

Abstract
Integrins are heterodimeric cell-adhesion receptors comprising α and β subunits that transmit signals allosterically in both directions across the membrane by binding to intra- and extracellular components. The human platelet antigen-1 (HPA-1) polymorphism in αIIbβ3 arises from a Leu → Pro exchange at residue 33 in the genu of the β3 subunit, resulting in Leu33 (HPA-1a) or Pro33 (HPA-1b) isoforms. Although clinical investigations have provided conflicting results, some studies have suggested that Pro33 platelets exhibit increased thrombogenicity. Under flow-dynamic conditions, the Pro33 variant displays prothrombotic properties, characterized by increased platelet adhesion, aggregate/thrombus formation, and outside-in signaling. However, the molecular events underlying this prothrombotic phenotype have remained elusive. As residue 33 is located >80 Å away from extracellular binding sites or transmembrane domains, we hypothesized that the Leu → Pro exchange allosterically shifts the dynamic conformational equilibrium of αIIbβ3 toward an active state. Multiple microsecond-long, all-atom molecular dynamics simulations of the ectodomain of the Leu33 and Pro33 isoforms provided evidence that the Leu → Pro exchange weakens interdomain interactions at the genu and alters the structural dynamics of the integrin to a more unbent and splayed state. Using FRET analysis of fluorescent proteins fused with αIIbβ3 in transfected HEK293 cells, we found that the Pro33 variant in its resting state displays a lower energy transfer than the Leu33 isoform. This finding indicated a larger spatial separation of the cytoplasmic tails in the Pro33 variant. Together, our results indicate that the Leu → Pro exchange allosterically shifts the dynamic conformational equilibrium of αIIbβ3 to a structural state closer to the active one, promoting the fully active state and fostering the prothrombotic phenotype of Pro33 platelets.
Keywords: molecular dynamics, conformational change, fluorescence resonance energy transfer, FRET, integrin, platelet, activation, polymorphism, structural dynamics, thrombus
Introduction
Integrins are heterodimeric cell-adhesion receptors formed of α and β subunits. Each subunit is divided into three parts: a large extracellular domain (ectodomain), a single-pass transmembrane domain, and a short cytoplasmic tail connecting the extracellular to the intracellular environment (1). In addition to their biomechanical role (2), integrins transmit signals allosterically (3) in both directions across the membrane (termed “outside-in” and “inside-out” signaling) by binding to intra- and extracellular components (4). In the present study, we focused on αIIbβ3 (5–8), which is expressed on the platelet surface and essential for platelet aggregation (8).
The ectodomain can be divided into two parts (9, 10) (Fig. 1). The “head” of the receptor is formed by the β-propeller and βA domains, and the “legs” are formed by the thigh and calf domains (αIIb subunit) as well as EGF domains together with the β-tail domain (β3 subunit) (4). The genu located between the thigh and calf-1 domain as well as the EGF-1 and EGF-2 domains in the αIIb and β3 subunits, respectively, forms a region of interdomain flexibility (11). Integrin structural dynamics is characterized by at least three states: a closed, bent, low-affinity state; a closed, extended, low-affinity state; and an open, extended, high affinity state (12). Although the magnitude of conformational changes has remained a matter of discussion (6, 10, 13), the majority of crystal structures of αvβ3 (14), αIIbβ3 (9), and αxβ2 (15) integrins show their ectodomain in a bent conformation (14, 16, 17). Here, the head is flexed toward the membrane at an angle of 135° (7) related to the legs, with the genu being the angle's vertex.
Figure 1.
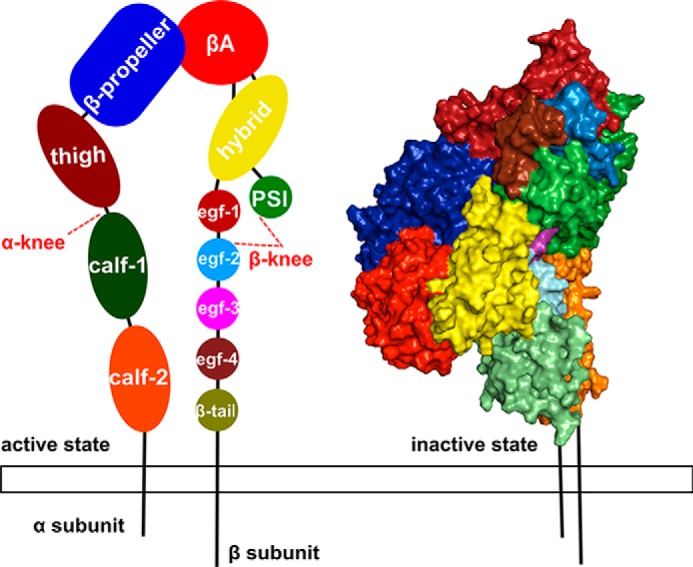
Representation of the domain organization of integrin αIIbβ3. On the left, the open conformation is depicted as a schematic with the α subunit divided into four different domains and the β subunit divided into eight domains. Each domain is colored and labeled. The knees of the protein are highlighted by red dashed lines and lie between the thigh and calf-1 domains and between the PSI and EGF-1/EGF-2 domains of the α and β subunits, respectively. On the right, the αIIbβ3 ectodomain starting structure (Protein Data Bank code 3FCS) in the bent conformation is depicted in surface representation (right) with domains colored as on the left.
According to current models, the genu plays a critical role in conformational transitions between the three structural states as a straightening in the genu region leads to a separation of the head from the legs and thus an unbending of the conformation. This motion is associated with reduced interactions between the two subunits, resulting in a spatial separation (“splaying”) of the α and β subunit legs (18). With respect to our study, the role of the plexin-semaphorin-integrin (PSI)4 domain, which is part of the β3 genu (5), is of particular interest in integrin activation. Located >80 Å away from the extracellular binding site and >90 Å away from the membrane (estimated from Protein Data Bank code 3FCS), the domain's involvement in integrin activation has been demonstrated (19, 20). Specifically, the domain is believed to have a biomechanical role in the allosteric signal transmission across the structure (21).
The human platelet antigen-1 (HPA-1) polymorphism of the β3 gene of αIIbβ3 arises from a Leu → Pro exchange at residue 33 of the mature β3 subunit (22, 23), resulting in Leu33 (HPA-1a) or Pro33 (HPA-1b) platelets. This amino acid exchange, located within the PSI domain, leads to an inherited dimorphism that can be of clinical relevance (22). For example, the HPA-1b allele was significantly more frequent among young patients with acute coronary syndrome than among age-matched healthy subjects (24). In the Ludwigshafen Risk and Cardiovascular Health (LURIC) trial, an association study including more than 4,000 individuals, we documented that patients with coronary artery disease (CAD), who are carriers of the HPA-1b allele, experience their myocardial infarction 5 years earlier in life than CAD patients who are HPA-1b–negative (22, 23). In a prospective study on CAD patients undergoing saphenous-vein coronary-artery bypass grafting, we demonstrated that HPA-1b is a hereditary risk factor for bypass occlusion, myocardial infarction, or death after coronary-artery bypass surgery (25). These results suggest that the Leu → Pro exchange may modulate functional properties of αIIbβ3, resulting in a prothrombotic integrin variant. Prothrombotic properties are also displayed by Pro33 platelets under flow-dynamic conditions (26). However, the molecular mechanism underlying the suggested prothrombotic phenotype of the Pro33 (HPA-1b) variant has remained elusive.
We hypothesized that the Leu → Pro exchange allosterically shifts the dynamic conformational equilibrium of αIIbβ3 toward an active state. This shift, in turn, would facilitate reaching the fully active state in the presence of integrin ligands. To examine this hypothesis, we performed multiple microsecond-long all-atom molecular dynamics (MD) simulations of the ectodomain and Förster resonance energy transfer (FRET) measurements of αIIbβ3-transfected HEK293 cells expressing either the Leu33 (HPA-1a) or Pro33 (HPA-1b) isoform. Our MD simulations provide evidence that the Leu → Pro exchange weakens interdomain interactions at the genu and alters the structural dynamics of the integrin to a more unbent and splayed state, resulting in overall conformational changes that have been linked to integrin activation (18, 27). In accord with these results, FRET analyses of αIIbβ3 transfectants reveal that the Pro33 (HPA-1b) variant in the resting state displays a significantly lower energy transfer than the Leu33 (HPA-1a) variant.
Results
Platelet thrombus size in relation to αIIbβ3 HPA-1 isoforms under flow conditions in vitro
Given the prothrombotic phenotype of Pro33 platelets, we initially focused on platelet thrombus formation under arterial flow conditions comparing Leu33 (HPA-1a) with Pro33 (HPA-1b) platelets. To study the dynamics of platelet thrombus formation, mepacrine-labeled citrated whole blood from healthy volunteers genotyped for HPA-1 of αIIbβ3 and α2C807T of α2β1 (see supporting methods) was perfused at shear rates >500 s−1 through a flow chamber coated with collagen type I. Image acquisition was achieved by a series of stacks corresponding to confocal sections from the bottom to the apex of forming platelet thrombi. For quantitation of thrombus formation in vitro, we applied a voxel-based method for 3D visualization of real time-resolved volume data using ECCET software (www.eccet.de)5 (28). As depicted in Fig. 2A, ECCET allows determination of the number, bottom area, height, and volume of single platelet thrombi formed in vitro.
Figure 2.

Dynamics and volumetric analysis of platelet thrombus formation under flow-dynamic conditions. A rectangular flow chamber coated with collagen type I (3 mg/ml) at the lower surface was perfused with mepacrine-labeled citrated whole blood for 10 min at an initial near-wall shear rate of 500 s−1, simulating arterial flow conditions. Fluorescence signals were detected by confocal laser scanning microscopy, and digital imaging was processed as described under “Experimental procedures”. Volumetry of forming platelet thrombi was assessed by real-time 3D visualization. A, a reconstruction of formed platelet thrombi obtained from a stack of 30 images by confocal laser scanning microscopy and subsequent data processing by ECCET. B and C, initial platelet thrombus formation and subsequent thrombus growth were recorded in 25-s intervals for each single thrombus. Addition of abciximab (4 μg/ml), an inhibitory antibody to αIIbβ3, abrogated any platelet thrombus formation. B shows the mean single platelet thrombus volume, and C shows the corresponding thrombus bottom area. D, schematic illustrating the narrowing of the flow path within the perfusion chamber with a resulting increase in shear rates upon apical thrombus growth. Blue diamonds, homozygous Leu33 (HPA-1a) platelets (n = 8); red squares, homozygous Pro33 (HPA-1b) platelets (n = 8); black rectangles, control in the presence of abciximab (n = 2). Error bars indicate mean ± S.E. Asterisks indicate statistical significance (*, p < 0.05).
