

Abstract
Transition metals have been recognized and studied primarily in the context of their essential roles as structural and metabolic cofactors for biomolecules that compose living systems. More recently, an emerging paradigm of transition-metal signaling, where dynamic changes in transitional metal pools can modulate protein function, cell fate, and organism health and disease, has broadened our view of the potential contributions of these essential nutrients in biology. Using copper as a canonical example of transition-metal signaling, we highlight key experiments where direct measurement and/or visualization of dynamic copper pools, in combination with biochemical, physiological, and behavioral studies, have deciphered sources, targets, and physiological effects of copper signals.
Keywords: copper signaling, neurobiology, cancer biology, immunology, lipolysis, copper transport, metal homeostasis
Introduction
Transition metals are indispensable for all living systems. These essential elements serve not only as structural cofactors for proteins and nucleic acids (1), but they also perform exquisite chemistry in their roles as metabolic cofactors in enzymes, facilitating essential processes such as oxygen transport, neurotransmitter synthesis and metabolism, nucleic acid repair and epigenetics, and construction of the extracellular matrix (1). Key transition metals that can undergo reduction-oxidation (redox) cycling under biological conditions (e.g. copper, iron, manganese, and nickel) also facilitate the electron transport necessary for photosynthesis, cellular respiration, and enzymatic redox reactions (2). In addition to these critical functions, transition-metal signaling has emerged as an exciting new field of study in living systems (3). In this expanded paradigm, transition metals not only serve as static structural and metabolic cofactors, but also are mobilized by cells to dynamically regulate enzymatic activity and communicate information both within cells and between cells. Indeed, recent advances in the observation of transition-metal signaling across multiple organ systems in both healthy and diseased states highlight its importance in mammalian biology. Here, we focus on copper as a canonical example of a transition metal signal, providing a summary of current understanding of copper signaling in neurobiology, immunity, cancer, and fat metabolism and future prospects for the field.
Acquisition and trafficking of copper in mammals
To set the stage for discussing copper signaling, we first provide an overview of copper homeostasis in mammalian systems. Copper is an essential mineral that must be acquired through the diet and trafficked to the organs, cells, and proteins that require copper for health (4). The majority (70%) of copper import into mammalian cells occurs through copper transporter 1 (CTR1) (5), from which copper is passed to intracellular chaperone proteins or small molecules, such as glutathione, which carry copper through the cytoplasm (6, 7). The copper chaperone Atox1 delivers copper to the copper-transporting ATPases ATP7A and ATP7B, copper exporters typically located on the Golgi apparatus and responsible for loading copper into cuproenzymes, including ceruloplasmin and dopamine β-hydroxylase (8, 9). Under conditions of high intracellular copper, ATP7A relocalizes to the plasma membrane to facilitate copper export (10). The copper chaperone for superoxide dismutase delivers copper to Cu,Zn-superoxide dismutase 1 (SOD1), an enzyme responsible for detoxifying superoxide by converting it to hydrogen peroxide (11). Additional unknown chaperones deliver copper to the mitochondrial matrix, where it is mobilized through various channels to metalate cytochrome c oxidase, an essential component of the electron transport pathway, and mitochondrial SOD2 (12). For a detailed discussion of copper homeostasis in eukaryotes, the reader is referred to an insightful review by Nevitt et al. (13).
These well-established copper acquisition and trafficking pathways highlight the importance of proper delivery of copper to essential cuproenzymes. However, mounting experimental evidence suggests the presence of kinetically accessible pools of copper that are not buried in enzyme-active sites but can be readily mobilized to the intra- or extracellular space with a cellular stimulus (14). Although the molecular mechanisms of copper mobilization remain poorly understood, we suggest that mobilization would involve the rapid reorganization of copper ions from one subset of protein and small molecule-binding sites to another, which may result in copper crossing membranes. On a practical level, this pool of accessible copper can be diminished by short-term treatment with copper chelators, leading to the term “labile” or “chelatable” copper pool. The choice of chelator is critical. On short time scales (<1 h), membrane-impermeable chelators deplete only extracellular copper and copper that may be rapidly moving through release-and-uptake cycles, whereas membrane-permeable chelators can access both intra- and extracellular copper, as well as potentially form ternary complexes with copper chaperones (15), preventing copper trafficking or mobilization. Additionally, copper ionophores, such as clioquinol and other 8-hydroxyquinoline (8HQ) derivatives, redistribute copper across cell and organelle membranes, altering the labile copper pool without adding or removing copper from the system (16, 17). Changes to the labile copper pool may be assessed by the use of fluorescent small-molecule indicators that can equilibrate with kinetically accessible copper in the cytosol, enabling one to distinguish labile copper from total copper, the latter of which is typically measured using direct techniques, including X-ray fluorescence microscopy (XFM) or mass spectrometry imaging (14, 18).
Augmentation or restriction of the labile copper pool alters the function of a number of enzymes, some through direct binding to copper, leading to the notion that copper itself may serve as a cellular signal that modulates the activity of proteins. Indeed, copper signaling has been identified through multiple independent studies centered on the brain, and it has now been expanded to encompass a broader range of organ systems, as we shall explore below. The reader is also referred to an excellent summary of copper as a key regulator of cell signaling pathways by Grubman and White (19).
Copper signaling in neurobiology
Early studies suggesting that copper could serve essential functions outside of enzyme-active sites came from the observation by Hartter and Barnea (20) that isolated synaptic termini (e.g. synaptosomes) preloaded with radioactive 67Cu could rapidly release copper upon depolarization. These in vitro data were corroborated by ex vivo studies by Gitlin and co-workers (21), who observed that depolarization of hippocampal neurons with KCl or stimulation through the N-methyl-d-aspartate (NMDA) receptor caused trafficking of the copper exporter ATP7A to the plasma membrane. NMDA stimulation also caused efflux of radioactive copper, except in neurons lacking ATP7A, suggesting that the mechanism of copper release from stimulated hippocampal neurons requires ATP7A (21, 22).
Work from our laboratory using XFM provided the first direct imaging evidence for selective and dynamic copper translocation upon neuronal stimulation, showing a redistribution of copper from the cell body to dendrites (23). Complementary visualization of copper fluxes was observed using the fluorescent small molecule Coppersensor-3 (CS3), which undergoes an increase in fluorescence intensity in the presence of copper and provides evidence that mobile neural copper involves labile copper pools. Furthermore, copper redistribution was dependent on calcium signaling, as neurons depolarized in the presence of the calcium chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) did not show a redistribution of copper, suggesting that canonical calcium signaling precedes copper signaling (23). Additional studies with Copper Fluor-3 (CF3), a copper sensor suitable for use in tissue slices, demonstrated that cultured hippocampal neurons and retinal tissue slices contain an endogenous labile pool of copper that can be depleted by treatment with copper chelators (24). Depletion of labile copper increased spontaneous activity in these neural models, establishing a fundamental role for copper in regulating normal neural function. Mechanistically, maintenance of the neural labile copper pool depends on the copper import protein CTR1, as neurons isolated from CTR1+/− mice contained lower levels of total and labile copper and exhibited higher spontaneous firing, similar to neurons acutely treated with copper chelators (24), leading to the speculation that CTR1 could serve in an “ion channel-like” role for copper fluxes in certain contexts (Fig. 1). Taken together, these data suggest that some neurons contain pools of labile copper that are required for spontaneous firing and are mobilized during neural depolarization, initiating a copper signal that is dependent on calcium signaling.
