

Abstract
It is well established that pregnancy early in life reduces the risk of breast cancer in women and that this effect is universal. This phenomenon of parity protection against mammary cancer is also observed in rodents. Earlier studies have demonstrated that short-term administration of estradiol (E) in combination with progesterone mimics the protective effect of parity in rats. In this study, the lowest effective E dosage for preventing mammary cancer was determined. Rats were injected with N-methyl-N-nitrosourea at 7 weeks of age; 2 weeks later, the rats were subjected to sustained treatment with 20 μg, 100 μg, 200 μg, or 30 mg of E in silastic capsules for 3 weeks. Treatments with 100 μg, 200 μg, and 30 mg of E resulted in serum levels of E equivalent to those of pregnancy and were highly effective in preventing mammary cancer. E treatment (20 μg) did not result in pregnancy levels of E and was not effective in reducing the mammary cancer incidence. In another set of experiments, we determined the effect of different durations of E with or without progesterone treatments on mammary carcinogenesis. These experiments indicate that a period as short as one-third the period of gestation is sufficient to induce protection against mammary carcinogenesis. The pioneering aspect of our study in contrast to long-term estrogen exposure, which is thought to increase the risk of breast cancer, is that short-term sustained treatments with pregnancy levels of E can induce protection against frank mammary cancer.
Breast cancer is the most common cancer among women worldwide. The incidence of breast cancer in the United States for the year 2000 was estimated to be ≈184,200 new cases (1). The epidemiological evidence provides strong support for the concept that environmental, hormonal, and genetic factors affect the risk for development of breast cancer (2–6). Among the various recognized reproductive risk factors are age at menarche, age at first pregnancy, and age at menopause, all of which seem to suggest that endogenous ovarian steroids may profoundly affect the process of carcinogenesis (3, 6, 7).
Various studies reveal that nulliparous women have a higher risk of breast cancer compared with women who have undergone single or multiple pregnancies early in life (8–10). The protective effect of pregnancy is universal, and pregnancy is the only normal physiological condition that consistently prevents breast cancers in all ethnic backgrounds in all countries without known adverse effects (8–10). The phenomenon of parity protection against mammary carcinogenesis is also observed in rats and mice. Moon (11) demonstrated that parous rats exposed to chemical carcinogens developed fewer cancers compared with virgin rats. Earlier Marchant (12) and very recently Medina and Smith (13) have demonstrated that pregnancy provides protection against mammary carcinogenesis in mouse models. Short-term administration with ovarian steroids [estrogens and progesterone (P)] or human chorionic gonadotropins either before or after carcinogen treatment decreases the incidence of mammary cancers in rodents (14–22). Although hormone treatments administered before or after carcinogen administration induce protection from mammary carcinogenesis, it is not established whether they work through similar mechanisms. Recently in our laboratory we have demonstrated that administration of pregnancy levels of estradiol (E) with P shortly before or after carcinogen treatment drastically reduces mammary cancer incidence and multiplicity (23). These studies demonstrated that protection against mammary carcinogenesis could be achieved by treatment with physiological levels of E and P for 21 days (rat gestation period) or less (23).
The actual mechanism involved in parity protection or hormone-induced protection against breast cancer is yet to be clearly defined. The most widely accepted explanation for pregnancy protection against mammary cancer is that the protective effect is attributable to the pregnancy-induced differentiation of the target structures, terminal-end buds, and terminal ducts for carcinogenesis, thus reducing the target cells for carcinogenesis (24). The other explanation for pregnancy protection against mammary cancer is that pregnancy primes the maternal immune system against pregnancy-related antigens which are expressed during the growth of mammary tumors (25–28). Furthermore, the splenocytes of parous rats have significantly higher cytotoxic activity against mammary tumor cells when compared with virgin controls (29). It has been reported that parous rats have decreased levels of mammogenic hormones, and this decrease has been attributed to the decreased incidence of mammary cancers in these animals (30, 31). In our laboratory, we had shown that differentiation of the mammary gland alone was not enough to confer protection against mammary cancer (23). Sivaraman et al. (32) and Medina et al. (22) suggest that protective hormone treatment results in persistent alterations in intracellular pathways governing proliferation responses to carcinogenesis, and also reported that complete differentiation of the mammary gland was not an obligatory prerequisite for protection against mammary carcinogenesis.