Using these tools, we detected that, upon perfusion over 10 min, platelets homozygous for Pro33 (HPA-1b) formed single thrombi that were significantly higher than those of platelets homozygous for Leu33 (HPA-1a) (Fig. 2B). This difference in mean single thrombus volume was due to an increased thrombus height, whereas the number and bottom area of thrombi (Fig. 2C) did not differ between the HPA-1 isoforms. Platelet adhesion and subsequent aggregate/thrombus formation under flow were abrogated by the monoclonal antibody LJ-Ib1 (a gift from Dr. Z. M. Ruggeri, La Jolla, CA) that completely inhibits binding of von Willebrand factor (VWF) to the platelet glycoprotein (GP) Ib-IX-V complex or by the monoclonal antibody 5C4 (a gift from Dr. M. Gawaz, Tübingen, Germany) that blocks the platelet collagen receptor GPVI (data not shown). As depicted in the schematic illustration (Fig. 2D), with increasing time, the flow path of the perfusion chamber becomes narrowed as the thrombi are growing. Consequently, shear rates gradually increase, and formed platelet thrombi, especially at their apex, are exposed to higher shear than initially applied. Thus, the difference in mean single thrombus volumes between the HPA-1 isoforms can be indicative of a higher thrombus stability of Pro33 (HPA-1b) than Leu33 (HPA-1a) platelets as reported before (29).
Structural variability of αIIbβ3 HPA-1 isoforms in MD simulations of the integrin ectodomain
To provide an atomistic view on the effect of the Leu → Pro exchange, the Leu33 (HPA-1a) and Pro33 (HPA-1b) isoforms were investigated by all-atom MD simulations using the respective integrin ectodomains in the bent conformation as starting structures. The quality of the crystal structure used as a starting structure for the Leu33 isoform and to model the Pro33 isoform was validated by MolProbity (30), yielding a percentile score of 1.70, equal to a 99th percentile rank, where a 0th percentile rank indicates the worst and a 100th percentile rank indicates the best structure among structures with comparable resolution (∼2.55 Å in the case of Protein Data Bank code 3FCS). For generating the Pro33 isoform from the crystal structure, the Leu33 side chain was mutated using the best rotamer of Pro at this position according to Swiss-PdbViewer (31). For this structure, the quality of the PSI domain in complex with the EGF-1 and EGF-2 domains (genu of the β3 subunit) was assessed by MolProbity, yielding a percentile score of 1.30, equal to a 98th percentile rank.
Three independent MD simulations of 1-μs length each were carried out. The convergence of the MD simulations was tested by computing the root-mean-square deviation (RMSD) average correlation as described previously (32) (Fig. S1). The root-mean-square average correlation is a measure of the time scales on which structural changes occur in MD simulations. From the bumps in the curves, we can estimate that observed structural changes occur within ∼50–200 ns. For time intervals >200 ns, the curves are smooth, suggesting that no large structural changes happen during the investigation period.
In addition, we analyzed the overlap of histograms of principal component (PC) projections obtained in a pairwise manner from each simulation for a given isoform as a function of time (Fig. S2). The PC analysis was performed on the whole protein after a mass-weighted fitting on the β-propeller and βA domains. The results reveal that, overall, the Kullback-Leibler divergence between histograms of the respective first three PCs becomes small (<0.02) after ∼600 ns of simulation time, whereas values in the first 100 ns can be as high as ∼0.1. Hence, the analyses indicate that, in the given simulation times, rather similar conformational spaces were sampled by MD simulations of one isoform. However, in some cases, a small increase in the Kullback-Leibler divergence is observed toward the end of the simulation time; this behavior is not unexpected because the MD simulations were started from bent conformations of the isoforms that can relax to more open conformations (33). Given that, in the absence of force, the timescale of integrin activation is on the order of 10−3 (34) to 1 s (35), one cannot expect that the MD simulations are converged with respect to the bent-open conformational equilibrium of αIIbβ3 integrin. In total, differences in structural parameters between both isoforms that we report below relate to differences in the tendency of the ectodomains to go from a bent to an open state. Unless stated otherwise, all results of the MD simulations are expressed as arithmetic means calculated over time, and we considered only uncorrelated instances for S.E. calculations (see “Experimental procedures”).
The βA domain contains three metal ion–binding sites (Fig. S3). To assess their structural integrity during the MD simulations, we monitored the time evolution of distances between the SyMBS, MIDAS, and ADMIDAS metal ions and the respective coordinating residues (Table S3 and Fig. S3). The results reveal that during the production runs the distances remain almost unchanged with S.E. <0.1 Å in almost all cases. Thus, the local geometry of the metal ion–binding sites is well preserved throughout the MD simulations.
The structural similarity of the conformations obtained by MD simulations with respect to the starting structure was explored in terms of the RMSD of Cα atoms after mass-weighted superimpositioning. Similar to our previous MD studies performed on integrin α5β1 (18, 27), the simulations revealed minor structural changes of the single domains as mirrored by RMSD values that were largely below 3 Å with the exception of the calf-2 and EGF domains and the β-tail (RMSD up to 5 Å) (Table S4 and Fig. S4). Although the β-tail has been characterized as highly flexible (14), the larger RMSD of the calf-2 domain, in part, is due to the presence of long flexible loops (9); furthermore, the larger RMSD may result from simulating the ectodomain only; i.e. at the C-terminal ends of each subunit, the transmembrane domains are missing. As to the EGF domains, visual inspection of the MD trajectories revealed that the larger RMSD resulted in part from motions of the domains relative to each other.
In contrast, when aligning only the head region, the mean RMSD increased up to ∼17 Å (Table S5 and Fig. S5) with the highest values found for the calf-2 and β-tail domains of the legs. Hence, these larger structural changes must arise from relative movements of the domains (or subunits) with respect to each other, considering that the single domains were structurally rather invariant. Comparing both isoforms of αIIbβ3, a larger mean RMSD (∼9.2 ± 0.34 Å) was found for Pro33 than Leu33 (∼6.6 ± 0.83 Å) (Fig. 3A). In accord with that, the mean radius of gyration (Rg) of the overall structure was larger for the Pro33 (∼40.3 ± 0.22 Å) than the Leu33 isoform (∼39.6 ± 0.09 Å) (Fig. 3A). Taken together, the sampled conformational space of both αIIbβ3 isoforms varied significantly with respect to the difference of the mean values of these structural parameters (Table S6). To conclude, the Pro33 variant displayed significantly larger structural deviations from the bent starting structure and became less compact than the Leu33 isoform during MD simulations.
Figure 3.

Conformational changes of the Leu33 and Pro33 isoforms of αIIbβ3 during MD simulations. A, two-dimensional histogram of the RMSD of Cα atoms of the entire ectodomain after mass-weighted fitting on the β-propeller and βA domains of the starting structure versus Rg for the ectodomains. Bluish colors represent the three MD simulations of the Leu33 isoform, and reddish colors represent those of the Pro33 variant. B–D, histograms of the kink, bending, and splaying angles averaged over three MD simulations with error bars showing the S.E. and asterisks indicating the statistical significance (*, p < 0.05; ***, p < 0.0001). Above the plots, the definitions of the angles are given: B, kink angles as black solid line connecting the three points (center of mass of Cα atoms of Lys112 and Ile118, center of mass of Cα atoms of Gln119 and Lys125, and center of mass of Cα atoms of Leu126 and Leu132) on both isoforms; C, bending angle as a black solid line connecting the three points (center of mass of the β-propeller and βA domains, center of mass of the PSI domain, and center of mass of the calf-2 and β-tail domains); D, splaying angle as a black solid line connecting the three points (center of mass of Cα atoms of Leu788 and Gly796 in the calf-2 domain, center of mass of the Cα atoms of Cys602 and Cys608 in the thigh domain, and center of mass of Cα atoms of Glu1557 and Val1561 in the β-tail domain).
Conformational changes of the ectodomains of αIIbβ3 HPA-1 isoforms toward a more open, extended conformation
To further characterize the structural differences between the αIIbβ3 isoforms, we monitored geometric parameters along the MD trajectories that have been linked with conformational changes of the ectodomain from an inactive to an active state (18, 27) (Fig. S6 and Table S7). First, we investigated possible variations in the region of the center of helix α1 and the N terminus of helix α7 (27, 37). This region was shown to form a “T-junction” upon activation (21, 27). We computed the kink angle of helix α1 (Fig. 3B), which revealed a mean value over three MD trajectories that is larger by 15° in the Pro33 (∼153 ± 1.5°) than the Leu33 isoform (∼138 ± 2.8°) (Fig. 3B). Hence, helix α1 straightens more in Pro33 and thus shows a stronger tendency to form the T-junction than in Leu33. The spread in the mean values found for Pro33 (Table S7) resulted from a rapid and pronounced increase of the kink angle, which was initially ∼143° (calculated from Protein Data Bank code 3FCS), within the first 200 ns in two of the three MD simulations (Fig. S6A).
Second, we evaluated the unbending of the structure in terms of the separation of the head region and the terminal part of the legs (calf-2 domain and β-tail) (Fig. 3C). Furthermore, we monitored the spatial separation (splaying) of the integrin's legs (Fig. 3D). Similar parameters were successfully used previously (18). The bending angle was ∼7° larger in the Pro33 (∼49 ± 1.8°) than in the Leu33 isoform (∼42 ± 1.4°) (Fig. 3C, Fig. S6B, and Table S7). The splaying angle was ∼3° larger in Pro33 (∼28 ± 0.5°) than in Leu33 (∼25 ± 0.2°) (Fig. 3D, Fig. S6C, and Table S7). In the latter case, in two MD simulations, the time evolution of the splaying angle revealed a decrease of ∼22° within the last 200 ns of the simulation (Fig. S6C). The differences between angles in the Leu33 and Pro33 isoforms were significant in all cases (Table S7). As additional indicators of structural changes, we evaluated the opening of the structure in terms of changes in internal distances between the N and C termini of each subunit and between the C termini of the two subunits (Fig. S6D). All evaluated distances were larger in the Pro33 than the Leu33 isoform, and the differences between respective distances were significant in all cases (Table S8). To conclude, our results revealed significant differences in the conformational states of both αIIbβ3 isoforms with the ectodomain of Pro33 displaying a stronger tendency to move toward the extended conformation with more splayed legs.