Figure 1.
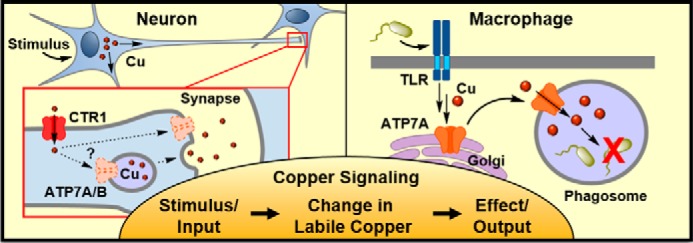
Observations of copper signaling in mammalian biology. Left (Neuron), upon depolarization, copper relocalizes from the cell body to dendrites. Copper can be released from neurons by an unknown process requiring ATP7A. Copper transporter 1 (CTR1) is an essential copper import channel for copper cycling in neurons. Right (Macrophage), upon stimulation of Toll-like receptors (TLRs), the copper exporter ATP7A moves from the Golgi to the phagosome in a copper-dependent manner. ATP7A pumps copper into the phagosome, leading to microbial death or requiring microbes to adapt to high copper conditions.
The effect of copper signaling on neurotransmission has been explored through studies of receptors exposed to exogenous copper. Included are γ-aminobutyric acid (GABA) (25–27), NMDA (28, 29), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) (30), and P2X (31, 32) receptors, as well as voltage-gated sodium, potassium, and calcium channels (33, 34). Beyond neurotransmitter receptors, an early prediction by Crabtree (35) that copper could bind thiols to enable olfactory function was validated in an elegant study by Zhuang and co-workers (36) who showed that copper is required for an odorant receptor in mice that senses a thiol-containing pheromone.
Ultimately, the effect of copper on one or more proteins must lead to a change in synaptic strength, activity, or plasticity in order for copper to impact neural output and behavior. In rat hippocampal slices, application of as little as 1 μm CuCl2 inhibited long-term potentiation (LTP) (37), a measure of the strength of a synaptic connection, an effect also observed in rats fed a high copper diet (38) or injected with copper (39). However, copper-mediated inhibition of LTP, measured at the cellular level, does not correlate with memory loss, as rats injected with copper show no defect in navigating a Morris Water Maze (39). Recently, LTP in mouse amygdala slices was shown to be suppressed by acute bath application of the extracellular copper chelator bathocuproine sulfonate (BCS), suggesting that endogenous extracellular copper is necessary for LTP (40). Additionally, copper has been shown to lower the firing threshold and increase spontaneous activity in the rat hippocampus CA1 region, whereas copper chelation decreases spontaneous activity (41). Finally, in addition to copper's roles in learning and memory, copper has also been shown to influence circadian rhythm. Chelation of copper with BCS or tetrathiomolybdate (TTM) from slices of circadian clock neurons caused ∼2.5–3-h phase delays in circadian neuronal activity, which depended on NMDA receptor activity (42). Interestingly, copper addition also altered circadian rhythm, but in a manner independent of NMDA receptor activity, suggesting a separate mechanism for this effect (42).
Beyond impacting normal neural function, copper directly interacts with cellular prion protein (PrPC), amyloid precursor protein (APP), SOD1, and huntingtin (Htt), proteins involved in spongiform encephalopathy, Alzheimer's disease, amyotrophic lateral sclerosis (ALS), and Huntington's disease (HD), respectively. PrPC and APP bind copper through their extracellular domains (43, 44), and PrPC may serve as a buffer for copper binding in the extracellular space, perhaps at the synapse (45). Indeed, expression levels of PrPC (43) and APP (46, 47) have been shown to influence neural copper levels, placing PrPC and APP in the network of proteins that regulate neural copper homeostasis. Interestingly, copper also alters the activity of glycogen synthase kinase 3β (GSK3β), an enzyme involved in regulating APP processing, linking copper to APP biology through multiple mechanisms (48, 49). Additionally, pathological aggregation of both APP and Htt may be mediated by copper binding (50, 51). Indeed, Zhou and co-workers (51) showed that when potential copper-binding residues on Htt were mutated, changes in neural copper homeostasis no longer influenced Htt aggregation, suggesting that copper levels directly impact HD progression. Finally, aggregation of mutant forms of SOD1, an enzyme that requires copper and zinc cofactors for catalytic function, leads to ALS (52, 53), and the disease course can be modulated by changes in dietary zinc and copper (54).
Copper homeostasis is intricately connected to health and disease in the brain, and these connections have been established on biochemical, physiological, and behavioral levels. For detailed discussions of the role of copper in neurobiology, the reader is referred to recent reviews by D'Ambrossi and Rossi (55), Gaier et al. (56), and Opazo et al. (57). Although neurobiology has long hinted at the prospects of copper signaling, the identification and study of copper signaling in other tissues with more detailed study of specific molecular targets are rapidly expanding. We now turn our attention to these systems.
Copper in immunology: Toxin or resource?
Besides the brain, copper is mobilized in other cells and tissues in response to a variety of stimuli, including infection. The antimicrobial properties of copper have been appreciated for centuries, and today, copper is used in medical devices to prevent fouling from microbial growth (58, 59). Appropriately, the mammalian immune system also uses copper's toxicity to prevent microbial growth. Innate immune cells, such as macrophages, respond to infection by internalizing microbes into vesicles called phagosomes, which rapidly acidify and are filled with reactive oxygen species (ROS) and lytic proteins that kill microbes (60). In a seminal study, Petris and co-workers (61) showed that in response to bacteria or immune stimuli the ATP7A copper exporter in macrophages moves to the phagosomal membrane in a copper-dependent manner, suggesting that copper is mobilized within immune cells during infection and may be pumped into phagosomes as part of an immune response (Fig. 1). Indeed, Escherichia coli cells lacking the bacterial copper exporter copA are less able to survive internalization within macrophages than wildtype E. coli, but this deficit is ameliorated when ΔcopA E. coli cells are internalized within ATP7A−/− macrophages (61). These data suggest that phagosomal copper negatively affects bacterial growth, such that bacteria possess copper export machinery able to repel a copper insult. A copper-sensing and export response has been observed in a variety of bacterial pathogens, including Mycobacterium tuberculosis (62), Streptococcus pneumoniae (63), and Salmonella enterica sv. typhimurium (64, 65), and copper accumulation has been measured by XFM imaging of macrophages infected by Mycobacterium species (66).
