

Abstract
The extracellular matrix protein collagen VII is part of the microenvironment of stratified epithelia and critical in organismal homeostasis. Mutations in the encoding gene COL7A1 lead to the skin disorder dystrophic epidermolysis bullosa (DEB), are linked to skin fragility and progressive inflammation-driven fibrosis that facilitates aggressive skin cancer. So far, these changes have been linked to mesenchymal alterations, the epithelial consequences of collagen VII loss remaining under-addressed. As epithelial dysfunction is a principal initiator of fibrosis, we performed a comprehensive transcriptome and proteome profiling of primary human keratinocytes from DEB and control subjects to generate global and detailed images of dysregulated epidermal molecular pathways linked to loss of collagen VII. These revealed downregulation of interaction partners of collagen VII on mRNA and protein level, but also increased abundance of S100 pro-inflammatory proteins in primary DEB keratinocytes. Increased TGF-β signaling because of loss of collagen VII was associated with enhanced activity of lysosomal proteases in both keratinocytes and skin of collagen VII-deficient individuals. Thus, loss of a single structural protein, collagen VII, has extra- and intracellular consequences, resulting in inflammatory processes that enable tissue destabilization and promote keratinocyte-driven, progressive fibrosis.
The cellular microenvironment encompasses structural components, the extracellular matrix (ECM)1, and soluble factors, e.g. growth factors (1). The ECM was initially regarded as purely structural scaffold keeping cells in place and ensuring tissue/organ integrity. Through the discovery of ECM-cell contact sites and underlying signaling events, the crucial role of the ECM for cell survival, proliferation and differentiation emerged (1, 2). Importantly, it is now clear that a dysregulated ECM can actively promote disease progression in humans (3). The skin contains a complex ECM, the dermal-epidermal junction zone (DEJZ), which binds the epidermis and the dermis and is generated by cell types of distinct epithelial and mesenchymal linages: keratinocytes, the main cell type of the epidermis, and fibroblasts, the main mesenchymal-derived cell type of the dermis. The DEJZ is vital for skin integrity. As part of the DEJZ the ECM protein collagen VII (C7) forms anchoring fibrils, which entrap dermal fibrils and establish stable dermal-epidermal adhesion (4).
Biallelic loss-of-function mutations of the gene COL7A1 encoding C7 cause dystrophic epidermolysis bullosa (DEB), an inherited skin fragility disorder characterized by a broad spectrum of clinical manifestations: skin blistering, abnormal wound healing, excessive scarring often resulting in aggressive skin cancer (5). However, the variety and multitude of the symptoms are not easily explained by the underlying COL7A1 mutations.
Toward this end “omics” approaches have been used to delineate the cellular and microenvironmental consequences of C7 loss. However so far, mainly the consequences of C7-deficiency on already transformed squamous cell carcinoma cells (6, 7), and on dermal fibroblasts were addressed (8, 9). Transcriptome analyses have suggested that the gene expression profile of DEB fibroblasts resembles that of cancer-associated fibroblasts (CAFs) and that loss of C7 may generate a permissive microenvironment for tumor development (9). Quantitative mass spectrometry (MS)-based proteomics studies using DEB fibroblasts focused on the extracellular consequences of the absence of C7 and revealed reduction of basement membrane proteins and increase of dermal matrix proteins, highlighting the multifaceted roles of C7 (8). Reduced crosslinking of dermal ECM proteins because of lower abundance of transglutaminase 2, an interaction partner of C7, indicated altered mechanical properties of the fibroblast microenvironment because of loss of C7 (10).
It has become apparent that progressive TGF-β-mediated soft tissue fibrosis plays a major role in disease evolution of DEB (11, 12). In other diseases epithelial dysfunction is known to be a major instigator of fibrosis (13). Known for DEB is that C7 as a master regulator of laminin-332 deposition may exert control over the laminin-332-integrin α6β4 signaling axis (38). However, the potential contribution of these and additional epithelial alterations to progression of DEB remain elusive. Therefore, we performed a global transcriptome and proteome profiling comparing primary DEB keratinocytes to normal human keratinocytes (NHK), delineating C7-dependent molecular pathways dysregulated in disease. We combined high-throughput sequencing using RNAseq with stable isotope labeling by amino acids in cell culture (SILAC)-based quantitative MS followed by bioinformatics analyses to address alterations in primary human keratinocytes and in skin of affected individuals. Loss of C7 did not only affect the composition of the cellular microenvironment, but it led to global changes in cell homeostasis on mRNA and on protein level. Altered abundance of ECM proteins and integrin receptors indicated dysregulated TGF-β activity caused by disruption of laminin-332-integrin α6β4 signaling. Indeed, TGF-β-dependent inflammatory and proteolytic processes were perturbed both in keratinocytes in vitro and in human DEB skin in vivo. Thus, we provide a detailed image of dysregulated molecular pathways in genetic skin fragility and show that loss of C7 leads to far more complex changes as anticipated. Importantly, our study reveals intrinsic epidermal alterations directly caused by C7 deficiency and increased TGF-β signaling in epidermal keratinocytes as instigators of dermal fibrosis.
EXPERIMENTAL PROCEDURES
More information about procedures can be found online in the supplemental Information.
Patients and DEB Diagnosis
Skin specimens were obtained from ten DEB patients (DEB1–10) for diagnostic purposes and the remaining material was used for research purposes after informed consent of the patients and approval of the Ethics Committee of the University of Freiburg. The study was conducted according to the Declaration of Helsinki. Specimens are stored in an internal collection. Immunofluorescence mapping revealed lack of C7 in the skin of all patients. Mutation analysis of the C7 gene, COL7A1 was performed as described before and disclosed mutations that lead to premature stop codons, thus confirming the diagnosis of severe generalized DEB (Table I) (14, 15). To elucidate disease mechanisms DEB keratinocytes were compared with primary human keratinocytes (Ctrl1–12) derived from age-matched healthy donors (Table II). Cells used for each experiment are indicated in the respective figure according to the designations introduced in Tables I and II.
Table I. Characteristics of primary DEB cells used in this study. n.d.: not detected.
| ID | Age at biopsy/gender | COL7A1 mutation | C7 levels (IF analysis) | References (report of patients) |
|---|---|---|---|---|
| DEB 1 | 8 years/m | c.1934delC | n.d. | (15) |
| c.1934delC | ||||
| DEB 2 | 1 month/f | c.425A>G | n.d. | (15) |
| c.682 + 1G>A | ||||
| DEB 3 | 1 month/m | c.425A>G | n.d. | (14) |
| c.5261dupC | ||||
| DEB 4 | 6 years/f | c.425A>G | n.d. | Patient not reported before |
| c.682 + 1G>A | ||||
| DEB 5 | 1 month/f | c.4119 + 1G>C | n.d. | Patient not reported before |
| c.8523_8536del | ||||
| DEB 6 | 3 years/f | c.682 + 1G>A | n.d. | Patient not reported before |
| c.5261dupC | ||||
| DEB 7 | 1 month/f | c.425A>G | n.d. | (15) |
| c.425A>G | ||||
| DEB 8 | 30 years/m | c.426 + 1G>A | n.d. | (15) |
| c.1474del11 | ||||
| DEB 9 | 53 years/m | c.425A>G | n.d. | Patient not reported before |
| c.425A>G | ||||
| DEB 10 | 47 years/m | c.425A>G | n.d. | Patient not reported before |
| c.425A>G |
Table II. Characteristics of primary control cells used in this study.
| ID | Age at biopsy/ gender |
|---|---|
| Ctrl 1 | 2 years/m |
| Ctrl 2 | 4 years/m |
| Ctrl 3 | 3 years/m |
| Ctrl 4 | 20 years/f |
| Ctrl 5 | 13 years/f |
| Ctrl 6 | 1 year/m |
| Ctrl 7 | 38 years/f |
| Ctrl 8 | 4 years/m |
| Ctrl 9 | 6 years/m |
| Ctrl 10 | 34 years/f |
| Ctrl 11 | 13 years/f |
| Ctrl 12 | 1 years/m |
Patient DEB9 had a severe generalized DEB with lack of C7 in the affected skin, as shown in a skin biopsy taken from scarred, not acutely blistered or inflamed skin next to the left knee (supplemental Fig. S6). A skin biopsy taken from a clinically unaffected skin patch on the left knee revealed C7 re-expression at around 70% of the biopsy's width. A subsequent analysis of the COL7A1 gene from keratinocytes of the unaffected skin patch confirmed the correction of the COL7A1 gene on one allele (data not shown).
