, UQCRFS1, VDAC1, VDAC2, VDAC3 | 6.57 | 37 |
| Oxidative phosphorylation | ATP5H, ATP5O, ATP5A1, ATP5B, ATP6VOD1, ATP6V0A1, NDUFA11, NDUFB6, TCIRG1, COX7A2, COX7A2L, AIFM1, NSDHL, PMVK, EBP, IDI1, MVK, UQCRC1, UQCRFS1, NDUFB4 | 3.28 | |
| Citrate cycle (TCA cycle) | MDH2, PCK2 | 1.43 | |
| Glycolysis/gluconeogenesis | GALM, HK1, LDHB, PCK2, PGM2 | 1.38 | |
| Pyruvate metabolism | LDHB, MDH2, ME2, PCK2 | 1.19 | |
| Fatty acid metabolism | ACADS, ECHS1, ACAA2 | 2.03 | |
| Fatty acid elongation in mitochondria | ECHS1, ACAA2 | 1.17 | |
| Fatty acid beta-oxidation | ACAA2, ECHS1, HSD17B4, SLC27A4, ACADS, HSD17B4 | 2.21 | |
| Superpathway of cholesterol biosynthesis | EBP, IDI1, MVK, NSDHL, PMVK, SQLE | 1.85 | |
| Cell Growth and Apoptosis | |||
| Apoptosis signaling pathway | AIFM1, CHP1, RIPK1, AKT1, AKT2 | 2.68 | 29 |
| MAPK signaling pathway | LAMTOR3, CHP1, NFKB2, RRAS2, RPS6KA4, STK4, AKT1, AKT2, CRKL | 3.26 | |
| ERBB signaling pathway | CAMK2D, ABL2, AKT1, AKT2, CRKL | 3.36 | |
| mTOR signaling pathway | AKT1, AKT2, FAU, PIK3C2A, RHOG, RPS2, RPS9, RPS13, RPS15, RPS18, RPS23, RPS26, RPS29, RPS6KA4, RPTOR, RRAS2 | 1.62 | |
| Insulin receptor signaling | AKT1, AKT2, CRKL, EIF2B2, PIK3C2A, RPTOR, RRAS2, STX4, SYNJ2 | 1.26 | |
| PI3K/AKT signaling pathway | AKT1, AKT2, CRKL, NFKB2, PIK3C2A | 2.02 | |
| JAK/STAT signaling pathway | AKT1, AKT2, NFKB2, PIK3C2A, RRAS2 | 2.11 | |
| Insulin signaling pathway | HK1, PCK2, PHKA1, RPTOR, AKT1, AKT2, CRKL | 4.71 | |
| Cell Cycle | |||
| G2/M DNA damage checkpoint regulation | AURKA, PKMYT1, CKS1B, AJUBA, ZC3HC1 | 1.85 | 21 |
| Mitosis | F2, DBI, APOE, CKAP2, GRN, AJUBA, KIFC1, ZC3HC1, AURKA, CDCA8, TAF6, ARRB1, AKT1, ERRFI1, UBE2C, NUDCD3, CUL7, AXL, BUB1B, PKMYT1, PPP1R12C | 1.43 | |
| Ploidy | SQSTM1, CKAP2, AURKA, AKT1, CKSlB, BUB1B | 1.32 |
Mitochondrial Function
Most drug-mediated protein responses relating to mitochondrial dysfunction and metabolism were unique to the combination group. The signaling pathways involved include oxidative phosphorylation, TCA cycle, glycolysis/gluconeogenesis, fatty acid and pyruvate metabolism, and cholesterol biosynthesis. We performed clustering analysis of the temporal expression of altered proteins, which can provide insights into the regulation of drug-mediated protein expression and response mechanisms (39). K-means clustering classified the 37 quantified proteins altered only by the drug combination into three clusters for each drug treatment group (Fig. S4). The functional description and quantitation of these molecules are shown in Table II.
Table II. Quantification and functional annotation of proteins altered exclusively by combined birinapant/paclitaxel and associated with mitochondrial function or metabolism.
| Gene Name | Uniprot Entry | Protein Name | GO Category | Log2 Ratio |
p value | Cluster Category | |||
|---|---|---|---|---|---|---|---|---|---|
| BP6 | BP24 | BP48 | BP72 | ||||||
| ATP5H | O75947 | ATP synthase subunit d | Oxidative phosphorylation | 0.1121 | 0.0717 | 0.2984 | 0.6768 | 0.0000569 | Cluster 1 |
| ATP5B | P06576 | ATP synthase subunit beta | Oxidative phosphorylation | 0.1282 | 0.1421 | 0.2880 | 0.5474 | 0.00000549 | Cluster 1 |
| ATP6V0D1 | P61421 | V-type proton ATPase subunit d 1 | Oxidative phosphorylation | 0.1190 | 0.1689 | 0.3550 | 0.6006 | 0.00024743 | Cluster 1 |
| ATP5A1 | P25705 | ATP synthase subunit alpha | Oxidative phosphorylation | 0.0844 | 0.1238 | 0.3664 | 0.6538 | 0.0000261 | Cluster 1 |
| ATP6V0A1 | Q93050 | V-type proton ATPase | Oxidative phosphorylation | 0.3076 | 0.5165 | 0.8411 | 1.2432 | 0.0000321 | Cluster 1 |
| ATP5O | P48047 | ATP synthase subunit O | Oxidative phosphorylation | 0.0616 | 0.0967 | 0.3680 | 0.5672 | 0.00000413 | Cluster 1 |
| TCIRG1 | Q13488 | V-type proton ATPase 116 kDa subunit a isoform 3 | Oxidative phosphorylation | 0.0009 | 0.0103 | 0.3546 | 0.5104 | 0.00380696 | Cluster 1 |
| COX7A2 | P14406 | Cytochrome c oxidase subunit 7A2 | Oxidative phosphorylation | 0.2916 | 0.3907 | 0.5357 | 0.6657 | 0.01293719 | Cluster 1 |
| COX7A2L | O14548 | Cytochrome c oxidase subunit 7A-related protein | Oxidative phosphorylation | 0.3812 | 0.2867 | 0.4464 | 0.7139 | 0.00011716 | Cluster 1 |
| NDUFB4 | O95168 | NADH dehydrogenase [ubiquinone]1 beta subcomplex subunit 4 | Oxidative phosphorylation | 0.2172 | 0.1688 | 0.2420 | 0.5068 | 0.00039813 | Cluster 1 |
| UQCRC1 | P31930 | Cytochrome b-c1 complex subunit 1 | Oxidative phosphorylation | 0.1066 | 0.1458 | 0.2864 | 0.5716 | 0.0000303 | Cluster 1 |