Pathogenic yeast, including Candida albicans and Cryptococcus neoformans, may harness mobilized phagosomal copper to augment virulence. Copper hyper-accumulation is necessary for yeast virulence in some systems (67), and metallothionein, a protein that sequesters and buffers copper, is up-regulated in yeast during infection (68). Additionally, C. albicans can switch from a Mn-SOD to a Cu,Zn-SOD under high copper conditions (69), and it expresses a copper-only SOD at the cell surface that may be metallated directly from copper in the phagosome (70). Thus, the copper that the host cell injects into the phagosome as a toxin becomes a resource that the pathogen uses to detoxify ROS.
As copper mobilization is an integral part of the innate immune response, manipulating labile copper within immune cells may be a viable therapeutic for microbial infections. For example, cellular treatment with the copper ionophore 8HQ increases bioavailable copper within cells and reduces microbial growth (17). Recently, a caged form of 8HQ, which may be subsequently uncaged in the presence of ROS, was developed to mobilize copper specifically within ROS-producing macrophages, resulting in higher microbial killing (71). As the need for new antimicrobial compounds continues to increase, copper may emerge as a valuable linchpin for gaining leverage over microbial pathogens.
Controlling cancer with labile copper
Metal dyshomeostasis and an oxidizing cellular environment are hallmarks of cancer cells (72, 73). As a result, substantial research has been dedicated to understanding and manipulating copper in cancer to prevent cancer progression. Delivery of copper to cancer cells, for example as a disulfiram complex (74) or bis(thiosemicarbazonato) complex (e.g. Cu(Gtsm) (75)), takes advantage of the oxidizing environment in cancer cells, which is less equipped to buffer increases in copper, to cause cell death (76). Additionally, systemic chelation of copper with TTM has shown positive results in phase II clinical trials for the prevention triple-negative breast cancer (TNBC) metastasis, a lethal step in TNBC progression (77, 78).
Here, we emphasize the growing importance of copper in cancer as a regulator of tumor cell proliferation. In cancer as well as healthy cells, the mitogen-activated protein kinase (MAPK) pathway is a signaling cascade that drives cellular proliferation and differentiation. Activating mutations in upstream components of the pathway, such as BRAFV600E, lead to constitutive MAPK signaling, causing tumorigenesis and tumor growth (79). At the terminus of the MAPK pathway, MEK1/2 phosphorylates the MAPK protein (ERK1/2), which in turn phosphorylates and activates various enzymes and transcription factors. BRAF inhibitors are used to treat BRAF-mutant cancers, but resistance commonly arises as cells acquire activating mutations downstream, for example in MEK1/2 (80). Foundational studies by Thiele and co-workers (81) in fly and mouse cell models led to the discovery that copper is required for MEK1/2 activity. Biochemical characterization demonstrated that copper binds directly to purified MEK1/2, facilitating phosphorylation of recombinant ERK1/2 in vitro; conversely, copper chelation inhibits ERK1/2 phosphorylation in vitro (81). Elegant work by Brady et al. (82) showed the physiological importance of this copper–MAPK connection in a cancer setting. In melanoma cells carrying the oncogenic BRAFV600E mutation, knockout of the copper importer CTR1 decreased phospho-ERK1/2 levels and inhibited cell proliferation. Restricting labile copper by oral administration of TTM to mice for 2 weeks inhibited the growth of tumors from injected BRAFV600E mouse embryonic fibroblasts or BRAF mutant DM440 melanoma cells. Notably, TTM treatment was effective against tumor cells expressing MEK1/2C121S, a mutation known to confer resistance to tumors treated with BRAF inhibitors (82). Together, these studies reveal that labile copper can play a key role in the growth of cancer and that restriction of the labile copper pool can inhibit cancer growth by limiting MEK1/2 activity, even in the presence of mutations known to activate MEK1/2. The requirement of copper for full MEK1/2 activity suggests that cells or tissues may regulate copper pools to tune the MAPK pathway, integrating a transition metal signal into traditional kinase signal transduction pathways (Fig. 2). Indeed, manipulation of copper levels through administration of copper chelators or ionophores may be an effective way to modulate MAPK signaling, particularly in the context of combination therapies for cancer.
Figure 2.
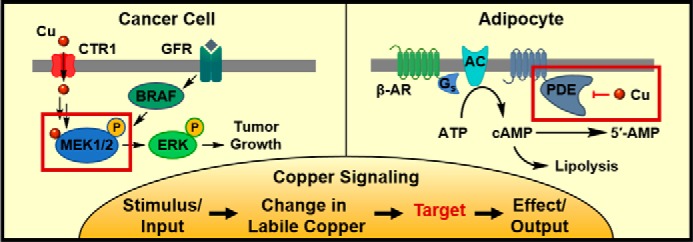
Copper-signaling pathways and molecular targets in mammalian biology. Left (Cancer Cell), stimulation of growth factor receptors (GFRs) or mutations in proto-oncogene protein B-Raf (BRAF) cause activation of the MAPK pathway, leading to phosphorylation of mitogen-activated protein kinase kinase (MAP2K1/2 or MEK1/2). MEK1/2 activation requires direct binding of cellular copper, which is supplied through the copper importer CTR1. Fully activated MEK1/2 phosphorylates MAPK or ERK, leading to tumor growth. Right (Adipocyte), activation of the β-adrenergic receptor (β-AR) causes recruitment of a G-protein (Gs) and activation of adenylyl cyclase (AC). Adenylyl cyclase converts adenosine triphosphate (ATP) to cyclic-adenosine monophosphate (cAMP), which activates downstream pathways leading to lipolysis. The pathway is shut down when cAMP is hydrolyzed by phosphodiesterase 3B (PDE). However, direct binding of copper to phosphodiesterase inhibits cAMP hydrolysis and maintains activation of the lipolysis pathway.
Copper regulation of fat metabolism
Dysregulation of lipid metabolism is a symptom of dietary copper deficiency or overload (83–85). Moreover, during the progression of diet-induced non-alcoholic fatty liver disease, liver copper levels decrease, and the expression of copper homeostasis proteins is altered (86, 87). In addition to such nutritional studies linking copper and fat metabolism, the mouse model of Wilson disease, which lacks the copper exporter ATP7B and accumulates copper in the liver, shows changes in lipid metabolism in the liver before traditional Wilson disease symptoms, such as inflammation and liver failure, appear (88). A liver-specific ATP7B knockout mouse develops lipid dysregulation without inflammation, suggesting that metabolic symptoms may be directly linked to copper levels and not a secondary symptom of liver inflammation observed in Wilson disease (89).
