

Abstract
Chronic hepatitis C virus (HCV) infection is characterized by dysregulated natural killer (NK) cell responses. NKs play a critical role in achieving sustained responses to interferon (IFN)‐α‐based therapy. Rapid sustained HCV‐RNA clearance is now achieved with direct‐acting antivirals (DAAs). Studies of patients receiving first‐wave DAAs suggest NK functional restoration. Here, we investigate the effect of mainstream DAA treatment on NKs. We collected a prospective cohort of male HCV genotype 1‐infected patients treated with ledipasvir/sofosbuvir (n = 22). Peripheral blood was obtained at treatment start, week 2 (W2), W4, W8, and W12 of treatment and 12 weeks posttreatment. Flow cytometry was used to characterize NK responses to therapy. Mean baseline viral load was 1.75 million IU/mL. All subjects rapidly cleared virus and remained HCV RNA‐negative posttreatment. No change was seen in total NK levels; however, the frequency of immature NKs (clusters of differentiation [CD]56bright) decreased by W2 and was maintained throughout the study. Phenotypic changes were evident by W2/W4, coincident with rapid viral clearance. At W2, T‐cell immunoglobulin and mucin‐domain containing‐3 and CD161 were significantly increased, returning to pretreatment levels by W12. Some changes were not evident until late (W12 or posttreatment). Down‐regulation of several activation markers, including NKp30 and tumor necrosis factor–related apoptosis‐inducing ligand, was observed at W12 and sustained posttreatment. No difference was observed in IFN‐γ production or cytokine‐mediated killing of NK‐sensitive cell line K562 posttreatment compared to pretreatment. Conclusion: Our phenotype data suggest transient activation followed by dampening of NK cell activity to pretreatment levels. The NK response to ledipasvir/sofosbuvir is not universal in a homogeneous patient cohort. More studies are needed to elucidate the roles of NK cells in IFN‐free regimens, which will have implications for protection from re‐infection and fibrosis progression. (Hepatology Communications 2018;2:364‐375)
Abbreviations
- CCL
chemokine [C‐C motif] ligand
- CD
clusters of differentiation
- DAA
direct‐acting antiviral
- DNAM
DNAX accessory molecule‐1
- HCV
hepatitis C virus
- IFN
interferon
- Ig
immunoglobulin
- IL
interleukin
- MIP‐1β
macrophage inflammatory protein‐1β
- NK
natural killer
- NKR
natural killer receptor
- PBMC
peripheral blood mononuclear cell
- Siglec‐7
sialic acid‐binding immunoglobulin‐like lectin 7
- SVR
sustained virologic response
- Tim‐3
T‐cell immunoglobulin and mucin‐domain containing‐3
- TRAIL
tumor necrosis factor‐related apoptosis‐inducing ligand
Introduction
Innate immune natural killer (NK) cells represent a critical component of infection control and antitumor immunity through production of cytokines, chemokines, and cytolytic activity.1, 2, 3 They are also intimately involved in immune regulation and surveillance and play a central antifibrotic role.4, 5, 6 Expression of neural cell adhesion molecule (clusters of differentiation [CD]56) identifies NK cells in humans, and relative expression of this antigen identifies functionally distinct immature/regulatory (CD56bright) and mature/effector (CD56dim) NK subsets. Effector cells account for the majority of circulating NKs7, 8; however, in chronic hepatitis C virus (HCV), the immature population is relatively expanded.9, 10 In addition to these conventional NK cell subsets, a highly dysfunctional subset (CD56negative [neg]CD16positive [pos]) has been described that appears to be terminally differentiated and has impaired cytokine production and cytolytic function.11 This dysfunctional subset is increased in chronic HCV infection. High baseline levels have been correlated with failure to achieve sustained virologic response (SVR) on treatment with interferon (IFN)‐α‐based therapy.12
Activation of NKs is controlled by a network of activating and inhibitory NK receptors (NKRs), with overall activation status determined by the balance of signals transduced by these receptors. The predominant inhibitory NKRs are the killer immunoglobulin‐like receptors, which recognize human leukocyte antigen class I on autologous cells. Other important NKRs include C‐type lectin‐like receptors of the CD94/NKG2 family, comprising inhibitory (NKG2A) and activating (NKG2D) isoforms, as well as the natural cytotoxicity receptors NKp30 and NKp46, DNAX accessory molecule‐1 (DNAM; CD226), FAS (CD95), and tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL) receptors that deliver signals mediating activation.13, 14, 15, 16 Several other receptors involved in inhibition of NK cells have been described, including immunoglobulin (Ig)‐like transcript 2 (CD85j), sialic acid‐binding Ig‐like lectin 7 (Siglec‐7; CD328), and T‐cell Ig and mucin‐domain containing‐3 (Tim‐3).17, 18, 19 Dysregulation of NKR expression toward an activated phenotype is a feature of chronic HCV infection, and different NK‐cell phenotypic features in patients treated with IFN‐α‐based therapy are observed between nonresponder patients versus those achieving an SVR.20, 21
Chronic HCV infection is characterized by dysregulated or exhausted NK cell responses, which are critical effectors to achieving SVR to IFN‐α‐based therapies.20, 22 Data with respect to the functionality of NK cells in the setting of chronic HCV infection favor a polarization model with overactive cytotoxic and inadequate IFN‐γ responses. Several groups have provided convincing evidence that activation of NK cells by IFN‐α is important to achieve treatment‐induced viral clearance.20, 23 IFN‐α is a potent activator of NK cells; therefore, it is not surprising that NK cell activation has been identified as a key factor associated with SVR responses to IFN‐α‐based therapies.24 However, treatment of chronic HCV has been revolutionized by the development of direct‐acting antivirals (DAAs) and no longer includes IFN‐α. Rapid and sustained HCV‐RNA clearance can now be achieved with DAAs in the vast majority of treated patients.25 Rapid control of virus and decreased endogenous IFN‐α induced by DAA therapy may impact NK cell activation. Indeed, pilot studies using first‐line DAA therapy (daclatasvir/asunaprevir) suggest that NK cells contribute to clearance of HCV during DAA therapy.26, 27, 28 The aim of this study was to investigate the effect of current mainstream DAA (ledipasvir/sofosbuvir) treatment on NK cells.
