

Abstract
The epithelial Na+ channel (ENaC) is a member of the ENaC/degenerin family of ion channels. In the structure of a related family member, the “thumb” domain's base interacts with the pore, and its tip interacts with the divergent “finger” domain. Between the base and tip, the thumb domain is characterized by a conserved five-rung disulfide ladder holding together two anti-parallel α helices. The ENaC α and γ subunits' finger domains harbor autoinhibitory tracts that can be proteolytically liberated to activate the channel and also host an ENaC-specific pair of cysteines. Using a crosslinking approach, we show that one of the finger domain cysteines in the α subunit (αCys-263) and both of the finger domain cysteines in the γ subunit (γCys-213 and γCys-220) lie near the dynamic finger–thumb domain interface. Our data suggest that the αCys-256/αCys-263 pair is not disulfide-bonded. In contrast, we found that the γCys-213/γCys-220 pair is disulfide-bonded. Our data also suggest that the γ subunit lacks the terminal rung in the thumb domain disulfide ladder, suggesting asymmetry between the subunits. We also observed functional asymmetry between the α and γ subunit finger–thumb domain interfaces; crosslinks bridging the α subunit finger–thumb interface only inhibited ENaC currents, whereas crosslinks bridging the γ subunit finger–thumb interface activated or inhibited currents dependent on the length of the crosslinker. Our data suggest that reactive cysteines lie at the dynamic finger–thumb interfaces of the α and γ subunits and may play a yet undefined role in channel regulation.
Keywords: allosteric regulation, cysteine-mediated crosslinking, epithelial sodium channel (ENaC), protein conformation, reactive oxygen species (ROS)
Introduction
The epithelial Na+ channel (ENaC)2 facilitates Na+ transport in the aldosterone-sensitive distal nephron, the respiratory tract, and the colon (1–4). ENaC-mediated Na+ reabsorption in the distal nephron is critical to K+ secretion and plays a key role in extracellular fluid volume homeostasis and blood pressure maintenance (1). ENaC also plays sensory roles in the taste buds, vascular endothelia and smooth muscle, and the brain (5). At the luminal surface in its varied roles, ENaC is exposed to dynamic extracellular environments and is responsive to stimuli in those environments, including Na+, Cl−, H+, and shear stress. These factors directly regulate the channel by influencing the channel's open probability (6).
ENaC is a member of the ENaC/degenerin family of ion channels, which are cation-selective channels that gate in response to extracellular cues (6). There are four ENaC subunits, α, β, γ, and δ, that assemble to form either αβγ or δβγ heterotrimers. Channel assemblies that include ENaC subunits and acid-sensing ion channel (ASIC) subunits have also been reported (7–9). Sequence comparisons between ENaC subunits and ASIC1, whose structure has been reported, suggest that the core folds of the proteins are similar (palm and β-ball domains in Fig. 1A), but that the peripheral helical domains (finger, thumb, and knuckle in Fig. 1A) are divergent and may underlie functional differences between the channels (10, 11). The sites of channel activating proteolytic cleavage in the α and γ subunits map to their respective finger domains (6, 12, 13), underlining the notion that the divergent domains are functionally important. We previously reported a model of the α subunit extracellular domains using ASIC1 homology complemented by constraints for the divergent finger domain derived from functional experiments (10).
Figure 1.

ENaC subunit cysteines. A, schematic of an ENaC subunit, adapted from Ref. 6. Cysteine residues are represented by yellow circles, and disulfide bonds observed in the ASIC1 structure are represented by black bars. Conserved aspartic acids αAsp-171 and γAsp-115 are represented by a red circle, and αTyr-474 is represented by a blue circle. B, sequence alignment of finger domain segment hosting conserved cysteines in ENaC subunits. C, sequence alignment of the thumb domain residues proximal to the finger domain in the ASIC1 structure. Identities are shaded gray. Cysteines are shaded black.
ENaC/degenerin proteins contain 14 conserved cysteines in the extracellular domains (11, 14). Based on functional experiments, Sheng et al. (14) proposed a model for the α subunit where 10 of the cysteines formed a disulfide bond ladder that defined one domain, and the remaining 4 cysteines formed two disulfide bonds elsewhere in the structure. The ASIC1 structure accorded with these results and showed that the disulfide bond ladder held two helices together in an antiparallel fashion and formed the core of the thumb domain (Fig. 1A) (11). Firsov et al. also functionally examined the extracellular domain cysteines, and proposed asymmetry between the subunits regarding the cysteine pair at the top of the thumb domain (15). The thumb domain of each subunit interacts with the finger domain at one end and the extracellular entry to the channel pore at the other. Each ENaC subunit finger domain contains two cysteines that are unique to ENaC subunits in the protein family (Fig. 1 (A and B); Cys-2 and Cys-3) and two additional cysteines that are unique to ENaC β subunits (Fig. 1B). Although we assumed the finger domain cysteines to be disulfide-bonded in our α subunit homology model, the disposition of these cysteines had not been experimentally determined.
Double cleavage of the ENaC α and γ subunits liberates autoinhibitory tracts from each subunit (13, 16). Synthetic peptides corresponding to each of the liberated tracts inhibit the channel (17, 18). We have shown that the α subunit-derived inhibitory peptide and the autoinhibitory tract from which it is derived probably bind the α subunit at overlapping sites (10, 19). One key feature of these sites is involvement of the finger–thumb domain interface. We hypothesized that α subunit cleavage activates the channel by releasing an interaction between the finger and thumb domains. We proposed that this conformational change propagates through the thumb domain to the transmembrane helices that form the channel pore (see Fig. 1A). Indeed, directly crosslinking the finger–thumb interface inhibited the channel in a crosslinker length–dependent manner (20).
Here, through crosslinking experiments, we explore the finger–thumb domain interfaces of the α and γ subunits. We show that both interfaces are dynamically linked to channel activity. We also demonstrate that the conserved cysteines in the finger domains of the α and γ subunits (Cys-2 and Cys-3) lie close to the respective finger–thumb domain interfaces. However, our data suggest that whereas the γ subunit finger domain cysteines are probably disulfide-bonded, the α subunit equivalents are not. Additionally, our data suggest that the γ subunit harbors a pair of thumb domain cysteines at the finger–thumb domain interface that are not likely to be disulfide-bonded. These data suggest that reactive cysteines lie at the dynamic finger–thumb interfaces of the α and γ subunits.
Results
Finger domain cysteines in the α subunit are near the finger–thumb domain interface
A peptide derived from the furin-released autoinhibitory fragment of the α subunit decreased channel currents by binding at the finger–thumb domain interface of the α subunit (19). Previously, we recapitulated this effect in the absence of the inhibitory peptide by constraining the distance between the finger domain (using residue 171 in helix 1; red circle in Fig. 1A) and the thumb domain (blue circle in Fig. 1A) of the α subunit using short methanethiosulfonate (MTS)-based crosslinkers (20). Inhibition by MTS crosslinkers was length-dependent. To further examine the dynamics at the finger–thumb domain interface, we aimed to induce a direct disulfide bridge between cysteines introduced at positions 171 and 474 in the α subunit using the oxidizing agent copper phenanthroline (Cu-Phe) (21). However, when we applied 4 μm Cu-Phe for 15 s to the αY474C single mutant co-expressed with wildtype β and γ subunits in Xenopus oocytes, we observed a modest inhibition of channel currents (Fig. 2). Cu2+ alone had no effect on αY474C currents (Fig. 2A, cyan traces), reducing currents by 5 ± 2% (n = 7, p = not significant by paired t test). Inhibition by Cu-Phe reversed only upon the addition of the reducing agent DTT (Fig. 2A), suggesting a covalent modification of the channel. This contrasted with effects on wildtype currents, which were unaffected by 4 μm Cu-Phe or 4 μm Cu2+ addition. The absence of an effect with Cu2+ alone is consistent with previous work (22). Notably, extracellular DTT treatment did not affect wildtype currents, consistent with previous studies (20, 23, 24). Wildtype channel insensitivity to DTT suggests either that native extracellular disulfide bonds are not susceptible to reduction by DTT or that reduction of those bonds does not affect channel function. Although Cu-Phe–induced disulfide bonds may also be DTT-insensitive, DTT reversal after oxidation in the case of αY474C suggests reduction of an induced disulfide bond. We also found that Cu-Phe had no effect on neighboring αK476C mutant channels, showing that the Cu-Phe effect is specific to αY474C (Fig. 2, A and C).