Consistent with dietary and genetic studies linking long-term changes in copper to changes in fat metabolism (90, 91), work from our laboratory identified copper as a newly recognized allosteric regulator of phosphodiesterase 3B (PDE3B), a phosphodiesterase that controls lipolysis in adipocytes (92). Lipolysis, the breakdown of triglycerides into glycerol and fatty acids, may be induced in tissues and cells by treatment with isoproterenol, which stimulates the β-adrenergic receptor and induces a signaling cascade mediated by the second messenger cyclic-adenosine monophosphate (cAMP). Elevated cAMP levels stimulate protein kinase A (PKA), leading to downstream events that culminate in lipid breakdown (93). Copper-deficient adipose tissue isolated from ATP7B knockout mice and stimulated with isoproterenol releases less glycerol and has lower cAMP levels than isoproterenol-stimulated adipose tissue isolated from healthy heterozygous siblings (92). In differentiated 3T3-L1 cells, a cell culture model of white fat, copper supplementation potentiated and copper chelation inhibited isoproterenol-induced glycerol release, demonstrating that fat metabolism is influenced by manipulation of the labile copper pool. Additionally, the fluorescent copper sensor copper silicon Rhodol 1 (CSR1), but not the control CSR1 analog lacking copper-responsive binding groups, revealed an acute decrease in labile copper after isoproterenol treatment within 5–10 min, before the onset of fatty acid release, suggesting that changes in labile copper are upstream of lipid breakdown (92).
Further characterization of the lipolysis signaling cascade identified PDE3B, the phosphodiesterase responsible for hydrolyzing cAMP to shut down lipolysis, as the molecular target of copper. Biochemical studies of PDE3B demonstrated direct inhibition of PDE3B by copper binding to Cys-768, a cysteine located away from the enzyme-active site, establishing Cys-768 as the first example of a molecularly characterized allosteric copper-binding site (92). Indeed, in cells expressing PDE3B with a C768S mutation, changes in copper levels no longer impacted isoproterenol-induced lipolysis. These data revealed a copper signal that acts through allosteric modulation of PDE3B to affect lipolysis in cells and animals, and the data demonstrated the intersection of copper signaling with signaling through the second messenger cAMP (Fig. 2). Together with dietary and genetic studies, these biochemical studies suggest that manipulation of copper levels may be a viable option for influencing molecular pathways controlling fat metabolism and treating diseases involving dysregulation of lipid homeostasis. Indeed, a subsequent study by our laboratory using the luciferin-based bioluminescent probe CCL-1 revealed a liver-localized copper deficiency in a diet-induced model of non-alcoholic fatty liver disease (87).
Conclusion
Copper signaling represents a canonical example of transition-metal signaling and illustrates the broad and growing influence of this new paradigm on the health and disease of diverse tissues and systems within mammalian organisms, spanning neurobiology, immunity, fat metabolism, and cancer. To date, multiple potential protein targets of copper signaling have been identified, and an allosteric binding site that confers dynamic copper responsiveness has been identified for the first time in PDE3B (92). This work is accompanied by the observation of copper–MEK interactions that presage a potentially rich copper kinome to mine (82). Efforts to rigorously identify the molecular mechanisms of copper signaling in other tissues, especially the brain, will advance the field of copper signaling by connecting intriguing observations about copper-related phenomena, such as learning and memory, to molecular targets that may be subjected to pharmacological and/or genetic manipulation.
As the field of copper signaling continues to mature, many questions remain, including identifying the origins of copper signals. In the brain, extracellular copper signals may be released directly from the synapse (20). Interestingly, vesicular storage and mobilization of copper have been observed in other systems, including the algae Chlamydomonas reinhardtii, which accumulate copper in acidocalcisomes and mobilize stored copper under conditions of nutritional copper restriction (94). In mammalian systems, intracellular copper trafficking is influenced by the cell's redox state (95), and changes in redox state may be able to trigger mobilization of copper signals. Other storage sites for copper, which may serve as the origin of copper signals, include the copper chaperones (96) or small molecules such as glutathione (6). Continued efforts to develop new technologies to image and characterize copper pools in living systems will aid in addressing these outstanding questions (14, 18, 97, 98).
In addition to exploring the origins of copper signals, the ability to manipulate copper signaling may prove an effective therapy, whether alone or in combination with existing therapies, for diseases such as neurodegeneration, infection, cancer, and obesity. Manipulation of copper levels may be achieved most simply by adjustment of dietary copper intake, whereas targeted delivery of copper supplements, chelators, or ionophores to specific organs could serve to mitigate potential toxicities associated with systemic changes to copper homeostasis. Finally, this short discussion has focused on copper, but this metal represents just one transition-metal signaling element in the periodic table of life, and opportunities spanning chemistry, biology, and engineering of metals will surely contribute to a better global understanding of health, aging, and disease.
This work was supported in part by National Institutes of Health Grant R01 GM079465. This is the third article in the Thematic Minireview series “Metals in Biology 2018: Copper homeostasis and utilization in redox enzymes.” The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supported by a Fannie and John Hertz Foundation Fellowship and National Institutes of Health Chemical Biology Interface Training Grant T32 GM066698.
- SOD
- superoxide dismutase
- 8HQ
- 8-hydroxyquinoline
- XFM
- X-ray fluorescence microscopy
- NMDA
- N-methyl-d-aspartate
- LTP
- long-term potentiation
- BCS
- bathocuproine sulfonate
- PrPC
- cellular prion protein
- APP
- amyloid precursor protein
- Htt
- huntingtin
- HD
- Huntington's disease
- ALS
- amyotrophic lateral sclerosis
- TTM
- tetrathiomolybdate
- MAPK
- mitogen-activated protein kinase
- ERK
- extracellular signal-regulated kinase
- MEK
- mitogen-activated protein kinase/extracellular signal-regulated kinase kinase
- ROS
- reactive oxygen species
- TNBC
- triple-negative breast cancer.