The current experiment was undertaken to determine whether a short-term sustained exposure to pregnancy levels of E alone will provide long-term protection against chemical carcinogen-induced mammary cancer in rats. If it can be achieved, it will provide first-hand evidence that estrogen can have a profound beneficial effect on the prevention of mammary cancer, depending on mode of administration and treatment duration. To address questions dealing with optimal dosing of E plus P or E alone that would result in parity-like protection against mammary carcinogenesis, we performed various studies. The specific questions included (i) What is the lowest dose of E required to confer protection against N-methyl-N-nitrosourea (MNU)-induced rat mammary carcinogenesis? (ii) Do the preventive effects of E relate to pregnancy levels of E? (iii) Is complete lobulo-alveolar differentiation of the mammary gland required for protection? (iv) What is the shortest treatment duration for the different treatments to confer protection?
Materials and Methods
Animals.
Virgin Lewis rats were purchased from Harlan Sprague–Dawley (Indianapolis and San Diego). The rats were housed in a temperature-controlled room with 12-h light/dark schedule. They were fed food (Teklad 8640; Teklad, Madison, WI) and water ad libitum. All of the procedures followed Univ. of California Animal Care and Use Committee guidelines.
Carcinogen Treatment.
A single i.p. injection of MNU (Ashe Stevens, Detroit) at a dose of 50 mg/kg of body weight was given to all rats at 7 weeks of age. MNU was dissolved in physiological saline that had been adjusted to pH 5.0 (33).
Hormone Treatment.
All of the hormone treatments in the following experiments were started 2 weeks after the administration of the carcinogen. The hormones were packed in individual silastic capsules (size 0.078 inch i.d. × 0.125 inch o.d., 2 cm in length; Baxter Health Care, Mundelein, IL). All doses of E 17β (Sigma) were packed in the silastic capsules in a cellulose matrix except for a 30-mg dose of E that was packed with the hormone alone. P (30 mg; Sigma) was packed into the silastic capsules without any matrix. Control animals received empty silastic capsules. All silastic capsules were dorsally implanted s.c. All capsules were primed before implantation by soaking in media 199 (GIBCO) overnight at 37°C.
Effect of Different Doses of E on Prevention of Mammary Carcinogenesis After Exposure to MNU.
When the rats were 9 weeks of age, they were divided into 5 groups, each group consisting of 12 rats and receiving one of the following treatments: (i) control, (ii) 20 μg of E, (iii) 100 μg of E, (iv) 200 μg of E, and (v) 30 mg of E. Each treatment was continued for 3 weeks and at the end of the treatment, the silastic capsules were removed from the animals. All of the hormone treatments were given for a 3-week period because the rat gestation period lasts 21 days.
Effect of Different Doses and Durations of E and P on Prevention of Mammary Carcinogenesis After Exposure to MNU.
Rats were divided into following groups: (i) control, (ii) 100 μg of E plus 30 mg of P administered for 2 weeks, (iii) 100 μg of E administered for 2 weeks, (iv) 200 μg of E plus 30 mg of P administered for 1 week, (v) 200 μg of E administered for 1 week, (vi) 100 μg of E plus 30 mg of P administered for 1 week, and (vii) 100 μg of E administered for 1 week. All of the hormone treatments were given by silastic capsules. Each group consisted of 14–16 rats.
Whole-Mount Analysis of Mammary Glands of the Treated Animals.
For morphological analysis of gland development, the anterior abdominal (no. 4) glands from either side were removed by normal surgical procedure immediately after the different treatments and were fixed in 10% neutral buffered formalin, defatted in acetone, hydrated, stained in alum carmine, washed in water, dehydrated in graded alcohols, and stored in histoclear (34). They were photographed to record development in terms of ductal growth and lobulo-alveolar differentiation.
Hormone Assay.
Blood was collected by cardiac puncture from animals of all of the different groups on the day the treatment was discontinued. Serum was separated, frozen, and assayed for E 17β by using the solid phase RIA kit purchased from Diagnostics Product (Los Angeles).
Mammary Carcinogenesis.