Figure 2.
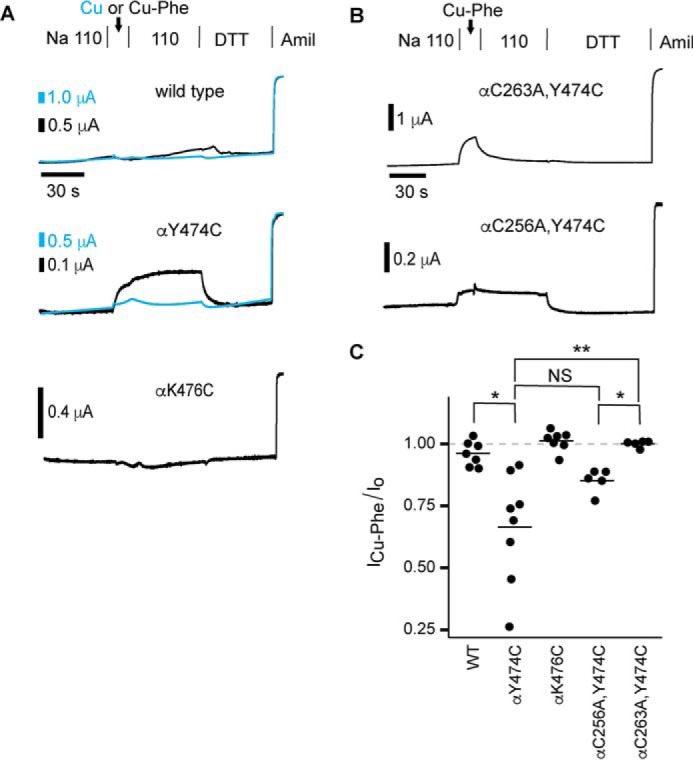
Conserved αCys-263 in the finger domain is proximal to the finger–thumb domain interface. Mutant or wildtype α subunits with wildtype β and γ subunits were expressed in Xenopus oocytes, and currents were measured by two-electrode voltage clamp at −100 mV. Oocytes were treated with 4 μm Cu-Phe for 15 s, followed by 110-Na buffer for 60 s and 10 mm DTT for 60–90 s. A and B, representative recordings. C, inhibition by Cu-Phe was determined by comparing the amiloride-sensitive current at the end of Cu-Phe washout with the amiloride-sensitive current before the addition of Cu-Phe. Bars, mean of each group. *, p < 0.05; **, p < 0.01 versus wildtype by Kruskal–Wallis rank sum test (p < 0.0001) followed by Dunn test for multiple comparisons with p values adjusted with the Benjamini–Hochberg method. NS, not significant.
We hypothesized that Y474C formed a disulfide bond with an endogenous cysteine in the presence of Cu-Phe. ENaC subunits have cysteine pairs that correspond to all seven disulfide-bridged pairs observed in the extracellular domains of the ASIC1 structure (Fig. 1). Functional experiments suggest that these cysteines may be similarly disulfide-bonded in ENaC subunits (14, 15). ENaC subunits have an additional pair of conserved cysteines in their finger domains (Fig. 1 (A and B); Cys-2 and Cys-3). The disulfide bonding status and position of the finger domain cysteine pair is unknown, although our α subunit homology model posited Cys-256 (αCys-2) and Cys-263 (αCys-3) in a disulfide bond and placed them more than 15 Å from Tyr-474 (10). To test whether Cu-Phe induced a bond between αY474C and either of the α subunit finger domain cysteines, we mutated each to alanine in the background of αY474C and treated oocytes expressing the double mutants with 4 μm Cu-Phe. We found that Cu-Phe inhibited αC256A,Y474C currents in a DTT-reversible manner, similar to what we observed with the αY474C single mutant and different from wildtype. In contrast, any Cu-Phe effect on αC263A,Y474C currents reversed upon washout of the oxidant, and DTT had no effect on measured currents. We note here that we frequently observed Cu-Phe inhibition that reversed upon washout prior to DTT addition in each of the groups tested. This suggests that an unstable conformational change may be induced by Cu-Phe, but this effect is difficult to interpret. These results show that DTT-reversible Cu-Phe inhibition of αY474C requires endogenous αCys-263. These results suggest that αCys-263 is in close proximity to the finger–thumb domain interface and is free to form a disulfide bond in the presence of αY474C.
Crosslinking across the γ subunit finger–thumb domain interface potentiates ENaC currents
Sequence homology suggests significant structural and functional symmetry between ENaC subunits (25), yet functional differences are clearly evident. For example, the α and γ subunits are subject to activating proteolysis, whereas the β and δ subunits are not (12, 26, 27). We hypothesized that the conformations of the channel pore and the γ subunit's finger–thumb domain interface were coupled, analogous to the α subunit (20). We introduced a cysteine at position 115 in the γ subunit (γD115C), which is equivalent to site 171 in helix 1 of the α subunit at the finger–thumb domain interface (see Fig. 1A). We previously reported evidence of crosslinking between this site and Y474C at the top of the thumb domain in the α subunit (20). When we perfused bifunctional MTS-2-MTS for 1 min, we observed an increase in channel currents that remained elevated after washout of the crosslinker (Fig. 3A). DTT appeared to slowly reverse the activation, although its slow kinetics made it difficult to distinguish it from channel run-down. Furthermore, the effect was dependent on the length of the crosslinker. Neither MTS-1-MTS nor MTS-3-MTS had an effect on γD115C currents (Fig. 3B). In contrast, 1-min treatment of each of these MTS crosslinkers irreversibly inhibited wildtype ENaC currents by an average of 24% and in each case was different from the mutant tested with the same crosslinker. We and others have previously reported this effect on wildtype ENaC and that it is attenuated by amiloride (20, 23, 28). Our data showing MTS-2-MTS activation of γD115C in contrast to inhibition of wildtype channels suggest covalent modification of γD115C. We hypothesized that the length-dependent activation and DTT reversibility observed with γD115C resulted from crosslinking between γD115C on helix 1 in the finger domain and an endogenous cysteine.
Figure 3.

Crosslinkers activate γD115C in a length-dependent manner. Wildtype or D115C γ subunits were co-expressed with wildtype α and β ENaC subunits in Xenopus oocytes, and currents were measured by two-electrode voltage clamp at −100 mV. Oocytes were treated with 10 μm MTS-n-MTS for 60 s, followed by 110-Na buffer for 60 s and 10 mm DTT for 90 s. A, representative recordings are shown. MTS-2-MTS inhibited currents of wildtype (p < 0.05 by paired Student's t test), and subsequent DTT addition had no effect. In contrast, MTS-2-MTS addition increased currents of γD115C (p < 0.0001 by paired Student's t test), and subsequent DTT addition reversed the activation (p < 0.001 by paired Student's t test). B, the change in current due to MTS-n-MTS was determined by comparing the amiloride-sensitive current at the end of MTS-n-MTS washout with the amiloride-sensitive current before MTS-n-MTS treatment. Bars, mean of each group. **, p < 0.01; ****, p < 0.0001 by one-way ANOVA followed by Tukey post hoc analysis.
To test our hypothesis, we examined endogenous cysteines that may be nearby. The γ subunit finger domain has 2 cysteines, γCys-213 (γCys-2) and γCys-220 (γCys-3), that are conserved among ENaC subunits (Fig. 1, A and B). To identify the putative endogenous cysteine required for γD115C activation by MTS-2-MTS, we tested γD115C channels where candidate cysteines were mutated to alanine (Fig. 4A). When we tested both γD115C,C213A and γD115C,C220A, we found that MTS-2-MTS activated both channels similarly to MTS-2-MTS activation of γD115C (Figs. 4, B and C). γD115C lies on helix 1 in the finger domain at the finger–thumb domain interface (see Figs. 1A and 4A). The thumb domain has 4 cysteines that may form two disulfide pairs near the finger–thumb interface (Figs. 1, A and C) (11, 14, 15). In the mouse ENaC γ subunit, the two thumb domain pairs are γCys-413/γCys-440 (γCys-10/Cys-13) and γCys-415/γCys-429 (γCys-11/Cys-12). Firsov et al. (15) suggested that γCys-415 and γCys-429 may not be disulfide-bonded. We tested γD115C channels bearing alanine mutations at each of these sites with MTS-2-MTS. We found that γD115C,C415A channels were not activated by MTS-2-MTS, which was different from MTS-2-MTS activation of γD115C (Fig. 4C). In contrast, γD115C,C429A and γD115C,C440A channels were activated by MTS-2-MTS, but that activation was attenuated compared with γD115C channels. We could not test γD115C,C413A due to poor functional expression. Our results show that MTS-2-MTS activation of γD115C requires endogenous γCys-415. The results of these experiments suggest that in the presence of γD115C, γCys-415 is free and in close proximity to γD115C.