Experimental Design and Statistical Rationale
In total, we analyzed 22 primary cell populations as listed in Tables I and II. Omics analyses were performed from two primary DEB and normal human keratinocyte populations, each. As DEB is a rare disease (less than 10 of one million newborns are affected), not more primary cells could be used for 'omics' analyses. For SILAC-based proteomics analyses two biological replicates each were analyzed using swapped labels.
All statistical tests were corrected for multiple testing as outlined in the respective paragraphs.
Cell Lysates and ECM Isolation of Keratinocytes for MS Analysis
A SILAC approach was used to compare DEB keratinocytes to normal controls by MS-based proteomics. 5 × 105 fully SILAC-labeled keratinocytes were seeded on 10 cm2 cell culture dishes. On the following day, the medium was exchanged and 50 μg/ml ascorbate was added to allow proper maturation and secretion of collagens (16). Keratinocytes were kept in culture for 6 days and fresh culture medium containing ascorbate was added daily. After 6 days, keratinocytes were washed 3 times with DPBS. The cells were removed from the underlying ECM through incubation with 0.5% Triton X-100 in DPBS for 10 s, followed by a washing step with DPBS and an incubation for 10 s with 20 mm NH4OH in DPBS (17). Cell lysates were concentrated by ultrafiltration using vivaspin columns (10 kDa MWCO). The remaining ECM was carefully washed 3 times with DPBS to eliminate intracellular contaminants and then solubilized with 4% SDS in 0.1 m Tris-HCl, pH 7.6 (18). Aliquots of the different ECM samples or cell lysates were used for WB analysis or mixed 1:1:1 and prepared for MS analysis to test accurate mixing ratios. Mixing was adjusted for actual MS analysis afterward.
MS Sample Preparation
In-gel Digestion
Samples were heated in SDS-PAGE loading buffer, reduced with 1 mm DTT for 5 min at 95 °C and alkylated using 5.5 mm iodoacetamide for 30 min at room temperature. Protein mixtures were separated on 4–12% gradient gels. The gel lanes were cut into 10 equal slices, the proteins were in-gel digested with trypsin and the resulting peptide mixtures were processed on STAGE tips and analyzed by LC-MS/MS (19, 20).
Mass Spectrometry
MS measurements were performed on an LTQ Orbitrap XL mass spectrometer coupled to an Agilent 1200 nanoflow-HPLC. HPLC-column tips (fused silica) with 75 μm inner diameter were self-packed with Reprosil-Pur 120 ODS-3 to a length of 20 cm. Samples were applied directly onto the column without a pre-column. A gradient of A (0.5% acetic acid in water) and B (0.5% acetic acid in 80% acetonitrile in water) with increasing organic proportion was used for peptide separation (loading of sample with 2% B; separation ramp: from 10–30% B within 80 min). The flow rate was 250 nL/min and for sample application 500 nL/min. The mass spectrometer was operated in the data-dependent mode and switched automatically between MS (max. of 1 × 106 ions) and MS/MS. Each MS scan was followed by a maximum of five MS/MS scans in the linear ion trap using normalized collision energy of 35% and a target value of 5000. Parent ions with a charge state from z = 1 and unassigned charge states were excluded for fragmentation. The mass range for MS was m/z = 370–2000. The resolution was set to 60,000. MS parameters were as follows: spray voltage 2.3 kV; no sheath and auxiliary gas flow; ion-transfer tube temperature 125 °C.
Identification of Proteins and Protein Ratio Assignment Using MaxQuant
The MS raw data files were uploaded into the MaxQuant software version 1.4.1.2 for peak detection, generation of peak lists of mass error corrected peptides, and for database searches (21). A full-length UniProt human database additionally containing common contaminants such as keratins and enzymes used for in-gel digestion (based on UniProt human FASTA version September 2013, 87,000 entries) was used as reference. Carbamidomethylcysteine was set as fixed modification and protein amino-terminal acetylation and oxidation of methionine were set as variable modifications. Triple SILAC was chosen as quantitation mode. Three missed cleavages were allowed, enzyme specificity was trypsin/P, mass tolerance for first search was 20 ppm, and the MS/MS tolerance was set to 0.5 Da. The average mass precision of identified peptides was in general less than 1 ppm after recalibration. Peptide lists were further used by MaxQuant to identify and relatively quantify proteins using the following parameters: peptide and protein false discovery rates, based on a forward-reverse database, were set to 0.01, minimum peptide length was set to 6, minimum number of peptides for identification and quantitation of proteins was set to one which must be unique, minimum ratio count was set to two, and identified proteins were requantified. The “match-between-run” option (2 min) was used. Annotated spectra of single peptide-based protein IDs are provided as Supplemental Files S1 and S2.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD005873 (22).
RESULTS
Disease-relevant Transcriptional Changes in Primary Human DEB Keratinocytes
DEB keratinocytes are characterized by a loss of C7 (Fig. 1A) and skin of affected individuals exhibits mechanical fragility, inflammation, excessive scarring and fibrosis (Fig. 1B). Whereas disease contributions of mesenchyme-derived cells have been extensively studied, the influence of epithelial cells remains obscure, which led us to investigate the epidermal responses to loss of C7 (see Tables I-II and Experimental Procedures for designation of used primary cells).
Fig. 1.
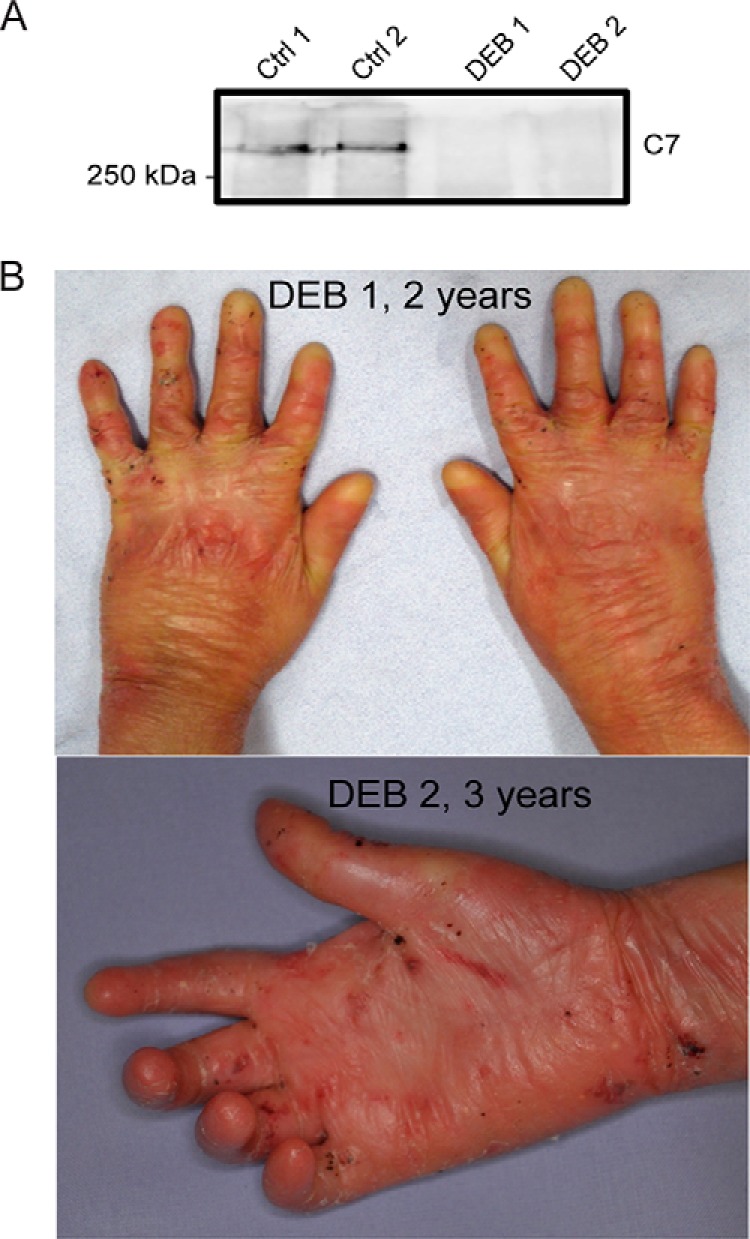
Loss of C7 in DEB is associated with skin fragility, inflammation and scarring. A, Anti-C7 Western blot. C7 is deficient in the ECM isolated from cultured primary DEB keratinocytes derived from depicted patients. B, Clinical phenotypes of DEB. Loss of C7 is associated with skin fragility, inflammation and scarring. The age of patient is indicated.