Rats were palpated once every week beginning 1 month after carcinogen exposure for 9 months to monitor for mammary cancer development. Histopathological examination was performed to confirm the carcinomatous nature of the palpable tumors.
Statistics.
The effects of the different hormonal treatments were analyzed by using the χ2 test for 2 × 2 contingency tables and Student's t test. Values with P < 0.05 were considered significant.
Results
Effect of Different Doses of E on the Mammary Gland Morphology (Fig. 1).
Figure 1.
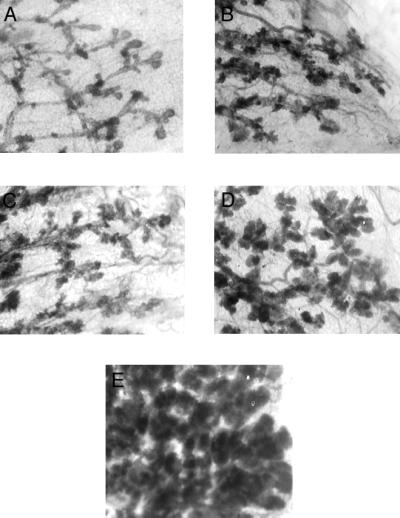
Effect of different doses of E in silastic capsules on the mammary gland morphology (×32). (A) Mammary gland of an untreated 12-week-old virgin Lewis rat. (B) Mammary gland of virgin rats treated with 20 μg of E. (C) Mammary gland of virgin rats treated with 100 μg of E. (D) Mammary gland of virgin rats treated with 200 μg of E. (E) Mammary gland of virgin rats treated with 30 mg of E. Note that treatment with doses of 200 μg or less of E does not result in full lobulo-alveolar development by the end of the treatment.
The 3-week treatment with 20 μg, 100 μg, 200 μg, or 30 mg of E resulted in morphological changes in the mammary gland compared with untreated control rats. Administration of 20, 100, or 200 μg of E resulted in mammary glands with lobulo-alveolar structures similar to those seen in early pregnancy. The mammary glands of rats treated with 30 mg of E were fully differentiated, similar to those seen in late pregnancy, with numerous lobules and alveoli filled with secretion and did not have any terminal-end buds.
Effect of Different Doses of E on Serum E Levels.
Blood was collected immediately after the 21-day treatment period for assay of E levels. Control animals had 16.2 ± 2.9 pg/ml of E in the serum. Treatment with 20 μg, 100 μg, 200 μg, and 30 mg of E resulted in serum E levels of 49, 67, 94, and 143 pg/ml, respectively. Three weeks of treatment with 20 μg of E resulted in serum levels of E lower than pregnancy levels, whereas treatments with 100 μg, 200 μg, or 30 mg of E resulted in levels of serum E equivalent to those found during pregnancy, and these values are in agreement with earlier observations on pregnancy levels of E (refs. 35–37; Table 1). Serum levels of E have been found to range from 55 to 630 pg/ml during pregnancy in Long Evans and Sprague–Dawley strains of rats (35–37).
Table 1.
Serum levels of E 17β after short-term E treatments
| Dose of E | E 17β, pg/ml |
|---|---|
| Control | 16.2 ± 2.9 |
| 20 μg of E | 49.9 ± 12.4 |
| 100 μg of E | 67.8 ± 13.9 |
| 200 μg of E | 94.9 ± 17.4 |
| 30 mg of E | 143.5 ± 23.9 |
Effect of Different Doses of E on the Mammary Cancer Incidence and Multiplicity.
The effect of 3 weeks of E treatment on mammary cancer incidence and multiplicity is shown in Table 2. Rats treated with 200 μg or 30 mg of E had a cancer incidence of 0% compared with 82% in control rats at 6 months after carcinogen treatment. Nine months after administration of the carcinogen, control animals had 100% mammary cancer incidence, whereas rats that had received 100 μg, 200 μg, or 30 mg of E for 3 weeks resulting in pregnancy levels of serum E had a greatly reduced incidence (17%) of mammary cancer. Treatment with 20 μg of E resulting in 73% incidence was not significantly different compared with controls. The control animals had a cancer multiplicity of 3.0 ± 0.5, whereas rats treated with 20 μg, 100 μg, 200 μg, and 30 mg of E had a drastically lowered multiplicity of 1.1, 0.6, 0.3, and 0.2, respectively. The dose of 100 μg of E was as effective as 30 mg of E in inducing protection from mammary carcinogenesis. The protective effect of these doses of E was highly significant compared with controls. Rats treated with 100 μg or 200 μg or 30 mg of E for a 3-week period after MNU treatment developed ≈90% fewer mammary carcinomas during the 9-month observation period compared with control rats that did not receive any hormone treatments. Rats treated with 20 μg of E treatment had 61% fewer mammary carcinomas compared with the control rats (Table 2). All doses of E used significantly increased the latency of mammary cancers compared with the controls (Fig. 2).