| UQCRFS1 | P47985 | Cytochrome b-c1 complex subunit Rieske | Oxidative phosphorylation | 0.1959 | 0.2386 | 0.3754 | 0.4951 | 0.00022715 | Cluster 1 |
| AIFM1 | O95831 | Apoptosis-inducing factor 1 | Oxidative phosphorylation | 0.2255 | 0.2125 | 0.4084 | 0.5765 | 0.0000706 | Cluster 1 |
| NSDHL | Q15738 | Sterol-4-alpha-carboxylate 3-dehydrogenase | Cholesterol biosynthesis | 0.1378 | 0.1713 | 0.4075 | 0.7025 | 0.0000135 | Cluster 1 |
| PMVK | Q15126 | Phosphomevalonate kinase | Cholesterol biosynthesis | 0.4368 | 0.4302 | 0.6558 | 0.8547 | 0.01003841 | Cluster 1 |
| EBP | Q15125 | 3-beta-hydroxysteroid-Delta(8),Delta(7)-isomerase | Cholesterol biosynthesis | 0.2148 | 0.2046 | 0.3413 | 0.5735 | 0.00928668 | Cluster 1 |
| ECHS1 | P30084 | Enoyl-CoA hydratase | Fatty acid beta-oxidation | 0.2127 | 0.1983 | 0.3093 | 0.5532 | 0.00014265 | Cluster 1 |
| SLC27A4 | Q6P1M0 | Long-chain fatty acid transport protein 4 | Fatty acid beta-oxidation | 0.1461 | 0.2644 | 0.4550 | 0.5508 | 0.0000956 | Cluster 1 |
| ACADS | P16219 | Short-chain specific acyl-CoA dehydrogenase | Fatty acid beta-oxidation | 0.3782 | 0.1762 | 0.4338 | 0.5436 | 0.02931929 | Cluster 1 |
| ACAA2 | P42765 | 3-ketoacyl-CoA thiolase | Fatty acid beta-oxidation | 0.1852 | 0.2393 | 0.3317 | 0.6119 | 0.00142667 | Cluster 1 |
| HSD17B4 | P51659 | Peroxisomal multifunctional enzyme type 2 | Fatty acid beta-oxidation | 0.2445 | 0.4326 | 0.6979 | 0.8934 | 0.0029735 | Cluster 1 |
| CAT | P04040 | Catalase | Mitochondrial function | 0.0133 | 0.0608 | 0.2327 | 0.4949 | 0.0000375 | Cluster 1 |
| CYB5A | P00167 | Cytochrome b5 | Mitochondrial function | 0.2283 | 0.3207 | 0.6140 | 0.7809 | 0.00010234 | Cluster 1 |
| SOD2 | P04179 | Superoxide dismutase [Mn] | Mitochondrial function | 0.1832 | 0.2115 | 0.5235 | 0.9563 | 0.00211055 | Cluster 1 |
| PRDX3 | P30048 | Thioredoxin-dependent peroxide reductase | Mitochondrial function | 0.1215 | 0.1434 | 0.2740 | 0.5253 | 0.000012 | Cluster 1 |
| VDAC1 | P21796 | Voltage-dependent anion-selective channel protein 1 | Mitochondrial function | 0.1031 | 0.1066 | 0.2365 | 0.5463 | 0.0000465 | Cluster 1 |
| VDAC2 | P45880 | Voltage-dependent anion-selective channel protein 2 | Mitochondrial function | 0.0855 | 0.1200 | 0.3238 | 0.6569 | 0.00000235 | Cluster 1 |
| VDAC3 | Q9Y277 | Voltage-dependent anion-selective channel protein 3 | Mitochondrial function | 0.0459 | 0.1259 | 0.2378 | 0.4924 | 0.00000768 | Cluster 1 |
| HK1 | P19367 | Hexokinase-1 | Glycolysis | 0.1734 | 0.1559 | 0.2759 | 0.4864 | 0.00208264 | Cluster 1 |
| PCK2 | Q16822 | Phosphoenolpyruvate carboxykinase | Gluconeogenesis | 0.3505 | 0.5556 | 0.5284 | 0.5958 | 0.00376596 | Cluster 1 |
| ME2 | P23368 | NAD-dependent malic enzyme | Pyruvate metabolism | 0.0674 | 0.1303 | 0.2912 | 0.4948 | 0.00060027 | Cluster 1 |
| MDH2 | P40926 | Malate dehydrogenase | Citrate cycle | 0.1241 | 0.1602 | 0.3691 | 0.6935 | 0.000015 | Cluster 1 |
| PGM2 | Q96G03 | Phosphoglucomutase-2 | Glycolysis | 0.0615 | −0.2167 | −0.3777 | −0.5587 | 0.00035251 | Cluster 2 |
| GALM | Q96C23 | Galactose mutarotase | Glycolysis | −0.4497 | −0.4652 | −0.6392 | −0.6742 | 0.00228728 | Cluster 2 |
| LDHB | P07195 | L-lactate dehydrogenase B chain | Pyruvate metabolism | −0.4277 | −0.4367 | −0.5084 | −0.5900 | 0.00048992 | Cluster 2 |
| SQLE | Q14534 | Squalene monooxygenase | Cholesterol biosynthesis | −0.0799 | 0.2452 | 0.6233 | 0.8730 | 0.00020105 | Cluster 3 |
| TXN2 | Q99757 | Thioredoxin | Mitochondrial function | 0.1716 | 0.2894 | 0.4029 | 0.6675 | 0.00318442 | Cluster 3 |
Cluster 1
Cluster 1 was enriched in proteins that were more strongly up-regulated by the drug combination compared with single-agent responses (32 of 37 proteins). The molecular function terms and quantitative data for these proteins are shown in Table II. Within the mitochondrial function group, the voltage-dependent anion channel (VDAC) regulates metabolites and ion flux between mitochondria and cytosol and is considered as a potential target for cancer therapy (40). Here, VDAC1, VDAC2, and VDAC3 were up-regulated significantly and most strongly by combined birinapant/paclitaxel. This response could reflect drug-induced activation of the mitochondrial permeability transition pore resulting from interruption of mitochondrial calcium homeostasis, which would generate reactive oxygen species (ROS) and release cytochrome c, thus initiating intrinsic apoptosis (41). Furthermore, overexpression of VDAC could promote homo- or hetero-oligomerization and create larger pores for cytochrome c release, enhancing the initiation of apoptosis (34).