To study the effects of C7 loss on the transcriptome, DEB keratinocytes from two donors and two age-matched controls were cultured to the same number of passages and the extracted RNAs were subjected to next-generation RNA sequencing. In total, expression of 39,293 gene variants was detected (supplemental Table S1) of which 300 were differentially regulated in DEB and control keratinocytes (BH-corrected q-value 0.1) (supplemental Table S2). To illustrate altered gene expression in DEB keratinocytes, we performed a cluster analysis of the differentially expressed genes and observed a greater variability between the two DEB cells than between the control cells (Fig. 2A).
Fig. 2.

Gene expression changes in primary DEB keratinocytes. A, Altered gene expression in DEB keratinocytes. Cluster analysis of 300 differentially regulated genes in DEB keratinocytes (BH corrected q-value 0.1) as determined by next generation sequencing. TPM values were log2-transformed and z-score normalized. Columns containing data from the different samples and rows containing gene entries were hierarchically clustered. Data separated into 5 clusters showing clear differences between DEB and control cells (Ctrl). B, Functional annotation of significantly regulated genes in DEB cells. Differentially expressed genes in DEB are significantly enriched in the shown processes (BH corrected q-value 0.05). Loss of C7 affects transcription of genes involved in cell migration and ECM organization. C, Examples of dysregulated gene expression in DEB. Bar graphs show transcript abundances (TPM values) of differentially expressed genes. Expression of genes encoding direct and indirect binding partners of C7 (LAMA3/LAMB3/LAMC2, ITGA6/ITGB4) were found to be downregulated in DEB keratinocytes whereas the transcript encoding LTBP1 was more abundant in DEB keratinocytes.
GO term analysis of differentially expressed genes revealed enrichment of GO terms like cell migration and ECM organization which are processes known to be affected by the loss of C7 (8, 10) (Fig. 2B). Expression of genes encoding laminin-332 (LAMA3/LAMB3/LAMC2) and integrin α6β4 (ITGA6/ITGB4), direct and indirect binding partners of C7, respectively, were downregulated in DEB keratinocytes. In contrast, the transcript encoding latent-transforming growth factor beta-binding protein 1 (LTBP1) was more abundant, indicating increased TGF-β secretion (23) and activation of a wound response program in cells devoid of C7 (Fig. 2C).
Taken together, next-generation RNA sequencing revealed distinct changes of mRNA levels highlighting a perturbed transcriptional regulation because of loss of C7 in primary keratinocytes. Expression of genes encoding direct and indirect binding partners of C7 were found to be downregulated, whereas expression of genes encoding for proteins involved in response to injury and TGF-β signaling were upregulated in DEB keratinocytes.
Altered ECM Proteome Composition Because of Loss of C7
To complement transcriptome analyses and directly assess the extracellular consequences of loss of C7, we performed quantitative proteome analyses of the keratinocyte ECM using MS. Cells were cultured in the presence of ascorbate to ensure correct post-translational modification and stable triple-helix formation of collagens. After 6 days, cells were removed carefully from culture dishes using Triton/PBS followed by NH4OH/PBS (Fig. 3A) (17). Remaining insoluble proteins on the culture plate were harvested with 4% SDS and were defined as ECM. Efficiency of the protocol and purity of the ECM isolation were tested by Western blot (Fig. 3B). Intracellular proteins corresponding to the cell compartments nucleus (TBP), Golgi apparatus (GOLGA1), endoplasmic reticulum (HSPA5) and cytosol (GAPDH) were only detected in cell lysates. ECM proteins such as the transmembrane protein collagen XVII (COL17A1) and C7, both components of the DEJZ, fibronectin (FN1) and tenascin C (TNC) were found in both, cell lysate and ECM. Thus, intracellular proteins were depleted and extracellular components were enriched in the ECM fraction.
Fig. 3.

Analysis of the extracellular proteome of DEB keratinocytes. A, Cell culture scheme. Keratinocytes were kept in culture for 6 days. Fresh culture medium containing ascorbate was added daily. The ECM was harvested at day 6 using 4% SDS. B, Analysis of ECM isolation procedure. Western blot analyses were performed to address the quality of ECM isolation. Intracellular proteins representing the cell compartments nucleus (TBP), Golgi apparatus (GOLGA1), endoplasmic reticulum (HSPA5) and cytosol (GAPDH) were only detected in whole cell lysate (WCL). ECM proteins such as collagen XVII (COL17A1), C7 as well as fibronectin (FN1) and tenascin C (TNC) were found in both, WCL and ECM. C, SILAC-based quantitative MS workflow. Medium and heavy SILAC-labeled cells were mixed with a light-labeled standard 1:1:1 and samples were processed as outlined. The resulting peptide mixtures were analyzed by LC-MS/MS. In total 2 biological replicates of 2 DEB and control cells, each, were analyzed. D, SOTA clustering of proteins quantified in ECM of DEB and control cells. ECM proteins were filtered bioinformatically and respective protein ratios were log2-transformed and z-score normalized. Proteins in each cluster were tested by DAVID for enriched GO terms (biological process (BP), cellular compartment (CC)). Selected terms are shown (full list in supplemental Table S5). E, Network analysis of significantly regulated proteins. STRING DB was used to identify protein-protein interactions of significantly regulated proteins (confidence score 0.4). Known C7 binding partners, such as integrin α6β4 and laminin-332 were significantly downregulated in ECM isolated from DEB keratinocytes, whereas proteins involved in fibrosis and inflammation were significantly upregulated (Welch's t test, Permutation-based FDR 0.05). The figure was produced using Servier Medical Art.
The same primary keratinocytes as used for gene expression analyses were SILAC labeled to assess the effect of C7 deficiency on the abundances of extracellular proteins. A Super-SILAC mix of light isotope-labeled keratinocyte ECM derived from all four donors was generated and spiked into differentially heavy isotope-labeled ECM of control and DEB keratinocytes (Fig. 3C) (24). Samples were fractionated by SDS-PAGE and proteins in-gel digested with trypsin (19). Generated peptides were identified and quantified by reverse-phase LC-MS/MS. 1386 proteins were identified in two biological replicates each, of which 1237 could be quantified in at least one sample (average correlation coefficient = 0.66; supplemental Fig. S1 and supplemental Table S3; see supplemental File S1 for annotated spectra of single peptide IDs). Because of the high sensitivity of modern mass spectrometers, even small amounts of contaminating intracellular proteins are detected in the ECM fraction. To distinguish them and to obtain a comprehensive list of potential true ECM proteins, the original protein list was filtered: proteins defined in the “matrisome” and those annotated by the GO terms “extracellular” and/or “cell adhesion” were defined as the “ECM proteome” (25). This led to a list of 209 identified ECM proteins of which 199 were quantified in minimally one sample (supplemental Table S4).