Table 2.
Effect of short-term E treatment on mammary carcinogenesis
| Treatment | No. of rats with cancer per no. of rats | Percent of rats with mammary cancer | Average no. of cancers per rat |
|---|---|---|---|
| Control | 11/11 | 100 | 3.0 ± 0.5 |
| 20 μg of E | 8/11 | 73 | 1.2 ± 0.4* |
| 100 μg of E | 2/12 | 17** | 0.6 ± 0.4** |
| 200 μg of E | 2/12 | 17** | 0.3 ± 0.2** |
| 30 mg of E | 2/12 | 17** | 0.2 ± 0.1** |
Effect of different doses of E on mammary carcinogenesis. Rats were treated with MNU at 7 weeks of age. At 9 weeks of age, the rats were treated with various doses of E for 3 weeks.
, P < 0.05;
, P < 0.01;
, P < 0.001 (compared to control).
Figure 2.

Effect of different doses of E treatment on mammary carcinogenesis in MNU-exposed rats. Rats treated with 100 μg, 200 μg, and 30 mg of E had a significant reduction in mammary cancer incidence compared with the controls. All of the different doses of E treatment used significantly increased the latency period and decreased the multiplicity of mammary cancers compared with the controls.
Effect of Different Duration of Treatment and Doses of E With or Without P on Mammary Cancer Incidence.
Treatment with different doses of E (100 and 200 μg) alone or in combination with 30 mg of P were given for 1 or 2 weeks after carcinogen administration to determine whether shorter duration treatments of E alone or in combination with P could prevent mammary carcinogenesis (Table 3). Control rats receiving MNU followed by empty silastic pellets had an 87% incidence of mammary cancers with an average of 2.1 cancers per rat at 9 months after the administration of carcinogen. Rats treated with 100 μg of E plus 30 mg of P for 2 weeks exhibited a mammary cancer incidence of only 20% (average no. of mammary cancers per rat = 0.3). Treatment with 200 μg of E plus 30 mg of P for 1 week decreased the mammary cancer incidence (14%) and reduced the number of cancers per rat (0.2) significantly, compared with the controls. A dose of 100 μg of E plus 30 mg of P given for 1 week significantly decreased the incidence of mammary cancers (21%) and the average number of cancers per rat (0.4) compared with the controls. A 1-week treatment with 200 μg of E drastically decreased the mammary cancer incidence (20%) and the average number of cancers per rat (0.2) when compared with the controls. E treatments of 100 μg for 1 or 2 weeks also decreased the mammary cancer incidence to 37% and 44%, respectively. The average number of cancers per rat was also significantly decreased compared with the controls after 1 and 2 weeks (0.6) of E treatment. Even the 1-week treatments with E alone or in combination with P were effective in increasing the latency period of mammary cancers compared with the control groups (Table 3). There was no significant difference between E or E plus P treatments in conferring protection against mammary carcinogenesis.
Table 3.