References
- 1. Lippard S. J., and Berg J. M. (1994) Principles of Bioinorganic Chemistry, University Science Books, Mill Valley, CA [Google Scholar]
- 2. Yruela I. (2013) Transition metals in plant photosynthesis. Metallomics 5, 1090–1109 10.1039/c3mt00086a [DOI] [PubMed] [Google Scholar]
- 3. Chang C. J. (2015) Searching for harmony in transition-metal signaling. Nat. Chem. Biol. 11, 744–747 10.1038/nchembio.1913 [DOI] [PubMed] [Google Scholar]
- 4. Kim B.-E., Nevitt T., and Thiele D. J. (2008) Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol. 4, 176–185 10.1038/nchembio.72 [DOI] [PubMed] [Google Scholar]
- 5. Lee J., Petris M. J., and Thiele D. J. (2002) Characterization of mouse embryonic cells deficient in the Ctr1 high affinity copper transporter. J. Biol. Chem. 277, 40253–40259 10.1074/jbc.M208002200 [DOI] [PubMed] [Google Scholar]
- 6. Maryon E. B., Molloy S. A., and Kaplan J. H. (2013) Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1. Am. J. Physiol. Cell Physiol. 304, C768–C779 10.1152/ajpcell.00417.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Robinson N. J., and Winge D. R. (2010) Copper metallochaperones. Annu. Rev. Biochem. 79, 537–562 10.1146/annurev-biochem-030409-143539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. La Fontaine S., and Mercer J. F. (2007) Trafficking of the copper-ATPases, ATP7A and ATP7B: role in copper homeostasis. Arch. Biochem. Biophys. 463, 149–167 10.1016/j.abb.2007.04.021 [DOI] [PubMed] [Google Scholar]
- 9. Linz R., and Lutsenko S. (2007) Copper-transporting ATPases ATP7A and ATP7B: cousins, not twins. J. Bioenerg. Biomembr. 39, 403–407 10.1007/s10863-007-9101-2 [DOI] [PubMed] [Google Scholar]
- 10. Pase L., Voskoboinik I., Greenough M., and Camakaris J. (2004) Copper stimulates trafficking of a distinct pool of the Menkes copper ATPase (ATP7A) to the plasma membrane and diverts it into a rapid recycling pool. Biochem. J. 378, 1031–1037 10.1042/bj20031181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Culotta V. C., Yang M., and O'Halloran T. V. (2006) Activation of superoxide dismutases: putting the metal to the pedal. Biochim. Biophys. Acta 1763, 747–758 10.1016/j.bbamcr.2006.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hamza I., and Gitlin J. D. (2002) Copper chaperones for cytochrome c oxidase and human disease. J. Bioenerg. Biomembr. 34, 381–388 10.1023/A:1021254104012 [DOI] [PubMed] [Google Scholar]
- 13. Nevitt T., Ohrvik H., and Thiele D. J. (2012) Charting the travels of copper in eukaryotes from yeast to mammals. Biochim. Biophys. Acta 1823, 1580–1593 10.1016/j.bbamcr.2012.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cotruvo J. J. Jr., Aron A. T., Ramos-Torres K. M., and Chang C. J. (2015) Synthetic fluorescent probes for studying copper in biological systems. Chem. Soc. Rev. 44, 4400–4414 10.1039/C4CS00346B [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alvarez H. M., Xue Y., Robinson C. D., Canalizo-Hernández M. A., Marvin R. G., Kelly R. A., Mondragón A., Penner-Hahn J. E., and O'Halloran T. V. (2010) Tetrathiomolybdate inhibits copper trafficking proteins through metal cluster formation. Science 327, 331–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ding W.-Q., and Lind S. E. (2009) Metal ionophores: An emerging class of anticancer drugs. IUBMB Life 61, 1013–1018 10.1002/iub.253 [DOI] [PubMed] [Google Scholar]
- 17. Anderson B. I., and Swaby R. J. (1951) Factors influencing the fungistatic action of 8-hydroxyquinoline (oxine) and its metal complexes. Aust. J. Sci. Res. B 4, 275–282 10.1071/BI9510275 [DOI] [PubMed] [Google Scholar]
- 18. Ackerman C. M., Lee S., and Chang C. J. (2017) Analytical methods for imaging metals in biology: from transition metal metabolism to transition-metal signaling. Anal. Chem. 89, 22–41 10.1021/acs.analchem.6b04631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grubman A., and White A. R. (2014) Copper as a key regulator of cell signalling pathways. Expert Rev. Mol. Med. 16, e11 10.1017/erm.2014.11 [DOI] [PubMed] [Google Scholar]
- 20. Hartter D. E., and Barnea A. (1988) Evidence for release of copper in the brain: depolarization-induced release of newly taken-up 67-copper. Synapse 2, 412–415 10.1002/syn.890020408 [DOI] [PubMed] [Google Scholar]
- 21. Schlief M. L., Craig A. M., and Gitlin J. D. (2005) NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J. Neurosci. 25, 239–246 10.1523/JNEUROSCI.3699-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schlief M. L., West T., Craig A. M., Holtzman D. M., and Gitlin J. D. (2006) Role of the Menkes copper-transporting ATPase in NMDA receptor-mediated neuronal toxicity. Proc. Natl. Acad. Sci. U.S.A. 103, 14919–14924 10.1073/pnas.0605390103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dodani S. C., Domaille D. W., Nam C. I., Miller E. W., Finney L. A., Vogt S., and Chang C. J. (2011) Calcium-dependent copper redistributions in neuronal cells revealed by a fluorescent copper sensor and X-ray fluorescence microscopy. Proc. Natl. Acad. Sci. U.S.A. 108, 5980–5985 10.1073/pnas.1009932108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dodani S. C., Firl A., Chan J., Nam C. I., Aron A. T., Onak C. S., Ramos-Torres K. M., Paek J., Webster C. M., Feller M. B., and Chang C. J. (2014) Copper is an endogenous modulator of neural circuit spontaneous activity. Proc. Natl. Acad. Sci. U.S.A. 111, 16280–16285 10.1073/pnas.1409796111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kardos J., Kovács I., Hajós F., Kálmán M., and Simonyi M. (1989) Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci. Lett. 103, 139–144 10.1016/0304-3940(89)90565-X [DOI] [PubMed] [Google Scholar]
- 26. Sharonova I. N., Vorobjev V. S., and Haas H. L. (1998) High-affinity copper block of GABA(A) receptor-mediated currents in acutely isolated cerebellar Purkinje cells of the rat. Eur. J. Neurosci. 10, 522–528 10.1046/j.1460-9568.1998.00057.x [DOI] [PubMed] [Google Scholar]
- 27. McGee T. P., Houston C. M., and Brickley S. G. (2013) Copper block of extrasynaptic GABAA receptors in the mature cerebellum and striatum. J. Neurosci. 33, 13431–13435 10.1523/JNEUROSCI.1908-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marchetti C., Baranowska-Bosiacka I., and Gavazzo P. (2014) Multiple effects of copper on NMDA receptor currents. Brain Res. 1542, 20–31 10.1016/j.brainres.2013.10.029 [DOI] [PubMed] [Google Scholar]