DEB and control ECMs were clearly distinguished by a principal component analysis (PCA) with DEB ECM being more heterogeneous than control ECM (supplemental Fig. S2). Thus, loss of C7 has global effects on the composition of keratinocyte-derived ECM. To identify groups of co-regulated proteins, proteins were clustered by a self-organizing tree algorithm (SOTA), the data set separating into seven clusters (Fig. 3D). Proteins in each cluster were tested for enriched GO terms using the program DAVID (complete list in supplemental Table S5) (26). Cluster 1 contained proteins that were consistently downregulated in ECM of DEB keratinocytes and that are involved in basement membrane organization, cell-cell and cell-matrix adhesion. In contrast, cluster 6 comprised proteins that showed a higher abundance in DEB ECM; they are related to inflammatory processes and wound healing. For the other clusters, especially clusters 3, 4 and 5 it was clear that ECM derived from DEB keratinocytes showed patient-specific effects and was more heterogeneous than control ECM. Analysis of significantly regulated ECM proteins between DEB and control ECM revealed a network of 36 proteins with known interactions (Fig. 3E; Welch's t test, Permutation-based FDR 0.05). Like the results obtained by transcriptome analysis, the direct and indirect binding partners of C7, laminin-332 and integrin α6β4 were downregulated on protein level. Proteins that are involved in tissue repair such as fibronectin and tenascin C were upregulated, changes being indicative of increased TGF-β activity caused by disruption of integrin α6β4 signaling (27, 28) and in line with increased LTBP1 expression (Fig. 2C).
Taken together, loss of C7 led to pronounced changes in the microenvironment of DEB keratinocytes. Especially, basement membrane components, direct and indirect binding partners of C7 were found to be downregulated whereas proteins that are involved in wound healing were upregulated in DEB keratinocyte-derived ECM, largely consistent with transcriptome analyses.
Loss of C7 Leads to an Increase of Inflammation Markers
To distinguish transcriptional from translational or post-translational regulation mechanisms, we directly analyzed the correlation between mRNA and protein abundance differences comparing DEB to control keratinocytes. The differences in gene expression and protein abundance of ECM proteins correlated only weakly (Fig. 4A, r = 0.25, Pearson). Transcripts and proteins that were both downregulated in DEB keratinocytes display GO terms such as “extracellular region” and “cell adhesion” and are highlighted in yellow (Fig. 4A). This group contains proteins associated with desmosomes like plakophilin 2 and integrin alpha-6, as well as protease inhibitors, i.e. cystatin A. Additional ECM proteins like laminin beta-1 and gamma-1 were identified as downregulated whereas respective mRNA levels did not correlate with the change in protein abundance, indicating perturbed post-transcriptional regulation mechanisms (supplemental Fig. S3). Downregulated transcripts of proteins that were upregulated in DEB are marked blue and are associated with GO terms “extracellular space,” “secreted,” and “calcium ion binding.” Several annexins and S100 proteins, as well as calreticulin and cathepsin D belong to this group. Fibronectin was found to be upregulated on transcriptome and proteome level, similarly to corneodesmosin and filaggrin-2 (Fig. 4A, marked red).
Fig. 4.

Gene expression and protein abundance correlate only weakly in primary keratinocytes. A, Correlation of gene transcription and ECM protein abundance. Gene expression and protein abundance only correlate weakly (r = 0.25). Genes showing a 2-fold change on RNA and protein level were highlighted and tested by DAVID for GO enrichment. Selected terms are shown. S100A8 and A9 were significantly regulated on protein level but not on mRNA level. B, Relative mRNA and protein abundances of S100A8 and S100A9. Relative values compared with the respective control 1 (Ctrl 1) are shown. Protein levels were significantly higher (Welch's t test, Permutation-based FDR 0.05) in ECM isolated of DEB cells compared with controls, whereas mRNA levels did not change significantly. C, In vivo accumulation of S100A9. Immunofluorescence staining of DEB skin reveals a strong increase of S100A9 in the epidermis (green signal). Blue = DAPI staining. D, Correlation of gene transcription and intracellular protein abundance. Gene expression and protein abundance correlated weakly (r = 0.19). Genes in each quadrant with a fold change > 2 on mRNA and protein level were colored and tested by DAVID for GO enrichment (biological process (BP), cellular compartment (CC), molecular function (MF) and SP_PIR_KEYWORDS (KW)). Selected terms are shown.
Interestingly, the proteins S100A8 and S100A9 displayed a marked increase in ECM of DEB keratinocytes compared with controls (log2 fold change >4) while showing only minimal regulation on mRNA level (Fig. 4A–4B, Welch's t test, Permutation-based FDR 0.05). S100A8 and S100A9 often appear as calcium-binding heterodimer named calprotectin that has been used as biomarker for acute and chronic inflammation (29, 30). Importantly, accumulation of S100A9 protein was also observed in vivo in the epidermis comparing immunofluorescence staining of DEB and control skin indicating that inflammation in DEB is promoted by keratinocytes (Fig. 4C).
As transcriptome and ECM proteome analyses revealed complex and far reaching changes because of the loss of C7, we also analyzed the intracellular proteome of respective cells comparing DEB to control keratinocytes. Applying SILAC-based MS analysis 3809 proteins were identified of which 2054 could be quantified in all four samples (supplemental Table S6, see supplemental File S2 for annotated spectra of single peptide IDs). Also in this setting correlation of gene transcription and protein abundance was rather weak (Fig. 4D; r = 0.19, Pearson), indicating that also in a disease model gene expression analyses cannot be employed to reliably infer protein abundances. Genes in each quadrant with a fold change >2 on protein and mRNA level were colored and selected enriched GO terms are shown. Again, loss of C7 had a dramatic influence on expression of genes involved in focal adhesion, hydroxylation and extracellular matrix. Intracellularly, S100A8 and S100A9 were less dysregulated, indicating that the extracellular abundance of both proteins may either be influenced through an increased translation coupled to an increased secretion, or a decreased extracellular degradation/increased stability in DEB (Fig. 4B).
Keratins are the major intermediate filament proteins of epithelia and vital for mechanical stability (31). As we identified a significantly reduced expression of several keratin-encoding genes in DEB keratinocytes (supplemental Fig. S4), we analyzed keratin protein abundance in detail. Interestingly, only keratin 19 protein levels were significantly reduced in DEB cells (supplemental Table S6). In contrast, intracellular proteins exhibiting known protein-protein interactions and being involved in regulation of apoptosis, ECM organization and collagen binding were significantly enriched in DEB cells indicating a perturbation on protein level of respective pathways (supplemental Fig. S5) (32, 33).
Taken together, fold changes of mRNA and protein levels correlated weakly in primary human DEB and control keratinocytes, regardless if mRNA levels were compared with intracellular or extracellular protein abundances. Several proteins that are involved in inflammatory processes, such as S100A8 and S100A9 were only found to be upregulated on protein level indicating so far unknown, keratinocyte-intrinsic, post-transcriptional regulation mechanisms and characterizing keratinocytes as actively promoting inflammation in DEB.
Increased Cathepsin B Abundance and Activity in C7 Deficient DEB Skin
To get a global impression of intracellular protein abundance differences induced by the loss of C7 we again performed a SOTA clustering and GO term enrichment analysis (Fig. 5A, full list of GO terms in supplemental Table S7). The GO terms “lysosome” and “endopeptidase activity” were found significantly enriched in a cluster containing highly abundant proteins in DEB cells. Indeed, several proteases were found to be significantly dysregulated in primary human DEB keratinocytes (supplemental Table S8), the lysosomal proteases cathepsin Z and cathepsin B being significantly upregulated (Fig. 5B–5C, t test, p < 0.05). Higher abundance of the active forms of cathepsin B, the single-chain (SC) and heavy-chain enzyme (HC), in DEB keratinocytes was also shown by Western blot analysis (Fig. 5D). In conditioned DEB keratinocyte medium the proenzyme (PE) and the active single-chain enzyme were found enriched, suggesting a role of extracellular cathepsin B in ECM degradation (Fig. 5E).
Fig. 5.