Effect of short-term treatments with E alone or with P on mammary carcinogenesis
| Treatment | No. of rats with cancer per no. of rats | % of rats with mammary cancer | Average number of cancers per rat | Mammary cancer latency† |
|---|---|---|---|---|
| Control | 13/15 | 87 | 2.1 ± 0.4 | 16.3 ± 4.1 |
| 100 μg of E + 30 mg of P (2 weeks) | 3/15** | 20 | 0.3 ± 0.1** | 24.7 ± 4.8 |
| 100 μg of E (2 weeks) | 7/16* | 44 | 0.6 ± 0.2* | 18.0 ± 3.2 |
| 200 μg of E + 30 mg of P (1 week) | 2/14** | 14 | 0.2 ± 0.1** | 26.0 ± 6.0 |
| 200 μg of E (1 week) | 3/15** | 20 | 0.2 ± 0.1** | 19.4 ± 3.0 |
| 100 μg of E + 30 mg of P (1 week) | 3/14** | 21 | 0.4 ± 0.3* | 28.0 ± 1.5 |
| 100 μg of E (1 week) | 6/16** | 37 | 0.6 ± 0.2* | 23.7 ± 2.6 |
Effect of short-term treatment with E with or without P treatments on MNU-induced mammary carcinogenesis. Rats were treated with MNU at 7 weeks of age and treated 2 weeks later with E alone or in combination with P for 2 weeks or 1 week.
, P < 0.05;
, P < 0.01;
, P < 0.001 (compared to control).
Average number of weeks to the appearance of the first palpable tumor.
Discussion
It is well established that an early full-term pregnancy induces a refractory state of the mammary gland against carcinogenesis in both humans (8–10) and rodents (11–13). This refractoriness can be attained by a single pregnancy with or without accompanying lactation (11, 38, 39). During pregnancy, many hormones act to cause proliferation and differentiation of the mammary gland. The mammary gland is exposed to high levels of ovarian (estrogens and P), pituitary (prolactin and growth hormone), and placental [human chorionic gonadotropin (hCG)] hormones. It is not known which of the myriad of hormonal changes during pregnancy is responsible for conferring parity protection. However, earlier studies have demonstrated that hormonal regimens comprised of the ovarian steroids E and P (14, 18, 23, 40) or human chorionic gonadotropins (20) can provide protection against rat mammary tumorigenesis similar to pregnancy.
We have also shown that physiological levels of ovarian steroids can provide a parity-like protection (23), and we now report the results of additional studies involving different doses and durations of these hormones. Treatment for 21 days with various doses of E resulted in a dose-dependent increase in blood levels of E. All doses of E tested, except for the 20 μg dose, resulted in levels of E equivalent to pregnancy levels and were capable of conferring protection against mammary carcinogenesis with respect to both incidence and multiplicity compared with controls. The lower dose of 20 μg of E, which did not result in pregnancy level of E, was also unavailable to reduce the incidence but was effective in decreasing the multiplicity and increasing the latency. These data suggest that there is a threshold level below which the treatment is not effective in conferring full protection. The higher doses of E resulting in pregnancy levels of E were highly effective in not only lowering the mammary cancer incidence but also in decreasing the multiplicity of mammary cancers and prolonging the latency of the cancers.
The Russos (24) have put forth the hypothesis that pregnancy protection against mammary cancer is caused by the pregnancy-induced differentiation of the target structures, terminal-end buds, and terminal ducts. In this study, different doses of E resulted in varying morphological changes in the mammary glands, ranging from incomplete to complete differentiation similar to that seen during late pregnancy and early lactation. The doses of E (100 and 200 μg) used in the present study did not result in complete differentiation of the mammary gland but were as effective as the higher-dose E (30 mg) treatment groups in preventing mammary carcinogenesis. The results of the experiments reported herein again demonstrate that complete morphological differentiation of the mammary gland induced by hormones is not a compulsory requirement for inducing the refractory state. These findings confirm our earlier result (23) that demonstrated complete differentiation of mammary glands by the use of perphenazine, a dopamine receptor inhibitor causing acute release of prolactin, did not result in parity-like protection. Other investigators have also demonstrated that complete morphological differentiation of the mammary gland induced by hormones is not an obligatory prerequisite for inducing the refractory state (22, 32, 39). Taken together, these results demonstrate that the parity protection is not the result of differentiation of the mammary gland per se. Rather, it is the increasing level of ovarian steroids, among a myriad of hormonal changes taking place during pregnancy, that are the major determinants of parity protection against mammary cancer. The results from exposure to different doses and durations of E with or without P demonstrate that sustained exposure to pregnancy levels of E even as short as 1 week are effective in reducing the incidence of mammary cancer. This finding suggests that a pregnancy level of E is a major factor in conferring a protective effect against mammary cancers. Epidemiological evidence demonstrates that a full-term pregnancy is required to confer protection against mammary cancer, whereas our result show a short-term sustained treatment for one-third the length of pregnancy is as effective as full-term pregnancy. This study suggests that sustained treatment with pregnancy levels of E for a short period is likely to be a highly effective means for conferring protection against frank mammary cancer.