Experimental evidence for spatial rearrangements of the cytoplasmic tails of αIIbβ3 upon Leu → Pro exchange
To investigate a possible influence of the Leu → Pro exchange on the spatial separation of α and β subunits, we performed FRET acceptor photobleaching (APB) analyses in individual cells transfected with αIIbmVenus and β3Leu33mCherry (HPA-1a) (Table S1) or β3Pro33mCherry (HPA-1b) (Table S2) plasmids, respectively. Using FRET, the spatial separation of the subunits is inferred from the amount of energy transferred between the fluorescent proteins mVenus and mCherry attached to the cytoplasmic tails of the subunits.
By fluorescence microscopy performed 48 h after transfection, we verified that both subunits were colocalized at the cell membrane (Fig. 4A). Concordant with the presence of the integrin at the plasma membrane, we detected the complete αIIbβ3 receptor (recognized by a complex-specific anti-αIIbβ3 antibody, anti-CD41, clone MEM-06) by flow cytometry. Functional integrity of both integrin isoforms and correct membrane insertion were documented by intact activation of αIIbβ3 in transfected cells upon phorbol 12-myristate 13-acetate–induced stimulation of protein kinase C and specific binding of Alexa Fluor 647-fibrinogen to αIIbβ3 upon inside-out activation (38). Notably, flow cytometry measurements of CD41 expression upon five independent transfection experiments indicated that the levels of αIIbβ3 expressing either the Leu33 (HPA-1a) or the Pro33 (HPA-1b) isoform did not differ more than 10% from each other (Fig. 4B).
Figure 4.

Transient expression of the complete αIIbβ3 receptor in HEK293 cells. A, phase-contrast and fluorescence microscopy images of a representative HEK293 cell transfected with αIIbmVenus and β3Leu33mCherry plasmids (upper panel) and a representative HEK293 cell transfected with αIIbmVenus and β3Pro33mCherry plasmids (lower panel). B, flow cytometric analyses of αIIbβ3 (CD41), expressing either isoform Leu33 (HPA-1a) or Pro33 (HPA-1b), performed 48 h after transfection in five independent experiments. Of note, the transfectants displayed less than 10% difference in αIIbβ3 expression of either Leu33 (HPA-1a) or Pro33 (HPA-1b) isoform. Values represent mean fluorescence intensity after staining of the transfectants with APC-conjugated CD41 antibody, a complex-specific anti-αIIbβ3 antibody. C, FRET-APB measurements in a representative HEK293 cell transfected with αIIbmVenus and β3Leu33mCherry plasmids. D, results of FRET efficiency of fused individual Leu33 (HPA-1a) or Pro33 (HPA-1b) cells and respective donor controls. To determine the efficiency of energy transfer, the fluorescence of mVenus was measured in a defined region of the membrane (red circled) before and after photobleaching of mCherry at 561 nm (39, 40). Details are given under “Experimental procedures.” The error bars indicate mean ± S.E.
Using these transfectants, photobleaching of mCherry at 561 nm on a defined cellular region (region of interest) encompassing part of the cell membrane led to a complete loss of energy transfer and, consequently, to an increase in mVenus fluorescence intensity (Fig. 4C). For a control, cells were transfected with αIIbmVenus and β3Leu33 or β3Pro33 plasmids (without mCherry), a condition that abrogated any energy transfer (data not shown). To focus on non-activated αIIbβ3 transfectants, as evidenced by absence of binding of Alexa Fluor 647-fibrinogen or PAC1, an activation-dependent anti-αIIbβ3 monoclonal antibody (data not shown), cells were left resting on chamber slides with culture medium for 24 h prior to FRET analyses, all of which were subsequently carried out with minimal manipulation of the cells to prevent any possible cellular activation.
FRET-APB analyses were performed in a total of 249 single cells: 91 Leu33 cells, 88 Pro33 cells, 35 Leu33 donor control cells, and 35 Pro33 donor control cells. FRET-APB efficiency was computed according to Equation 2 (see “Experimental procedures” and Refs. 39 and 40). Notably, FRET-APB efficiency between mVenus and mCherry in Leu33 cells (mean ± S.E., 18.20 ± 0.276) was significantly higher (p < 0.0001) than in HPA-1b cells (15.74 ± 0.395) (Fig. 4D). This difference in energy transfer upon photobleaching of both αIIbβ3 isoforms suggested a larger spatial separation in the Pro33 than the in Leu33 isoform when both isoforms were examined in their bent conformation. This observation is indicative of a state more prone to activation as a consequence of the Leu → Pro exchange at residue 33 in the ectodomain of the β subunit of αIIbβ3.
Short- and mid-range structural, dynamics, and stability changes induced by the Leu → Pro exchange
The two-dimensional (2D) RMSD of Cα atoms of the EGF-1/EGF-2/EGF-3 domains along the MD trajectories was computed after mass-weighted superimposition onto the respective starting structures of the domains. The 2D RMSD values indicated that the domains showed larger differences from the initial starting structure in the Pro33 than in the Leu33 isoform (see also Table S4) but also that the two isoforms adopted conformational states that largely deviated from each other (RMSD up to 8 Å) (Fig. 5A). Next, we computed the residue-wise root-mean-square fluctuations (RMSFs) of the PSI domain, a measure of atomic mobility, to identify differences in the conformational variations associated with the Leu → Pro exchange. The results revealed a marked increase in atomic mobility for residues Glu29–Pro37 of the loop between strands A and B in the PSI domain (Fig. 5B) with a ∼0.6-Å difference found at residue 33 (Leu or Pro). Beyond this region, the amino acid exchange did not affect the atomic mobility (Fig. 5B). Likewise, we did not detect significant differences in the secondary structure propensity of the AB loop residues between the Leu33 and Pro33 isoform except for a small decrease of the α-helix propensity in the helix C-terminal to the loop (Fig. S7). To conclude, in both isoforms, the PSI domain did not undergo marked changes in structure (see also Table S4) as a consequence of the polymorphism at residue 33 of the β3 subunit. This was in contrast to the EGF domains, which revealed marked structural changes in Pro33. However, the structural dynamics of the AB loop of the PSI domain increased in the Pro33 variant.
Figure 5.

Short- and mid-range structural and dynamics changes induced by the Leu → Pro exchange. A, 2D RMSD plot calculated for the Cα atoms of the EGF-1, EGF-2, and EGF-3 domains after superimposing onto the respective domains. All trajectories accounting for an aggregate simulation time of 2 × 3 μs were considered together in the analysis, excluding the first 200 ns of each trajectory and extracting frames at intervals of 100 ns. B, residue-wise mean backbone RMSF of the PSI domain after a mass-weighted fitting onto the starting structure. Error bars denote S.E. Blue and red curves represent the Leu33 and Pro33 isoforms, respectively; the two black lines delineate the AB loop (residues Glu29–Pro37). C, αIIbβ3 is shown in schematic representation and colored in light gray (with the exception of the PSI domain (green), EGF-1 domain (firebrick), and EGF-2 domain (marine)). Domains are labeled. The enlargement shows the location of the Leu → Pro exchange in the PSI domain within the genu interface of the β3 subunit. Black dashed lines indicate distances computed in D with the distance values of the starting structure reported next to them. Residues Leu33 (PSI domain), Ser469 (EGF-1 domain), and Gln481 (EGF-2 domain) are depicted in ball-and-stick representation. D, mean distances between the Cα atoms of Leu33/Pro33 as well as Ser469 (EGF-1) and Gln481 (EGF-2) calculated for the Leu33 isoform (blue boxes) and the Pro33 variant (red boxes) and measured in the crystal structure (Protein Data Bank code 3FCS; gray boxes). Error bars indicate mean ± S.E., and asterisks denote a significant difference (*, p < 0.05; ***, p < 0.0001) between the two isoforms of αIIbβ3.
As this loop faces the EGF-1 and EGF-2 domains (41), the Leu → Pro exchange may also impact the structure, interactions, and stability of this interface. Therefore, we monitored the time evolution of the distance between the Cα atoms of residue Leu33 or Pro33 and Ser469 and Gln481 to investigate the level of compactness of the interface between the PSI domain and the EGF-1/EGF-2 domains (Fig. 5C). In the bent conformation of αIIbβ3, the Cα atom at residue 33 is separated by 9.4 and 15.8 Å from the Cα atoms of Ser469 and Gln481 (calculated from Protein Data Bank code 3FCS), respectively.
Comparing both isoforms of αIIbβ3, we found a mean value for the Leu/Pro33···Ser469 distance that is smaller by ∼3.7 Å in Leu33 (∼8.1 ± 0.40 Å) than in Pro33 (∼11.8 ± 0.79 Å). A mean value smaller by ∼5.8 Å in Leu33 (∼6.6 ± 0.37 Å) than in Pro33 (∼12.4 ± 1.02 Å) was found for the Leu/Pro33···Gln481 distance. The differences between distances in the Leu33 and Pro33 isoforms were significant in all cases (Fig. 5D and Table S9). The pronounced decrease from the initial structure observed in the Leu33 isoform (∼9 Å) for the Leu33···Gln481 distance is in line with the description of a contact area between these two domains in the closed, low-affinity, bent state (41). This contact is lost in the extended conformation (41). These results indicated that the interface between the PSI domain and the EGF-1/EGF-2 domains is more tightly packed in the Leu33 than in the Pro33 isoform.