Cluster 1 also was enriched in proteins relating to metabolism and generation of energy. The Warburg effect is a tumor metabolic response that involves drastically increased glucose uptake and defective mitochondrial oxidative phosphorylation (42). The switch to glycolysis supplies the metabolic requirements for proliferation and is an adaptation to hypoxia in the cancer microenvironment (43). H+-ATP synthase is a key component in the second stage of OXPHOS, but its expression and activity is reduced in tumors, which promotes the Warburg effect (44). Exposure to the birinapant/paclitaxel combination resulted in increased expression of four ATP synthase subtypes (ATP5B, ATP5O, ATP51A1, ATP5H) within 48h, whereas changes mediated by either drug alone were minimal (Fig. S4), suggesting the restoration of OXPHOS in mitochondria and potential alleviation of the Warburg effect. Furthermore, H+-ATP synthases are critical for the pro-apoptotic function of Bax (45), and a sufficient supply of ATP favors apoptosome formation to trigger the cascade of caspase activation, chromatin condensation, and apoptosis initiation (46). Restoration of OXPHOS would also accelerate reactive oxygen species generation and trigger cell death as a result of high metabolic stress (47, 48). Consequently, (H+)-ATP synthase can function as a tumor suppressor, with the restoration of mitochondrial respiration and oxidation of substrates acting to reduce malignant growth.
Fatty acid elongation and β-oxidation were also elevated by exposure to birinapant/paclitaxel to an extent greater than by single-agent treatment (Fig. S4), as reflected by increased ACAA2, ECHS1, HSD17B4, SLC27A4, and ACADS (Table II). SLC27A4 is a key long-chain fatty acid transporter, and ECHS1, ACADS, and ACAA2 are required for several steps in fatty acid oxidation (12, 49). Enhanced mitochondrial β-oxidation of fatty acids has been reported to promote apoptosis via release of ATP and reactive oxygen species (50) and suggests increased mitochondrial activity and reduction in the Warburg effect.
Cluster 2
Cluster 2 proteins underwent progressive decrease with combined birinapant/paclitaxel exposure. Phosphoglycerate mutase (Table II) is required for glycolysis, and its overexpression in cancer cells enhances glycolytic flux-induced proliferation (51). Galactose mutarotase also participates in aerobic glycolysis in cancer cells (52). Progressive down-regulation of these proteins by combined birinapant/paclitaxel would contribute to a reduction in aerobic glycolysis and thereby the Warburg effect.
Cluster 3
Cluster 3 proteins increased only with the drug combination (Fig. S4, Table II). Squalene monooxygenase is required for cholesterol biosynthesis (53), and thioredoxin (TXN2) facilitates the elevation of mitochondrial activity (50).
Overall, the functional analysis of proteins associated with mitochondrial function and metabolism suggested that the birinapant/paclitaxel combination may counter the Warburg effect in pancreatic cancer cells via enhanced mitochondrial activity, oxidative phosphorylation, cholesterol biosynthesis, and fatty acid β-oxidation, and reduced glycolysis. This hypothesis was investigated below (“Cell Energy Phenotype Analysis”).
Cell Growth and Apoptosis
Proteins uniquely altered by combined birinapant/paclitaxel exposure also suggested effects upon cell growth- and apoptosis-related signaling pathways, including the insulin signaling/receptor pathways, ERBB, mTOR, PI3K/AKT, and JAK/STAT (Table I). The network of protein interactions, including upstream/downstream regulators, was analyzed using IPA to explore the broader, systemwide drug effects suggested by the observed changes (Fig. 5A). Altered proteins shared between the PI3K/AKT and JAK/STAT signaling pathways that were downregulated to a greater extent by the drug combination included AKT1, AKT2, and PI3K-complex proteins (Fig. S5A), and these changes suggested reduced signaling and proliferation (54). The observed increase in expression of NFκB would accelerate the transcription of tumor suppressor genes and inhibit cell growth (55). The data suggested the birinapant/paclitaxel combination decreased P38/MAPK-ERK1/2 signal transduction based upon up-regulated AIFM1, and downregulated MSK1/2 and AKT. Up-regulation of RHOG and RIPK1 was also observed with combined birinapant/paclitaxel, which would activate downstream JNK, promoting apoptosis by activating pro-apoptotic proteins such as caspases.
Fig. 5.

Functional proteins involved in cell proliferation and cell cycle progression that were uniquely altered in the combination treatment group. (A) IPA prediction of upstream regulators of the uniquely altered proteins in cell growth and cell death. The predicted key regulators included Jnk, p38/MAPK, Insulin, PI3K complex, Pkcs, ERK1/2, NFκB. Red and green colors, respectively, represent the up- and down-regulated molecules, the expression ratio is shown under each dysregulated molecule, treated/control. The quantitative data is from samples with combined drugs treatment for 72 h; white represents the predicted upstream regulators of these uniquely altered proteins. Those with purple circles were further validated by immunoassay. (B) STRING network of altered molecules that were exclusively induced by combined birinapant/paclitaxel and associated with cell cycle progression. Proteins are represented as nodes in the figure. The confidence of protein-protein interactions is represented by the thickness of lines linking molecules. All nodes involved in cell cycle progression were included in interactions and most were of high confidence (>0.700).
Cell Cycle Progression
Proteins altered by combined birinapant/paclitaxel were also enriched in signaling pathways related to cell cycle progression, including G2/M DNA damage checkpoint regulation, mitosis, and ploidy, and the network of related protein interactions was investigated using STRING analysis (Fig. 5B). K-means clustering yielded two clusters of proteins that were either up-regulated or downregulated only by the drug combination (Fig. S5B). Cluster 1 (downregulated proteins) included CKS1B, mitotic checkpoint serine/threonine kinase B (BUB1B), ubiquitin-conjugating enzyme E2C (UBE2C), and AKT1. CKS1B plays a key role in cell cycle regulation as the essential accessory protein to the SCFSKP2-CKS1 ubiquitin ligase complex. CKS1B enhances SKP2-mediated degradation of p27Kip1 (a cyclin-dependent kinase inhibitor) and induces G1/S phase transition (56). With prolonged exposure (48–72 h), the drug combination downregulated CKS1B to a greater extent than either single agent, leading to G1/S arrest based upon alleviated p27 degradation. BUB1B delays the onset of anaphase to ensure all chromosomes are properly attached to the mitotic spindle (57), and thus a decrease in BUB1B mediated by combined birinapant/paclitaxel could promote the generation of polyploid cells, which would further promote apoptosis. UBE2C, which encodes a member of the E2 ubiquitin-conjugating enzyme family, also was decreased by the birinapant/paclitaxel combination. UBE2C is required for mitotic cyclin destruction and, as an essential component of the anaphase-promoting complex/cyclosome (APC), promotes M phase progression (24). Overexpression of UBE2C causes chromosome mis-segregation and contributes to tumorigenesis and is considered a potential biomarker for cancer prognosis (58). The significant decrease in UBE2C mediated by the birinapant/paclitaxel combination would suggest a further inhibition of progression through M phase. Cluster 2 (up-regulated proteins) involved APOE, CKAP2, F2, GRN, AJUBA, SQSTM1, and DBI (Fig. S5B). For example, greater up-regulation of APOE was discovered with longer exposure to combined birinapant and paclitaxel, leading to G1/S arrest based on p27 (59). CKAP2, as a substrate of APC/C, was up-regulated with birinapant/paclitaxel combination through restricted ubiquitination and degradation (60). This result is consistent with the observed down-regulation of UBE2C, a component of functional APC/C, caused by combined birinapant and paclitaxel. Thus, the proteomic data suggest the combination of birinapant and paclitaxel could cause cell cycle delay and arrest in both G1/S and G2/M phases.