Recently it has been reported that several apoptotic genes like testosterone-repressed prostate message 2 (TRPM2), IL-1-beta-converting enzyme (CE), p53, c-myc, and bcl-XS were activated in rats which were administered with human chorionic gonadotropin (hCG) for protection before carcinogen treatment (41). This report also has stated that hCG induced the synthesis of inhibin, a protein with tumor-suppressor activity. Thordarson et al. (30, 31) and Abrams et al. (42) reported that protection against mammary cancers in parous rats was not permanent but plastic. When parous rats were injected with MNU and the mammary glands were observed at autopsy for the total number of occult tumors, they did not show any significant difference compared with the controls (43). It has also been demonstrated that the levels of growth hormone and prolactin are reduced in parous rats compared with their age-matched controls (30, 44). Parous women have persistently decreased levels of prolactin compared with nulliparous women (45), and this finding has been correlated with the decrease in the risk of breast cancer. These results suggest that parity-induced protection against mammary cancer may result from a reduced promotional environment for cancer development. Although there are various mechanisms and reasons attributed to the phenomenon of pregnancy protection or hormone-induced protection against breast cancers, none of them have been proven.
In summary, our results demonstrate that short-term sustained treatment with any dose of E resulting in pregnancy levels of circulatory E, soon after carcinogen administration, is highly effective in inducing refractoriness to carcinogen-induced rat mammary carcinogenesis. We have consistently observed that a combination of E plus P has a better protective effect compared with the corresponding treatments of E alone. From the morphology of the mammary glands of the protected rats, it is clearly evident that complete differentiation is not a prerequisite for conferring protection. Our treatment duration studies show that treatment as short as 1 week corresponding to one-third the duration of rat pregnancy can induce protection against mammary carcinogenesis. Recent findings demonstrate estrogen can alter the adult brain neuroanatomy and reorganize neuronal connections (46). Hence, we speculate that the protective effect may be the result of a persistent alteration in the hypothalamo-hypophyseal axis, resulting in reduced circulating levels of mammogenic hormones such as growth hormone and prolactin, which provide the promotional environment for mammary carcinogenesis. This alteration may also explain the persistent decrease in prolactin levels in women who have undergone a full-term pregnancy. Unlike the long-term estrogen exposure, which is thought to increase the risk of breast cancer, our short short-term sustained treatments with pregnancy levels of E can induce protection against mammary carcinogenesis.
Acknowledgments
We thank Joanne Leung and Nate Rogers for technical assistance, Shyam Laxminarayan for his help in preparation of the manuscript, and Carol Slatten and Judith Yee for administrative assistance. This work was supported by National Cancer Institute Grants CA 62598, CA 63369, CA 71590, and CA 72598.