Patients and Methods
SAMPLE COLLECTION AND STORAGE
We collected a prospective cohort of male patients with HCV genotype 1 and treated with ledipasvir/sofosbuvir per American Association for the Study of Liver Diseases–Infectious Diseases Society of America guidelines for 8/12 weeks (n = 22). The vast majority, all except 2, of subjects recruited for the study were male patients. We therefore decided to perform the assays using only male individuals to remove possible sex bias. The study protocol was approved by the Institutional Review Board at the University of Colorado Denver and the Denver Veteran's Affairs medical center. Both written and oral consent was obtained before samples were acquired. Peripheral blood was obtained at treatment start (pretreatment), week 2 (W2), W4, W8, and W12 of treatment and 12 weeks after treatment end (posttreatment). Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood by cellular preparation tubes (anticoagulant sodium citrate; Becton‐Dickinson, Franklin Lakes, NJ). PBMCs were viably frozen in 80% fetal bovine serum (BioWhittaker, Walkersville, MD), 10% dimethyl sulfoxide, and 10% Roswell Park Memorial Institute 1640 Media (Life Technologies, Grand Island, NY) and stored in liquid nitrogen for subsequent analyses.
ANTIBODIES FOR DETECTION AND FLUORESCENCE‐ACTIVATED CELL SORTING ANALYSIS OF ANTIGEN EXPRESSION
Fluorochrome‐labeled monoclonal antibodies specific for CD3 (V500) and CD56 (V450) (BD Biosciences, San Jose, CA) were used to identify NK (CD3negCD56pos) cells within the overall lymphocyte population. Anti‐NKR antibodies CD16, CD161, CD122, CD158a, CD158e, CD94, CD69, CD95 (FAS), CD226 (DNAM), CD253 (TRAIL), and CD314 (NKG2D) were purchased from BD Biosciences. Anti‐CD335 (NKp46), CD337 (NKp30), and CD159a (NKG2A) were purchased from Beckman Coulter (Indianapolis, IN). Anti‐CD158b and CD328 (Siglec‐7) were purchased from eBioscience (San Diego, CA). Anti‐Tim‐3 was purchased from R&D Systems (Minneapolis, MN).
Thawed PBMCs (1‐2 × 106) were stained for cell‐surface antigen expression at 4°C in the dark for 30 minutes. Samples were then washed twice in 2 mL phosphate‐buffered saline containing 1% bovine serum albumin and 0.01% sodium azide (Facs Wash; MilliporeSigma, St. Louis, MO) and subsequently fixed in 200 μL 1X BD stabilization fixative. Isotype‐matched control antibodies were used to determine background levels of staining. Multicolor flow cytometry was performed using a BD FACSCanto II instrument (BD Biosciences), compensated with single fluorochromes and analyzed using Diva software (BD Biosciences). Lymphocyte populations were identified by their characteristic forward scatter/side scatter properties.
CYTOKINE AND CHEMOKINE ASSAYS
PBMCs were cultured for 24 hours at a concentration of 1 million/mL in the presence or absence of interleukin (IL)‐12 (25 ng/mL; R&D Systems) and IL‐18 (10 ng/mL, R&D Systems). Intracellular flow cytometric staining was used to measure NK IFN‐γ responses. Four hours before the end of the culture, brefeldin A (10 μg/mL; MilliporeSigma) was added to the wells. Cells were harvested, washed, surface stained for CD3/CD56, then fixed and incubated with anti‐IFN‐γ (BD Biosciences) monoclonal antibody for 1 hour at room temperature using the Invitrogen eBioscience Intracellular Fixation and Permeabilization Buffer Set (Thermo Fisher). For the detection of chemokine production, duplicate cultures were performed without the addition of brefeldin A. Supernatants were collected and chemokine levels were measured using the LEGENDplex Human Proinflammatory Chemokine Panel (Biolegend, San Diego, CA) according to the manufacturer's instructions.
CYTOTOXICITY ASSAYS
Thawed mononuclear cell suspensions were enriched for NK cells using the NK Isolation Kit II from Miltenyi Biotec (Gladbach, Germany) following the manufacturer's instructions. The purity of isolated NKs was >90% in all cases as assessed by flow cytometry. Following isolation, the NKs were cultured in the presence or absence of IL‐2 (25 ng/mL; R&D Systems) or IL‐15 (100 ng/mL; R&D Systems) for 48 hours at 37°C and 5% CO2. Following culture, carboxy fluorescein succinimidyl ester‐labeled NK‐sensitive target cells (K562s; MilliporeSigma) were added to the NKs at effector:target concentrations of 0:1 (negative control) and 10:1 (test) and incubated at 37°C for 4 hours. After incubation, cytotoxicity was measured using the flow‐cytometry based Total Cytotoxicity & Apoptosis Detection Kit from Immunochemistry (Bloomington, MN). Immediately before acquisition, 7‐aminoactinomycin D was added to effector:target populations and incubated for 15 minutes on ice.