In addition, we computed the number of contacts present in the starting structure (“native contacts”) and those formed over the course of the MD simulations (“non-native contacts”). Contacts were evaluated between the nine residues of the AB loop and residues of the adjacent EGF-1 and EGF-2 domains, applying a distance cutoff of 7 Å between the side-chain atoms. In all three MD simulations of the Pro33 variant, the total number of contacts was significantly lower than in the Leu33 isoform (Fig. 6A and Table S10). This difference became even more pronounced when only non-native contacts were considered (2-fold decrease). The same holds true for specific interactions (hydrogen bonds and salt bridges) that were conserved in the Leu33 isoform only (Fig. S8). In the segment connecting the EGF-1 domain with the EGF-2 domain, Gln481 is hydrogen-bonded to Ser469 with a high occupancy (∼70% along the MD trajectories) and/or with Gln470 (∼27%). Additional stable intradomain hydrogen bond interactions (>60%) were found within the EGF-2 domain that involve Cys492, which also forms a disulfide bridge with Cys473 of the EGF-1 domain (Fig. S8). To conclude, the Leu → Pro exchange leads to a less compact interface between the PSI domain and EGF-1/EGF-2 domains. Moreover, fewer interactions across the interface and within the EGF-1/EGF-2 domains were found in the Pro33 variant compared with the Leu33 isoform.
Figure 6.

Changes within the PSI/EGF domain interface and in the structural stability between the Leu33 and Pro33 isoforms. A, shown are the active contacts (left) and non-native contacts (right) formed between the AB loop (PSI domain) and all the side chains located within a distance range of 7 Å. Mean values were computed over three MD simulations of the Leu33 isoform (blue histograms) and Pro33 variant (red histograms). Asterisks denote a significant difference (***, p < 0.0001) between the two isoforms of αIIbβ3. B, difference stability map generated by CNA and averaged over three MD simulations showing the difference in structural stability between both isoforms, focusing on the β3 genu region. The color gradient indicates residues with lower structural stability in the Leu33 (blue) or Pro33 isoform (red). C, enlargements of three areas highlighted within the difference stability map by black boxes (B) and corresponding to the AB loop (PSI domain), residues Ser469–Asp484 (loop connecting the EGF-1 domain to the EGF-2 domain), and residues Gly519–Cys536 (EGF-3 domain), exemplifying changes in structural stability due to the Leu → Pro exchange. The results for the latter two regions are also displayed on the structure of the hybrid (yellow), PSI (green), EGF-1 (firebrick)/EGF-2 (light blue)/EGF-3 (purple) domains of αIIbβ3 (green sphere, Cα atom of residue 33) in terms of lines connecting residues whose mutual stability has decreased in the Pro33 isoform (Δrcij > −1.5 kcal mol−1).
Changes in structural stability of the EGF domains occur at long range from residue 33
To analyze changes in the structural stability of the interface between the PSI domain and EGF-1/EGF-2 domains resulting from the Leu → Pro exchange, we performed Constraint Network Analysis (CNA) on the β3 leg (hybrid domain/PSI and EGF domains) of both αIIbβ3 isoforms, Leu33 and Pro33. In CNA (42), a molecular system is represented as a network of nodes (atoms) connected by constraints (non-covalent bonds). This network is analyzed applying rigidity theory (43), revealing rigid (i.e. structurally stable) clusters and flexible links in between (44). By rigidity analysis, long-range effects on the stability of distant structural parts due to a local structural change can be detected (45). Performing a constraint dilution simulation (46), a stability map (42), rcij (where i and j are residue numbers), is obtained that reports on the hierarchy of structural stability of the molecular system. The difference stability map calculated as rcij (Leu33) − rcij (Pro33) then reports on the influence on structural stability due to the Leu → Pro exchange (blue (red) colors in Fig. 6, B and C, indicate residues that were less stable in the Leu33 (Pro33) isoform, respectively). The AB loop showed a local increase in structural stability, which results from the overconstrained five-membered ring of Pro33 compared with the flexible side chain of Leu33 (48) (Fig. 6, B and C). By contrast, the loop connecting the EGF-1 to the EGF-2 domain and pointing toward the AB loop (21), which is >15 Å apart from residue 33, became less stable in the Pro33 variant (Fig. 6, B and C; the segment formed by residues Ser469–Gln481 is highlighted). The EGF-3 domain, although not directly interacting with the PSI domain, has been suggested to be important for keeping the integrin in its bent conformation (20). Residues Gly519–Cys536 of the EGF-3 domain >30 Å apart from residue 33 became less structurally stable in the Pro33 variant. To conclude, the Leu → Pro exchange leads to long-range decreases in the structural stability of the EGF domains.
Discussion
In this study, we provide evidence that indicates that the Pro33 variant of αIIbβ3 allosterically shifts the dynamic conformational equilibrium of the integrin toward a more active state. This finding can provide an explanation for the prothrombotic phenotype of Pro33 platelets that has been suggested in several clinical association studies (22–25) but also in experimental settings (26, 38, 49).
Both clinical and laboratory data regarding a possible impact of the HPA-1 polymorphism of αIIbβ3 on modulating platelet function have been discussed controversially. Specifically, it has been debated whether or not the Leu → Pro exchange at residue 33 of the β3 subunit induces an increased thrombogenicity of Pro33 platelets. We therefore initially studied the dynamics of platelet thrombus formation using a collagen type I matrix in an established perfusion system, simulating arterial flow conditions. Quantitation of thrombus growth in vitro demonstrated that the mean volume of single thrombi formed by Pro33 platelets is significantly higher than that of the Leu33 platelets (Fig. 2). The initial adhesion of circulating platelets with a collagen matrix is complex, involving platelet capture (“tethering”) by immobilized VWF via GPIbα of the platelet GPIb-IX-V complex, subsequent GPIb-IX-V–dependent signaling, and direct interaction with collagen via α2β1 and GPVI, the platelet collagen receptors, inducing platelet activation (50, 51). To block some of these interactions, we therefore used specific monoclonal antibodies such as LJ-Ib1 that completely inhibits VWF binding to the platelet GPIb-IX-V complex or 5C4 that blocks the platelet GPVI receptor (data not shown). The expression of α2β1 on the platelet surface is genetically controlled and modulated by nucleotide polymorphisms in the α2 gene (52). Moreover, because the α2807TT genotype of α2β1 has also been suggested to be a prothrombotic integrin variant (22), volunteers of this series of experiments were carefully selected by recruiting only carriers of the α2807CC genotype.
A specific feature of the experiments summarized in Fig. 2 is that the difference in single thrombus volumes between Pro33 and Leu33 platelets is due to differences in apical thrombus growth (Fig. 2B). This is remarkable, especially because apical thrombus segments become exposed to increasing shear over time, exceeding an initial near-wall shear rate of 500 s−1 (Fig. 2D). Our finding is indicative of a higher thrombus stability of Pro33 than Leu33 platelets as reported before (29). By contrast, considering the fact that neither the number nor the bottom area of formed thrombi differs between both isoforms of αIIbβ3, it appears rather unlikely that the initial adhesive interactions between the collagen matrix and platelets under flow had a significant effect on the results.
Assuming that the difference in thrombus volumes between both αIIbβ3 isoforms is indeed due to increased thrombus stability in the Pro33 variant, it would be an attractive assumption that the Leu → Pro exchange has an impact on the mechanotransduction mediated by the integrin. Such a contention is in line with previous observations documenting a significantly more stable adhesion of Pro33 than of Leu33 platelets onto immobilized fibrinogen at shear rates ranging from 500 to 1,500 s−1 (26). Moreover, it has been shown that the Pro33 variant displays increased outside-in signaling (53). These findings suggest that the HPA-1 polymorphism of αIIbβ3 modulates the function and activity of the integrin.
However, the molecular nature underlying this modulation has remained elusive so far. In this context, a marked concern in the past has been that the point mutation at residue 33 of the β3 subunit is located >80 Å away from relevant functional domains of αIIbβ3 such as extracellular binding sites or transmembrane domains. Conversely, due to its distant location, it appears quite appropriate to exclude that the Leu → Pro exchange would directly influence interactions with ligands at the extracellular or even intracellular binding sites. It is more likely that an increased activity of αIIbβ3 results from a change in the structural dynamics of the integrin. To probe this assumption, we performed microsecond-long MD simulations on the ectodomains of both αIIbβ3 isoforms, Leu33 and Pro33. The ectodomains of either isoform initially only differed in the side chains of residue 33.
Ectodomains of integrins have been successfully used by us (18, 27) and others (34, 54) in previous studies as model systems to explore possible influences of structure and solvent on integrin activation. For the MD simulations, we used established parameterizations for the solvent (55) and the protein (56, 57), which we had applied successfully in other integrin simulations (18, 27, 37), although we note that more recent protein force fields have become available (58, 59). The impact of force field deficiencies on our results is expected to be small due to cancellation of errors when comparatively assessing the two isoforms. Furthermore, we expect the deficiency of the ff99SB force field (56, 57) to destabilize helical structures (60) to not have a decisive influence on our results because the mutation site (residue 33) is located in a loop region. Finally, ff99SB was also shown to have some issues with side-chain torsions (61). As Leu33 is located at the vertex of the AB loop, with the side chain facing away from the β3 subunit, we do not expect imperfect leucine torsions to impact structural properties markedly however.
The present simulations were started from the bent conformation with closed legs as present in the crystal structure (62), representing a low-affinity, inactive state of the integrin (63). As depicted, our simulation findings reveal that the Pro33 variant displays significantly larger structural deviations from the bent starting structure and becomes less compact than the Leu33 isoform (Fig. 3). Furthermore, we evaluated geometric parameters within the βA domain (“T-junction formation” between helices α1 and α7; Fig. 3B) and variables characterizing the bending and splaying of the structure (Fig. 3, C and D), which had been used successfully in related studies to characterize inactive-to-active transitions (27, 37, 64). The results display significant differences in the conformational states of both isoforms of αIIbβ3 with the ectodomain of the Pro33 variant showing a stronger tendency to move toward an open, extended conformation with more splayed legs than the Leu33 isoform. We performed triplicate MD simulations for both isoforms, which allows probing for the influence of the starting conditions and determining the significance of the computed results by statistical testing and rigorous error estimation. As to the latter, we paid close attention to only consider uncorrelated instances for the S.E. calculations (Equations 1 and 2).
The results are consistent across three independent MD simulations for each isoform. This demonstrates the robustness of our approach. We are aware that the magnitudes of the changes of the bending or splaying angles do not correspond to those described for a fully open, extended ectodomain conformation (8). However, in consideration of the simulation times used here, this finding is in complete accord with the timescale of integrin activation in the absence of biomechanical forces, ranging from microseconds to seconds (34, 35).