Biological Validation
Metabolic Transition from Glycolysis to Oxidative Phosphorylation
Based upon functional analysis of protein alterations mediated by combined birinapant/paclitaxel in multiple metabolic pathways, we hypothesized that the drug combination alleviates the Warburg effect in PDAC cells by promoting a transition from glycolysis to mitochondrial oxidative phosphorylation. We tested this hypothesis by analyzing drug effects upon mitochondrial respiration and glycolytic rate. A high glycolysis rate correlates with proton production and a higher extracellular acidification rate and, in parallel, a low oxygen consumption rate. OXPHOS and glycolysis were monitored using a metabolic analyzer, thus characterizing the metabolic programming, stage of transformation, and metabolic potential of the cancer cells. Under both baseline and stressed respiratory conditions, treatment with combined birinapant/paclitaxel increased oxygen utilization to a greater extent than either single agent (Fig. S6) and, in parallel, decreased ECAR. Metabolic stressors included oligomycin, a proton channel inhibitor that inhibits mitochondrial ATP production, resulting in a compensatory increase in glycolytic ATP production, and the proton ionophore FCCP, which depolarizes the mitochondrial membrane and drives OCR higher via a compensatory mitochondrial response to restore membrane potential. Under stressed conditions, the effects of combined birinapant/paclitaxel to increase OCR and decrease ECAR were greater than for either drug alone. Because the OCR and ECAR baselines and magnitude varied at different time points, the results are expressed as the OCR/ECAR ratio normalized to the control groups, which represents the predominant metabolic pattern satisfying energy demand (61). Under nonstressed conditions, the OCR/ECAR ratio increased 1.65-fold after 6 h of exposure to the birinapant/paclitaxel combination and 2.93-fold with 72 h of exposure, thus revealing the temporal transition from glycolysis to oxidative phosphorylation mediated by the drug combination. Under stressed conditions, the OCR/ECAR ratio increased to 6.93-fold over 72 h of exposure to the drug combination, a greater increase than for the drugs alone (Fig. 6A).
Fig. 6.

Cell energy phenotype analysis of the treatment groups under baseline and stressed conditions. (A) Ratio of OCR/ECAR in each group was calculated under both baseline and stressed conditions. Oligomycin and FCCP were used as metabolic stressors. All ratios at different time points were normalized to control groups. Detailed data without normalization is shown in Fig. S6. Under baseline conditions, the OCR/ECAR ratio increased to 2.93-fold after 72 h exposure to the combined drugs and under stressed conditions, the OCR/ECAR ratio increased to 6.93 after 72 h exposure to the combination, indicating the metabolic transition from glycolysis to oxidative phosphorylation was induced by the birinapant/paclitaxel combination. (B) Metabolic potentials of different groups were investigated based upon the ratio of stressed OCR/baseline OCR and stressed ECAR/baseline OCR. The metabolic potential of OCR was increased with time in the birinapant/paclitaxel combination group to a greater extent compared with single agents, whereas the metabolic potential of ECAR was not significantly altered with combined treatment, suggesting that with birinapant/paclitaxel treatment, oxidative phosphorylation became the dominant metabolic pattern to satisfy energy demand, which relieved Warburg effect. Statistical significance in A and B was evaluated with student's t test and is denoted by asterisks (* p < 0.05, ** p < 0.01).
The metabolic potential, defined as the percentage increase of stressed OCR/baseline OCR or stressed ECAR/baseline ECAR (62), was used to evaluate cells' ability to meet energy demand via respiration versus glycolysis under stress. Although ECAR was not altered significantly under these conditions, the metabolic potential of OCR increased with time for the birinapant/paclitaxel combination to greater extent compared with the single agents (Fig. 6B). The results suggest that oxidative phosphorylation is the dominant metabolic pattern to meet energy demand during birinapant/paclitaxel exposure, and that the drug combination relieves the Warburg effect.
Cell Growth Inhibition and Apoptosis Induction
PDAC cell exposure to combined birinapant/paclitaxel resulted in proteomic changes in MAPK/ERK, PI3K/AKT, and JAK/STAT signaling pathways, but to a much lesser extent than observed for either drug alone, suggesting the drug combination caused inhibition of cell proliferation and induction of apoptosis. These hypothesized signaling pathway effects were investigated using an orthogonal approach, Western blot analysis (Fig. 7A).
Fig. 7.

Immunoblotting analysis of regulatory proteins related to cell growth, apoptosis, and cell cycle arrest pathways implicated by proteomic changes with birinapant/paclitaxel combination. (A) Important predicted upstream molecules (ERK, AKT, STAT3, NFkB, JNK) based on proteomic analysis and related to cell growth were validated at both protein abundance and phosphorylation levels. (B) The abundance of apoptotic proteins such as PARP, caspase3, and the anti-apoptotic protein Bcl-2 were evaluated. Cleaved-PARP and caspase3 increased with time, particularly at 48 h and 72 h, in cells exposed to combined birinapant/paclitaxel. Bcl-2 decreased, indicating apoptosis induction by the drug combination. (C) The abundance and phosphorylation of Rb, CDK1, cyclin B1, and Wee1 were examined. The downregulated phosphorylation of Rb with time in the combined birinapant/paclitaxel group induced G1/S arrest to a greater extent compared with the single drugs alone. The temporal increase in phosphorylation of CDK1 and decrease in cyclin B1 and phosphorylated Wee1 mediated by combined birinapant/paclitaxel suggested arrest at the G2/M cell cycle transition that was more marked than in the single-agent paclitaxel group.
ERK phosphorylation was increased drastically after 48 and 72 h of exposure to either birinapant or paclitaxel alone (Fig. 7A), implying activation of pathways conferring drug resistance. The increase was most marked with longer-term exposure to single-agent paclitaxel. However, the combined drugs mediated a significant dampening of the drug-mediated increase in p-ERK, particularly after 48 h of exposure (Fig. 7A, Fig. S7).