Increased cathepsin B abundance and activity in DEB keratinocytes. A, Cluster analysis of intracellular protein abundances. SOTA clustering of intracellular proteins that were quantified in DEB keratinocytes compared with controls. Protein ratios were log2-transformed and z-score normalized. Proteins in each cluster were tested by DAVID for enriched GO terms (biological process (BP), cellular compartment (CC) and molecular function (MF). Selected terms are shown (full list in supplemental Table S7). B–C, Protein abundance measurements. Protein abundances of cathepsin B (B) and cathepsin Z (C) were significantly higher in DEB compared with control keratinocytes as determined by SILAC-based, quantitative MS (t test, two-tailed, two-sample equal variance, p < 0.05). a and b: biological replicates of indicated primary cells analyzed by swapped SILAC labels. D–E, Western blot analysis revealed higher amounts of cathepsin B in DEB keratinocytes. Higher abundance of the cathepsin B single-chain enzyme (SC) and the heavy chain of the two-chain enzyme (HC) were detected in DEB whole cell lysates of independent biological replicates by Western blot (D). In conditioned keratinocyte medium increased amounts of proenzyme (PE) and active cathepsin B (HC form) were detected under DEB conditions (E). F, Cathepsin B activity assays revealed more active cathepsin B in DEB keratinocytes. Bands corresponding to active cathepsins B and Z are highlighted by arrows and triangles, respectively. Biotin-DCG-04 was detected using fluorescently labeled streptavidin. DCG-04: substrate of cysteine cathepsins, E-64: cysteine protease inhibitor, Ca074: cathepsin B inhibitor, 95 °C (neg. control): samples were denaturated prior DCG-04 incubation in order to reveal unspecific binding. G, Quantification of activity blots. Four independently performed activity assays as shown in (F) were quantified by imageJ revealing significantly more active cathepsin B in DEB keratinocytes (t test, two-tailed, two-sample equal variance: p < 0.001). ACTB values (AU) were used for normalization. H–I, Increased autophagic flux in DEB keratinocytes. Regulation of lipidated MAP1LC3B (LC3-II), an autophagosomal marker protein, was analyzed in control and DEB keratinocytes plus/minus concanamycin A (ConA), an inhibitor of the lysosomal V-ATPase, by Western blot (H). Quantification of ConA treated samples compared with untreated samples revealed an increased autophagic flux in DEB keratinocytes (*: p < 0.05, t test, n = 3). J, Increased cathepsin B levels in DEB skin. Immunofluorescence staining of DEB skin revealed a strongly increased cathepsin B signal (green) at the blister roof and in the upper dermis. In healthy control skin cathepsin B occurred mainly at the dermal-epidermal junction and the upper dermis, showing a weaker signal. Blue = DAPI staining.
Higher protease abundance does not necessarily reflect higher protease activity, as this can be modulated by protease inhibitors (8). Interestingly the cathepsin B inhibitor cystatin A (CSTA) was found significantly depleted in DEB ECM, indicating an increased cathepsin B activity in DEB (Fig. 3E). To directly measure the proteolytic activity of cathepsin B in cultured keratinocytes, cells were lysed and labeled with the substrate DCG-04, which targets active cysteine cathepsins (34). To distinguish cathepsin B from other active cysteine cathepsins the pan-cathepsin inhibitor E-64 and the cathepsin B-specific inhibitor Ca074 were used in control experiments prior to DCG-04 incubation to block the active center of proteases. Indeed, Western blot analyses detecting protein-bound DCG-04 revealed significantly more active cathepsin B in DEB keratinocytes (Fig. 5F–5G). Orlowsi et al. conducted a similar cathepsin activity assays and characterized a Western blot signal just above cathepsin B as cathepsin Z (35). Thus, also cathepsin Z appeared to be more active in DEB keratinocytes than in controls and might as well be involved in ECM degradation in DEB (Fig. 5F).
Increased lysosomal protease abundance and activity might indicate a general increase in lysosomal protein degradation. Therefore, we assessed the activity of autophagy, an intracellular degradation pathway targeting proteins and organelles for lysosomal degradation (36). By assaying accumulation of the lipidated autophagosomal marker protein MAP1LC3B (LC3-II) in the presence and absence of concanamycin A (ConA), an inhibitor of the lysosomal V-ATPase, we detected an increased autophagic capacity in DEB keratinocytes (Fig. 5H–5I), which is in line with increased cathepsin activity in DEB.
Finally, in agreement with the in vitro data, increased cathepsin B immunofluorescence staining was also observed in DEB skin compared with respective controls (Fig. 5J). DEB skin revealed a particularly strong cathepsin B signal (green) in the blister roof and in the upper dermis. Taken together, an increase in cathepsin B abundance and activity as well as in autophagy was detected in DEB keratinocytes and skin, indicating a potential role of cathepsin B in DEB pathology as a mediator of both inflammation and activation of tissue-destabilizing proteolytic activities.
Increased Cathepsin B Levels Depend on TGF-β-signaling and C7
TGF-β can induce CTSB expression in vitro in mouse cells (37) and was identified as increased in DEB skin and fibroblasts (8, 9, 11, 38). To test if increased cathepsin B abundance in DEB keratinocytes also depends on TGF-β signaling, we analyzed TGFB1 expression levels and activation of the TGF-β target proteins SMAD2/3. Indeed, TGFB1 expression was significantly increased in DEB compared with control keratinocytes (Fig. 6A) leading to increased SMAD2/3 phosphorylation (Fig. 6B). Treatment of control keratinocytes with TGF-β led to a significant increase in cathepsin B levels (Fig. 6C–6D), and dual inhibition of TGF-β receptor type I/II with LY2109761 reduced cathepsin B levels in DEB keratinocytes (Fig. 6E–6F). Thus, increased TGF-β signaling contributes to increased cathepsin B levels and activity in DEB.
Fig. 6.

TGF-β-dependent increase of cathepsin B levels. A, Increased expression of TGFB1 in primary DEB keratinocytes. qPCRs testing TGFB1 expression levels were performed on mRNA isolated from primary DEB and Ctrl keratinocytes (n = 9). Expression levels were normalized to GAPDH expression. **: p < 0.01, T-Test. B, Increased activity of TGF-β1 in primary DEB keratinocytes. TGF-β1-responsive phosphorylation events of SMAD2/3 were analyzed by phosphosite-specific antibodies (SMAD2 phospho-Ser465/467; SMAD3 phospho-Ser423/425). Actin (ACTB) was used as loading control. C–D, TGF-β treatment led to increased cathepsin B levels. Control keratinocytes (immortalized using the HPV E6/E7 genes) were incubated with 10 ng/ml TGF-β1 for 48 h and cathepsin B levels were assessed by Western blot (C). TGF-β1 treatment led to a significant increase in cathepsin B levels (D) (paired t test, p < 0.05; n = 4). Beta tubulin (TUBB) was used as loading control and increased SMAD2/3 phosphorylation as positive control for increased TGF-β1 stimulation. E–F, TGF-β inhibition led to decreased cathepsin B levels in DEB cells. DEB keratinocytes (immortalized using the HPV E6/E7 genes) were incubated with 4.4 μg/ml LY2109761 for 48 h and cathepsin B levels were assessed by Western blot (E). Inhibition of TGF-β receptor type I/II led to a significant decrease in cathepsin B levels (F) (paired t test, p < 0.05; n = 4). Beta tubulin was used as loading control and decreased SMAD2/3 phosphorylation as positive control for inhibition of TGF-β signaling. G, C7 leads to decreased cathepsin B levels. Conditioned medium (CM) and cell lysates of primary reverted DEB cells showing a natural expression of C7 were analyzed by Western blot on cathepsin B level. TUBB served as loading control for cell lysates. SC: Cathepsin B single-chain enzyme; HC: heavy chain of the two-chain enzyme; PE: proenzyme (PE).
Finally, to test if increased cathepsin B abundance directly depends on the loss of C7, we made use of the fact that spontaneous restoration of COL7A1 expression occurs in skin patches in a subset of DEB patients because of genetic reversion of the disease-causing mutation (so called revertant mosaicism (39)). In a biopsy specimen obtained from a reverted skin patch, C7 was present (supplemental Fig. S6). Critically, restoration of endogenous expression of C7 led to a pronounced reduction in cathepsin B levels, indicating that C7 is sufficient to decrease cathepsin B levels (Fig. 6G).