As an independent approach to explore the impact of the Leu → Pro exchange on the structural dynamics of full-length αIIbβ3 integrin, FRET measurements on αIIbmVenus and β3Leu33mCherry or β3Pro33-mCherry transiently cotransfected in HEK293 cells were performed (Fig. 4, A–C). HEK293 cells have previously been shown to be a suitable cellular model for functional studies involving αIIbβ3 (65, 66). The transfectants display a significantly higher efficiency of energy transfer between the α and β subunits in the Leu33 than in the Pro33 isoform. This difference is indicative of a smaller spatial separation between the cytoplasmic tails of the Leu33 isoform in its resting state. Conversely, the lower energy transfer obtained in the Pro33 variant reflects a larger spatial separation of its cytoplasmic domains that is already present in the resting state (Fig. 4D). A limitation of the FRET method is that it furnishes indirect information. However, the level of evidence is consistent, and the observation is in good agreement with the findings of the MD simulations. A direct study comparing activity and stability of both receptor isoforms using purified protein would provide complementary information about receptor conformations but was beyond the scope of the present work.
Taken together, both the MD simulations and FRET experiments reveal structural changes in the ectodomain of αIIbβ3 or the full-length integrin for the Pro33 variant that relate to a conformational change from a closed, bent structural state with closed legs to a more open, extended state with splayed legs. According to current models (14, 16, 33), such a conformational change is required for integrin activation. Considering that in both the MD simulations and FRET measurements the integrin has been examined in the resting state, our results provide evidence that the Leu → Pro exchange can shift the dynamic conformational equilibrium of αIIbβ3 in such a way that a structural state more similar to the active conformation is present.
The effect of the Leu → Pro exchange appears to have some similarity to stimulatory monoclonal antibodies, which have been suggested to shift the dynamic conformational equilibrium in favor of those forms that lead to an increase in the proportion of a high-affinity integrin (62). As the effect induced by the amino acid substitution manifests in regions far away from the mutation site, the influence of the Leu → Pro exchange must be allosteric. Our results clearly go beyond a previous study (67) that used MD simulations of the β3 subunit only to investigate possible effects of the HPA-1 polymorphism on the structure of the β3 subunit.
To explore a possible mechanism of how the Leu → Pro exchange exerts an allosteric effect, applying MD simulations and rigidity analyses, we examined short- and mid-range structural, dynamics, and stability changes in the PSI domain and neighboring domains. Although the overall architecture of the PSI domain remains largely unchanged by the amino acid substitution, particularly the EGF domains show marked structural alterations in the Pro33 variant (Fig. 5). The EGF-1 and EGF-2 domains, although sequentially distant from the mutation located at residue 33, are spatially close to the AB loop of the PSI domain in the bent state, which carries the HPA-1 polymorphism (5, 19, 21). Parts of the AB loop are markedly more mobile in the Pro33 variant (Fig. 5B). Related to these changes, our analyses reveal that the Leu → Pro exchange leads to a less compact interface between the PSI domain and EGF-1/EGF-2 domains (Fig. 5, C and D). Specifically, fewer native and non-native contacts are formed across the interface and within the EGF-1/EGF-2 domains in the Pro33 variant than in the Leu33 isoform (Fig. 6A). These conformational and dynamic alterations are related to a change in the structural stability of the EGF-1 and EGF-2 domains that percolates from the interface region through these domains (Fig. 6, B and C).
Notably, similar changes in these regions have been related to integrin activation before. For example, the displacement of the PSI domain of about 70 Å, described to act as a mechanical lever upon outside-in signaling (21), alters the interface formed with the EGF-1 and EGF-2 domains (68). Furthermore, activating mutations have been identified in the N-terminal part of the PSI domain, the EGF-2 domain, and parts of the EGF-3 domain of αxβ2 integrin (20). These regions are thought to form the area of contact between α and β subunits in the bent conformation. Finally, when generating an integrin chimera by combining α and β subunits from different species, direct interactions between the subunits could not be formed, and the integrin no longer appeared locked in the closed conformation (20).
The results of this study provide an explanation for the prothrombotic phenotype of the Pro33 variant of αIIbβ3. Specifically, the shift of the dynamic conformational equilibrium of the Pro33 variant toward an active state may promote a fully active state in the presence of immobilized adhesive ligands such as fibrinogen or VWF and, consequently, favor outside-in signaling. This, in turn, may facilitate and accelerate platelet aggregation and subsequent formation of stable platelet thrombi. Thus, our results lend support to previous clinical (22–25, 29, 49) and experimental findings (26, 29, 38, 49) suggesting that the Leu → Pro exchange confers prothrombotic properties to αIIbβ3.
Experimental procedures
Blood collection
Blood was collected through a 21-gauge needle from 15 healthy, medication-free volunteers into vacutainer tubes (BD Biosciences) containing sodium citrate (0.38%, w/v). The volunteers were recruited by the Düsseldorf University Blood Donation Center. Written informed consent was obtained from the volunteers according to the Helsinki Declaration. The Ethics Committee of the Faculty of Medicine, Heinrich Heine University Düsseldorf, approved the study (study number 1864).
Parallel plate flow chamber
A custom-made rectangle flow chamber was used (flow-channel width, 5 mm; height, 80 μm; length, 40 mm). Glass coverslips forming the lower surface of the chamber were flame-treated, cooled, and coated with 0.04 μml/mm2 collagen type S (concentration, 3 mg/ml) containing 95% type I and 5% type III collagen (Roche). The perfusion system was flushed and filled with PBS buffer (pH 7.3) containing 2% BSA to block unspecific adhesion onto the glass slides. A syringe pump (Harvard Apparatus Inc., Holliston, MA) was used to aspirate mepacrine-labeled citrated whole blood through the flow chamber for 10 min at a constant flow rate of 9.6 ml h−1, producing an initial near-wall shear rate of 500 s−1.
Labeling of platelets
Platelets were stained in whole blood by direct incubation with the fluorescent dye mepacrine (quinacrine dihydrochloride; 10 μm final concentration). Although this dye also labels leukocytes, these cells could be readily distinguished from platelets by their relatively large size and sparsity; moreover, leukocyte attachment to the surface tested was negligible under the conditions used. Mepacrine accumulates in the dense granules of platelets and had no effect on normal platelet function at the concentration used. Platelet secretion after adhesion did not prevent their visualization. Furthermore, mepacrine did not affect platelet adhesion or platelet aggregate/thrombus formation.
Picture acquisition and digital image processing
The fluorescence signal of mepacrine-stained platelets was detected by a Zeiss Axiovert 100 M/LSM 510 confocal laser scanning microscope (Jena, Germany). During the flow period of 10 min, 25 stacks of images were acquired. One stack consisted of 30 slices with a height of 30 μm. Digitized images had a standard size of 512 × 512 pixels and an optical resolution of 1 μm.
Volumetry of single platelet thrombi
The stacks were reconstructed three-dimensionally and analyzed with the custom-made software package ECCET (www.eccet.de).5 The software integrated the slices of every stack and divided the three-dimensional space into multiple “voxels” (three-dimensional equivalent to a pixel). All fluorescence signals were smoothed by a separate linear Gaussian filter in all three planes (filter σ2). Voxels with a gray value >10 were marked as thrombus; voxels with lower gray values were disregarded. Thus, background noise of fluorescence signals from adjacent focus planes and single platelets was suppressed. Thrombi were then categorized by volume, and only platelet aggregates exceeding the cutoff volume of 100 μm3 were assessed to avoid interference by non-stationary objects, e.g. moving platelets.
Starting structures for molecular dynamics simulations
The starting structure for MD simulations of αIIbβ3 in the bent, closed form representing the inactive state of the Leu33 isoform was obtained from the coordinates of the X-ray structure of the ectodomain of αIIbβ3 integrin (Protein Data Bank code 3FCS) (9). In the Protein Data Bank entry, the αIIb subunit contains two unresolved regions within the calf-2 domain (residues 764–774 (AB loop) and 840–873 (XY loop)), and the β3 subunit has two unresolved regions within the EGF domains (residues 75–78 and 477–482). Residues unresolved in the αIIb subunit were not included in the starting structures, consistent with our previous studies on integrin (18, 27). The apparently high flexibility of these residues implies that they will not contribute significantly to stabilizing the bent conformation of the αIIbβ3 integrin. The short regions of unresolved residues of the β3 subunit were modeled and refined using the automatic loop refinement server ModLoop (69). The structure was finally refined by reverting the engineered residues Cys598 and Cys688 to the natural residues Leu598 and Pro688, respectively. MODELLER version 9.9 (70) was applied, allowing the modeling of the two Cys residues only. The Pro33 variant was obtained by mutating residue Leu33 to Pro33, using Swiss-PdbViewer (31), without changing the coordinates of any of the other amino acids. As a final step, we capped the charges at the N-terminal residues Glu764 and Gly840 and the C-terminal residues Asp774 and Gln873 using the leap module of Amber 12 (71). All structural ions present in the protein were modeled as Mg2+ ions. Integrin sequence numbers used throughout this study are according to UniProt.
Molecular dynamics simulations
Each starting structure of the two HPA-1 isoforms, Leu33 and Pro33, was subjected to three replicates of all-atom MD simulations of 1-μs length each in explicit solvent summing up to 6 μs of aggregate simulation time for production. MD simulations were performed with the Amber 12 suite of programs (71) using the force field ff99SB, initially described by Cornell et al. (56) and modified according to Simmerling and co-workers (57). Parameters for the Mg2+ ions were taken from Aqvist (72). The total charge of the system was neutralized by adding eight Na+ counter-ions with the leap module of Amber 12 (71), and the solutes were placed into an octahedral period box of TIP3P water molecules (55). The distance between the edges of the water box and the closest atom of the protein was at least 11 Å, resulting in systems of ∼200,000 atoms. The particle mesh Ewald method (73) was used to treat long-range electrostatic interactions, and bond lengths involving bonds to hydrogen atoms were constrained using the SHAKE algorithm (74). The time step for integrating Newton's equations of motion was 2 fs with a direct-space, non-bonded cutoff of 8 Å. Applying harmonic restraints with force constants of 5 kcal mol−1 Å−2 to all solute atoms, MD simulations in the NVT (constant number of particles, volume, and temperature) ensemble was carried out for 50 ps, during which the system was heated from 100 to 300 K. Subsequent MD simulations in the NPT (constant number of particles, pressure, and temperature) ensemble were used for 150 ps to adjust the solvent density. Finally, the force constant of the harmonic restraints on solute atom positions was gradually reduced to zero during 100 ps of NVT MD simulations. Subsequently, we performed a 1-μs unrestrained simulation; the first 200 ns were discarded, and the following 800 ns were used for analysis with the programs ptraj/cpptraj (75) with conformations extracted every 20 ps. The production MD simulations were performed with the graphics processing unit (GPU) version of the program pmemd (76).