Figure 4.

Endogenous γC415A is required for crosslinker activation of γD115C. A, schematic of γ subunit finger–thumb interface depicting locations of D115C on helix 1 (rectangle), and endogenous cysteines that may be nearby. Dashed lines, possible disulfide bonds. B, smaller schematics indicate which endogenous cysteine was mutated to alanine (open circle). Mutant γ subunits were co-expressed with wildtype α and β subunits in Xenopus oocytes, and currents were measured by two-electrode voltage clamp at −100 mV. Oocytes were treated with 10 μm MTS-2-MTS for 60 s, followed by 110-Na buffer for 60 s and then 10 mm DTT for 90 s. Representative recordings are shown. C, inhibition by MTS-2-MTS was quantified by comparing the amiloride-sensitive current at the end of MTS-2-MTS washout with the amiloride-sensitive current before MTS-2-MTS treatment. Bars, mean of each group. Dashed lines, mean values for the effect of MTS-2-MTS on wildtype and γD115C channels from Fig. 3. *, p < 0.05; ***, p < 0.001; ****, p < 0.0001 versus γD115C by one-way ANOVA followed by Tukey post hoc analysis.
Endogenous γ subunit finger domain cysteines are near the finger–thumb domain interface
In the course of performing the experiments shown in Fig. 4, we observed that both the γC213A (γCys-2) and γC220A (γCys-3) single mutants responded to bifunctional MTS crosslinkers differently than wildtype (Fig. 5). Whereas wildtype currents were consistently inhibited by each of the MTS crosslinkers tested and were not DTT-sensitive, currents from γC213A and γC220A channels both responded in a crosslinker length–dependent manner (Fig. 5 and Table 1). MTS-1-MTS had little effect on γC213A and γC220A channels. MTS-2-MTS stimulated currents from both mutant channels, which reversed upon DTT addition. This potentiation was in stark contrast to MTS-2-MTS inhibition of wildtype channels. MTS-4-MTS had no effect on either mutant channel; this non-effect was also different from MTS-4-MTS inhibition of wildtype channels. In contrast, MTS-8-MTS inhibited both γC213A and γC220A in a DTT-insensitive manner, reminiscent of the effect of MTS-8-MTS on wildtype channels. Clearly, the effects of the shorter MTS crosslinkers were length-dependent for γC213A and γC220A (see Table 1) and slowly reversed with DTT. These data suggest that the shorter MTS compounds bridged the remaining γ subunit finger domain cysteine and a nearby endogenous cysteine, whereas MTS-8-MTS may have inhibited mutant channel currents analogously to wildtype channels. Our data suggest that γCys-213 and γCys-220 are ordinarily disulfide-bonded but that when either is mutated to alanine, the other becomes capable of crosslinking to an endogenous cysteine. In support of this notion, when we mutated both γCys-213 and γCys-220 to alanine, the current response to both MTS-2-MTS and MTS-8-MTS reverted to wildtype (Fig. 5).
Figure 5.

Endogenous γ subunit finger domain cysteines are near the finger–thumb domain interface. Mutant γ subunits were co-expressed with wildtype α and β subunits in oocytes, and currents were measured by two-electrode voltage clamp at −100 mV. ENaC-expressing oocytes were treated with 10 μm MTS-n-MTS as indicated for 60 s, followed by 110-Na buffer for 60 s and then 10 mm DTT for 90 s. A, representative recordings are shown for wildtype and each mutant with an accompanying schematic illustrating the endogenous cysteine(s) removed (open circles). B, the effect of MTS-n-MTS was determined by comparing the amiloride-sensitive current at the end of MTS-n-MTS washout to the amiloride-sensitive current before MTS-n-MTS treatment. Bars, mean of each group. Data were analyzed by two-way ANOVA, which indicated differences between channels and between crosslinkers (p < 0.0001). We compared individual pairs using Tukey's multiple comparison test. Indicated in B are comparisons between γC213A or γC220A and wildtype channels with a given MTS compound (left column) and between double mutants bearing γC415A or γC429A and the corresponding single mutants, γC213A or γC220A, with a given MTS compound (across the top or center rows, respectively). *, p < 0.05; ****, p < 0.0001. Comparisons of the effects of different crosslinkers on a given channel are shown in Table 1.
Table 1.
Crosslinker length–dependent (MTS-n-MTS) effects on γ subunit cysteine mutants near the finger–thumb domain
For each channel in Fig. 5, we compared the effect of using a crosslinker of a given length with all other crosslinkers tested. The number of carbon atoms between MTS functional groups for the tested pair is indicated. Pairwise comparisons were performed by two-way ANOVA followed by Tukey's post hoc test. *, p < 0.05, **, p < 0.01, ***, p < 0.001, ****, p < 0.0001.

We hypothesized that like the α subunit finger domain cysteines, the γ subunit finger domain cysteines lie near the finger–thumb domain interface and that MTS-2-MTS activation of both γC213A and γC220A mutant channels may require residue γCys-415 or possibly γCys-429. In the background of each single mutant, we mutated either γCys-415 or γCys-429 to alanine. We then tested the four double mutants with each of the bifunctional MTS crosslinkers (Fig. 5). MTS-2-MTS did not activate currents from γC213A,C415A, in clear contrast to MTS-2-MTS activation of the corresponding single mutant. None of the four double mutants exhibited differences between the effects of MTS-1-MTS, MTS-2-MTS, and MTS-4-MTS, in contrast to the γC213A and γC220A single mutants (Fig. 5 and Table 1). Further, γC213A,C415A and γC220A,C415A double mutants had attenuated responses to MTS-8-MTS compared with the corresponding single mutant. These data suggest that although MTS-8-MTS inhibition of γC213A and γC220A was not DTT-reversible, the effect requires endogenous γCys-415. On its own, mutation of γCys-415 to alanine did not change the effects of MTS-2-MTS or MTS-8-MTS on the channel (data not shown). Altogether, our results show that the crosslinker effects on the endogenous γ subunit finger domain cysteine mutants (γCys-2/Cys-3) depend on the presence of endogenous γCys-415 (γCys-11) at the top of the thumb domain.
MTS compound reduction of wildtype ENaC currents and effects on finger–thumb interface mutants are separate phenomena
If inhibition of wildtype ENaC by MTS crosslinkers was due to modification of a pore cysteine that blocks the pore, as we and others have proposed (20, 23, 28), we would not expect that mutants far from the pore could negate that inhibition. Introduction of a free cysteine at the γ subunit finger–thumb domain interface does exactly that, either through introduction of a novel cysteine (γD115C) or through mutation of one-half of a putative disulfide pair (γC213A or γC220A) (Figs. 3 and 5). This suggests that an alternate mechanism underlies bifunctional MTS compound inhibition of wildtype ENaC. To test whether residues that are involved in MTS compound-mediated inhibition of wildtype ENaC overlap with residues involved in activation in the cases of γC213A and γC220A, we performed a protection experiment (Fig. 6). Before a 1-min treatment with MTS-2-MTS, we treated wildtype and mutant channels with monofunctional MTS-2 for 1 min or alternatively with vehicle. Whereas mock-pretreated mutants were activated by MTS-2-MTS as seen before in Fig. 5, MTS-2 pretreatment attenuated activation by MTS-2-MTS for both γC213A and γC220A by 61 and 81%, respectively. In contrast, MTS-2 pretreatment of wildtype channels had no effect on subsequent inhibition by MTS-2-MTS (Fig. 6). These data show that MTS-2 protects the γC213A and γC220A mutants from reaction with MTS-2-MTS but that MTS-2 cannot protect wildtype ENaC from irreversible inhibition by MTS-2-MTS. These data suggest that the mechanisms underlying wildtype channel inhibition and γC213A or γC220A activation by MTS compounds are distinct.