DISCUSSION
Next to its important structural scaffold functions, the ECM is critical for cell homeostasis, proliferation and differentiation. Deficiency of the ECM protein C7 in DEB leads to substantial dermal changes: impaired skin regeneration and TGF-β-driven fibrosis that facilitates aggressive squamous cell carcinoma (SCC) (8–10, 12, 38, 40). However, C7 is both a dermal and an epidermal product and so far the direct cellular changes that loss of C7 evokes in epidermal keratinocytes remained limitedly studied (7, 38). A proteomic analysis of the skin of DEB mice suggested a cell-intrinsic injury-independent progression of fibrosis in DEB, the epidermis being a prime suspect for promoting this activity (11). In addition, dysfunctioning epithelia have emerged as instigators of fibrosis in other fibrotic diseases (13). The above led us to use DEB as a model to study the consequences of genetic loss of the structural protein C7 on epidermal cell homeostasis by comprehensive “omics approaches.”
Intriguingly, the present study revealed dramatic changes in keratinocyte derived ECM as a consequence of loss of C7. Abundances of integrin α6β4, collagen XVII and laminin-332, indirect and direct ligands of C7, were reduced (Fig. 7). Integrin α6β4 and collagen XVII are transmembrane proteins that bind laminin-332 in the basement membrane (BM). Laminin-332 and collagen XVII form anchoring filaments that are attached to C7-containing anchoring fibrils (41, 42), which provide functional integrity of the dermal-epidermal junction zone (DEJZ) by connecting the collagen IV network of the BM to dermal collagen fibrils (Fig. 7) (43–45). Our findings suggest that loss of epidermal C7 leads to a global modification and destabilization of the DEJZ (46). Fibronectin was found to be strongly increased in ECM of DEB keratinocytes indicating a wound-like phenotype, at least in vitro (38, 47). Importantly, the integrin α6β4 receptor was shown to be a negative regulator of TGF-β expression and inversely increased TGF-β activity in keratinocytes may cause reduction of laminin-332 and integrin α6β4 and upregulation of proteins associated with tissue damage (27, 28). Thus, increased TGF-β signaling because of the loss of repression may lead to many of the changes observed in our global analysis.
Fig. 7.

Model of perturbed protein homeostasis in DEB. In normal skin, C7 is present and active in the extracellular environment. It may also be sensed intracellularly. In DEB skin, the lack of extracellular C7 leads to the loss of direct and indirect extracellular binding partners, like laminin-332 and integrin α6β4, contributing to increased TGF-β signaling by a loss of repression. Increased TGF-β leads to increased abundance and secretion of cathepsin B, which in turn may further release extracellular TGF-β in a positive feedback mechanism. Increased cathepsin B levels also depend on the presence of C7 by a so far unknown mechanism. In parallel, the levels of inflammatory proteins, like S100A8 and S100A9, are increased. Extracellular cathepsin B may further contribute to DEB pathology by degrading the ECM at the dermal-epidermal junction, e.g. laminin beta-1 and gamma-1 present in laminin 311 and laminin 511, and collagen IV (8), weakening the basement membrane.
To identify potential transcriptional or post-transcriptional regulation of protein abundances, we compared fold changes of mRNA to fold changes of protein abundance in DEB and control keratinocytes. Protein and mRNA abundances correlated rather weakly, corroborating the finding that under steady-state conditions protein abundances are mainly controlled by post-transcriptional mechanisms such as rate of protein translation and protein degradation (48, 49). Surprisingly intra- and extracellular protein levels correlated similarly with respective mRNA levels indicating that in keratinocytes - once an extracellular protein is synthesized - protein secretion follows swiftly and is regulated on a global level for most proteins. Underlying molecular mechanisms will have to be addressed in detail in future studies.
The above analyses identified hitherto unknown dysregulated pathways in DEB keratinocytes. Several proteins were found to be regulated in the cellular microenvironment without showing significant changes of respective mRNA levels (Fig. 7). For example, S100A8 and S100A9, forming the calcium-binding heterodimer calprotectin, were identified as upregulated keratinocyte-products in DEB. Both proteins have been previously found upregulated in non-healing human skin wounds (50), and in acute mouse and human wounds, keratinocytes being the expressing cell type (51). Also, in inflammatory conditions, such as rheumatoid arthritis and abscesses, both marker proteins were identified as increased (52). The strongly upregulated keratinocyte-derived S100A8 and S100A9 might be interesting druggable targets to interfere with the persisting inflammation of DEB skin. Synthetic inhibitors called peptibodies (peptide-Fc fusion proteins) directed against S100A8 and S100A9 have been shown to interfere with their activity in mice (53).
Intracellular proteome changes because of the loss of C7 were extensive. Proteins carrying the GO-terms vesicle-mediated transport, lysosome and endopeptidase activity were significantly enriched in DEB keratinocytes indicating a dysregulation of proteolytic processes. In line, autophagy was identified as significantly upregulated in DEB keratinocytes, which is in stark contrast to DEB fibroblasts (10). Of the significantly enriched proteases, cathepsins B and Z are of high interest as they can cleave ECM proteins, interfere with cell-matrix contacts and activate TGF-β signaling (54–57). Thus, they may play a role in the detachment of epidermis and dermis, and contribute to fibrosis in DEB. Importantly, spontaneously gene-corrected mosaic keratinocytes from a DEB patient demonstrated that cathepsin B levels correlated inversely with C7 levels. The fact that cathepsin B levels were regulated by TGF-β suggests a detrimental positive feedback loop: loss of C7 leads to increased TGF-β signaling, increasing cathepsin B levels, which in turn may potentiate TGF-β signals (Fig. 7). The causality, kinetic regulation of events, as well as the regulation in human skin in vivo will have to be addressed in future studies.
In situ validation revealed a clear increase of cathepsin B in DEB skin, supporting an in vivo role of cathepsin B in the blister phenotype in DEB. Active cathepsin B might also contribute to post-transcriptional regulation of ECM proteins in DEB, such as the decrease of laminins beta-1 and gamma-1 (Fig. 7). Increased activity of matrix metalloprotease 1 (MMP1), produced by skin fibroblasts, has been suggested to play a role in the blister phenotype of DEB (58). Here we show that also keratinocytes may contribute to excessive ECM degradation via an increased cathepsin abundance and activity.
Secreted cathepsins have been implicated in keratinocyte migration (59) and wound healing (60). In line with this, cathepsins B and Z have also been shown to promote tumor progression in various mouse models of human cancers (61). Consequently, various approaches for cathepsin inhibition in cancers are currently investigated, as “stand alone” or part of combinatory therapy approaches (for review: (62)). As DEB patients often develop aggressive SCC, cathepsins B and Z might also be promising druggable targets in DEB.
Taken together, by applying a comprehensive approach combining SILAC-based quantitative MS with next generation RNA sequencing followed by bioinformatics analyses we generated a global, yet detailed picture of dysregulated molecular consequences of C7-deficiency. Our study revealed that loss of C7 from epidermal keratinocytes evokes cell-intrinsic wound healing-like responses. These epidermal activities establish a TGF-β-driven, pro-inflammatory and tissue destabilizing micromilieu enabling development of keratinocyte-promoted fibrosis in the dermis. Thus, by uncovering epidermal dysregulation as a catalyst of dermal fibrosis our study identifies potential new drug targets for design of new treatment strategies for DEB. Importantly, it may also provide insights into the pathogenesis of other conditions dependent on chronic, uninhibited TGF-β signaling, associated with scarring and fibrosis.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD005873 (22).
Supplementary Material
Acknowledgments
We thank the Freiburg Galaxy Platform for bioinformatics support (https://galaxy.uni-freiburg.de/).