Analysis of the trajectories
For the analysis of the trajectories, ptraj/cpptraj (75) of the AmberTools suite of programs (71) were applied. For investigating structural deviations along the MD trajectories, the RMSD of all Cα atoms was computed after minimizing the mass-weighted RMSD of the Cα atoms of the βA and β-propeller domains with respect to the starting structure. In addition, to investigate the structural changes of a domain, the Cα atom RMSD of each domain was computed after superimposing the respective domain. To evaluate the level of compactness of the structure, the Rg was calculated with respect to the complete ectodomain. To examine atomic mobility, RMSFs were computed for the backbone atoms of the PSI domain. An analysis of the secondary structure of the PSI domain was carried out to monitor variations in the content of the two helices bordering the AB loop. Structural changes in the ectodomain were characterized as reported previously (18, 27). The kinking of the helix α1 was measured by the three points (center of mass of Cα atoms of Lys112 and Ile118, center of mass of Cα atoms of Gln119 and Lys125, and center of mass of Cα atoms of Leu126 and Leu132). The unbending of the structure was evaluated using the angle formed by the centers of mass of the β-propeller, βA, and PSI domains, and the splaying of the two legs was evaluated using the angle formed by the centers of mass of the calf-2, thigh, and β-tail domains. Changes in the β3 genu region were first quantified by computing the distances between the Cα atom of residue 33 and the Cα atom of Ser469 (EGF-1 domain) and with the Cα atom of Gln481 (EGF-2 domain). To identify a network of interactions keeping the interdomain interface stable, a maximal distance of 3.5 Å and a minimal angle of 120° were used as exclusion criteria to identify hydrogen bond formation.
The CNA software package was used to provide a link between structure and rigidity/flexibility of the HPA-1 isoforms (77). To derive information of the effect of Pro33 on a local level, we first generated an ensemble of 400 equally distributed structures from the 200–1,000-ns intervals of each MD simulation, considering only the hybrid, PSI, and EGF block domains. Thermal unfolding simulations of the Leu33 and Pro33 isoforms were then carried out with CNA to identify differences in the structural stability within the β3 genu region following established protocols (77). For each isoform, we generated three different stability maps and three different neighbor stability maps; from them we calculated the mean values used to build a final stability map and neighbor stability map for Leu33 and Pro33. Finally, a difference stability map was calculated as rcij (Leu33 isoform) − rcij (Pro33 isoform).
Statistical analysis
Results from three independent MD simulations are expressed as arithmetic means ± S.E. calculated over the time. The overall S.E. for each simulated system was calculated according to the law of error propagation (Equation 1),
| (1) |
where the subscripts i = {1, 2, 3} indicate the three trajectories. S.E.i was computed following Ref. 78 and applying the multiple Bennett acceptance ratio method (79), which allows detecting the decorrelation time of an investigated variable along each MD simulation. From it, the effective sample size is established, and the S.E.i is derived.
In the case of hydrogen bond and contact analyses, S.E. is calculated from the S.D. of the three means of the three MD simulations according to Equation 2, assuming that the three MD simulations are statistically independent.
| (2) |
Differences between mean values are considered statistically significant if p < 0.05 and p < 0.001 (indicated as * and **, respectively, in figures and tables) and highly statistically significant if p < 0.0001 (indicated as ***) according to the Student's t test for parametric testing. The statistical analysis was performed using R software (80) and the pymbar module for multiple Bennett acceptance ratio (79).
The FRET efficiency results obtained performing the FRET-AB experiments are expressed as means ± S.E. For statistical analysis, the unpaired t test was applied using GraphPad Prism version 6.00 for Windows (GraphPad Software, La Jolla, CA).
Figure preparation
The crystal structure of the αIIbβ3 integrin (Protein Data Bank code 3FCS) was used to represent the protein together with conformations extracted from the MD trajectories. PyMOL was used to generate molecular figures (36), and graphs were prepared using Gnuplot (47).
Live-cell imaging of αIIbβ3-transfected HEK 293 cells expressing either isoform Leu33 or Pro33
Live-cell imaging was performed to examine the cellular distribution of αIIbβ3-transfected HEK 293 cells expressing either isoform Leu33 or Pro33. 24 h after transfection, 3.7 × 104 cells in complete culture medium were allowed to settle for more 24 h in individual chambers in a μ-slide 4-well ibiTreat chamber slide (Ibidi, Martinsried, Germany) previously coated with 50 μg/ml fibrinogen from human plasma (Sigma-Aldrich) in PBS without Ca2+ and Mg2+ for 1 h at 37 °C. Live-cell imaging was performed with an Axiovert S100 inverted fluorescence microscope (Zeiss) equipped with a 12.0 Monochrome without IR-18 monochromatic camera (Diagnostic Instruments, Inc, Sterling Heights, MI) and an LEJ EBQ 100 isolated lamp (Leistungselektronik Jena GmbH, Jena, Germany). Images were obtained with a 63× oil immersion objective lens using a 5,000-ms exposure time for mVenus, 100 ms for mCherry, and 300 ms for brightfield. Image acquisition was performed with MetaMorph software (version 7.7.7.0). Background subtraction and image processing were performed using Adobe Photoshop CS3 software (Adobe, San Jose, CA).
Flow cytometry
Transfected cells at 70–80% confluence were harvested 24, 48, and 72 h after transfection. Subsequently, cells were pelleted by centrifugation at 400 × g for 7 min and suspended again in 100 μl of Dulbecco's phosphate-buffered saline (PBS). Staining with allophycocyanin (APC)-conjugated mouse anti-human CD41 monoclonal antibody (clone MEM-06; Exbio, Praha, Czech Republic; 0.15 μg/ml) was performed for 30 min at room temperature protected from light. After staining, cells were washed once in Dulbecco's PBS and analyzed on a FACS Canto II flow cytometer (BD Biosciences) equipped with 488 and 633 nm lasers for excitation and FITC, phycoerythrin, and APC filters for detection of mVenus, mCherry, and APC, respectively. The collected data were analyzed with FACSDiva software version 6.1.3 (BD Biosciences). PAC1 was obtained from BD Biosciences, and Alexa Fluor 647-fibrinogen was from Thermo Fisher Scientific (Dreieich, Germany).
FRET measurements using APB
24 h after transfection, cells were harvested and seeded in a μ-slide 8-well ibiTreat chamber slide (Ibidi). Subsequently, 24 h later (48 h after transfection) and before measuring FRET efficiency, the culture medium was substituted by identical medium but containing phenol red–free FluorobriteTM DMEM (Thermo Fisher, formerly Life Technologies).
Live cells were examined with an LSM 780 (Zeiss) inverted microscope equipped with a C-Apochromat 40×/1.20 W Corr (from correction ring) M27 water-immersion objective lens, an AxioCam camera, and an HPX 120C lamp. FRET acceptor photobleaching experiments including image acquisition, definition of regions of interest for bleaching, and data readout were performed using the LSM software package ZEN 2012 (Zeiss). The chamber slide containing the live cells was mounted on a heating frame within a large incubation chamber (PeCon, Erbach, Germany) set to 37 °C. mVenus was excited with the 488 nm line of an argon multiline laser and detected between 513 and 558 nm using a gallium arsenide phosphide detector, whereas mCherry was excited at 561 nm using a diode-pumped solid-state laser and detected between 599 and 696 nm. The beam splitter was MBS 488/561/633. In total, a time series of 20 frames (128 × 128 pixels; pixel size, 0.33 μm) at a pixel time of 2 μs/pixel was acquired for each FRET experiment. The entire measurement including bleaching of mCherry was finished within 3.5 s. After the fifth frame, an area corresponding to half of a cell, with a constant dimension of 42 × 42 pixels (region of interest), was bleached by 30 iterations of the mCherry excitation wavelength (561 nm) using 100% laser power. After bleaching, 15 additional frames were recorded. The mean intensity of mVenus fluorescence at the cell membrane within the bleached area was extracted and analyzed according to Equation 3,
| (3) |
where Ibefore (intensity of mVenus before bleaching) and Iafter (intensity of mVenus after bleaching) correspond to the mean intensity values of mVenus fluorescence of five images before and after bleaching within the bleached area at the cell membrane (39, 40).
Author contributions
G. P. and J. P. P. data curation; G. P., J. P. P., V. R. S., R. E. S., and H. G. formal analysis; G. P., J. P. P., and V. R. S. validation; G. P. and J. P. P. investigation; G. P. and J. P. P. visualization; G. P., J. P. P., and V. R. S. writing-original draft; G. P., J. P. P., R. E. S., and H. G. writing-review and editing; V. R. S., R. E. S., and H. G. supervision; A. B. software; A. B. methodology; R. E. S. and H. G. conceptualization; R. E. S. and H. G. funding acquisition; R. E. S. and H. G. project administration; H. G. resources.
Supplementary Material
Acknowledgments
We are grateful for computational support and infrastructure provided by the Zentrum für Informations und Medientechnologie (ZIM) at the Heinrich Heine University Düsseldorf and the computing time provided by the John von Neumann Institute for Computing (NIC) to H. G. on the supercomputers JUROPA and JURECA at Jülich Supercomputing Center (JSC) (Projects 6710, 8348, and 10307; user, HDD11). We thank Dr. Rainer B. Zotz and Beate Maruhn-Dembowski for genotyping HPA-1 of αIIbβ3 and α2C807T of α2β1. The assistance of Bianca Maassen-Weingart and Jan Peveling-Oberhag in the platelet experiments is also gratefully acknowledged. The FRET measurements were performed at the Center of Advanced Imaging at Heinrich Heine University Düsseldorf. We thank Dr. Stefanie Weidtkamp-Peters for scientific assistance.