Figure 6.
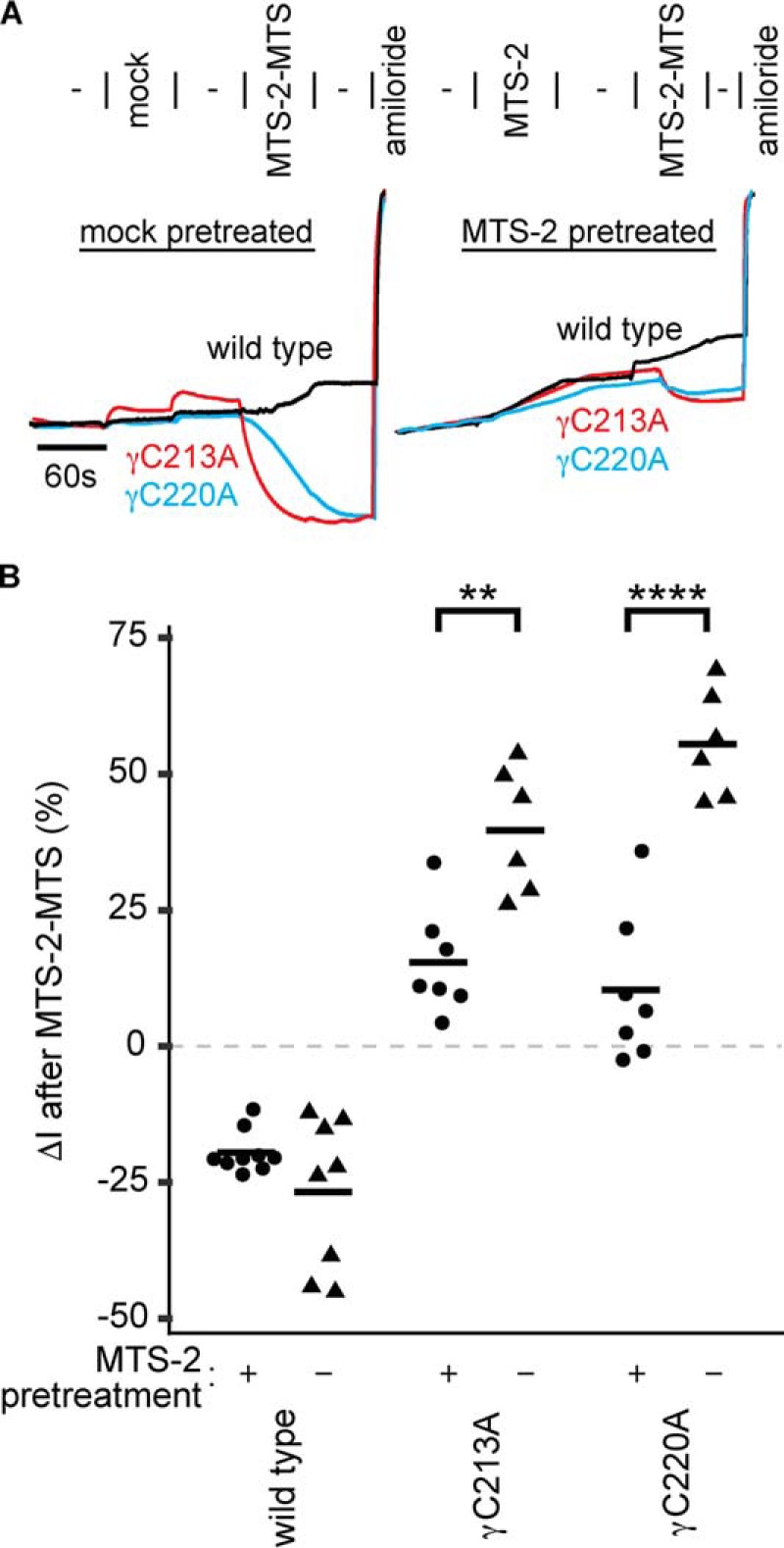
Monofunctional MTS-2 protects mutant channels but not wildtype channels from bifunctional MTS-2-MTS. Mutant or wildtype γ subunits were co-expressed with wildtype α and β ENaC subunits in Xenopus oocytes. A, ENaC currents were measured using two-electrode voltage clamp at −100 mV. Oocytes were mock-treated or treated with 10 μm MTS-2 for 60 s followed by washout with 110-Na buffer for 60 s. Oocytes were then treated with 10 μm MTS-2-MTS for 60 s followed by washout with 110-Na buffer for 60 s. B, the effect of MTS-2-MTS treatment after mock or MTS-2 treatment was determined by comparing current after washout of MTS-2-MTS with current just before MTS-2-MTS addition. Bars, mean. **, p < 0.01; ****, p < 0.0001, by Student's t test.
Mutation of finger–thumb domain interface cysteines reduces channel expression at the cell surface
We observed reduced currents for channels with mutated finger–thumb domain interface cysteines. Changes in whole-cell currents may reflect changes in the number of channels at the cell surface and/or the biophysical characteristics of those channels. To determine whether these mutations affected the number of channels at the cell surface, we incorporated a β-subunit with an extracellular FLAG tag to perform a chemiluminescence-based assay. One day after cRNA injection of oocytes, we measured amiloride-sensitive currents by two-electrode voltage clamp at −100 mV (Fig. 7A). Currents were greatly reduced for αC256A,Y474C and αC263A,Y474C, moderately reduced for γC415A and γC429A, and modestly reduced for γC220A. Two days after cRNA injection, we used an anti-FLAG antibody to quantify channels on the cell surface (Fig. 7B). Surface expression measurements largely paralleled current measurements. Similar to the effect on currents, αC256A,Y474C and αC263A,Y474C exhibited the greatest reductions in surface expression. Surface expression of γC213A was modestly reduced where currents were unchanged, and surface expression of γC429A was unchanged where currents were moderately reduced. These data suggest that changes in cell-surface expression largely account for the differences in currents that we observed but that there are probably differences at the single-channel level as well.
Figure 7.

Mutation of finger–thumb domain interface cysteines reduces cell-surface expression. Extracellular FLAG-epitope tagged β subunits (βF) were co-expressed with wildtype and mutant ENaC subunits, as indicated. A, 1 day after cRNA injection of oocytes, amiloride-sensitive (10 μm) currents were measured using a two-electrode voltage clamp at −100 mV. B, 2 days after injection, the remaining oocytes were placed on ice, and anti-FLAG antibodies were used to detect channels at the cell surface by chemiluminescence. Bars, mean for each group. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 versus αβFγ by one-way ANOVA followed by Tukey post hoc analysis.
Finger–thumb domain interface cysteines do not mediate inhibition by SIN-1, a peroxynitrite generator
Our data suggest that neither αCys-256/αCys-263 (αCys-2/Cys-3) in the finger domain nor γCys-415/γCys-429 (γCys-11/Cys-12) in the thumb domain are disulfide-bonded. That their modification can modulate channel activity raises the possibility that oxidants or reactive oxygen species can directly regulate ENaC through these sites. Reactive oxygen species have been shown to regulate ENaC activity (29–31). DuVall et al. reported that peroxynitrite produced from SIN-1 inhibited currents from ENaC expressed in oocytes (32). SIN-1 reacts at physiological pH to produce nitric oxide and superoxide, which subsequently react to form peroxynitrite. SIN-1 treatment of oocytes expressing ENaC rapidly inhibited amiloride-sensitive currents (76 ± 16%) (Fig. 8, A and B). Subsequent removal of SIN-1 from the perfusate rapidly restored currents to 77 ± 12% of their starting values (Fig. 8, A and C). We then tested alanine mutants of αCys-256, αCys-263, γCys-415, and γCys-429 to determine whether SIN-1–dependent inhibition required cysteine residues at any of these sites. For each of the mutants tested, the reduction of current in the presence of SIN-1 was similar to wildtype. Nor did any of the mutants tested reduce the sustained inhibition observed after SIN-1 removal. On the contrary, αC256A increased inhibition observed after SIN-1 removal, compared with wildtype. Our data suggest that SIN-1–dependent ENaC inhibition does not require any of the cysteines tested here.
Figure 8.