STATISTICS
Results are expressed as mean (range or SEM). Paired and unpaired t tests were used as appropriate to compare differences in levels, activity, and marker expression. Correlations were assessed using Spearman's rank correlation coefficient. Significance was defined as P < 0.05. Adjustment for multiple comparisons was not performed, and we report all uncorrected P values <0.05 and include all nonsignificant comparisons in http://onlinelibrary.wiley.com/doi/10.1002/hep4.1166/full. GraphPad Prism 7 (La Jolla, CA) statistical software package was used for analysis and graphing.
Results
The average age of the all‐male HCV genotype 1‐infected cohort (n = 22) was 58 years (range 30‐67 years). Eleven of the subjects were treated for 8 weeks, and the remaining 50% were treated for 12 weeks. The mean baseline viral load for the 8‐week group was 1.29 million IU/mL and 2.20 million IU/mL for the 12‐week group. The viral load was not statistically different between the two treatment groups. Patient characteristics are shown in Table 1. All subjects rapidly cleared virus and remained HCV RNA‐negative 12 weeks posttreatment (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1166/full). There were no statistical differences between the two treatment groups. To establish actual changes in NK cell‐level phenotype and function in response to DAA therapy, all data collected during the course of treatment and posttreatment were compared to the pretreatment time point.
Table 1.
Subject Baseline Characteristics
| Pre‐Tx HCV RNA IU/mL × 106 | Weeks TX | ALT | AST | Calc Fib‐4 |
|---|---|---|---|---|
| 2.56 | 8 | 91 | 111 | 3.24 |
| 0.35 | 8 | 200 | 87 | 1.71 |
| 0.25 | 8 | 70 | 36 | 0.59 |
| 3.84 | 8 | 39 | 19 | 1.12 |
| 1.72 | 8 | 69 | 43 | 1.24 |
| 1.12 | 8 | 89 | 38 | 0.61 |
| 1.00 | 8 | 37 | 39 | 2.38 |
| 0.58 | 8 | 50 | 26 | 0.77 |
| 0.22 | 8 | 62 | 42 | 1.28 |
| 2.28 | 8 | 41 | 27 | 1.04 |
| 0.24 | 8 | 183 | 109 | 2.47 |
| 1.09 | 12 | 59 | 33 | 1.53 |
| 0.44 | 12 | 67 | 33 | 1.38 |
| 5.72 | 12 | 26 | 23 | 1.04 |
| 0.73 | 12 | 40 | 27 | 1.27 |
| 0.59 | 12 | 214 | 189 | 6.34 |
| 4.65 | 12 | 34 | 37 | 1.80 |
| 1.20 | 12 | 63 | 28 | 0.79 |
| 1.02 | 12 | 32 | 21 | 1.06 |
| 5.88 | 12 | 133 | 48 | 1.19 |
| 1.52 | 12 | 35 | 34 | 1.6 |
| 1.41 | 12 | 47 | 28 | 1.91 |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; Calc Fib‐4, calculated fibrosis 4.
Flow cytometry was used to investigate the levels and phenotype of total (CD3negCD56pos), immature (CD3negCD56bright), and mature effector (CD3negCD56dim) NK cell populations before, during, and after treatment with ledipasvir/sofosbuvir per American Association for the Study of Liver Diseases–Infectious Diseases Society of America guidelines. No change was seen in total NK levels; however, immature NK cell (CD56bright) levels decreased by W2 and continued at this lower level throughout the study (Fig. 1). There was a concomitant increase in mature NK cell levels (data not shown). In addition, there was no change in the levels of the highly dysfunctional CD56negCD16pos NK cell subset (data not shown).
Figure 1.
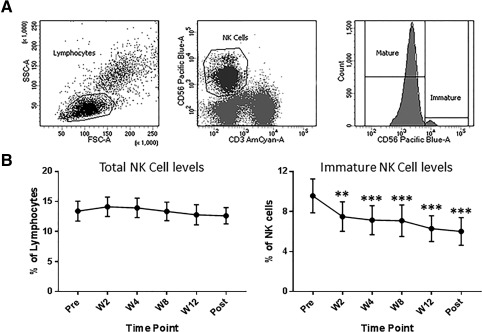
Immature (CD56bright) NK cell levels are decreased by week 2 of treatment. NK cells are defined as CD3‐negative CD56‐positive cells with low forward scatter and side scatter properties. (A) Mature and immature subsets of NK cells are identified by the expression level of CD56. (B) Total NK cell levels remained unchanged; however, immature NK cell levels were decreased by 2 weeks on DAA therapy and were at this lower level throughout the study period. **P < 0.01, ***P < 0.005, compared to pretreatment levels. Abbreviations: FSC, forward scatter; SSC, side scatter.