AKT phosphorylation was active in drug-free controls and in cells exposed to birinapant or paclitaxel alone for up to 6 h but was much less active with the combined treatment (Fig. 7A). Moreover, p-AKT declined substantially within 6 h of exposure to the birinapant/paclitaxel combination, whereas either drug alone was delayed in suppressing p-AKT, suggesting more rapid inhibition of AKT activation by the combination. This response was quantified and confirmed by the proteomic analysis, which demonstrated the drug combination reduced p-AKT to nadir levels within 6 h of exposure, more rapidly than for either drug alone (Fig. S7).
In the JAK/STAT signaling pathway, STAT3 plays a pivotal role in cytoplasmic signal transduction. STAT3 was activated by paclitaxel alone with exposures ≥24 h, consistent with changes observed in the MAPK/EKR pathway (Fig. 7A). With the drugs in combination, the paclitaxel-mediated increase in STAT3 phosphorylation was reduced nearly to control levels (Fig. S7). Another regulator of cell growth and the apoptosis network, NFκB, is activated in cancers and controls the transcription of genes for proliferation and survival and represents a target for chemotherapy (63). NFκB was activated by birinapant exposure (Fig. 7A) but not by paclitaxel. The birinapant/paclitaxel combination reduced birinapant-mediated phosphorylation of NFκB, which could reduce cell proliferation and increase sensitivity to apoptosis.
The JNK/stress-activated protein kinases, another MAPK subfamily kinase (64), plays a key role in both extrinsic and intrinsic apoptotic pathways (65) JNK phosphorylation was enhanced significantly by combined birinapant/paclitaxel and to a lesser extent by paclitaxel alone (Fig. 7A, Fig. S7), suggesting enhancement of apoptosis by the drug combination via this pathway.
Proteomic evidence for the activation of apoptosis was also investigated by orthogonal approaches (Fig. 7B). PARP repairs DNA strand breaks, and caspase-3 cleavage of PARP into 85- and 25-kDa fragments prevents its DNA repair function during apoptosis (66). The 85-kDa fragment also has been reported to modulate pro-apoptotic molecules such as p53 (67). PARP cleavage was much greater in cells exposed to combined birinapant/paclitaxel compared with either drug alone (Fig. 7B, Fig. S8A), as was caspase-3 cleavage, thereby confirming enhanced activation of apoptotic cascades (35). The anti-apoptotic protein Bcl2 interrupts apoptosis induction by preventing the release of mitochondrial apoptogenic factors such as cytochrome c (68). Bcl2 was elevated in cells exposed to either birinapant or paclitaxel alone, compared with the vehicle control, but was reduced drastically by the birinapant/paclitaxel combination, especially at 48 and 72 h (Fig. 7B, Fig. S8A). Thus, the combined drug treatment not only inhibited cell growth but also induced apoptosis by blocking survival-promoting signaling pathways, and activating pro-apoptotic mediators.
Cell Cycle Progression
Several key checkpoint regulators of G1/S and G2/M transition were confirmed by Western blot analysis (Fig. 7C). The retinoblastoma (Rb) tumor suppressor plays a pivotal role in negative regulation of cell cycle progression and carcinogenesis by arresting cells in G1 phase (69). It complexes with E2F1 transcription factor and inhibits gene transcription required for transition from G1 to S phase (70). Phosphorylation of Rb inhibits this interaction and allows cell cycle progression. Consistent with the quantitative proteomic data suggesting greater inhibition of cell cycle progression by the combined drugs, single-agent birinapant or paclitaxel inhibited the Rb phosphorylation at Ser807/811 to a moderate extent (Fig. 7C, Fig. S8B), whereas phosphorylation decreased earlier and to a greater extent with the drug combination, suggesting onset of G1/S arrest.
G2/M arrest would be expected for paclitaxel-containing regimens, based upon its mechanism of action (71). Several markers for G2/M progression were examined by immunoassay, including phosphorylated CDK1, cyclin B1, myt1, and phosphorylated Wee1 (Fig. 7C, Fig. S8B). CDK1 interacts with cyclin B1 to form an active heterodimer driving G2/M transition (72). In addition to regulation by cyclin partners, Wee1 (73) negatively regulates CDK1 activity by phosphorylation at Tyr15/Thr14, and cdc25 phosphatase positively regulates CDK1 by dephosphorylation. Birinapant and paclitaxel, alone or combined, did not affect CDK1 protein levels (Fig. 7C, Fig. S8B). However, the drug combination did increase phosphorylated CDK1 with time to a greater extent than either drug alone (Fig. 7C), thus decreasing the drive for G2/M progression. Cyclin B1 down-regulation was also observed for the drug combination with prolonged exposure (Fig. 7C), which would reduce formation of the cdc2-cyclin B1 complex that is required for entry into mitosis. Wee1 is regulated negatively by phosphorylation, and combined birinapant/paclitaxel decreased its phosphorylation, which accounts for the increased phosphorylation and inactivation of CDK1 observed here. Thus, the results together suggest that the birinapant/paclitaxel combination could interrupt cell cycle progression by inducing G2/M arrest, whereas the single-drug treatments would not.
DISCUSSION
Development of effective chemotherapeutic regimens for pancreatic cancer, one of the most lethal cancers, remains a challenge. Here we investigated the interaction of birinapant and paclitaxel on pancreatic cancer cells, and observed synergistic inhibition of cell proliferation. Using a comprehensive, well-controlled and high-throughput proteomic approach (IonStar) suitable for large-cohort studies, we investigated underlying molecular mechanisms of drug action and interaction in a moderately large (n = 40) set of biological samples. Low missing values and excellent quantitative accuracy and precision were achieved. From this sample set, 4,061 proteins were quantified without missing data in any of the 40 samples, and 541 were altered significantly by drug treatments. The altered proteins mapped to numerous cellular response pathways, predominant among which were mitochondrial dysfunction, apoptosis, and cell cycle progression. The drug combination exerted effects in numerous processes that were not observed for either drug alone. Drug responses suggested by proteomics analysis were investigated by immunohistochemistry and metabolic analysis. Fig. 8 shows a schematic of the major protein interactions and functional alterations mediated by the drug combination. Metabolic effects of combined birinapant and paclitaxel were consistent with a reduction in the Warburg effect and increased OXPHOS activity, changes that were not significant with either drug alone. The drug combination also inhibited signaling pathways that facilitate the survival and proliferation of cancer cells, such as the PI3K/AKT, JAK/STAT, and MAPK/ERK pathways. Combined birinapant/paclitaxel also established a pro-apoptotic state that resulted in enhanced cell death. In terms of cell cycle progression, the combined drugs inhibited both G1/S and G2/M progression, as evidenced by hallmark changes such as decreased p-Rb and elevated p-CDK1. These interpretations of the proteomic data were all successfully confirmed by either energy phenotype profiling or immunoassay. These results provide global insights into the molecular mechanisms of birinapant/paclitaxel interaction in pancreatic cancer cells and implicate mechanisms that may mediate enhanced sensitivity to paclitaxel by the combination. With establishment of this proof-of-concept work, in vivo studies are warranted in animal model systems that more truthfully recapitulate the biological complexity of human pancreatic cancer. For example, the therapeutic effects and toxicity, as well as the characteristics of drug delivery, metabolism, and related molecular mechanisms of the birinapant/paclitaxel combination, will be investigated further in vivo with our proteomics platform. Such studies could provide new insights not only into complex cellular interactions of combination chemotherapy but also into interindividual differences in drug responses, which could provide a basis for individualization of combination chemotherapy regimens.