SIN-1–mediated inhibition of ENaC does not require free cysteines at finger–thumb interface of the α or γ subunits. Mutant ENaC subunits were co-expressed with wildtype cognate subunits in Xenopus oocytes, and currents were measured by two-electrode voltage clamp at −100 mV. Solutions of 1 mm SIN-1 in 110-Na buffer were perfused for 10 min, preceded and followed by brief pulses with 10 μm amiloride. A, representative recordings are shown. B, rapid inhibition by SIN-1 was calculated by comparing the steady-state current in the presence of SIN-1 with the amiloride-sensitive current before adding SIN-1. C, sustained inhibition was calculated by comparing the steady-state current after washing out SIN-1 with the amiloride-sensitive current before adding SIN-1. *, p < 0.05 by one-way ANOVA followed by Dunnett's post hoc test.
Discussion
Two groups previously identified several disulfide pairs in the extracellular domains of the α, β, and γ subunits using a double-mutant cycle approach (14, 15). In accordance with these results, the structure of homologous ASIC1 showed that the 14 extracellular cysteines that are conserved between ENaC and ASIC subunits formed seven disulfide bonds (11). ENaC subunits have a unique pair of cysteines in the divergent finger domain. Experiments by Sheng et al. (14) to determine whether the conserved finger domain cysteines are disulfide-bonded to each other were either inconclusive or unsupportive of the presence of such a bond. Regarding the γCys-415/γCys-429 (γCys-11/Cys-12) pair, previous experiments suggested a disulfide bond for the equivalent pairs in the α (14, 15) and β subunits (15). However, neither study concluded that γCys-415 and γCys-429 are disulfide-bonded. Indeed, Firsov et al. (15) noted functional asymmetry regarding this cysteine pair in the γ subunit.
For the α subunit, our results suggest that αCys-263 (αCys-3) is near the finger–thumb domain interface and is free to form a disulfide bond with an introduced cysteine. We cannot rule out the possibility that αCys-256 (αCys-2) and αCys-263 (αCys-3) are a disulfide pair in the absence of an introduced cysteine in the thumb domain (e.g. αY474C). In contrast to the endogenous cysteines in the α subunit finger domain, our results suggest that γCys-213 (γCys-2) and γCys-220 (γCys-3) are disulfide-bonded. The removal of one by mutagenesis renders channel currents susceptible to cysteine-reactive MTS reagents. Furthermore, our data suggest that γCys-415 (γCys-11) and γCys-429 (γCys-12) are not disulfide-bonded, at least in the context of the γD115C, γC213A, or γC220A mutants, in accord with earlier studies (14, 15). This was somewhat surprising because earlier studies were ambiguous on the status of the γCys-415/γCys-429 pair, these residues are conserved across the protein family, and the equivalent sites in ASIC1 are disulfide-bonded (11). This finding suggests that thiol-reactive molecules could directly regulate the channel via an available cysteine near the finger–thumb interface of either the α or γ subunits. We tested SIN-1, a nitric oxide and peroxynitrite generator that has been reported to inhibit ENaC currents in oocytes (32, 33). We found that inhibition rapidly reversed when we removed SIN-1, suggesting either a non-covalent interaction or an unstable modification of the channel by SIN-1 or its decomposition products. We also found that none of the cysteines at the finger–thumb domain interface that our data suggest are free were required for SIN-1–dependent inhibition.
We also observed functional asymmetry between the α and γ subunits. We previously reported that constraining the finger–thumb interface of the α subunit using short bifunctional MTS crosslinkers inhibited ENaC (20). In contrast, here we show that constraining the finger–thumb interface through a number of different sites on the γ subunit finger domain using a short bifunctional MTS crosslinker activated ENaC. The disparity may reflect important differences in the experiment (e.g. involvement of endogenous cysteines in the γ thumb domain that are close but do not align with the thumb domain site we used in the α subunit). Alternatively, equivalent movements at the finger–thumb interface in the α and γ subunits may not have equivalent effects on the conformation of the channel pore. For example, the γ subunit's TM1–β1 linker, which may transduce conformational changes from the thumb domain to the pore (see Fig. 1A), is 2 residues shorter than those of either the α or β subunits. We previously used normal mode analysis to examine motions in the ectodomains that resonated with gating-like motions in the pore (10). Despite using a symmetric α3 ENaC homology model with or without the autoinhibitory tract in all subunits, we observed asymmetry in the finger and thumb domains. Plainly, a channel composed of distinct but related subunits could give rise to functional asymmetry. Other ENaC functional properties demonstrate asymmetry as well, including proteolytic activation (27), Cl− inhibition (34), Na+ self-inhibition (35, 36), and those relating to the pore (37, 38).
We have revised our model of the ENaC α subunit extracellular domains to account for our data. Our results suggest that αCys-263 and αCys-256 are not disulfide-bonded. Our data also suggest that αCys-263 (αCys-2) lies close to αTyr-474 in the finger–thumb domain interface, in contrast to our previous model. Our earlier model was based on homology to ASIC1 with the exception of the finger domain (10). We constrained the finger domain using predicted secondary structure and constraints derived from functional experiments examining inhibition by an α subunit-derived inhibitory peptide. These included scanning mutagenesis and double-mutant cycle experiments. We have subsequently described crosslinking experiments here (Fig. 2) and in previous reports (10, 20, 24). These allow for a straightforward derivation of new structural constraints. We have included these (Table 2) and generated a new model of the ENaC α subunit extracellular domains using Modeler (39). Several of the new constraints provide more specificity than those based on scanning mutagenesis. As a result, we removed previously derived constraints that included residues Glu-174, Ser-269, and Ser-270, leaving a total of 49 experimentally derived constraints. We generated 20 models (Fig. 9), and scored each by summing the violation distances of experimentally derived constraints (Fig. 9B). The best scoring models satisfy nearly all of the constraints that we imposed, with nine models exhibiting <1 Å of cumulative violations. Superimposing the models using backbone Cα atoms outside of the finger domain illustrates that the structure of the finger domain is better constrained near the finger–thumb interface that defines the N-terminal end of the inhibitory peptide binding pocket as compared with the C-terminal end of the peptide-binding pocket (Fig. 9A) or residues near either of the furin cleavage sites. Our best scoring model places αCys-263 close to αTyr-474 (Fig. 9, C and D), consistent with our data (see Fig. 2).
Table 2.
Experimentally derived constraints added to the ENaC α subunit model
Additional constraints consisted of residues (site 1) with a set maximum distance to another residue(s) (site 2). Constraints were added to the α ENaC model based on the results of Refs. 10, 20, and 24 and Fig. 2.
| Residue 1 | Residue 2 | Upper bound | Experimental evidence (reference) |
|---|---|---|---|
| Å | |||
| 168 | 211–218 | ≤11.7 | Mutagenesis (10) |
| 171 | 210 | ≤13.8 | MTS-3-MTS crosslink (20) |
| 171 | 474 | ≤11.0 | MTS-1-MTS crosslink (20) |
| 172 | 211–218 | ≤11.7 | Mutagenesis (10) |
| 174 | 210 | ≤15.1 | MTS-4-MTS crosslink (10) |
| 176 | 369 | ≤7.1 | DTT activation (24) |
| 263 | 474 | ≤7.1 | Cu-Phe–induced disulfide (Fig. 2) |
| 269 | 219 | ≤17.5 | MTS-6-MTS crosslink (20) |
| 270 | 219 | ≤15.1 | MTS-4-MTS crosslink (20) |
| 284 | 210 | ≤15.1 | MTS-4-MTS crosslink (20) |
| 473 | 210 | ≤12.5 | MTS-2-MTS crosslink (20) |
| 474 | 210 | ≤12.5 | MTS-2-MTS crosslink (20) |
| 475 | 210 | ≤15.1 | MTS-4-MTS crosslink (20) |
| 476 | 210 | ≤15.1 | MTS-4-MTS crosslink (20) |
| 477 | 210 | ≤15.1 | MTS-4-MTS crosslink (20) |
Figure 9.