Phenotypic changes were evident as early as W2/W4, which coincided with a rapid decrease of virus from serum. At W2 and W4, there was a significant increase in the expression of NK activation markers Tim‐3 and CD161, predominantly on the mature NK cell population, which returned to pretreatment levels by W8. Conversely, CD69, expressed by activated NKs, was decreased on total and effector NKs at W2 through W12 and on immature NKs posttreatment (Fig. 2). Other changes in NK cell phenotype were not evident until several weeks after clearance of virus (W12 or posttreatment). Down‐regulation of NK cytotoxicity receptors, NKp30 on all NK cell subsets, and TRAIL on the immature NK cell population was observed at W12 and sustained posttreatment. Another cytotoxicity receptor, NKp46, was down‐regulated posttreatment on all NK cell subsets (Fig. 3). Phenotypic changes were also detected at an intermediate time point. FAS (CD95), which plays a central role in the physiologic regulation of programmed cell death and is up‐regulated on NK cells in response to cytokines,29 was down‐regulated at W8 on total and effector NKs. Siglec‐7, an NK cell inhibitory receptor,30 was up‐regulated on these populations at the same time (Fig. 4). Several other receptors that have been implicated in the NK cell response to HCV and IFN‐based therapy, including killer immunoglobulin‐like receptors (CD158a/b/e), NKG2A/D, and DNAM, were unaffected by treatment with ledipasvir/sofosbuvir (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1166/full).
Figure 2.

Changes in NK cell phenotype early during the course of treatment. Flow cytometric analysis of the expression levels of NK receptors on total NKs and immature (CD56bright) and mature (CD56dim) subsets demonstrated up‐regulation of activation markers Tim‐3 and CD161 at W2 and W4 of treatment; markers returned to pretreatment levels by W8. Conversely, CD69, another activation marker expressed at low levels on peripheral NKs, was down‐regulated by W2. *P < 0.05, **P < 0.01, ***P < 0.005, compared to pretreatment levels. Abbreviations: MFI, mean fluorescence intensity; Post, posttreatment.
Figure 3.
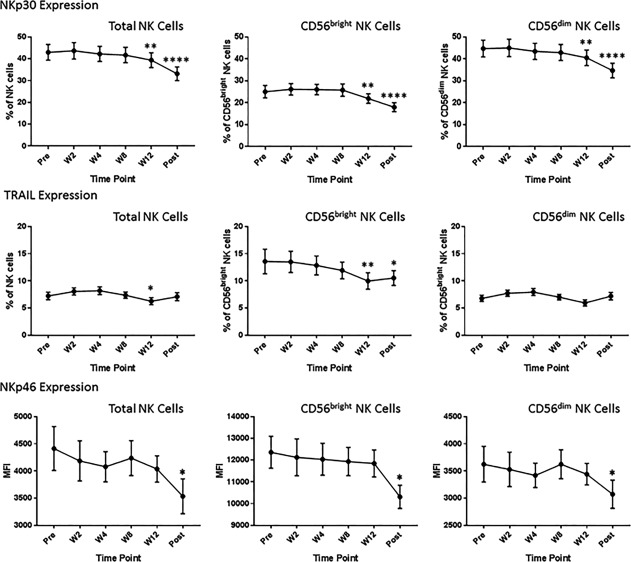
Changes in NK cell phenotype late during the course of treatment. Flow cytometric analysis demonstrated decreased expression of NK receptors involved in cytotoxicity late in the course of treatment (W12) and 12 weeks posttreatment. These changes occurred several weeks after clearance of virus. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.0001, compared to pretreatment levels. Abbreviations: MFI, mean fluorescence intensity; Post, posttreatment.
Figure 4.
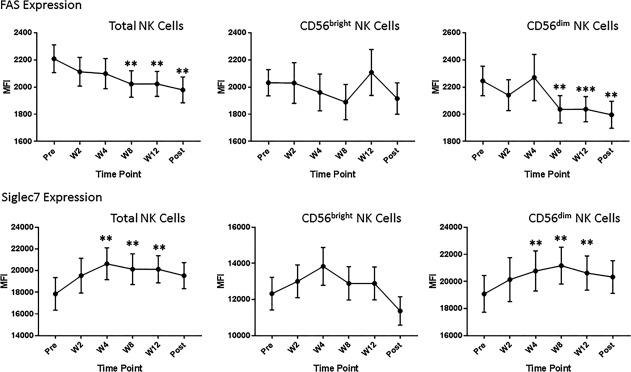
Changes in NK cell phenotype at an intermediate time point during the course of treatment. At around 8 weeks of treatment, we observed down‐regulation of the activation receptor FAS and up‐regulation of the inhibitory receptor Siglec‐7 on total and effector (CD56dim) NK cells. **P < 0.01, ***P < 0.005, compared to pretreatment levels. Abbreviations: MFI, mean fluorescence intensity; Post, posttreatment.
We also performed an analysis of the two treatment groups to determine if there was a difference in NK response to DAA therapy between patients with 8‐week or 12‐week therapy. We analyzed total NK cells and CD56bright and CD56dim subsets between the two treatment groups. We found no difference in the levels or in the phenotype of the NK cells/subsets for the markers tested and for all time points except for the expression of NKG2A (% positive) on total and CD56dim NK cells. NKG2A was lower for the 12‐week treatment group but was not changed during the course of treatment. In addition, NKG2A expression on total NK cells does not correlate with baseline viral load (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1166/full).
To assess the possible functional implications of the above observed phenotypic changes in NK cells, intranuclear staining was used to look at the expression of transcription factors important for NK cell cytokine production.31 Decreased expression of T‐cell‐specific T‐box transcription factor (T‐bet), which is involved in control of IFN‐γ expression and serves as an NK cell maturation marker, was only identified posttreatment. No change was detected in the expression of eomesodermin (Eomes) (Fig. 5).