Footnotes
* This work was supported by the German Research Foundation (DFG) through the CRC 992 (RB), CRC 850 (LBT, TR, JD), DE 1757/3–2 (JD), NY90/2–1, NY90/3–2 (AN), KI1795/1–1 (DK), by the German Federal Ministry of Education and Research (BMBF) grant 031A538A (RB), under the frame of Erare-4, by the ERA-Net for Research on Rare Diseases (EBThera; LBT), by the Excellence Initiative of the German Federal and State Governments through EXC 294 BIOSS (TR, RB, LBT, JD), and by the Swiss National Science Foundation, grant 31003A-166482/1 (JD).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- ECM
- extracellular matrix
- BH
- Benjamini-Hochberg
- BM
- basement membrane
- C7
- collagen VII
- CAF
- cancer-associated fibroblasts
- DEJZ
- dermal-epidermal junction zone
- DEB
- dystrophic epidermolysis bullosa
- NHK
- normal human keratinocytes
- PCA
- principal component analysis
- SCC
- squamous cell carcinoma
- SILAC
- stable isotope labelling by amino acids in cell culture
- SOTA
- self-organizing tree algorithm.
REFERENCES
- 1. Hynes R. O. (2009) The extracellular matrix: not just pretty fibrils. Science 326, 1216–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pozzi A., Yurchenco P. D., and Iozzo R. V. (2017) The nature and biology of basement membranes. Matrix Biol. 57–58, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bonnans C., Chou J., and Werb Z. (2014) Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 15, 786–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Has C., and Nystrom A. (2015) Epidermal basement membrane in health and disease. Curr. Top. Membr. 76, 117–170 [DOI] [PubMed] [Google Scholar]
- 5. Fine J. D., Eady R. A., Bauer E. A., Bauer J. W., Bruckner-Tuderman L., Heagerty A., Hintner H., Hovnanian A., Jonkman M. F., Leigh I., et al. (2008) The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J. Am. Acad. Dermatol. 58, 931–950 [DOI] [PubMed] [Google Scholar]
- 6. Dayal J. H., Cole C. L., Pourreyron C., Watt S. A., Lim Y. Z., Salas-Alanis J. C., Murrell D. F., McGrath J. A., Stieger B., Jahoda C., et al. (2014) Type VII collagen regulates expression of OATP1B3, promotes front-to-rear polarity and increases structural organisation in 3D spheroid cultures of RDEB tumour keratinocytes. J. Cell Sci. 127, 740–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Watt S. A., Pourreyron C., Purdie K., Hogan C., Cole C. L., Foster N., Pratt N., Bourdon J. C., Appleyard V., Murray K., et al. (2011) Integrative mRNA profiling comparing cultured primary cells with clinical samples reveals PLK1 and C20orf20 as therapeutic targets in cutaneous squamous cell carcinoma. Oncogene 30, 4666–4677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kuttner V., Mack C., Rigbolt K. T., Kern J. S., Schilling O., Busch H., Bruckner-Tuderman L., and Dengjel J. (2013) Global remodelling of cellular microenvironment due to loss of collagen VII. Mol. Syst. Biol. 9, 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ng Y. Z., Pourreyron C., Salas-Alanis J. C., Dayal J. H., Cepeda-Valdes R., Yan W., Wright S., Chen M., Fine J. D., Hogg F. J., et al. (2012) Fibroblast-derived dermal matrix drives development of aggressive cutaneous squamous cell carcinoma in patients with recessive dystrophic epidermolysis bullosa. Cancer Res. 72, 3522–3534 [DOI] [PubMed] [Google Scholar]
- 10. Kuttner V., Mack C., Gretzmeier C., Bruckner-Tuderman L., and Dengjel J. (2014) Loss of collagen VII is associated with reduced transglutaminase 2 abundance and activity. J. Invest. Dermatol. 134, 2381–2389 [DOI] [PubMed] [Google Scholar]
- 11. Nystrom A., Thriene K., Mittapalli V., Kern J. S., Kiritsi D., Dengjel J., and Bruckner-Tuderman L. (2015) Losartan ameliorates dystrophic epidermolysis bullosa and uncovers new disease mechanisms. EMBO Mol. Med. 7, 1211–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Odorisio T., Di Salvio M., Orecchia A., Di Zenzo G., Piccinni E., Cianfarani F., Travaglione A., Uva P., Bellei B., Conti A., et al. (2014) Monozygotic twins discordant for recessive dystrophic epidermolysis bullosa phenotype highlight the role of TGF-beta signalling in modifying disease severity. Hum. Mol. Genet. 23, 3907–3922 [DOI] [PubMed] [Google Scholar]
- 13. Sakai N., and Tager A. M. (2013) Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim. Biophys. Acta 1832, 911–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kern J. S., Gruninger G., Imsak R., Muller M. L., Schumann H., Kiritsi D., Emmert S., Borozdin W., Kohlhase J., Bruckner-Tuderman L., et al. (2009) Forty-two novel COL7A1 mutations and the role of a frequent single nucleotide polymorphism in the MMP1 promoter in modulation of disease severity in a large European dystrophic epidermolysis bullosa cohort. Br. J. Dermatol. 161, 1089–1097 [DOI] [PubMed] [Google Scholar]
- 15. Kern J. S., Kohlhase J., Bruckner-Tuderman L., and Has C. (2006) Expanding the COL7A1 mutation database: novel and recurrent mutations and unusual genotype-phenotype constellations in 41 patients with dystrophic epidermolysis bullosa. J. Invest. Dermatol. 126, 1006–1012 [DOI] [PubMed] [Google Scholar]
- 16. Myllyharju J., and Kivirikko K. I. (2004) Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 20, 33–43 [DOI] [PubMed] [Google Scholar]
- 17. Vlodavsky I. (2001) Preparation of extracellular matrices produced by cultured corneal endothelial and PF-HR9 endodermal cells. Curr. Protoc. Cell Biol. Chapter 10, Unit 10 14 [DOI] [PubMed] [Google Scholar]
- 18. Wisniewski J. R., Zougman A., Nagaraj N., and Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 19. Shevchenko A., Tomas H., Havlis J., Olsen J. V., and Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 20. Rappsilber J., Mann M., and Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 21. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 22. Vizcaino J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., et al. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saharinen J., and Keski-Oja J. (2000) Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Mol. Biol. Cell 11, 2691–2704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Geiger T., Cox J., Ostasiewicz P., Wisniewski J. R., and Mann M. (2010) Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat. Methods 7, 383–385 [DOI] [PubMed] [Google Scholar]
- 25. Naba A., Clauser K. R., Hoersch S., Liu H., Carr S. A., and Hynes R. O. (2012) The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteomics 11, M111 014647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang da W, Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 27. Rodius S., Indra G., Thibault C., Pfister V., and Georges-Labouesse E. (2007) Loss of alpha6 integrins in keratinocytes leads to an increase in TGFbeta and AP1 signaling and in expression of differentiation genes. J. Cell. Physiol. 212, 439–449 [DOI] [PubMed] [Google Scholar]
- 28. Tsuruta D., Kobayashi H., Imanishi H., Sugawara K., Ishii M., and Jones J. C. (2008) Laminin-332-integrin interaction: a target for cancer therapy? Curr. Med. Chem. 15, 1968–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Herrera O. R., Christensen M. L., and Helms R. A. (2016) Calprotectin: Clinical Applications in Pediatrics. J. Pediatr. Pharmacol. Ther. 21, 308–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burri E., and Beglinger C. (2012) Faecal calprotectin – a useful tool in the management of inflammatory bowel disease. Swiss Med. Wkly. 142, w13557. [DOI] [PubMed] [Google Scholar]
- 31. Moll R., Divo M., and Langbein L. (2008) The human keratins: biology and pathology. Histochem. Cell Biol. 129, 705–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Knaup J., Verwanger T., Gruber C., Ziegler V., Bauer J. W., and Krammer B. (2012) Epidermolysis bullosa - a group of skin diseases with different causes but commonalities in gene expression. Exp. Dermatol. 21, 526–530 [DOI] [PubMed] [Google Scholar]