Abbreviations
- MNU
N-methyl-N-nitrosourea
- E
estradiol
- P
progesterone
References
- 1.Greenlee R T, Murray T, Bolden S, Wingo P A. CA Cancer J Clin. 2000;50:7–33. doi: 10.3322/canjclin.50.1.7. [DOI] [PubMed] [Google Scholar]
- 2.Henderson I C. Cancer (Philadelphia) 1993;71:2127–2140. doi: 10.1002/1097-0142(19930315)71:6+<2127::aid-cncr2820711602>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 3.Pike M C, Spicer D V, Dahmoush L, Press M F. Epidemiol Rev. 1993;15:17–35. doi: 10.1093/oxfordjournals.epirev.a036102. [DOI] [PubMed] [Google Scholar]
- 4.Harris R E, Namboodiri K K, Wynder E L. J Natl Cancer Inst. 1992;84:1575–1582. doi: 10.1093/jnci/84.20.1575. [DOI] [PubMed] [Google Scholar]
- 5.Dupont W D, Page D L. N Engl J Med. 1985;312:146–151. doi: 10.1056/NEJM198501173120303. [DOI] [PubMed] [Google Scholar]
- 6.Bernstein L, Ross R K. Epidemiol Rev. 1993;15:48–65. doi: 10.1093/oxfordjournals.epirev.a036116. [DOI] [PubMed] [Google Scholar]
- 7.Mauvais-Jarvis P, Kuttenn F, Gompel A. Ann NY Acad Sci. 1986;464:152–167. doi: 10.1111/j.1749-6632.1986.tb16002.x. [DOI] [PubMed] [Google Scholar]
- 8.MacMahon B, Cole P, Lin T M, Lowe C R, Mirra A P, Ravnihar B, Salber E J, Valaoras V G, Yuasa S. Bull WHO. 1970;43:209–221. [PMC free article] [PubMed] [Google Scholar]
- 9.Henderson B E, Powell D, Rosario I, Keys C, Hanisch R, Young M, Casagrande J, Gerkins V, Pike M C. J Natl Cancer Inst. 1974;53:609–614. doi: 10.1093/jnci/53.3.609. [DOI] [PubMed] [Google Scholar]
- 10.Kelsey J L. Epidemiol Rev. 1993;15:256–263. doi: 10.1093/oxfordjournals.epirev.a036112. [DOI] [PubMed] [Google Scholar]
- 11.Moon R C. Int J Cancer. 1969;4:312–317. doi: 10.1002/ijc.2910040308. [DOI] [PubMed] [Google Scholar]
- 12.Marchant J. J Pathol Bacteriol. 1955;70:415–418. doi: 10.1002/path.1700700218. [DOI] [PubMed] [Google Scholar]
- 13.Medina D, Smith G H. J Natl Cancer Inst. 1999;91:967–969. doi: 10.1093/jnci/91.11.967. [DOI] [PubMed] [Google Scholar]
- 14.Huggins C, Moon R C, Morii S. Proc Natl Acad Sci USA. 1962;48:379–386. doi: 10.1073/pnas.48.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCormick G M, Moon R C. Eur J Cancer. 1973;9:483–486. doi: 10.1016/0014-2964(73)90131-x. [DOI] [PubMed] [Google Scholar]
- 16.Medina D. J Natl Cancer Inst. 1974;53:213–221. doi: 10.1093/jnci/53.1.213. [DOI] [PubMed] [Google Scholar]
- 17.Lemon H M. Lancet. 1973;10:546–547. doi: 10.1016/s0140-6736(73)90353-x. [DOI] [PubMed] [Google Scholar]
- 18.Grubbs C J, Farnell D R, Hill D L, McDonough K C. J Natl Cancer Inst. 1985;74:927–931. [PubMed] [Google Scholar]
- 19.Grubbs C J, Peckham J C, McDonough K D. Carcinogenesis. 1983;4:495–497. doi: 10.1093/carcin/4.4.495. [DOI] [PubMed] [Google Scholar]
- 20.Russo J, Russo I H. Lab Invest. 1987;57:112–137. [PubMed] [Google Scholar]
- 21.Swanson S M, Whitaker L M, Stockard C R, Myers R B, Oelschlager D, Grizzle W E, Juliana M M, Grubbs C J. Anticancer Res. 1997;17:1202–1208. [PubMed] [Google Scholar]
- 22.Medina D, Peterson L E, Moraes R, Gay J. Cancer Lett. 2001;169:1–6. doi: 10.1016/s0304-3835(01)00507-9. [DOI] [PubMed] [Google Scholar]