Figure 5.

NK cell transcription factor expression. Intranuclear staining was used to examine the expression of transcription factors important for the function of NK cells. Down‐regulation of T‐bet was highly significant at 12 weeks posttreatment. No changes were observed for EOMES expression levels. *P < 0.05, ****P < 0.0001, compared to pretreatment levels. Abbreviations: MFI, mean fluorescence intensity; Post, posttreatment.
Overall, our phenotypic data suggested early (W2) transient activation of NK cells after treatment start with ledipasvir/sofosbuvir, followed by dampening of NK cell activity to below pretreatment levels by 12 weeks after cessation of treatment. Therefore, we assessed the functionality of NK cells at an early time point (W2) that coincided with rapid viral decline and at 12 weeks after the end of treatment when virus had not been circulating for a considerable amount of time. No difference was seen in IFN‐γ production in response to stimulation with IL‐12 and IL‐18 at the early time point. Even at 12 weeks after treatment end, there was no evidence of restoration of IFN‐γ responses (data not shown). NK cells are also important producers of chemokines,32 and several serum cytokines, including IFN‐inducible protein 10 (chemokine [C‐X‐C motif] ligand 10) and macrophage inflammatory protein‐1β (MIP‐1β; chemokine [C‐C motif] ligand 4 [CCL4]), are dysregulated after successful DAA treatment for HCV.28, 33, 34 We therefore asked if chemokine production by NK cells was altered by ledipasvir/sofosbuvir therapy. The majority of chemokines tested did not change significantly during the course of treatment (http://onlinelibrary.wiley.com/doi/10.1002/hep4.1166/full). There was a trend toward decreased MIP‐3α (CCL20) production at W2 of treatment and 12 weeks posttreatment. Production of MIP‐1α (CCL3), involved in recruitment and activation of polymorphonuclear leukocytes,35 increased slightly at W2 of treatment and reached significance posttreatment.
We also assessed the cytotoxic function of NK cells at the same time points we examined IFN‐γ and chemokine production. Natural cytotoxicity against the NK‐sensitive cell line K562 was unchanged during treatment. Fifty percent of subjects showed improved and the other 50% demonstrated unchanged or reduced IL‐2‐ and IL‐15‐mediated killing of K562 posttreatment compared to pretreatment (Fig. 6B,C). There was no positive or negative correlation between IFN‐γ production and cytotoxic activity.
Figure 6.
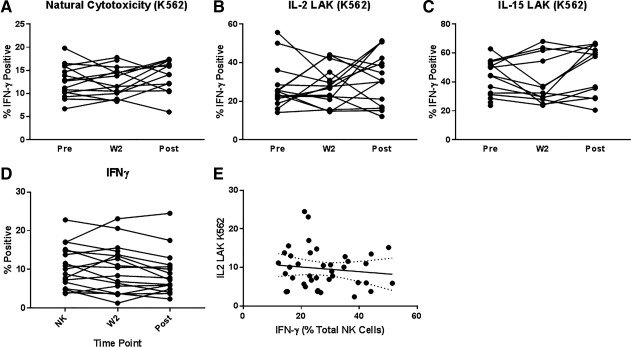
Cytotoxicity and interferon‐γ production. The cytolytic activity of isolated NK cells at the same time points we examined. IFN‐γ and chemokine production was measured using a flow cytometry assay. (A) Natural cytotoxicity against the NK‐sensitive cell line K562 was unchanged during treatment and posttreatment. (B,C) Improved LAK (IL‐2 or IL‐15) of K562 was evident for 50% of subjects posttreatment compared to pretreatment. (D,E) There was no positive or negative correlation between IFN‐γ production and cytotoxic activity.
Discussion
Altered NK cell subset distribution and activation marker expression and function are hallmarks of chronic HCV infection.20 IFN‐α is a potent activator of NK cells; therefore, it is not surprising that NK cell activation has been identified as a key factor associated with SVR responses to IFN‐α‐based therapies.24 In contrast, DAAs would not be expected to have a direct effect on NK cell phenotype and function; however, rapid control of virus could result in decreased endogenous IFN‐α, which may result in decreased activation. Indeed, pilot studies on first‐line DAA therapy (daclatasvir/asunaprevir) suggest that NK cells may contribute to clearance of HCV during DAA therapy.26, 27, 28, 36 These early studies focused mainly on differences between responders and nonresponders or comparisons with external healthy control groups without liver disease. In the present study, we evaluated the levels, phenotype, and function of peripheral NK cells in a fairly homogeneous cohort of all‐male HCV genotype 1‐infected subjects treated with ledipasvir/sofosbuvir, a current mainstream treatment for HCV. Our study was designed to assess the effects of highly successful DAA therapy on NK cells. To achieve this, we used the baseline pretreatment NK cell profile of each subject as our reference control.