- 33. Maier K., He Y., Wolfle U., Esser P. R., Brummer T., Schempp C., Bruckner-Tuderman L., and Has C. (2016) UV-B-induced cutaneous inflammation and prospects for antioxidant treatment in Kindler syndrome. Hum. Mol. Genet. 25, 5339–5352 [DOI] [PubMed] [Google Scholar]
- 34. Greenbaum D., Medzihradszky K. F., Burlingame A., and Bogyo M. (2000) Epoxide electrophiles as activity-dependent cysteine protease profiling and discovery tools. Chem. Biol. 7, 569–581 [DOI] [PubMed] [Google Scholar]
- 35. Orlowski G. M., Colbert J. D., Sharma S., Bogyo M., Robertson S. A., and Rock K. L. (2015) Multiple cathepsins promote Pro-IL-1beta synthesis and NLRP3-mediated IL-1beta activation. J. Immunol. 195, 1685–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gretzmeier C., Eiselein S., Johnson G. R., Engelke R., Nowag H., Zarei M., Kuttner V., Becker A. C., Rigbolt K. T. G., Hoyer-Hansen M., et al. (2017) Degradation of protein translation machinery by amino acid starvation-induced macroautophagy. Autophagy 13, 1064–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang Y., Woosley A. N., Sivalingam N., Natarajan S., and Howe P. H. (2016) Cathepsin-B-mediated cleavage of Disabled-2 regulates TGF-beta-induced autophagy. Nat. Cell Biol. 18, 851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nystrom A., Velati D., Mittapalli V. R., Fritsch A., Kern J. S., and Bruckner-Tuderman L. (2013) Collagen VII plays a dual role in wound healing. J. Clin. Invest. 123, 3498–3509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kiritsi D., Valari M., Michos A., Karakosta V., and Has C. (2016) The mysteries of mosaicism: phenotypic variability in a family with incontinentia pigmenti. Eur. J. Dermatol. 26, 504–506 [DOI] [PubMed] [Google Scholar]
- 40. Watt S. A., Dayal J. H., Wright S., Riddle M., Pourreyron C., McMillan J. R., Kimble R. M., Prisco M., Gartner U., Warbrick E., et al. (2015) Lysyl hydroxylase 3 localizes to epidermal basement membrane and is reduced in patients with recessive dystrophic epidermolysis bullosa. PLoS ONE 10, e0137639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. de Pereda J. M., Ortega E., Alonso-Garcia N., Gomez-Hernandez M., and Sonnenberg A. (2009) Advances and perspectives of the architecture of hemidesmosomes: lessons from structural biology. Cell Adh. Migr. 3, 361–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Walko G., Castanon M. J., and Wiche G. (2015) Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 360, 529–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Villone D., Fritsch A., Koch M., Bruckner-Tuderman L., Hansen U., and Bruckner P. (2008) Supramolecular interactions in the dermo-epidermal junction zone: anchoring fibril-collagen VII tightly binds to banded collagen fibrils. J. Biol. Chem. 283, 24506–24513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chung H. J., and Uitto J. (2010) Type VII collagen: the anchoring fibril protein at fault in dystrophic epidermolysis bullosa. Dermatol. Clin. 28, 93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nystrom A., Bornert O., and Kuhl T. (2017) Cell therapy for basement membrane-linked diseases. Matrix Biol. 57–58, 124–139 [DOI] [PubMed] [Google Scholar]
- 46. Bremer J., Bornert O., Nystrom A., Gostynski A., Jonkman M. F., Aartsma-Rus A., van den Akker P. C., and Pasmooij A. M. (2016) Antisense Oligonucleotide-mediated Exon Skipping as a Systemic Therapeutic Approach for Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. Nucleic Acids 5, e379. [DOI] [PubMed] [Google Scholar]
- 47. Fritsch A., Loeckermann S., Kern J. S., Braun A., Bosl M. R., Bley T. A., Schumann H., von Elverfeldt D., Paul D., Erlacher M., et al. (2008) A hypomorphic mouse model of dystrophic epidermolysis bullosa reveals mechanisms of disease and response to fibroblast therapy. J. Clin. Invest. 118, 1669–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vogel C., and Marcotte E. M. (2012) Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schwanhausser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., and Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 50. Eming S. A., Koch M., Krieger A., Brachvogel B., Kreft S., Bruckner-Tuderman L., Krieg T., Shannon J. D., and Fox J. W. (2010) Differential proteomic analysis distinguishes tissue repair biomarker signatures in wound exudates obtained from normal healing and chronic wounds. J. Proteome Res. 9, 4758–4766 [DOI] [PubMed] [Google Scholar]
- 51. Thorey I. S., Roth J., Regenbogen J., Halle J. P., Bittner M., Vogl T., Kaesler S., Bugnon P., Reitmaier B., Durka S., et al. (2001) The Ca2+-binding proteins S100A8 and S100A9 are encoded by novel injury-regulated genes. J. Biol. Chem. 276, 35818–35825 [DOI] [PubMed] [Google Scholar]
- 52. Yui S., Nakatani Y., and Mikami M. (2003) Calprotectin (S100A8/S100A9), an inflammatory protein complex from neutrophils with a broad apoptosis-inducing activity. Biol. Pharm. Bull. 26, 753–760 [DOI] [PubMed] [Google Scholar]
- 53. Qin H., Lerman B., Sakamaki I., Wei G., Cha S. C., Rao S. S., Qian J., Hailemichael Y., Nurieva R., Dwyer K. C., et al. (2014) Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat. Med. 20, 676–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Akkari L., Gocheva V., Kester J. C., Hunter K. E., Quick M. L., Sevenich L., Wang H. W., Peters C., Tang L. H., Klimstra D. S., et al. (2014) Distinct functions of macrophage-derived and cancer cell-derived cathepsin Z combine to promote tumor malignancy via interactions with the extracellular matrix. Genes Dev. 28, 2134–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bengsch F., Buck A., Gunther S. C., Seiz J. R., Tacke M., Pfeifer D., von Elverfeldt D., Sevenich L., Hillebrand L. E., Kern U., et al. (2014) Cell type-dependent pathogenic functions of overexpressed human cathepsin B in murine breast cancer progression. Oncogene 33, 4474–4484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gogineni V. R., Gupta R., Nalla A. K., Velpula K. K., and Rao J. S. (2012) uPAR and cathepsin B shRNA impedes TGF-beta1-driven proliferation and invasion of meningioma cells in a XIAP-dependent pathway. Cell Death Dis. 3, e439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Moles A., Tarrats N., Fernandez-Checa J. C., and Mari M. (2012) Cathepsin B overexpression due to acid sphingomyelinase ablation promotes liver fibrosis in Niemann-Pick disease. J. Biol. Chem. 287, 1178–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Colombi M., Gardella R., Zoppi N., Moro L., Marini D., Spurr N. K., and Barlati S. (1992) Exclusion of stromelysin-1, stromelysin-2, interstitial collagenase and fibronectin genes as the mutant loci in a family with recessive epidermolysis bullosa dystrophica and a form of cerebellar ataxia. Hum. Genet. 89, 503–507 [DOI] [PubMed] [Google Scholar]
- 59. Buth H., Wolters B., Hartwig B., Meier-Bornheim R., Veith H., Hansen M., Sommerhoff C. P., Schaschke N., Machleidt W., Fusenig N. E., et al. (2004) HaCaT keratinocytes secrete lysosomal cysteine proteinases during migration. Eur. J. Cell Biol. 83, 781–795 [DOI] [PubMed] [Google Scholar]
- 60. Buth H., Luigi Buttigieg P., Ostafe R., Rehders M., Dannenmann S. R., Schaschke N., Stark H. J., Boukamp P., and Brix K. (2007) Cathepsin B is essential for regeneration of scratch-wounded normal human epidermal keratinocytes. Eur. J. Cell Biol. 86, 747–761 [DOI] [PubMed] [Google Scholar]
- 61. Reinheckel T., Peters C., Kruger A., Turk B., and Vasiljeva O. (2012) Differential impact of cysteine cathepsins on genetic mouse models of de novo carcinogenesis: cathepsin B as emerging therapeutic target. Front. Pharmacol. 3, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Olson O. C., and Joyce J. A. (2015) Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 15, 712–729 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD005873 (22).