- 23.Guzman R C, Yang J, Rajkumar L, Thordarson G, Chen X, Nandi S. Proc Natl Acad Sci USA. 1999;96:2520–2525. doi: 10.1073/pnas.96.5.2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russo I, Russo J. Environ Health Perspect. 1996;104:938–967. doi: 10.1289/ehp.96104938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarcione E J, Zloty M, Delluomo D S, Mizejewski G, Jacobson H. Cancer Res. 1983;43:3739–3741. [PubMed] [Google Scholar]
- 26.Grudzinskas J G, Teisner B, Al-Ani A T, Folkersen J, Westergaard J G, Chard T. Clin Chim Acta. 1980;106:1–7. doi: 10.1016/0009-8981(80)90368-x. [DOI] [PubMed] [Google Scholar]
- 27.Bremner R D, Nisbet A D, Herriot R, Horne C H, McArdle C, Crawford D, Bohn H. Oncodev Biol Med. 1981;2:55–62. [PubMed] [Google Scholar]
- 28.Monteiso J, Biswas A, Awquati M A, Greening W P, McKinn J A, Neville A M. Br J Cancer. 1981;46:279–282. doi: 10.1038/bjc.1982.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chakravarty P K, Ghosh S K, Sinha D K. Oncology. 1991;48:425–430. doi: 10.1159/000226973. [DOI] [PubMed] [Google Scholar]
- 30.Thordarson G, Jin E, Guzman R C, Swanson S M, Nandi S, Talamantes F. Carcinogenesis. 1995;16:2847–2853. doi: 10.1093/carcin/16.11.2847. [DOI] [PubMed] [Google Scholar]
- 31.Thordarson G, Van Horn K, Guzman R C, Nandi S, Talamantes F. Carcinogenesis. 2001;22:1027–1033. doi: 10.1093/carcin/22.7.1027. [DOI] [PubMed] [Google Scholar]
- 32.Sivaraman L, Stephens L C, Markaverich B M, Clark J A, Krnacik S, Conneely O M, O'Malley B W, Medina D. Carcinogenesis. 1998;19:1573–1582. doi: 10.1093/carcin/19.9.1573. [DOI] [PubMed] [Google Scholar]
- 33.McCormick D L, Adamowski C B, Fiks A, Moon R C. Cancer Res. 1981;41:1690–1694. [PubMed] [Google Scholar]
- 34.Thompson H L, McGinley J N, Rothhammer K, Singh M. Carcinogenesis. 1995;16:2407–2411. doi: 10.1093/carcin/16.10.2407. [DOI] [PubMed] [Google Scholar]
- 35.de Lauzon S, Uhrich F, Vandel S, Cittanova N, Jayle M F. Steroids. 1974;24:31–40. doi: 10.1016/0039-128x(74)90043-9. [DOI] [PubMed] [Google Scholar]
- 36.Shaikh A A. Biol Reprod. 1971;5:297–307. doi: 10.1093/biolreprod/5.3.297. [DOI] [PubMed] [Google Scholar]
- 37.Watson J, Anderson F B, O'Grady J E, Heald P J. J Endocrinol. 1975;65:7–17. doi: 10.1677/joe.0.0650007. [DOI] [PubMed] [Google Scholar]
- 38.Russo J, Russo I H. Cancer Res. 1980;40:2677–2687. [PubMed] [Google Scholar]
- 39.Sinha D K, Pazik J E, Dao T L. Br J Cancer. 1988;57:390–394. doi: 10.1038/bjc.1988.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grubbs C J, Juliana M M, Whitaker L M. Anticancer Res. 1988;8:113–117. [PubMed] [Google Scholar]
- 41.Russo I H, Russo J. J Cell Biochem. 2000;34:1–6. [PubMed] [Google Scholar]
- 42.Abrams T J, Guzman R C, Swanson S M, Thordarson G, Talamantes F, Nandi S. Anticancer Res. 1998;18:4115–4122. [PubMed] [Google Scholar]
- 43.Yang J, Yoshizawa K, Nandi S, Tsubura A. Carcinogenesis. 1999;20:623–628. doi: 10.1093/carcin/20.4.623. [DOI] [PubMed] [Google Scholar]
- 44.Bridges R S, Felicio L F, Pellerin L J, Stier A M, Mann P E. Life Sci. 1993;53:439–445. doi: 10.1016/0024-3205(93)90648-m. [DOI] [PubMed] [Google Scholar]
- 45.Musey V C, Collins D C, Musey P I, Martino-Saltzman D, Preedy R K. N Engl J Med. 1987;316:229–234. doi: 10.1056/NEJM198701293160501. [DOI] [PubMed] [Google Scholar]
- 46.Yankova M, Hart S A, Woolley C S. Proc Natl Acad Sci USA. 2001;98:3525–3530. doi: 10.1073/pnas.051624598. . (First Published February 20, 2001; 10.1073/pnas.051624598) [DOI] [PMC free article] [PubMed] [Google Scholar]