Fig. 8.

Proposed mechanisms underlying the synergy of birinapant/paclitaxel combination. A network of three biological processes, including cell growth and apoptosis, cell cycle progression, and mitochondrial activity are depicted before and after combined drug treatment, along with the alterations of the molecules involved. Purple-labeled ellipses and rectangles indicate the activated molecules and activated biological processes, respectively. Green-labeled ellipses and rectangles indicate the inactivated molecules and inactivated biological process respectively.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited at to the ProteomeXchange Consortium via the PRIDE (32) partner repository with the dataset identifier PXD007890 and 10.6019/PXD007890.
Supplementary Material
Footnotes
* This work is supported, in part, by NIH grants AI129518 (JQ), GM121174 (JQ), AI125746(JQ), CA168454 (RMS), CA198096 (RMS), and HD071594 (JQ), a Center of Protein Therapeutics Industrial Award (JQ), and by an NIH CTSA award UL1TR001412 (RMS, JQ). This work utilized shared core resources supported by NIH grant P30CA016056 to Roswell Park Comprehensive Cancer Center.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- PDAC
- pancreatic adenocarcinoma
- SMAC
- second mitochondrial-derived activator of caspases
- IAP
- inhibitor of apoptosis proteins
- BIR
- baculovirus IAP repeat
- TRAF
- TNF receptor associated factor
- EN
- experimental null
- CV
- coefficient of variation
- FDR
- false discovery rate
- OCR
- oxygen consumption rate
- ECAR
- extracellular acidification rate
- SpC
- spectral counting
- OXPHOS
- oxidative phosphorylation
- VDAC
- voltage-dependent anion channel
- BUB1B
- mitotic checkpoint serine/threonine kinase B
- UBE2C
- ubiquitin-conjugating enzyme E2C
- PRIDE
- PRoteomics IDEntifications.
REFERENCES
- 1. Siegel R. L., Miller K. D., and Jemal A. (2016) Cancer statistics, 2016. CA: A Cancer Journal for Clinicians 66, 7–30 [DOI] [PubMed] [Google Scholar]
- 2. Rahib L., Smith B. D., Aizenberg R., Rosenzweig A. B., Fleshman J. M., and Matrisian L. M. (2014) Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 74, 2913–2921 [DOI] [PubMed] [Google Scholar]
- 3. Garrido-Laguna I., and Hidalgo M. (2015) Pancreatic cancer: From state-of-the-art treatments to promising novel therapies. Nat. Rev. Clin. Oncol. 12, 319–334 [DOI] [PubMed] [Google Scholar]
- 4. Ryan D. P., Hong T. S., and Bardeesy N. (2014) Pancreatic adenocarcinoma. New Eng. J. Med. 371, 1039–1049 [DOI] [PubMed] [Google Scholar]
- 5. Saif M. W. (2011) Pancreatic neoplasm in 2011: An update. J. Pancreas 12, 316–321 [PubMed] [Google Scholar]
- 6. Valsecchi M. E., Díaz-Canton E., de la Vega M., and Littman S. J. (2014) Recent treatment advances and novel therapies in pancreas cancer: A review. J. Gastrointest. Cancer 45, 190–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Witkiewicz A. K., Balaji U., Eslinger C., McMillan E., Conway W., Posner B., Mills G. B., O'Reilly E. M., and Knudsen E. S. (2016) Integrated patient-derived models delineate individualized therapeutic vulnerabilities of pancreatic cancer. Cell Rep. 16, 2017–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Teague A., Lim K. H., and Wang-Gillam A. (2015) Advanced pancreatic adenocarcinoma: A review of current treatment strategies and developing therapies. Therap. Adv. Med. Oncol. 7, 68–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thota R., Pauff J. M., and Berlin J. D. (2014) Treatment of metastatic pancreatic adenocarcinoma: A review. Oncology 28, 70–74 [PubMed] [Google Scholar]
- 10. Varbiro G., Veres B., Gallyas F. Jr, and Sumegi B. (2001) Direct effect of Taxol on free radical formation and mitochondrial permeability transition. Free Radical Biol. Med. 31, 548–558 [DOI] [PubMed] [Google Scholar]
- 11. Kampan N. C., Madondo M. T., McNally O. M., Quinn M., and Plebanski M. (2015) Paclitaxel and its evolving role in the management of ovarian cancer. BioMed Res. Int. 2015, 413076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carracedo A., Cantley L. C., and Pandolfi P. P. (2013) Cancer metabolism: Fatty acid oxidation in the limelight. Nature Rev. Cancer 13, 227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yardley D. A. (2013) nab-Paclitaxel mechanisms of action and delivery. J. Control. Release 170, 365–372 [DOI] [PubMed] [Google Scholar]
- 14. Goldstein D., El-Maraghi R. H., Hammel P., Heinemann V., Kunzmann V., Sastre J., Scheithauer W., Siena S., Tabernero J., Teixeira L., Tortora G., Van Laethem J. L., Young R., Penenberg D. N., Lu B., Romano A., and Von Hoff D. D. (2015) nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: long-term survival from a phase III trial. J. Natl. Cancer Inst. 107, dju413. [DOI] [PubMed] [Google Scholar]
- 15. Benetatos C. A., Mitsuuchi Y., Burns J. M., Neiman E. M., Condon S. M., Yu G., Seipel M. E., Kapoor G. S., Laporte M. G., Rippin S. R., Deng Y., Hendi M. S., Tirunahari P. K., Lee Y. H., Haimowitz T., Alexander M. D., Graham M. A., Weng D., Shi Y., McKinlay M. A., and Chunduru S. K. (2014) Birinapant (TL32711), a bivalent SMAC mimetic, targets TRAF2-associated cIAPs, abrogates TNF-induced NF-kappaB activation, and is active in patient-derived xenograft models. Mol. Cancer Therap. 13, 867–879 [DOI] [PubMed] [Google Scholar]
- 16. LaCasse E. C., Mahoney D. J., Cheung H. H., Plenchette S., Baird S., and Korneluk R. G. (2008) IAP-targeted therapies for cancer. Oncogene 27, 6252–6275 [DOI] [PubMed] [Google Scholar]
- 17. Marivin A., Berthelet J., Plenchette S., and Dubrez L. (2012) The inhibitor of apoptosis (IAPs) in adaptive response to cellular stress. Cells 1, 711–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hamacher R., Schmid R. M., Saur D., and Schneider G. (2008) Apoptotic pathways in pancreatic ductal adenocarcinoma. Mol. Cancer 7, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fulda S., and Vucic D. (2012) Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 11, 109–124 [DOI] [PubMed] [Google Scholar]