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 612, Project B2) (to R. E. S. and V. R. S.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supporting methods, Figs. S1–S8, and Tables S1–S10.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party-hosted site.
- PSI
- plexin-semaphorin-integrin
- APB
- acceptor photobleaching
- APC
- allophycocyanin
- CAD
- coronary artery disease
- CNA
- Constraint Network Analysis
- GP
- glycoprotein
- HPA-1
- human platelet antigen-1
- MD
- molecular dynamics
- NPT
- constant number of particles, pressure, and temperature
- NVT
- constant number of particles, volume, and temperature
- RMSD
- root-mean-square deviation
- RMSF
- root-mean-square fluctuation
- Rg
- radius of gyration
- VWF
- von Willebrand factor
- PC
- principal component
- SyMBS
- synergistic metal-binding site
- MIDAS
- metal ion–dependent adhesion site
- ADMIDAS
- adjacent to MIDAS.
References
- 1. Hynes R. O. (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 10.1016/S0092-8674(02)00971-6 [DOI] [PubMed] [Google Scholar]
- 2. Coppolino M. G., and Dedhar S. (2000) Bi-directional signal transduction by integrin receptors. J. Biochem. Cell Biol. 32, 171–188 [DOI] [PubMed] [Google Scholar]
- 3. Mould A. P. (1996) Getting integrins into shape: recent insights into how integrin activity is regulated by conformational changes. J. Cell Sci. 109, 2613–2618 [DOI] [PubMed] [Google Scholar]
- 4. Arnaout M. A. (2002) Integrin structure: new twists and turns in dynamic cell adhesion. Immunol. Rev. 186, 125–140 10.1034/j.1600-065X.2002.18612.x [DOI] [PubMed] [Google Scholar]
- 5. Arnaout M. A., Goodman S. L., and Xiong J. P. (2007) Structure and mechanics of integrin-based cell adhesion. Curr. Opin. Cell Biol. 19, 495–507 10.1016/j.ceb.2007.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Luo B. H., Carman C. V., and Springer T. A. (2007) Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25, 619–647 10.1146/annurev.immunol.25.022106.141618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Askari J. A., Buckley P. A., Mould A. P., and Humphries M. J. (2009) Linking integrin conformation to function. J. Cell Sci. 122, 165–170 10.1242/jcs.018556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bennett J. S., Berger B. W., and Billings P. C. (2009) The structure and function of platelet integrins. J. Thromb. Haemost. 7, Suppl. 1, 200–205 10.1111/j.1538-7836.2009.03378.x [DOI] [PubMed] [Google Scholar]
- 9. Zhu J., Luo B. H., Xiao T., Zhang C., Nishida N., and Springer T. A. (2008) Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol. Cell 32, 849–861 10.1016/j.molcel.2008.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Springer T. A., and Dustin M. L. (2012) Integrin inside-out signaling and the immunological synapse. Curr. Opin. Cell Biol. 24, 107–115 10.1016/j.ceb.2011.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bledzka K., Smyth S. S., and Plow E. F. (2013) Integrin αIIbβ3: from discovery to efficacious therapeutic target. Circ. Res. 112, 1189–1200 10.1161/CIRCRESAHA.112.300570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Luo B. H., and Springer T. A. (2006) Integrin structures and conformational signaling. Curr. Opin. Cell Biol. 18, 579–586 10.1016/j.ceb.2006.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arnaout M. A., Mahalingam B., and Xiong J. P. (2005) Integrin structure, allostery, and bidirectional signaling. Annu. Rev. Cell Dev. Biol. 21, 381–410 10.1146/annurev.cellbio.21.090704.151217 [DOI] [PubMed] [Google Scholar]
- 14. Xiong J. P., Stehle T., Diefenbach B., Zhang R., Dunker R., Scott D. L., Joachimiak A., Goodman S. L., and Arnaout M. A. (2001) Crystal structure of the extracellular segment of integrin αVβ3. Science 294, 339–345 10.1126/science.1064535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xie C., Zhu J., Chen X., Mi L., Nishida N., and Springer T. A. (2010) Structure of an integrin with an αI domain, complement receptor type 4. EMBO J. 29, 666–679 10.1038/emboj.2009.367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takagi J., Petre B. M., Walz T., and Springer T. A. (2002) Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 110, 599–611 10.1016/S0092-8674(02)00935-2 [DOI] [PubMed] [Google Scholar]
- 17. Beglova N., Blacklow S. C., Takagi J., and Springer T. A. (2002) Cysteine-rich module structure reveals a fulcrum for integrin rearrangement upon activation. Nat. Struct. Biol. 9, 282–287 10.1038/nsb779 [DOI] [PubMed] [Google Scholar]
- 18. Reinehr R., Gohlke H., Sommerfeld A., Vom Dahl S., and Häussinger D. (2010) Activation of integrins by urea in perfused rat liver. J. Biol. Chem. 285, 29348–29356 10.1074/jbc.M110.155135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xiong J. P., Stehle T., Goodman S. L., and Arnaout M. A. (2004) A novel adaptation of the integrin PSI domain revealed from its crystal structure. J. Biol. Chem. 279, 40252–40254 10.1074/jbc.C400362200 [DOI] [PubMed] [Google Scholar]
- 20. Zang Q., and Springer T. A. (2001) Amino acid residues in the PSI domain and cysteine-rich repeats of the integrin β2 subunit that restrain activation of the integrin αXβ2. J. Biol. Chem. 276, 6922–6929 10.1074/jbc.M005868200 [DOI] [PubMed] [Google Scholar]
- 21. Xiao T., Takagi J., Coller B. S., Wang J. H., and Springer T. A. (2004) Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature 432, 59–67 10.1038/nature02976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zotz R. B., Winkelmann B. R., Müller C., Boehm B. O., März W., and Scharf R. E. (2005) Association of polymorphisms of platelet membrane integrins αIIbβ3 (HPA-1b/Pl) and α2β1 (α807TT) with premature myocardial infarction. J. Thromb. Haemost. 3, 1522–1529 10.1111/j.1538-7836.2005.01432.x [DOI] [PubMed] [Google Scholar]
- 23. Zotz R. B., Winkelmann B. R., Nauck M., Giers G., Maruhn-Debowski B., März W., and Scharf R. E. (1998) Polymorphism of platelet membrane glycoprotein IIIa: human platelet antigen 1b (HPA-1b/PIA2) is an inherited risk factor for premature myocardial infarction in coronary artery disease. Thromb. Haemost. 79, 731–735 [PubMed] [Google Scholar]
- 24. Weiss E. J., Bray P. F., Tayback M., Schulman S. P., Kickler T. S., Becker L. C., Weiss J. L., Gerstenblith G., and Goldschmidt-Clermont P. J. (1996) A polymorphism of a platelet glycoprotein receptor as an inherited risk factor for coronary thrombosis. N. Engl. J. Med. 334, 1090–1094 10.1056/NEJM199604253341703 [DOI] [PubMed] [Google Scholar]
- 25. Zotz R. B., Klein M., Dauben H. P., Moser C., Gams E., and Scharf R. E. (2000) Prospective analysis after coronary-artery bypass grafting: platelet GP IIIa polymorphism (HPA-1b/PIA2) is a risk factor for bypass occlusion, myocardial infarction, and death. Thromb. Haemost. 83, 404–407 [PubMed] [Google Scholar]
- 26. Loncar R., Stoldt V., Hellmig S., Zotz R. B., Mihalj M., and Scharf R. E. (2007) HPA-1 polymorphism of αIIbβ3 modulates platelet adhesion onto immobilized fibrinogen in an in-vitro flow system. Thromb. J. 10.1186/1477-9560-1185-1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gohlke H., Schmitz B., Sommerfeld A., Reinehr R., and Häussinger D. (2013) α5β1-integrins are sensors for tauroursodeoxycholic acid in hepatocytes. Hepatology 57, 1117–1129 10.1002/hep.25992 [DOI] [PubMed] [Google Scholar]
- 28. Aurich V., and Beck A. (2002) ECCET: Ein System zur 3D-Visualisierung von Volumendaten mit Echtzeitnavigation, in Bildverarbeitung für die Medizin 2002 (Meiler M., Saupe D., Kruggel F., Handels H., and Lehmann T. M., eds) pp. 389–392, Springer, Berlin [Google Scholar]
- 29. Scharf R. E., Hasse M., Reiff E., Gyenes M., and Stoldt V. R. (2009) CD40 ligand (CD40L) increases thrombus stability and outside-in signaling through integrin αIIbβ3. In Abstracts of the XXII Congress of the International Society of Thrombosis and Haemostasis, Boston, MA, July 11–16, 2009, Suppl. 2, AS-TH-060 10.1111/j.1538-7836.2009.03473_1.x [DOI] [Google Scholar]
- 30. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guex N., and Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723 10.1002/elps.1150181505 [DOI] [PubMed] [Google Scholar]
- 32. Galindo-Murillo R., Roe D. R., and Cheatham T. E. 3rd. (2015) Convergence and reproducibility in molecular dynamics simulations of the DNA duplex d(GCACGAACGAACGAACGC). Biochim. Biophys. Acta 1850, 1041–1058 10.1016/j.bbagen.2014.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li J., Su Y., Xia W., Qin Y., Humphries M. J., Vestweber D., Cabañas C., Lu C., and Springer T. A. (2017) Conformational equilibria and intrinsic affinities define integrin activation. EMBO J. 36, 629–645 10.15252/embj.201695803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen W., Lou J., Hsin J., Schulten K., Harvey S. C., and Zhu C. (2011) Molecular dynamics simulations of forced unbending of integrin αVβ3. PLoS Comput. Biol. 7, e1001086 10.1371/journal.pcbi.1001086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Puklin-Faucher E., and Sheetz M. P. (2009) The mechanical integrin cycle. J. Cell Sci. 122, 179–186 10.1242/jcs.042127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.5.0.1, Schroedinger, LLC, New York [Google Scholar]
- 37. Donner L., Fälker K., Gremer L., Klinker S., Pagani G., Ljungberg L. U., Lothmann K., Rizzi F., Schaller M., Gohlke H., Willbold D., Grenegard M., and Elvers M. (2016) Platelets contribute to amyloid-β aggregation in cerebral vessels through integrin αIIbβ3-induced outside-in signaling and clusterin release. Sci. Signal. 9, ra52 10.1126/scisignal.aaf6240 [DOI] [PubMed] [Google Scholar]