In the present study, all subjects had a rapid response to DAA therapy, becoming nonviremic by 2 or 4 weeks on treatment and remaining HCV RNA‐negative 12 weeks posttreatment. Several changes were evident in the NK cell population coinciding with the rapid disappearance of virus from the serum. The proportion of CD56bright immature NKs, reported by several groups to be increased in chronic HCV, decreased at W2 and was maintained at a lower level throughout the study time frame. Concomitantly, CD56dim effector NK cells increased and demonstrated higher expression of activation markers Tim‐3 and CD161 at 2 and 4 weeks on therapy compared to pretreatment levels. Conversely, at this early time point, CD69, an activation marker, was decreased. Several other NK cell receptors, both inhibitory and activating, were unchanged. Taken together, these data suggest early activation of effector NK cells and support a potential role for NK cells in the clearance of HCV during DAA therapy, as suggested by first‐line DAA treatment regimens.26, 27, 28, 36 After viral clearance, it is likely that abatement of chronic inflammatory signals and their effect on immune cells lag behind and may not return to normal healthy levels due to the presence of underlying liver disease. The changes in NK phenotype that we observed late (predominantly 12 weeks after the end of treatment), comprising decreased expression of activation (CD69, CD161) and cytotoxicity (NKp30, NKp46, and TRAIL) receptors, suggest that the chronically activated NK phenotype characteristic of chronic HCV is reversed by ledipasvir/sofosbuvir therapy but that normalization of the activation status takes several weeks after the disappearance of virus. The expression patterns of FAS (CD95) and Siglec‐7 (CD328), which are altered at intermediate time points (W4‐W12), suggest that down‐regulation of the activation status of NK cells is a gradual process.29, 30
Data on NK cell dysfunction in the setting of chronic HCV favor a polarization model with overactive cytotoxic and inadequate IFN‐γ responses.23, 37, 38, 39 Successful antiviral therapy, including DAAs, may result in normalization of NK cell function.27, 28, 36, 38 The study by Spaan and colleagues28 did not include function analysis of NK cells; however, down‐regulation of NKp46, NKp30, and TRAIL, observed after daclatasvir/asunaprevir treatment, suggests that cytotoxic activity is reduced. Our study showed similar results for these cytotoxicity receptors with ledipasvir/sofosbuvir treatment. The study by Serti and colleagues27 also demonstrated down‐regulation of NKp46 and TRAIL and in addition providing convincing functional data that suggest NK cell IFN‐γ production is enhanced and cytotoxicity as assessed by CD107a degranulation is decreased by successful therapy with daclatasvir/asunaprevir. After SVR, one might expect reconstitution of immune cell function, and these studies suggest that rapid and sustained viral load decline induced by successful DAA therapy normalizes NK cell function.24 Our phenotype data suggested early transient activation of NK cells and a reduction in cytotoxic activity 12 weeks after the end of treatment. We found no evidence to support improvement in IFN‐γ production by NK cells or a reduction in cytotoxicity in our cohort at an early time point (W2) or 12 weeks after the end of treatment. There was no correlation between IFN‐γ production and natural cytotoxicity or lymphokine‐activated killing (LAK) activity. Of interest, 50% of subjects showed improved and the other half demonstrated unchanged or reduced IL‐2‐ and IL‐15‐mediated killing of NK‐sensitive cell line K562 posttreatment compared to pretreatment. Despite the fairly homogeneous nature of our cohort, this demonstrates differential responses to successful DAA therapy. Although it is difficult to extrapolate the findings from previous studies to ours because of differences in the cohorts, assays performed, and treatment, our phenotype data are in agreement, suggesting dampened NK cell activation after successful DAA treatment.27, 28, 36 While the phenotype data suggest normalization of NK cell function, in the present study we did not find functional evidence to support this. Mucosa‐associated invariant T cells, another innate immune cell population, were not functionally restored after DAA therapy, suggesting that reconstitution of immune cell function may not always be the case.40 Another important factor likely to influence the extent of immune reconstitution after successful DAA therapy is the severity of cirrhosis.33 Only 1 of our subjects had a relatively high fibrosis‐4, indicating the presence of cirrhosis, which precluded analysis of the relationship between NK cell phenotype and function with cirrhosis.
It remains unclear if NK restoration is needed for cure, and reported changes in this population may simply result from the disappearance of virus. Overall, our study suggests that disappearance of the virus correlates with phenotypic changes in the NK cell population. However, several changes are not evident until several weeks after clearance. In addition, our functional data suggest that NK cell restoration (at least for the parameters tested) is not essential for DAA‐mediated cure. It should also be noted that we do not have data on hepatic NK cells for this cohort. Changes seen in the peripheral NK cell populations may not mirror hepatic NK cell populations, and we cannot preclude an essential role for NKs at the site of viral replication.
Future studies using well‐defined larger cohorts of subjects treated with standard DAA regimens are needed to fully elucidate the extent, mechanisms, and kinetics of recovery of dysfunctional NK cell responses. Restoration of NK cell function has several potential clinical implications, such as protection from reinfection, reactivation of other viruses,41, 42 hepatocellular carcinoma development,33 and possibly reversal of fibrosis.43, 44
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1166/full.
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Table 1
Supporting Information Figure Caption
Potential conflict of interest: Nothing to report.
Supported by the National Institute of Allergy and Infectious Diseases (RO1AI120622 to H.R.R.) and the National Institute of Diabetes and Digestive and Kidney Diseases (RO1DK106491 to L.G‐M).