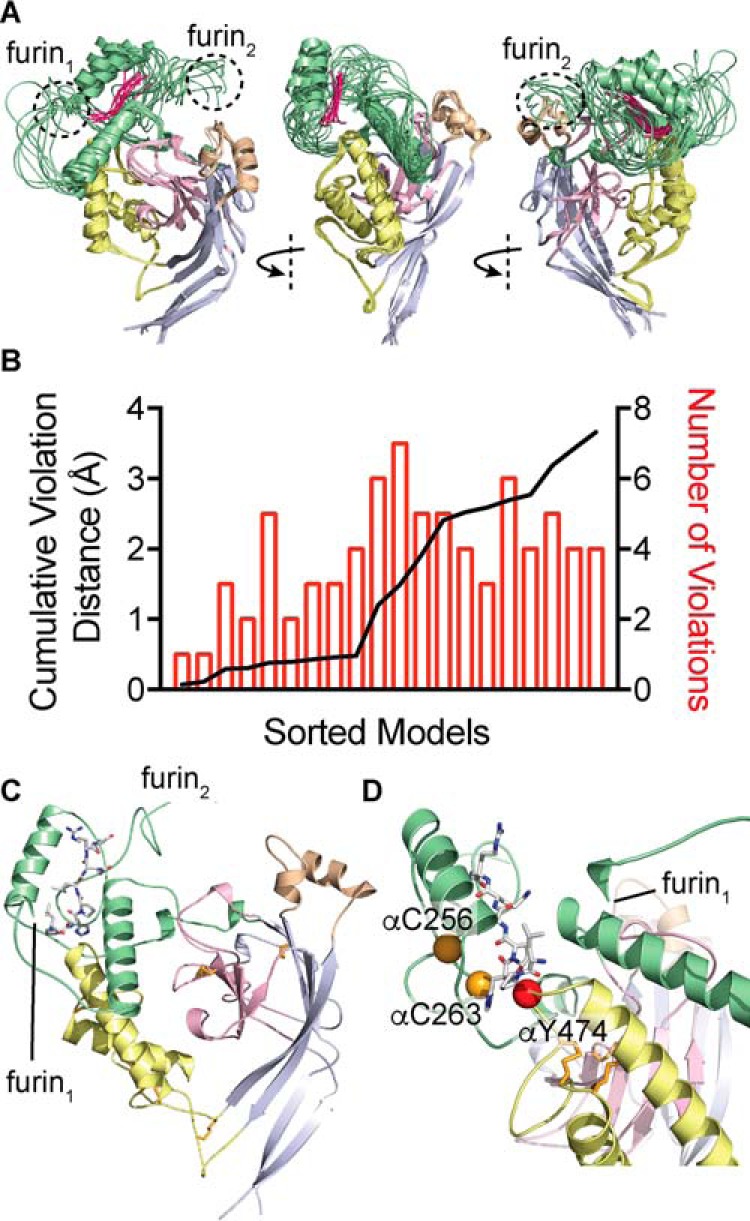
Model of the α subunit extracellular domains. Modeler was used to create a homology model of the ENaC α subunit extracellular domains, using cASIC1 (PDB code 4NYK) as a template. Distance constraints derived from functional data and predicted secondary structure were used to constrain the finger domain (green), which shares 8% sequence identity with cASIC1. A, overlay of nine models with <1 Å of cumulative distance violations of constraints derived from functional data, shown as a schematic. Models were superposed using non-finger domain residues: palm (gray), β-ball (pink), thumb (yellow), and knuckle (tan). The overlay shows that the highest-scoring models were better constrained at the finger–thumb domain interface near the N terminus of the bound inhibitory peptide (fuchsia, see center panel) and less well constrained near the C terminus of the bound inhibitory peptide and near the furin cleavage sites. B, plot of models sorted by cumulative violation of distance constraints derived from functional data. 9 of 20 models constructed showed less than 1 Å of cumulative violations. C, best scoring model is shown as a schematic, bound peptide is shown as sticks, and disulfide bonds are shown as orange sticks. D, close-up of best scoring model finger–thumb interface shown as a schematic with positions of key residues highlighted and shown as spheres.
In summary, our data suggest that the finger domain cysteines in both the α and γ subunits lie close to the finger–thumb domain interface. In the α subunit, they are probably free. In the γ subunit, they are probably disulfide-bonded. In the γ subunit, the terminal pair in the thumb domain cysteine ladder is probably not disulfide-bonded. These free cysteines lie at dynamic domain interfaces and may play a yet undefined role in channel regulation. In contrast to constraining the α subunit finger–thumb domain interface, constraining the γ subunit finger–thumb interface activated the channel, suggesting important dynamic differences between the subunits.
Experimental procedures
Plasmids and site-directed mutagenesis
pBluescriptSK (−) (Stratagene, San Diego, CA) vectors containing α, β, or γ mouse ENaC subunits were previously described (40). Site-directed mutagenesis was performed with the QuikChange II XL site-directed mutagenesis kit (Agilent, Santa Clara, CA). Primers were designed using QuikChange Primer Design (Agilent) and were obtained from Integrated DNA Technologies (Coralville, IA) to modify mouse ENaC subunits. Direct sequencing was used to confirm constructs.
ENaC functional expression in Xenopus oocytes
Plasmids were linearized with restriction endonucleases (New England Biolabs, Ipswich, MA) at 37 °C overnight and purified using the QIAquick PCR purification kit (Qiagen, Hilden, Germany). Linearized plasmids were transcribed into RNA using the mMessage mMachine T3 transcription kit (Invitrogen) and purified using the RNeasy MiniElute cleanup kit (Qiagen). Oocytes from Xenopus laevis were harvested and defolliculated using type II collagenase (Sigma-Aldrich). 1–4 ng of RNA per ENaC subunit was injected into stage V or VI Xenopus oocytes using a Nanoject II (Drummond, Broomall, PA). Xenopus oocytes were stored in modified Barth's saline (MBS) at 18 °C. The protocol for harvesting oocytes from X. laevis was approved by the University of Pittsburgh's institutional animal care and use committee.
Measurement of ENaC currents
ENaC current was measured 20–30 h postinjection using the two-electrode voltage clamp technique using a GeneClamp 500B voltage clamp amplifier (Molecular Devices, Sunnyvale, CA) and pClamp version 10.2 software (Molecular Devices). A 20-μl recording chamber (AutoMate Scientific, Berkeley, CA) was used with perfusion (3–5 ml/min) controlled by an eight-channel pinch valve system (AutoMate Scientific). Measurements of ENaC currents were made in Na-110 buffer (110 mm NaCl, 2 mm KCl, 2 mm CaCl2, and 10 mm HEPES (pH 7.4)). At the end of each experiment, currents were measured in 10 μm amiloride (stock solution of 100 mm amiloride in DMSO stored at room temperature diluted into Na-110 buffer) to determine the amiloride-sensitive component of the current. This was defined as the ENaC-mediated current.
Crosslinking during ENaC current measurements
Stock solutions of MTS reagents (Toronto Research Chemicals, North York, Canada) were prepared in DMSO at 10 mm and stored at −20 °C under desiccant for up to 6 months. MTS stock solutions were diluted into Na-110 buffer to 10 μm immediately preceding each recording. Stock solutions of 4 mm CuSO4 (Sigma-Aldrich) in H2O and 16 mm 1,10-phenanthroline (Sigma-Aldrich) in ethanol were stored at −20 °C. Cu-Phe solution was prepared on the day of the experiment by diluting CuSO4 and 1,10-phenanthroline stocks into Na-110 buffer for a final concentration of 4 μm CuSO4 and 16 μm 1,10-phenanthroline. CuSO4 solution was prepared by diluting CuSO4 stock into Na-110 buffer for a final concentration of 4 μm CuSO4. Stock solutions of 100 mm DTT (Sigma-Aldrich) in Na-110 buffer (stored at −20 °C) were diluted to 10 mm DTT in Na-110 buffer on the day experiments were performed.
ENaC cell-surface expression
ENaC cell-surface expression in oocytes was measured as described previously (41–43). Mutant α or γ subunits were co-expressed with a β subunit with an extracellular FLAG tag. One day following cRNA injection of oocytes, amiloride-sensitive currents were measured. Two days following cRNA injection of oocytes, oocytes were placed on ice and incubated for 30 min in antibiotic-free MBS and 1% bovine serum albumin (MBS/BSA) and then for 1 h with MBS/BSA with 1 μg/ml mouse anti-FLAG antibody (M2; Sigma). After wash with MBS/BSA, cells were incubated for 1 h with 1 μg/ml horseradish peroxidase–conjugated goat anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA). After wash, oocytes were transferred to a 96-well plate. SuperSignal ELISA Femto maximum sensitivity substrate (100 μl; Thermo Scientific, Rockford, IL) was added to each well. Chemiluminescence was quantified using a GloMax-Multi+ detection system (Promega, Madison, WI). The mean value measured from four wells with no cells was subtracted from the measurements of cells with ENaC-injected oocytes.