- 20. Dineen S. P., Roland C. L., Greer R., Carbon J. G., Toombs J. E., Gupta P., Bardeesy N., Sun H., Williams N., Minna J. D., and Brekken R. A. (2010) SMAC mimetic increases chemotherapy response and improves survival in mice with pancreatic cancer. Cancer Res. 70, 2852–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Greenbaum D., Colangelo C., Williams K., and Gerstein M. (2003) Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 4, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huber M., Bahr I., Krätzschmar J. R., Becker A., Muller E. C., Donner P., Pohlenz H. D., Schneider M. R., and Sommer A. (2004) Comparison of proteomic and genomic analyses of the human breast cancer cell line T47D and the antiestrogen-resistant derivative T47D-r. Mol. Cell. Proteomics 3, 43–55 [DOI] [PubMed] [Google Scholar]
- 23. Nouri-Nigjeh E., Sukumaran S., Tu C., Li J., Shen X., Duan X., DuBois D. C., Almon R. R., Jusko W. J., and Qu J. (2014) Highly multiplexed and reproducible ion-current-based strategy for large-scale quantitative proteomics and the application to protein expression dynamics induced by methylprednisolone in 60 rats. Anal. Chem. 86, 8149–8157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chou C. P., Huang N. C., Jhuang S. J., Pan H. B., Peng N. J., Cheng J. T., Chen C. F., Chen J. J., and Chang T. H. (2014) Ubiquitin-conjugating enzyme UBE2C is highly expressed in breast microcalcification lesions. PloS One 9, e93934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fisk J. C., Li J., Wang H., Aletta J. M., Qu J., and Read L. K. (2013) Proteomic analysis reveals diverse classes of arginine methylproteins in mitochondria of trypanosomes. Mol. Cell. Proteomics 12, 302–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shen S., Jiang X., Li J., Straubinger R. M., Suarez M., Tu C., Duan X., Thompson A. C., and Qu J. (2016) Large-scale, ion-current-based proteomic investigation of the rat striatal proteome in a model of short- and long-term cocaine withdrawal. J. Proteome Res. 15, 1702–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shen X., Hu Q., Li J., Wang J., and Qu J. (2015) Experimental null method to guide the development of technical procedures and to control false-positive discovery in quantitative proteomics. J. Proteome Res. 14, 4147–4157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vichai V., and Kirtikara K. (2006) Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 1, 1112–1116 [DOI] [PubMed] [Google Scholar]
- 29. Au J. L., Kumar R. R., Li D., and Wientjes M. G. (1999) Kinetics of hallmark biochemical changes in paclitaxel-induced apoptosis. AAPS PharmSci. 1, E8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shen X., Shen S., Li J., Hu Q., Nie L., Tu C., Wang X., Orsburn B., Wang J., and Qu J. (2017) An IonStar experimental strategy for MS1 ion current-based quantification using ultrahigh-field Orbitrap: Reproducible, in-depth, and accurate protein measurement in large cohorts. J. Proteome Res. 16, 2445–2456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sadygov R. G., Maroto F. M., and Hühmer A. F. (2006) ChromAlign: A two-step algorithmic procedure for time alignment of three-dimensional LC-MS chromatographic surfaces. Anal. Chem. 78, 8207–8217 [DOI] [PubMed] [Google Scholar]
- 32. Vizcaíno J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ariens E. J., Van Rossum J. M., and Simonis A. M. (1957) Affinity, intrinsic activity and drug interactions. Pharmacol. Rev. 9, 218–236 [PubMed] [Google Scholar]
- 34. Keinan N., Tyomkin D., and Shoshan-Barmatz V. (2010) Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell. Biol. 30, 5698–5709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McIlwain D. R., Berger T., and Mak T. W. (2013) Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 5, a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. An B., Zhang M., Johnson R. W., and Qu J. (2015) Surfactant-aided precipitation/on-pellet-digestion (SOD) procedure provides robust and rapid sample preparation for reproducible, accurate and sensitive LC/MS quantification of therapeutic protein in plasma and tissues. Anal. Chem. 87, 4023–4029 [DOI] [PubMed] [Google Scholar]
- 37. Qu J., Lesse A. J., Brauer A. L., Cao J., Gill S. R., and Murphy T. F. (2010) Proteomic expression profiling of Haemophilus influenzae grown in pooled human sputum from adults with chronic obstructive pulmonary disease reveal antioxidant and stress responses. BMC Microbiol. 10, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tu C., Mammen M. J., Li J., Shen X., Jiang X., Hu Q., Wang J., Sethi S., and Qu J. (2014) Large-scale, ion-current-based proteomics investigation of bronchoalveolar lavage fluid in chronic obstructive pulmonary disease patients. J. Proteome Res. 13, 627–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Washburn M. P., Koller A., Oshiro G., Ulaszek R. R., Plouffe D., Deciu C., Winzeler E., and Yates J. R. 3rd (2003) Protein pathway and complex clustering of correlated mRNA and protein expression analyses in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 100, 3107–3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McCommis K. S., and Baines C. P. (2012) The role of VDAC in cell death: Friend or foe? Biochim. Biophys. Acta 1818, 1444–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mathupala S. P., and Pedersen P. L. (2010) Voltage dependent anion channel-1 (VDAC-1) as an anti-cancer target. Cancer Biol. Therapy 9, 1053–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen X., Qian Y., and Wu S. (2015) The Warburg effect: Evolving interpretations of an established concept. Free Radical Biol. Med. 79, 253–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vander Heiden M. G., Cantley L. C., and Thompson C. B. (2009) Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 324, 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sanchez-Arago M., Formentini L., and Cuezva J. M. (2013) Mitochondria-mediated energy adaption in cancer: The H(+)-ATP synthase-geared switch of metabolism in human tumors. Antioxidants Redox Signal. 19, 285–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Matsuyama S., Xu Q., Velours J., and Reed J. C. (1998) The mitochondrial F0F1-ATPase proton pump is required for function of the proapoptotic protein Bax in yeast and mammalian cells. Mol. Cell 1, 327–336 [DOI] [PubMed] [Google Scholar]