REFERENCES
- 1. Lanier LL. Evolutionary struggles between NK cells and viruses. Nat Rev Immunol 2008;8:259‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol 2008;9:503‐510. [DOI] [PubMed] [Google Scholar]
- 3. Biron CA. Yet another role for natural killer cells: cytotoxicity in immune regulation and viral persistence. Proc Natl Acad Sci U S A 2012;109:1814‐1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sharma P, Kumar P, Sharma R. Natural killer cells ‐ their role in tumour immunosurveillance. J Clin Diagn Res 2017;11:BE01‐BE05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tosello‐Trampont A, Surette FA, Ewald SE, Hahn YS. Immunoregulatory role of NK cells in tissue inflammation and regeneration. Front Immunol 2017;8:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gao B, Radaeva S. Natural killer and natural killer T cells in liver fibrosis. Biochim Biophys Acta 2013;1832:1061‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer‐cell subsets. Trends Immunol 2001;22:633‐640. [DOI] [PubMed] [Google Scholar]
- 8. Chan A, Hong DL, Atzberger A, Kollnberger S, Filer AD, Buckley CD, et al. CD56bright human NK cells differentiate into CD56dim cells: role of contact with peripheral fibroblasts. J Immunol 2007;179:89‐94. [DOI] [PubMed] [Google Scholar]
- 9. Golden‐Mason L, Madrigal‐Estebas L, McGrath E, Conroy MJ, Ryan EJ, Hegarty JE, et al. Altered natural killer cell subset distributions in resolved and persistent hepatitis C virus infection following single source exposure. Gut 2008;57:1121‐1128. [DOI] [PubMed] [Google Scholar]
- 10. Bonorino P, Ramzan M, Camous X, Dufeu‐Duchesne T, Thelu MA, Sturm N, et al. Fine characterization of intrahepatic NK cells expressing natural killer receptors in chronic hepatitis B and C. J Hepatol 2009;51:458‐467. [DOI] [PubMed] [Google Scholar]
- 11. Mavilio D, Lombardo G, Benjamin J, Kim D, Follman D, Marcenaro E, et al. Characterization of CD56‐/CD16+ natural killer (NK) cells: a highly dysfunctional NK subset expanded in HIV‐infected viremic individuals. Proc Natl Acad Sci U S A 2005;102:2886‐2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gonzalez VD, Falconer K, Bjorkstrom NK, Blom KG, Weiland O, Ljunggren HG, et al. Expansion of functionally skewed CD56‐negative NK cells in chronic hepatitis C virus infection: correlation with outcome of pegylated IFN‐alpha and ribavirin treatment. J Immunol 2009;183:6612‐6618. [DOI] [PubMed] [Google Scholar]
- 13. Lanier LL. NK cell recognition. Annu Rev Immunol 2005;23:225‐274. [DOI] [PubMed] [Google Scholar]
- 14. Moretta A, Biassoni R, Bottino C, Moretta L. Surface receptors delivering opposite signals regulate the function of human NK cells. Semin Immunol 2000;12:129‐138. [DOI] [PubMed] [Google Scholar]
- 15. Bozzano F, Picciotto A, Costa P, Marras F, Fazio V, Hirsch I, et al. Activating NK cell receptor expression/function (NKp30, NKp46, DNAM‐1) during chronic viraemic HCV infection is associated with the outcome of combined treatment. Eur J Immunol 2011;41:2905‐2914. [DOI] [PubMed] [Google Scholar]
- 16. Smyth MJ, Cretney E, Kelly JM, Westwood JA, Street SE, Yagita H, et al. Activation of NK cell cytotoxicity. Mol Immunol 2005;42:501‐510. [DOI] [PubMed] [Google Scholar]
- 17. Colonna M, Navarro F, Bellon T, Llano M, Garcia P, Samaridis J, et al. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J Exp Med 1997;186:1809‐1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nicoll G, Ni J, Liu D, Klenerman P, Munday J, Dubock S, et al. Identification and characterization of a novel siglec, siglec‐7, expressed by human natural killer cells and monocytes. J Biol Chem 1999;274:34089‐34095. [DOI] [PubMed] [Google Scholar]
- 19. Xu L, Huang Y, Tan L, Yu W, Chen D, Lu C, et al. Increased Tim‐3 expression in peripheral NK cells predicts a poorer prognosis and Tim‐3 blockade improves NK cell‐mediated cytotoxicity in human lung adenocarcinoma. Int Immunopharmacol 2015;29:635‐641. [DOI] [PubMed] [Google Scholar]
- 20. Golden‐Mason L, Rosen HR. Natural killer cells: multifaceted players with key roles in hepatitis C immunity. Immunol Rev 2013;255:68‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oliviero B, Mele D, Degasperi E, Aghemo A, Cremonesi E, Rumi MG, et al. Natural killer cell dynamic profile is associated with treatment outcome in patients with chronic HCV infection. J Hepatol 2013;59:38‐44. [DOI] [PubMed] [Google Scholar]
- 22. Bi J, Tian Z. NK cell exhaustion. Front Immunol 2017;8:760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oliviero B, Varchetta S, Paudice E, Michelone G, Zaramella M, Mavilio D, et al. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology 2009;137:1151‐1160, 1160.e1151‐e1157. [DOI] [PubMed] [Google Scholar]
- 24. Mondelli MU. Direct‐acting antivirals cure innate immunity in chronic hepatitis C. Gastroenterology 2015;149:25‐28. [DOI] [PubMed] [Google Scholar]
- 25. Spengler U. Direct antiviral agents (DAAs) ‐ A new age in the treatment of hepatitis C virus infection. Pharmacol Ther 2017; doi:10.1016/j.pharmthera.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 26. Burchill MA, Golden‐Mason L, Wind‐Rotolo M, Rosen HR. Memory re‐differentiation and reduced lymphocyte activation in chronic HCV‐infected patients receiving direct‐acting antivirals. J Viral Hepat 2015;22:983‐991. [DOI] [PubMed] [Google Scholar]