SIN-1–generated peroxynitrite treatment
We prepared 100 mm 3-morpholino-sydnonimine (SIN-1) (Millipore, Darmstadt, Germany) stock in 10 mm phosphate buffer. We diluted SIN-1 into Na-110 buffer 1 h before recording currents.
Statistical analyses
Comparisons between groups were performed by ANOVA followed by a Tukey multiple-comparison test, by Student's t test, or by a Kruskal–Wallis rank sum test followed by the Dunn test for multiple comparisons with p values adjusted with the Benjamini–Hochberg method, as indicated. We used R (R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria) or Prism version 7 (GraphPad, Inc., La Jolla, CA) to perform statistical calculations. Adjusted values of p < 0.05 were considered significant.
Homology modeling
Modeling was performed using MODELLER version 9.13 (39). We used the ENaC α subunit-cASIC1 alignment described previously (10) and the cASIC1 structural model (PDB code 4NYK). In addition to the constraints we previously used, we added new constraints derived from crosslinking experiments reported here and in previous work (see Table 2) (10, 20, 24). We constrained distances of Cα–Cα pairs using an upper bound constraint dependent on the crosslinking agent used: Cu-Phe, 7.1 Å; MTS-1-MTS, 11.01 Å; MTS-2-MTS, 12.46 Å; MTS-3-MTS, 13.79 Å; MTS-4-MTS, 15.05 Å; MTS-6-MTS, 17.46 Å; MTS-8-MTS, 19.61 Å, calculated using the Molecule Calculator (44). Using our previous model (10) as an initial input, we generated 20 models, which were scored by summing deviations greater than the set distance of the upper bound constraint. We visually inspected the highest scoring models for structural violations (e.g. knots) and selected a high-scoring model with minimal violations. Superpositioning models and root mean square deviation calculations were performed using Swiss pdb viewer version 4.1.0_OSX (45). Molecular figures were generated using PyMOL (Schroedinger, LLC, New York, NY).
Author contributions
B. M. B. and O. B. K. conceptualized the study, interpreted results, prepared figures, and drafted and revised the manuscript. B. M. B. performed homology modeling. B. M. B., X.-P. W., and O. B. K. performed experiments, analyzed data, and approved the final version of the manuscript.
Acknowledgment
The Pittsburgh Center for Kidney Research was supported by Grant P30 DK079307 from the National Institutes of Health.
This work was supported by NIDDK, National Institutes of Health, Grants R01 DK098204 (to O. B. K.) and T32 DK061296 (to B. M. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ENaC
- epithelial Na+ channel
- ASIC
- acid-sensing ion channel
- Cu-Phe
- copper phenanthroline
- MTS
- methanethiosulfonate
- SIN-1
- 3-morpholinosydnonimine
- MBS
- modified Barth's saline
- PDB
- Protein Data Bank
- ANOVA
- analysis of variance.
References
- 1. Pearce D., Soundararajan R., Trimpert C., Kashlan O. B., Deen P. M., and Kohan D. E. (2015) Collecting duct principal cell transport processes and their regulation. Clin. J. Am. Soc. Nephrol. 10, 135–146 10.2215/CJN.05760513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Haq I. J., Gray M. A., Garnett J. P., Ward C., and Brodlie M. (2016) Airway surface liquid homeostasis in cystic fibrosis: pathophysiology and therapeutic targets. Thorax 71, 284–287 10.1136/thoraxjnl-2015-207588 [DOI] [PubMed] [Google Scholar]
- 3. Köckerling A., Sorgenfrei D., and Fromm M. (1993) Electrogenic Na+ absorption of rat distal colon is confined to surface epithelium: a voltage-scanning study. Am. J. Physiol. 264, C1285–C1293 10.1152/ajpcell.1993.264.5.C1285 [DOI] [PubMed] [Google Scholar]
- 4. Mall M., Bleich M., Kuehr J., Brandis M., Greger R., and Kunzelmann K. (1999) CFTR-mediated inhibition of epithelial Na+ conductance in human colon is defective in cystic fibrosis. Am. J. Physiol. 277, G709–G716 [DOI] [PubMed] [Google Scholar]
- 5. Ben-Shahar Y. (2011) Sensory functions for degenerin/epithelial sodium channels (DEG/ENaC). Adv. Genet. 76, 1–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kashlan O. B., and Kleyman T. R. (2011) ENaC structure and function in the wake of a resolved structure of a family member. Am. J. Physiol. Renal Physiol. 301, F684–F696 10.1152/ajprenal.00259.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kapoor N., Lee W., Clark E., Bartoszewski R., McNicholas C. M., Latham C. B., Bebok Z., Parpura V., Fuller C. M., Palmer C. A., and Benos D. J. (2011) Interaction of ASIC1 and ENaC subunits in human glioma cells and rat astrocytes. Am. J. Physiol. Cell Physiol. 300, C1246–C1259 10.1152/ajpcell.00199.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jeggle P., Smith E. S., Stewart A. P., Haerteis S., Korbmacher C., and Edwardson J. M. (2015) Atomic force microscopy imaging reveals the formation of ASIC/ENaC cross-clade ion channels. Biochem. Biophys. Res. Commun. 464, 38–44 10.1016/j.bbrc.2015.05.091 [DOI] [PubMed] [Google Scholar]
- 9. Trac P. T., Thai T. L., Linck V., Zou L., Greenlee M., Yue Q., Al-Khalili O., Alli A. A., Eaton A. F., and Eaton D. C. (2017) Alveolar non-selective channels are ASIC1a/α-ENaC channels and contribute to AFC. Am. J. Physiol. Lung Cell Mol. Physiol. 312, L797–L811 10.1152/ajplung.00379.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kashlan O. B., Adelman J. L., Okumura S., Blobner B. M., Zuzek Z., Hughey R. P., Kleyman T. R., and Grabe M. (2011) Constraint-based, homology model of the extracellular domain of the epithelial Na+ channel α subunit reveals a mechanism of channel activation by proteases. J. Biol. Chem. 286, 649–660 10.1074/jbc.M110.167098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jasti J., Furukawa H., Gonzales E. B., and Gouaux E. (2007) Structure of acid-sensing ion channel 1 at 1.9 Å resolution and low pH. Nature 449, 316–323 10.1038/nature06163 [DOI] [PubMed] [Google Scholar]
- 12. Hughey R. P., Bruns J. B., Kinlough C. L., Harkleroad K. L., Tong Q., Carattino M. D., Johnson J. P., Stockand J. D., and Kleyman T. R. (2004) Epithelial sodium channels are activated by furin-dependent proteolysis. J. Biol. Chem. 279, 18111–18114 10.1074/jbc.C400080200 [DOI] [PubMed] [Google Scholar]
- 13. Bruns J. B., Carattino M. D., Sheng S., Maarouf A. B., Weisz O. A., Pilewski J. M., Hughey R. P., and Kleyman T. R. (2007) Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the γ-subunit. J. Biol. Chem. 282, 6153–6160 10.1074/jbc.M610636200 [DOI] [PubMed] [Google Scholar]
- 14. Sheng S., Maarouf A. B., Bruns J. B., Hughey R. P., and Kleyman T. R. (2007) Functional role of extracellular loop cysteine residues of the epithelial Na+ channel in Na+ self-inhibition. J. Biol. Chem. 282, 20180–20190 10.1074/jbc.M611761200 [DOI] [PubMed] [Google Scholar]
- 15. Firsov D., Robert-Nicoud M., Gruender S., Schild L., and Rossier B. C. (1999) Mutational analysis of cysteine-rich domains of the epithelium sodium channel (ENaC): identification of cysteines essential for channel expression at the cell surface. J. Biol. Chem. 274, 2743–2749 10.1074/jbc.274.5.2743 [DOI] [PubMed] [Google Scholar]
- 16. Carattino M. D., Sheng S., Bruns J. B., Pilewski J. M., Hughey R. P., and Kleyman T. R. (2006) The epithelial Na+ channel is inhibited by a peptide derived from proteolytic processing of its α subunit. J. Biol. Chem. 281, 18901–18907 10.1074/jbc.M604109200 [DOI] [PubMed] [Google Scholar]