- 29. Trombley P. Q., and Shepherd G. M. (1996) Differential modulation by zinc and copper of amino acid receptors from rat olfactory bulb neurons. J. Neurophysiol. 76, 2536–2546 10.1152/jn.1996.76.4.2536 [DOI] [PubMed] [Google Scholar]
- 30. Weiser T., and Wienrich M. (1996) The effects of copper ions on glutamate receptors in cultured rat cortical neurons. Brain Res. 742, 211–218 10.1016/S0006-8993(96)01009-8 [DOI] [PubMed] [Google Scholar]
- 31. Acuña-Castillo C., Morales B., and Huidobro-Toro J. P. (2000) Zinc and copper modulate differentially the P2X4 receptor. J. Neurochem. 74, 1529–1537 [DOI] [PubMed] [Google Scholar]
- 32. Punthambaker S., and Hume R. I. (2014) Potent and long-lasting inhibition of human P2X2 receptors by copper. Neuropharmacology 77, 167–176 10.1016/j.neuropharm.2013.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Horning M. S., and Trombley P. Q. (2001) Zinc and copper influence excitability of rat olfactory bulb neurons by multiple mechanisms. J. Neurophysiol. 86, 1652–1660 10.1152/jn.2001.86.4.1652 [DOI] [PubMed] [Google Scholar]
- 34. Castelli L., Tanzi F., Taglietti V., and Magistretti J. (2003) Cu2+, Co2+, and Mn2+ modify the gating kinetics of high-voltage-activated Ca2+ channels in rat palaeocortical neurons. J. Membr. Biol. 195, 121–136 10.1007/s00232-003-0614-2 [DOI] [PubMed] [Google Scholar]
- 35. Crabtree R. H. (1978) Copper (I): A possible olfactory binding site. J. Inorg. Nucl. Chem. 40, 1453 10.1016/0022-1902(78)80071-2 [DOI] [Google Scholar]
- 36. Duan X., Block E., Li Z., Connelly T., Zhang J., Huang Z., Su X., Pan Y., Wu L., Chi Q., Thomas S., Zhang S., Ma M., Matsunami H., Chen G. Q., and Zhuang H. (2012) Crucial role of copper in detection of metal-coordinating odorants. Proc. Natl. Acad. Sci. U.S.A. 109, 3492–3497 10.1073/pnas.1111297109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Doreulee N., Yanovsky Y., and Haas H. L. (1997) Suppression of long-term potentiation in hippocampal slices by copper. Hippocampus 7, 666–669 [DOI] [PubMed] [Google Scholar]
- 38. Goldschmith A., Infante C., Leiva J., Motles E., and Palestini M. (2005) Interference of chronically ingested copper in long-term potentiation (LTP) of rat hippocampus. Brain Res. 1056, 176–182 10.1016/j.brainres.2005.07.030 [DOI] [PubMed] [Google Scholar]
- 39. Leiva J., Palestini M., Infante C., Goldschmidt A., and Motles E. (2009) Copper suppresses hippocampus LTP in the rat, but does not alter learning or memory in the morris water maze. Brain Res. 1256, 69–75 10.1016/j.brainres.2008.12.041 [DOI] [PubMed] [Google Scholar]
- 40. Gaier E. D., Rodriguiz R. M., Zhou J., Ralle M., Wetsel W. C., Eipper B. A., and Mains R. E. (2014) In vivo and in vitro analyses of amygdalar function reveal a role for copper. J. Neurophysiol. 111, 1927–1939 10.1152/jn.00631.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maureira C., Letelier J. C., Alvarez O., Delgado R., and Vergara C. (2015) Copper enhances cellular and network excitabilities, and improves temporal processing in the rat hippocampus. Eur. J. Neurosci. 42, 3066–3080 10.1111/ejn.13104 [DOI] [PubMed] [Google Scholar]
- 42. Yamada Y., and Prosser R. A. (2014) Copper chelation and exogenous copper affect circadian clock phase resetting in the suprachiasmatic nucleus in vitro. Neuroscience 256, 252–261 10.1016/j.neuroscience.2013.10.033 [DOI] [PubMed] [Google Scholar]
- 43. Brown D. R., Qin K., Herms J. W., Madlung A., Manson J., Strome R., Fraser P. E., Kruck T., von Bohlen A., Schulz-Schaeffer W., Giese A., Westaway D., and Kretzschmar H. (1997) The cellular prion protein binds copper in vivo. Nature 390, 684–687 10.1038/37783 [DOI] [PubMed] [Google Scholar]
- 44. Acevedo K. M., Hung Y. H., Dalziel A. H., Li Q.-X., Laughton K., Wikhe K., Rembach A., Roberts B., Masters C. L., Bush A. I., and Camakaris J. (2011) Copper promotes the trafficking of the amyloid precursor protein. J. Biol. Chem. 286, 8252–8262 10.1074/jbc.M110.128512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kretzschmar H. A., Tings T., Madlung A., Giese A., and Herms J. (2000) Function of PrP(C) as a copper-binding protein at the synapse. Arch. Virol. Suppl. 16, 239–249 [DOI] [PubMed] [Google Scholar]
- 46. White A. R., Reyes R., Mercer J. F., Camakaris J., Zheng H., Bush A. I., Multhaup G., Beyreuther K., Masters C. L., and Cappai R. (1999) Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice. Brain Res. 842, 439–444 10.1016/S0006-8993(99)01861-2 [DOI] [PubMed] [Google Scholar]
- 47. Maynard C. J., Cappai R., Volitakis I., Cherny R. A., White A. R., Beyreuther K., Masters C. L., Bush A. I., and Li Q.-X. (2002) Overexpression of Alzheimer's disease amyloid-β opposes the age-dependent elevations of brain copper and iron. J. Biol. Chem. 277, 44670–44676 10.1074/jbc.M204379200 [DOI] [PubMed] [Google Scholar]
- 48. Crouch P. J., Hung L. W., Adlard P. A., Cortes M., Lal V., Filiz G., Perez K. A., Nurjono M., Caragounis A., Du T., Laughton K., Volitakis I., Bush A. I., Li Q. X., Masters C. L., et al. (2009) Increasing Cu bioavailability inhibits A oligomers and τ phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 106, 381–386 10.1073/pnas.0809057106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Acevedo K. M., Opazo C. M., Norrish D., Challis L. M., Li Q.-X., White A. R., Bush A. I., and Camakaris J. (2014) Phosphorylation of amyloid precursor protein at threonine 668 is essential for its copper-responsive trafficking in SH-SY5Y neuroblastoma cells. J. Biol. Chem. 289, 11007–11019 10.1074/jbc.M113.538710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Atwood C. S., Scarpa R. C., Huang X., Moir R. D., Jones W. D., Fairlie D. P., Tanzi R. E., and Bush A. I. (2000) Characterization of copper interactions with Alzheimer amyloid β peptides: identification of an attomolar-affinity copper binding site on amyloid β1–42. J. Neurochem. 75, 1219–1233 [DOI] [PubMed] [Google Scholar]
- 51. Xiao G., Fan Q., Wang X., and Zhou B. (2013) Huntington disease arises from a combinatory toxicity of polyglutamine and copper binding. Proc. Natl. Acad. Sci. U.S.A. 110, 14995–15000 10.1073/pnas.1308535110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Karch C. M., Prudencio M., Winkler D. D., Hart P. J., and Borchelt D. R. (2009) Role of mutant SOD1 disulfide oxidation and aggregation in the pathogenesis of familial ALS. Proc. Natl. Acad. Sci. U.S.A. 106, 7774–7779 10.1073/pnas.0902505106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sheng Y., Chattopadhyay M., Whitelegge J., and Valentine J. S. (2012) SOD1 aggregation and ALS: role of metallation states and disulfide status. Curr. Top. Med. Chem. 12, 2560–2572 [DOI] [PubMed] [Google Scholar]