- 46. Green D. R., and Reed J. C. (1998) Mitochondria and apoptosis. Science 281, 1309–1312 [DOI] [PubMed] [Google Scholar]
- 47. Orrenius S., Gogvadze V., and Zhivotovsky B. (2007) Mitochondrial oxidative stress: Implications for cell death. Annu. Rev. Pharmacol. Toxicol. 47, 143–183 [DOI] [PubMed] [Google Scholar]
- 48. Finkel T. (2012) Signal transduction by mitochondrial oxidants. J. Biol. Chem. 287, 4434–4440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Currie E., Schulze A., Zechner R., Walther T. C., and Farese R. V. Jr. (2013) Cellular fatty acid metabolism and cancer. Cell Metabolism 18, 153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huang Q., Zhou H. J., Zhang H., Huang Y., Hinojosa-Kirschenbaum F., Fan P., Yao L., Belardinelli L., Tellides G., Giordano F. J., Budas G. R., and Min W. (2015) Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation 131, 1082–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kondoh H., Lleonart M. E., Gil J., Wang J., Degan P., Peters G., Martinez D., Carnero A., and Beach D. (2005) Glycolytic enzymes can modulate cellular life span. Cancer Res. 65, 177–185 [PubMed] [Google Scholar]
- 52. Yeh C. S., Wang J. Y., Chung F. Y., Lee S. C., Huang M. Y., Kuo C. W., Yang M. J., and Lin S. R. (2008) Significance of the glycolytic pathway and glycolysis related-genes in tumorigenesis of human colorectal cancers. Oncology Rep. 19, 81–91 [PubMed] [Google Scholar]
- 53. Nguyen V. T., Barozzi I., Faronato M., Lombardo Y., Steel J. H., Patel N., Darbre P., Castellano L., Györffy B., Woodley L., Meira A., Patten D. K., Vircillo V., Periyasamy M., Ali S., Frige G., Minucci S., Coombes R. C., and Magnani L. (2015) Differential epigenetic reprogramming in response to specific endocrine therapies promotes cholesterol biosynthesis and cellular invasion. Nature Commun. 6, 10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Franke T. F., Hornik C. P., Segev L., Shostak G. A., and Sugimoto C. (2003) PI3K/Akt and apoptosis: Size matters. Oncogene 22, 8983–8998 [DOI] [PubMed] [Google Scholar]
- 55. Hoesel B., and Schmid J. A. (2013) The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 12, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shi L., Wang S., Zangari M., Xu H., Cao T. M., Xu C., Wu Y., Xiao F., Liu Y., Yang Y., Salama M., Li G., Tricot G., and Zhan F. (2010) Over-expression of CKS1B activates both MEK/ERK and JAK/STAT3 signaling pathways and promotes myeloma cell drug-resistance. Oncotarget 1, 22–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Myslinski E., Gérard M. A., Krol A., and Carbon P. (2007) Transcription of the human cell cycle regulated BUB1B gene requires hStaf/ZNF143. Nucleic Acids Res. 35, 3453–3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hao Z., Zhang H., and Cowell J. (2012) Ubiquitin-conjugating enzyme UBE2C: Molecular biology, role in tumorigenesis, and potential as a biomarker. Tumour Biol. 33, 723–730 [DOI] [PubMed] [Google Scholar]
- 59. Kothapalli D., Castagnino P., Rader D. J., Phillips M. C., Lund-Katz S., and Assoian R. K. (2013) Apolipoprotein E-mediated cell cycle arrest linked to p27 and the Cox2-dependent repression of miR221/222. Atherosclerosis 227, 65–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Seki A., and Fang G. W. (2007) CKAP2 is a spindle-associated protein degraded by APC/C-cdh1 during mitotic exit. J. Biol. Chem. 282, 15103–15113 [DOI] [PubMed] [Google Scholar]
- 61. Arora R., Schmitt D., Karanam B., Tan M., Yates C., and Dean-Colomb W. (2015) Inhibition of the Warburg effect with a natural compound reveals a novel measurement for determining the metastatic potential of breast cancers. Oncotarget 6, 662–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jones R. G., and Thompson C. B. (2009) Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 23, 537–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dolcet X., Llobet D., Pallares J., and Matias-Guiu X. (2005) NF-kB in development and progression of human cancer. Virchows Arch. 446, 475–482 [DOI] [PubMed] [Google Scholar]
- 64. Cui J., Zhang M., Zhang Y. Q., and Xu Z. H. (2007) JNK pathway: Diseases and therapeutic potential. Acta Pharmacol. Sinica 28, 601–608 [DOI] [PubMed] [Google Scholar]
- 65. Dhanasekaran D. N., and Reddy E. P. (2008) JNK signaling in apoptosis. Oncogene 27, 6245–6251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ko H. L., and Ren E. C. (2012) Functional aspects of PARP1 in DNA repair and transcription. Biomolecules 2, 524–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wiman K. G. (2013) p53 talks to PARP: the increasing complexity of p53-induced cell death. Cell Death Differ. 20, 1438–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tsujimoto Y. (1998) Role of Bcl-2 family proteins in apoptosis: Apoptosomes or mitochondria? Genes Cells 3, 697–707 [DOI] [PubMed] [Google Scholar]
- 69. Harbour J. W., and Dean D. C. (2000) The Rb/E2F pathway: Expanding roles and emerging paradigms. Genes Dev. 14, 2393–2409 [DOI] [PubMed] [Google Scholar]
- 70. Giacinti C., and Giordano A. (2006) RB and cell cycle progression. Oncogene 25, 5220–5227 [DOI] [PubMed] [Google Scholar]
- 71. Das G. C., Holiday D., Gallardo R., and Haas C. (2001) Taxol-induced cell cycle arrest and apoptosis: dose-response relationship in lung cancer cells of different wild-type p53 status and under isogenic condition. Cancer Lett. 165, 147–153 [DOI] [PubMed] [Google Scholar]
- 72. Castedo M., Perfettini J. L., Roumier T., and Kroemer G. (2002) Cyclin-dependent kinase-1: Linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 9, 1287–1293 [DOI] [PubMed] [Google Scholar]
- 73. Lindqvist A., Rodríguez-Bravo V., and Medema R. H. (2009) The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J. Cell Biol. 185, 193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited at to the ProteomeXchange Consortium via the PRIDE (32) partner repository with the dataset identifier PXD007890 and 10.6019/PXD007890.