- 17. Passero C. J., Carattino M. D., Kashlan O. B., Myerburg M. M., Hughey R. P., and Kleyman T. R. (2010) Defining an inhibitory domain in the γ subunit of the epithelial sodium channel. Am. J. Physiol. Renal Physiol. 299, F854–F861 10.1152/ajprenal.00316.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carattino M. D., Passero C. J., Steren C. A., Maarouf A. B., Pilewski J. M., Myerburg M. M., Hughey R. P., and Kleyman T. R. (2008) Defining an inhibitory domain in the α-subunit of the epithelial sodium channel. Am. J. Physiol. Renal Physiol. 294, F47–F52 10.1152/ajprenal.00399.2007 [DOI] [PubMed] [Google Scholar]
- 19. Kashlan O. B., Boyd C. R., Argyropoulos C., Okumura S., Hughey R. P., Grabe M., and Kleyman T. R. (2010) Allosteric inhibition of the epithelial Na+ channel through peptide binding at peripheral finger and thumb domains. J. Biol. Chem. 285, 35216–35223 10.1074/jbc.M110.167064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kashlan O. B., Blobner B. M., Zuzek Z., Carattino M. D., and Kleyman T. R. (2012) Inhibitory tract traps the epithelial Na+ channel in a low activity conformation. J. Biol. Chem. 287, 20720–20726 10.1074/jbc.M112.358218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kobashi K. (1968) Catalytic oxidation of sulfhydryl groups by o-phenanthroline copper complex. Biochim. Biophys. Acta 158, 239–245 10.1016/0304-4165(68)90136-0 [DOI] [PubMed] [Google Scholar]
- 22. Chen J., Myerburg M. M., Passero C. J., Winarski K. L., and Sheng S. (2011) External Cu2+ inhibits human epithelial Na+ channels by binding at a subunit interface of extracellular domains. J. Biol. Chem. 286, 27436–27446 10.1074/jbc.M111.232058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Collier D. M., Tomkovicz V. R., Peterson Z. J., Benson C. J., and Snyder P. M. (2014) Intersubunit conformational changes mediate epithelial sodium channel gating. J. Gen. Physiol. 144, 337–348 10.1085/jgp.201411208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kashlan O. B., Blobner B. M., Zuzek Z., Tolino M., and Kleyman T. R. (2015) Na+ inhibits the epithelial Na+ channel by binding to a site in an extracellular acidic cleft. J. Biol. Chem. 290, 568–576 10.1074/jbc.M114.606152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Canessa C. M., Schild L., Buell G., Thorens B., Gautschi I., Horisberger J. D., and Rossier B. C. (1994) Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367, 463–467 10.1038/367463a0 [DOI] [PubMed] [Google Scholar]
- 26. Haerteis S., Krueger B., Korbmacher C., and Rauh R. (2009) The δ-subunit of the epithelial sodium channel (ENaC) enhances channel activity and alters proteolytic ENaC activation. J. Biol. Chem. 284, 29024–29040 10.1074/jbc.M109.018945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hughey R. P., Mueller G. M., Bruns J. B., Kinlough C. L., Poland P. A., Harkleroad K. L., Carattino M. D., and Kleyman T. R. (2003) Maturation of the epithelial Na+ channel involves proteolytic processing of the α- and γ-subunits. J. Biol. Chem. 278, 37073–37082 10.1074/jbc.M307003200 [DOI] [PubMed] [Google Scholar]
- 28. Snyder P. M., Olson D. R., and Bucher D. B. (1999) A pore segment in DEG/ENaC Na+ channels. J. Biol. Chem. 274, 28484–28490 10.1074/jbc.274.40.28484 [DOI] [PubMed] [Google Scholar]
- 29. Xu H., and Chu S. (2007) ENaC α-subunit variants are expressed in lung epithelial cells and are suppressed by oxidative stress. Am. J. Physiol. Lung Cell Mol. Physiol. 293, L1454–L1462 10.1152/ajplung.00248.2007 [DOI] [PubMed] [Google Scholar]
- 30. Ma H. P. (2011) Hydrogen peroxide stimulates the epithelial sodium channel through a phosphatidylinositide 3-kinase-dependent pathway. J. Biol. Chem. 286, 32444–32453 10.1074/jbc.M111.254102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Downs C. A., Kumar A., Kreiner L. H., Johnson N. M., and Helms M. N. (2013) H2O2 regulates lung epithelial sodium channel (ENaC) via ubiquitin-like protein Nedd8. J. Biol. Chem. 288, 8136–8145 10.1074/jbc.M112.389536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DuVall M. D., Zhu S., Fuller C. M., and Matalon S. (1998) Peroxynitrite inhibits amiloride-sensitive Na+ currents in Xenopus oocytes expressing αβγ-rENaC. Am. J. Physiol. 274, C1417–C1423 10.1152/ajpcell.1998.274.5.C1417 [DOI] [PubMed] [Google Scholar]
- 33. Chen L., Fuller C. M., Kleyman T. R., and Matalon S. (2004) Mutations in the extracellular loop of α-rENaC alter sensitivity to amiloride and reactive species. Am. J. Physiol. Renal Physiol. 286, F1202–F1208 10.1152/ajprenal.00352.2003 [DOI] [PubMed] [Google Scholar]
- 34. Collier D. M., and Snyder P. M. (2011) Identification of epithelial Na+ channel (ENaC) intersubunit Cl− inhibitory residues suggests a trimeric αγβ channel architecture. J. Biol. Chem. 286, 6027–6032 10.1074/jbc.M110.198127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sheng S., Bruns J. B., and Kleyman T. R. (2004) Extracellular histidine residues crucial for Na+ self-inhibition of epithelial Na+ channels. J. Biol. Chem. 279, 9743–9749 10.1074/jbc.M311952200 [DOI] [PubMed] [Google Scholar]
- 36. Maarouf A. B., Sheng N., Chen J., Winarski K. L., Okumura S., Carattino M. D., Boyd C. R., Kleyman T. R., and Sheng S. (2009) Novel determinants of epithelial sodium channel gating within extracellular thumb domains. J. Biol. Chem. 284, 7756–7765 10.1074/jbc.M807060200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li J., Sheng S., Perry C. J., and Kleyman T. R. (2003) Asymmetric organization of the pore region of the epithelial sodium channel. J. Biol. Chem. 278, 13867–13874 10.1074/jbc.M300149200 [DOI] [PubMed] [Google Scholar]
- 38. Schild L., Schneeberger E., Gautschi I., and Firsov D. (1997) Identification of amino acid residues in the α, β, and γ subunits of the epithelial sodium channel (ENaC) involved in amiloride block and ion permeation. J. Gen. Physiol. 109, 15–26 10.1085/jgp.109.1.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- 40. Ahn Y. J., Brooker D. R., Kosari F., Harte B. J., Li J., Mackler S. A., and Kleyman T. R. (1999) Cloning and functional expression of the mouse epithelial sodium channel. Am. J. Physiol. 277, F121–F129 [DOI] [PubMed] [Google Scholar]
- 41. Chen J., Kleyman T. R., and Sheng S. (2013) Gain-of-function variant of the human epithelial sodium channel. Am. J. Physiol. Renal Physiol. 304, F207–F213 10.1152/ajprenal.00563.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ray E. C., Chen J., Kelly T. N., He J., Hamm L. L., Gu D., Shimmin L. C., Hixson J. E., Rao D. C., Sheng S., and Kleyman T. R. (2016) Human epithelial Na+ channel missense variants identified in the GenSalt study alter channel activity. Am. J. Physiol. Renal Physiol. 311, F908–F914 10.1152/ajprenal.00426.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Carattino M. D., Hill W. G., and Kleyman T. R. (2003) Arachidonic acid regulates surface expression of epithelial sodium channels. J. Biol. Chem. 278, 36202–36213 10.1074/jbc.M300312200 [DOI] [PubMed] [Google Scholar]
- 44. Jensen J. H., and Kromann J. C. (2013) The molecule calculator: a web application for fast quantum mechanics-based estimation of molecular properties. J. Chem. Educ. 90, 1093–1095 10.1021/ed400164n [DOI] [Google Scholar]
- 45. Guex N., and Peitsch M. C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723 10.1002/elps.1150181505 [DOI] [PubMed] [Google Scholar]