- 54. Ermilova I. P., Ermilov V. B., Levy M., Ho E., Pereira C., and Beckman J. S. (2005) Protection by dietary zinc in ALS mutant G93A SOD transgenic mice. Neurosci. Lett. 379, 42–46 10.1016/j.neulet.2004.12.045 [DOI] [PubMed] [Google Scholar]
- 55. D'Ambrosi N., and Rossi L. (2015) Copper at synapse: release, binding and modulation of neurotransmission. Neurochem. Int. 90, 36–45 10.1016/j.neuint.2015.07.006 [DOI] [PubMed] [Google Scholar]
- 56. Gaier E. D., Eipper B. A., and Mains R. E. (2013) Copper signaling in the mammalian nervous system: synaptic effects. J. Neurosci. Res. 91, 2–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Opazo C. M., Greenough M. A., and Bush A. I. (2014) Copper: from neurotransmission to neuroproteostasis. Front. Aging Neurosci. 6, 143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Irene G., Georgios P., Ioannis C., Anastasios T., Diamantis P., Marianthi C., Philippe W., and Maria S. (2016) Copper-coated textiles: armor against MDR nosocomial pathogens. Diagn. Microbiol. Infect. Dis. 85, 205–209 10.1016/j.diagmicrobio.2016.02.015 [DOI] [PubMed] [Google Scholar]
- 59. Espírito Santo C., Lam E. W., Elowsky C. G., Quaranta D., Domaille D. W., Chang C. J., and Grass G. (2011) Bacterial killing by dry metallic copper surfaces. Appl. Environ. Microbiol. 77, 794–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Canton J. (2014) Phagosome maturation in polarized macrophages. J. Leukocyte Biol. 96, 729–738 10.1189/jlb.1MR0114-021R [DOI] [PubMed] [Google Scholar]
- 61. White C., Lee J., Kambe T., Fritsche K., and Petris M. J. (2009) A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J. Biol. Chem. 284, 33949–33956 10.1074/jbc.M109.070201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shi X., Festa R. A., Ioerger T. R., Butler-Wu S., Sacchettini J. C., Darwin K. H., and Samanovic M. I. (2014) The copper-responsive RicR regulon contributes to Mycobacterium tuberculosis virulence. mBio 5, e00876–00813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Johnson M. D., Kehl-Fie T. E., Klein R., Kelly J., Burnham C., Mann B., and Rosch J. W. (2015) Role of copper efflux in pneumococcal pathogenesis and resistance to macrophage-mediated immune clearance. Infect. Immun. 83, 1684–1694 10.1128/IAI.03015-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Osman D., Waldron K. J., Denton H., Taylor C. M., Grant A. J., Mastroeni P., Robinson N. J., and Cavet J. S. (2010) Copper homeostasis in Salmonella is atypical and copper-CueP Is a major periplasmic metal complex. J. Biol. Chem. 285, 25259–25268 10.1074/jbc.M110.145953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ladomersky E., Khan A., Shanbhag V., Cavet J. S., Chan J., Weisman G. A., and Petris M. J. (2017) Host and pathogen copper-transporting P-type ATPases function antagonistically during Salmonella infection. Infect. Immun. 85, e00351–00317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wagner D., Maser J., Lai B., Cai Z., Barry C. E. 3rd., Höner zu Bentrup K., Russell D. G., and Bermudez L. E. (2005) Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell's endosomal system. J. Immunol. 174, 1491–1500 10.4049/jimmunol.174.3.1491 [DOI] [PubMed] [Google Scholar]
- 67. Raja M. R., Waterman S. R., Qiu J., Bleher R., Williamson P. R., and O'Halloran T. V. (2013) A copper hyperaccumulation phenotype correlates with pathogenesis in Cryptococcus neoformans. Metallomics 5, 363–371 10.1039/c3mt20220h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ding C., Festa R. A., Chen Y.-L., Espart A., Palacios Ò., Espín J., Capdevila M., Atrian S., Heitman J., and Thiele D. J. (2013) Cryptococcus neoformans copper detoxification machinery is critical for fungal virulence. Cell Host Microbe 13, 265–276 10.1016/j.chom.2013.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li C. X., Gleason J. E., Zhang S. X., Bruno V. M., Cormack B. P., and Culotta V. C. (2015) Candida albicans adapts to host copper during infection by swapping metal cofactors for superoxide dismutase. Proc. Natl. Acad. Sci. U.S.A. 112, E5336–E5342 10.1073/pnas.1513447112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gleason J. E., Galaleldeen A., Peterson R. L., Taylor A. B., Holloway S. P., Waninger-Saroni J., Cormack B. P., Cabelli D. E., Hart P. J., and Culotta V. C. (2014) Candida albicans SOD5 represents the prototype of an unprecedented class of Cu-only superoxide dismutases required for pathogen defense. Proc. Natl. Acad. Sci. U.S.A. 111, 5866–5871 10.1073/pnas.1400137111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Festa R. A., Helsel M. E., Franz K. J., and Thiele D. J. (2014) Exploiting innate immune cell activation of a copper-dependent antimicrobial agent during infection. Chem. Biol. 21, 977–987 10.1016/j.chembiol.2014.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fouani L., Menezes S. V., Paulson M., Richardson D. R., and Kovacevic Z. (2017) Metals and metastasis: Exploiting the role of metals in cancer metastasis to develop novel anti-metastatic agents. Pharmacol. Res. 115, 275–287 10.1016/j.phrs.2016.12.001 [DOI] [PubMed] [Google Scholar]
- 73. Raza M. H., Siraj S., Arshad A., Waheed U., Aldakheel F., Alduraywish S., and Arshad M. (2017) ROS-modulated therapeutic approaches in cancer treatment. J. Cancer Res. Clin. Oncol. 143, 1789–1809 10.1007/s00432-017-2464-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Safi R., Nelson E. R., Chitneni S. K., Franz K. J., George D. J., Zalutsky M. R., and McDonnell D. P. (2014) Copper signaling axis as a target for prostate cancer therapeutics. Cancer Res. 74, 5819–5831 10.1158/0008-5472.CAN-13-3527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cater M. A., Pearson H. B., Wolyniec K., Klaver P., Bilandzic M., Paterson B. M., Bush A. I., Humbert P. O., La Fontaine S., Donnelly P. S., and Haupt Y. (2013) Increasing intracellular bioavailable copper selectively targets prostate cancer cells. ACS Chem. Biol. 8, 1621–1631 10.1021/cb400198p [DOI] [PubMed] [Google Scholar]