- 38. El Khattouti A., Stoldt R. V., and Scharf R. E. (2010) The HPA-1b (Pro33) isoform of platelet integrin αIIbβ3 is a prothrombotic variant: characterization by fluorescence resonance energy transfer. Blood 116, 2028 [Google Scholar]
- 39. Bleckmann A., Weidtkamp-Peters S., Seidel C. A., and Simon R. (2010) Stem cell signaling in Arabidopsis requires CRN to localize CLV2 to the plasma membrane. Plant Physiol. 152, 166–176 10.1104/pp.109.149930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karpova T. S., Baumann C. T., He L., Wu X., Grammer A., Lipsky P., Hager G. L., and McNally J. G. (2003) Fluorescence resonance energy transfer from cyan to yellow fluorescent protein detected by acceptor photobleaching using confocal microscopy and a single laser. J. Microsc. 209, 56–70 10.1046/j.1365-2818.2003.01100.x [DOI] [PubMed] [Google Scholar]
- 41. Shi M., Foo S. Y., Tan S. M., Mitchell E. P., Law S. K., and Lescar J. (2007) A structural hypothesis for the transition between bent and extended conformations of the leukocyte β2 integrins. J. Biol. Chem. 282, 30198–30206 10.1074/jbc.M701670200 [DOI] [PubMed] [Google Scholar]
- 42. Pfleger C., Radestock S., Schmidt E., and Gohlke H. (2013) Global and local indices for characterizing biomolecular flexibility and rigidity. J. Comput. Chem. 34, 220–233 10.1002/jcc.23122 [DOI] [PubMed] [Google Scholar]
- 43. Jacobs D. J., and Thorpe M. F. (1995) Generic rigidity percolation: the pebble game. Phys. Rev. Lett. 75, 4051–4054 10.1103/PhysRevLett.75.4051 [DOI] [PubMed] [Google Scholar]
- 44. Hermans M. A., Pfleger C., Nutschel C., Hanke C. A., and Gohlke H. (2017) Rigidity theory for biomolecules: concepts, software, and applications. Wiley Interdiscip. Rev. Comput. Mol. Sci. 7, e1311 10.1002/wcms.1311 [DOI] [Google Scholar]
- 45. Thorpe M., Jacobs D., and Djordjevic B. (1996) Generic rigidity percolation. J. Phys. Condens. Matter 11, 407–424 [Google Scholar]
- 46. Radestock S., and Gohlke H. (2008) Constraint network analysis: exploiting the link between protein rigidity and thermostability. Eng. Life Sci. 8, 507–522 10.1002/elsc.200800043 [DOI] [Google Scholar]
- 47. Williams T., and Kelley C. (2011) Gnuplot 4.5: an Interactive Plotting Program, SourceForge, La Jolla, CA [Google Scholar]
- 48. Whiteley W. (2005) Counting out to the flexibility of molecules. Phys. Biol. 2, S116–S126 10.1088/1478-3975/2/4/S06 [DOI] [PubMed] [Google Scholar]
- 49. Stoldt V. R., Peveling J., Loncar R., Beck A., Aurich V., and Scharf R. E. (2005) Evaluation of platelet thrombus formation under flow. Blood 106, 3964 [Google Scholar]
- 50. Savage B., and Ruggeri Z. M. (2013) The basis for platelet adhesion, in Hemostasis and Thrombosis. Basic Principles and Clinical Practice (Marder V. J., Aird W., Bennett J. S., Schulman S., and White G. C., eds) 6th Ed., pp. 400–409, Lippincott Williams & Wilkins, Philadelphia [Google Scholar]
- 51. Watson S. P., Farndale R. W., Moroi M., and Jung S. M. (2013) Platelet collagen receptors, in Haemostasis and Thrombosis. Basic Principles and Clinical Practice (Marder V. J., Aird W., Bennett J. S., Schulman S., and White G. C., eds) 6th Ed., pp. 420–430, Lippincott Williams & Wilkins, Philadelphia [Google Scholar]
- 52. Kritzik M., Savage B., Nugent D. J., Santoso S., Ruggeri Z. M., and Kunicki T. J. (1998) Nucleotide polymorphisms in the α2 gene define multiple alleles that are associated with differences in platelet α2β1 density. Blood 92, 2382–2388 [PubMed] [Google Scholar]
- 53. Vijayan K. V., Liu Y., Sun W., Ito M., and Bray P. F. (2005) The Pro33 isoform of integrin β3 enhances outside-in signaling in human platelets by regulating the activation of serine/threonine phosphatases. J. Biol. Chem. 280, 21756–21762 10.1074/jbc.M500872200 [DOI] [PubMed] [Google Scholar]
- 54. Puklin-Faucher E., Gao M., Schulten K., and Vogel V. (2006) How the headpiece hinge angle is opened: new insights into the dynamics of integrin activation. J. Cell Biol. 175, 349–360 10.1083/jcb.200602071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 10.1063/1.445869 [DOI] [Google Scholar]
- 56. Cornell W. D., Cieplak P., Bayly C. I., Gould I. R., Merz K. M., Ferguson D. M., Spellmeyer D. C., Fox T., Caldwell J. W., and Kollman P. A. (1995) A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J. Am. Chem. Soc. 117, 5179–5197 10.1021/ja00124a002 [DOI] [Google Scholar]
- 57. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., and Simmerling C. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 10.1002/prot.21123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Perez A., MacCallum J. L., Brini E., Simmerling C., and Dill K. A. (2015) Grid-based backbone correction to the ff12SB protein force field for implicit-solvent simulations. J. Chem. Theory. Comput. 11, 4770–4779 10.1021/acs.jctc.5b00662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., and Simmerling C. (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Best R. B., and Hummer G. (2009) Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J. Phys. Chem. B 113, 9004–9015 10.1021/jp901540t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J. L., Dror R. O., and Shaw D. E. (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 10.1002/prot.22711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Campbell I. D., and Humphries M. J. (2011) Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 3, a004994 10.1101/cshperspect.a004994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Xiong J. P., Stehle T., Goodman S. L., and Arnaout M. A. (2003) New insights into the structural basis of integrin activation. Blood 102, 1155–1159 10.1182/blood-2003-01-0334 [DOI] [PubMed] [Google Scholar]
- 64. Puklin-Faucher E., and Vogel V. (2009) Integrin activation dynamics between the RGD-binding site and the headpiece hinge. J. Biol. Chem. 284, 36557–36568 10.1074/jbc.M109.041194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vijayan K. V., Goldschmidt-Clermont P. J., Roos C., and Bray P. F. (2000) The Pl(A2) polymorphism of integrin β3 enhances outside-in signaling and adhesive functions. J. Clin. Investig. 105, 793–802 10.1172/JCI6982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Abraham D. G., Nutt E. M., Bednar R. A., Bednar B., Gould R. J., and Duong L. T. (1997) Arginine-glycine-aspartic acid mimics can identify a transitional activation state of recombinant αIIbβ3 in human embryonic kidney 293 cells. Mol. Pharmacol. 52, 227–236 10.1124/mol.52.2.227 [DOI] [PubMed] [Google Scholar]
- 67. Jallu V., Poulain P., Fuchs P. F., Kaplan C., and de Brevern A. G. (2012) Modeling and molecular dynamics of HPA-1a and -1b polymorphisms: effects on the structure of the β3 subunit of the αIIbβ3 integrin. PLoS One 7, e47304 10.1371/journal.pone.0047304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bennett J. S. (2005) Structure and function of the platelet integrin αIIbβ3. J. Clin. Investig. 115, 3363–3369 10.1172/JCI26989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fiser A., and Sali A. (2003) ModLoop: automated modeling of loops in protein structures. Bioinformatics 19, 2500–2501 10.1093/bioinformatics/btg362 [DOI] [PubMed] [Google Scholar]
- 70. Webb B., and Sali A. (2014) Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 47, 5.6.1–32 10.1002/0471250953.bi0506s47 [DOI] [PubMed] [Google Scholar]
- 71. Case D. A., Cheatham T. E. 3rd, Darden T., Gohlke H., Luo R., Merz K. M. Jr., Onufriev A., Simmerling C., Wang B., and Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 10.1002/jcc.20290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Aqvist J. (1992) Modeling of ion ligand interactions in solutions and biomolecules. J. Mol. Struct. 88, 135–152 [Google Scholar]
- 73. Cheatham T. E., Miller J. L., Fox T., Darden T. A., and Kollman P. A. (1995) Molecular dynamics simulations on solvated biomolecular systems: the particle mesh Ewald method leads to stable trajectories of DNA, RNA, and proteins. J. Am. Chem. Soc. 117, 4193–4194 10.1021/ja00119a045 [DOI] [Google Scholar]
- 74. Ryckaert J. P., Ciccotti G., and Berendsen H. J. C. (1977) Numerical integration of cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 10.1016/0021-9991(77)90098-5 [DOI] [Google Scholar]
- 75. Roe D. R., and Cheatham T. E. 3rd (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 10.1021/ct400341p [DOI] [PubMed] [Google Scholar]
- 76. Salomon-Ferrer R., Götz A. W., Poole D., Le Grand S., and Walker R. C. (2013) Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 9, 3878–3888 10.1021/ct400314y [DOI] [PubMed] [Google Scholar]
- 77. Pfleger C., Rathi P. C., Klein D. L., Radestock S., and Gohlke H. (2013) Constraint Network Analysis (CNA): a Python software package for efficiently linking biomacromolecular structure, flexibility, (thermo-)stability, and function. J. Chem. Inf. Model. 53, 1007–1015 10.1021/ci400044m [DOI] [PubMed] [Google Scholar]
- 78. Chodera J. D. (2016) A simple method for automated equilibration detection in molecular simulations. J. Chem. Theory Comput. 12, 1799–1805 10.1021/acs.jctc.5b00784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shirts M. R., and Chodera J. D. (2008) Statistically optimal analysis of samples from multiple equilibrium states. J. Chem. Phys. 129, 124105 10.1063/1.2978177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. R Development Core Team (2010) R: a Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.