- 27. Serti E, Chepa‐Lotrea X, Kim YJ, Keane M, Fryzek N, Liang TJ, et al. Successful interferon‐free therapy of chronic hepatitis C virus infection normalizes natural killer cell function. Gastroenterology 2015;149:190‐200.e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Spaan M, van Oord G, Kreefft K, Hou J, Hansen BE, Janssen HL, et al. Immunological Analysis during interferon‐free therapy for chronic hepatitis C virus infection reveals modulation of the natural killer cell compartment. J Infect Dis 2016;213:216‐223. [DOI] [PubMed] [Google Scholar]
- 29. Medvedev AE, Johnsen AC, Haux J, Steinkjer B, Egeberg K, Lynch DH, et al. Regulation of Fas and Fas‐ligand expression in NK cells by cytokines and the involvement of Fas‐ligand in NK/LAK cell‐mediated cytotoxicity. Cytokine 1997;9:394‐404. [DOI] [PubMed] [Google Scholar]
- 30. Shao JY, Yin WW, Zhang QF, Liu Q, Peng ML, Hu HD, et al. Siglec‐7 defines a highly functional natural killer cell subset and inhibits cell‐mediated activities. Scand J Immunol 2016;84:182‐190. [DOI] [PubMed] [Google Scholar]
- 31. Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, et al. T‐bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med 2014;211:563‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eberlein J, Nguyen TT, Victorino F, Golden‐Mason L, Rosen HR, Homann D. Comprehensive assessment of chemokine expression profiles by flow cytometry. J Clin Invest 2010;120:907‐923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Childs K, Merritt E, Considine A, Sanchez‐Fueyo A, Agarwal K, Martinez‐Llordella M, et al. Immunological predictors of nonresponse to directly acting antiviral therapy in patients with chronic hepatitis C and decompensated cirrhosis. Open Forum Infect Dis 2017;4:ofx067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hengst J, Falk CS, Schlaphoff V, Deterding K, Manns MP, Cornberg M, et al. Direct‐acting antiviral‐induced hepatitis C virus clearance does not completely restore the altered cytokine and chemokine milieu in patients with chronic hepatitis C. J Infect Dis 2016;214:1965‐1974. [DOI] [PubMed] [Google Scholar]
- 35. Wolpe SD, Davatelis G, Sherry B, Beutler B, Hesse DG, Nguyen HT, et al. Macrophages secrete a novel heparin‐binding protein with inflammatory and neutrophil chemokinetic properties. J Exp Med 1988;167:570‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Serti E, Park H, Keane M, O'Keefe AC, Rivera E, Liang TJ, et al. Rapid decrease in hepatitis C viremia by direct acting antivirals improves the natural killer cell response to IFNalpha. Gut 2017;66:724‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ahlenstiel G, Titerence RH, Koh C, Edlich B, Feld JJ, Rotman Y, et al. Natural killer cells are polarized toward cytotoxicity in chronic hepatitis C in an interferon‐alfa‐dependent manner. Gastroenterology 2010;138:325‐335.e321‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dessouki O, Kamiya Y, Nagahama H, Tanaka M, Suzu S, Sasaki Y, et al. Chronic hepatitis C viral infection reduces NK cell frequency and suppresses cytokine secretion: reversion by anti‐viral treatment. Biochem Biophys Res Commun 2010;393:331‐337. [DOI] [PubMed] [Google Scholar]
- 39. Mondelli MU, Oliviero B, Mele D, Mantovani S, Gazzabin C, Varchetta S. Natural killer cell functional dichotomy: a feature of chronic viral hepatitis? Front Immunol 2012;3:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hengst J, Strunz B, Deterding K, Ljunggren HG, Leeansyah E, Manns MP, et al. Nonreversible MAIT cell‐dysfunction in chronic hepatitis C virus infection despite successful interferon‐free therapy. Eur J Immunol 2016;46:2204‐2210. [DOI] [PubMed] [Google Scholar]
- 41. Perello MC, Fernandez‐Carrillo C, Londono MC, Arias‐Loste T, Hernandez‐Conde M, Llerena S, et al. Reactivation of Herpesvirus in Patients With Hepatitis C Treated With Direct‐Acting Antiviral Agents. Clin Gastroenterol Hepatol 2016;14:1662‐1666.e1661. [DOI] [PubMed] [Google Scholar]
- 42. Collins JM, Raphael KL, Terry C, Cartwright EJ, Pillai A, Anania FA, et al. Hepatitis B virus reactivation during successful treatment of hepatitis C virus with sofosbuvir and simeprevir. Clin Infect Dis 2015;61:1304‐1306. [DOI] [PubMed] [Google Scholar]
- 43. Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D‐dependent and tumor necrosis factor‐related apoptosis‐inducing ligand‐dependent manners. Gastroenterology 2006;130:435‐452. [DOI] [PubMed] [Google Scholar]
- 44. Kramer B, Korner C, Kebschull M, Glassner A, Eisenhardt M, Nischalke HD, et al. Natural killer p46High expression defines a natural killer cell subset that is potentially involved in control of hepatitis C virus replication and modulation of liver fibrosis. Hepatology 2012;56:1201‐1213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep4.1166/full.
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Figure 3
Supporting Information Table 1
Supporting Information Figure Caption