- 76. Denoyer D., Pearson H. B., Clatworthy S. A., Smith Z. M., Francis P. S., Llanos R. M., Volitakis I., Phillips W. A., Meggyesy P. M., Masaldan S., and Cater M. A. (2016) Copper as a target for prostate cancer therapeutics: copper-ionophore pharmacology and altering systemic copper distribution. Oncotarget 7, 37064–37080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jain S., Cohen J., Ward M. M., Kornhauser N., Chuang E., Cigler T., Moore A., Donovan D., Lam C., Cobham M. V., Schneider S., Hurtado Rúa S. M., Benkert S., Mathijsen Greenwood C., Zelkowitz R., et al. (2013) Tetrathiomolybdate-associated copper depletion decreases circulating endothelial progenitor cells in women with breast cancer at high risk of relapse. Ann. Oncol. 24, 1491–1498 10.1093/annonc/mds654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chan N., Willis A., Kornhauser N., Ward M. M., Lee S. B., Nackos E., Seo B. R., Chuang E., Cigler T., Moore A., Donovan D., Vallee Cobham M., Fitzpatrick V., Schneider S., Wiener A., et al. (2017) Influencing the tumor microenvironment: a phase II study of copper depletion using tetrathiomolybdate in patients with breast cancer at high risk for recurrence and in preclinical models of lung metastases. Clin. Cancer Res. 23, 666–676 10.1158/1078-0432.CCR-16-1326 [DOI] [PubMed] [Google Scholar]
- 79. Davies H., Bignell G. R., Cox C., Stephens P., Edkins S., Clegg S., Teague J., Woffendin H., Garnett M. J., Bottomley W., Davis N., Dicks E., Ewing R., Floyd Y., Gray K., et al. (2002) Mutations of the BRAF gene in human cancer. Nature 417, 949–954 10.1038/nature00766 [DOI] [PubMed] [Google Scholar]
- 80. Wagle N., Emery C., Berger M. F., Davis M. J., Sawyer A., Pochanard P., Kehoe S. M., Johannessen C. M., Macconaill L. E., Hahn W. C., Meyerson M., and Garraway L. A. (2011) Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 29, 3085–3096 10.1200/JCO.2010.33.2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Turski M. L., Brady D. C., Kim H. J., Kim B. E., Nose Y., Counter C. M., Winge D. R., and Thiele D. J. (2012) A novel role for copper in Ras/Mitogen-activated protein kinase signaling. Mol. Cell. Biol. 32, 1284–1295 10.1128/MCB.05722-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Brady D. C., Crowe M. S., Turski M. L., Hobbs G. A., Yao X., Chaikuad A., Knapp S., Xiao K., Campbell S. L., Thiele D. J., and Counter C. M. (2014) Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 509, 492–496 10.1038/nature13180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Morrell A., Tallino S., Yu L., and Burkhead J. L. (2017) The role of insufficient copper in lipid synthesis and fatty-liver disease. IUBMB Life 69, 263–270 10.1002/iub.1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Santos E. M., Ball J. S., Williams T. D., Wu H., Ortega F., van Aerle R., Katsiadaki I., Falciani F., Viant M. R., Chipman J. K., and Tyler C. R. (2010) Identifying health impacts of exposure to copper using transcriptomics and metabolomics in a fish model. Environ. Sci. Technol. 44, 820–826 10.1021/es902558k [DOI] [PubMed] [Google Scholar]
- 85. Burkhead J. L., and Lutsenko S. (2013) in The Role of Copper as a Modifier of Lipid Metabolism (Valenzuela Baez R, ed) InTech, London, UK [Google Scholar]
- 86. Aigner E., Strasser M., Haufe H., Sonnweber T., Hohla F., Stadlmayr A., Solioz M., Tilg H., Patsch W., Weiss G., Stickel F., and Datz C. (2010) A role for low hepatic copper concentrations in nonalcoholic fatty liver disease. Am. J. Gastroenterol. 105, 1978–1985 10.1038/ajg.2010.170 [DOI] [PubMed] [Google Scholar]
- 87. Heffern M. C., Park H. M., Au-Yeung H. Y., Van de Bittner G. C., Ackerman C. M., Stahl A., and Chang C. J. (2016) In vivo bioluminescence imaging reveals copper deficiency in a murine model of nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. U.S.A. 113, 14219–14224 10.1073/pnas.1613628113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Huster D., Purnat T. D., Burkhead J. L., Ralle M., Fiehn O., Stuckert F., Olson N. E., Teupser D., and Lutsenko S. (2007) High copper selectively alters lipid metabolism and cell cycle machinery in the mouse model of Wilson disease. J. Biol. Chem. 282, 8343–8355 10.1074/jbc.M607496200 [DOI] [PubMed] [Google Scholar]
- 89. Muchenditsi A., Yang H., Hamilton J. P., Koganti L., Housseau F., Aronov L., Fan H., Pierson H., Bhattacharjee A., Murphy R., Sears C., Potter J., Wooton-Kee C. R., and Lutsenko S. (2017) Targeted inactivation of copper transporter Atp7b in hepatocytes causes liver steatosis and obesity in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 313, G39–G49 10.1152/ajpgi.00312.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liggi M., Murgia D., Civolani A., Demelia E., Sorbello O., and Demelia L. (2013) The relationship between copper and steatosis in Wilson's disease. Clin. Res. Hepatol. Gastroenterol. 37, 36–40 10.1016/j.clinre.2012.03.038 [DOI] [PubMed] [Google Scholar]
- 91. Lutsenko S. (2014) Modifying factors and phenotypic diversity in Wilson's disease. Ann. N.Y. Acad. Sci. 1315, 56–63 10.1111/nyas.12420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Krishnamoorthy L., Cotruvo J. A. Jr., Chan J., Kaluarachchi H., Muchenditsi A., Pendyala V. S., Jia S., Aron A. T., Ackerman C. M., Wal M. N., Guan T., Smaga L. P., Farhi S. L., New E. J., Lutsenko S., and Chang C. J. (2016) Copper regulates cyclic-AMP-dependent lipolysis. Nat. Chem. Biol. 12, 586–592 10.1038/nchembio.2098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Carmen G.-Y., and Víctor S.-M. (2006) Signalling mechanisms regulating lipolysis. Cell. Signal. 18, 401–408 10.1016/j.cellsig.2005.08.009 [DOI] [PubMed] [Google Scholar]
- 94. Hong-Hermesdorf A., Miethke M., Gallaher S. D., Kropat J., Dodani S. C., Chan J., Barupala D., Domaille D. W., Shirasaki D. I., Loo J. A., Weber P. K., Pett-Ridge J., Stemmler T. L., Chang C. J., and Merchant S. S. (2014) Subcellular metal imaging identifies dynamic sites of Cu accumulation in Chlamydomonas. Nat. Chem. Biol. 10, 1034–1042 10.1038/nchembio.1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hatori Y., and Lutsenko S. (2016) The role of copper chaperone Atox1 in coupling redox homeostasis to intracellular copper distribution. Antioxidants 5, 25 10.3390/antiox5030025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Field L. S., Luk E., and Culotta V. C. (2002) Copper chaperones: personal escorts for metal ions. J. Bioenerg. Biomembr. 34, 373–379 10.1023/A:1021202119942 [DOI] [PubMed] [Google Scholar]
- 97. Fahrni C. J. (2013) Synthetic fluorescent probes for monovalent copper. Curr. Opin. Chem. Biol. 17, 656–662 10.1016/j.cbpa.2013.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chang C. J. (2017) Bioinorganic life and neural activity: toward a chemistry of consciousness? Acc. Chem. Res. 50, 535–538 10.1021/acs.accounts.6b00531 [DOI] [PMC free article] [PubMed] [Google Scholar]