

Abstract
Immature mucosal defenses contribute to increased susceptibility of newborn infants to pathogens. Sparse knowledge of age-dependent changes in mucosal immunity has hampered improvements in neonatal morbidity due to infections. Here, we report that exposure of neonatal mice to commensal bacteria immediately after birth is required for a robust host defense against bacterial pneumonia, the leading cause of death in newborn infants. This crucial window was characterized by an abrupt influx of interleukin (IL)-22 producing group 3 innate lymphoid cells (IL22+ILC3) into the lungs of newborn mice. This influx was dependent on sensing of commensal bacteria by intestinal mucosal dendritic cells. Disruption of postnatal commensal colonization or selective depletion of dendritic cells interrupted the migratory program of lung IL-22+ILC3 and made the newborn mice more susceptible to pneumonia, which was reversed by transfer of commensal bacteria after birth. Thus, the resistance of newborn mice to pneumonia relied on commensal bacteria-directed ILC3-influx into the lungs, which mediated IL-22-dependent host resistance to pneumonia during this developmental window. These data establish that postnatal colonization by intestinal commensal bacteria is pivotal in the development of lung defenses in mice.
Graphical abstract
Introduction
Development of the immune system requires a sequential series of timed and coordinated events that begin early in fetal life and continue through the early postnatal period (1). Disruption of immune development during the early neonatal period results in abnormal postnatal immune responses that are more dramatic and persistent than those after disruption during adult life, highlighting the importance of the neonatal period as a critical developmental window (2). While several host genetic and environmental factors modulate the development of the immune system during fetal and early postnatal life (3), few are as important as the continued interaction with commensal bacteria, which is not only the most intimate environmental exposure (4, 5), but also represents a challenge to the developing immune system (6, 7).
Commensal colonization, which begins at birth, progresses through a choreographed succession of bacterial species and evolves rapidly during the first month of life (8). These evolving microbial signals are hypothesized to play a critical role in the functional programming of immune cells. Modern childbirth practices like caesarean deliveries (9) and increased use of antibiotics in early life (10) not only alter the pattern of intestinal commensal colonization in the newborn, but are also associated with increased risk of sepsis and pneumonia (10–14), suggesting that intestinal commensal bacteria can promote the resistance of newborn infants to pneumonia. The interaction between host and the intestinal commensal bacteria extends beyond the local enteric environment and influences immune homeostasis at peripheral sites, exemplified by intestinal complications during respiratory disease and vice versa (15, 16). Nevertheless, the mechanistic basis of cross talk between the intestinal commensal bacteria and innate lung defense, the so-called gut-lung axis, remains poorly defined (17) and the developmental pathways underlying the association between commensal colonization in the early postnatal period and development of lung immunity in newborns remain unexplored.
Here, we show that interactions between host and the intestinal commensal bacteria shape the repertoires of immune cells in the newborn mouse lung and importantly directs the postnatal ontogeny of IL-22 producing type 3 innate lymphoid cells (ILC3), a group of sentinel cells that maintain homeostasis at mucosal barrier sites. This postnatal influx of IL-22+ILC3 promotes the resistance of neonatal mice to pneumonia. This crosstalk is mediated by mucosal dendritic cells (DC), which capture signals from intestinal commensal bacteria. Disruption of commensal bacteria interrupted the migratory program of ILC3, impairing their ability to traffic to the lungs and rendering the newborn mice more susceptible to pneumonia, which was reversed by exogenous IL-22 or through adoptive transfer of ILC3. Reconstitution of intestinal commensal bacteria restored the expression of CCR4 on the ILC3, restored the ability of ILC3 to migrate into the lungs and promoted IL-22 dependent resistance to pneumonia in newborn mice.
Results
Postnatal colonization by commensal bacteria promotes resistance to pneumonia in newborn mice
Prior epidemiological studies show that human infants whose mothers received frequent antibiotics before birth, or who were delivered by Caesarean section, not only had altered intestinal commensal bacteria (18, 19), but also had increased risk of developing pneumonia (20, 21). This led us to hypothesize that early life exposure to commensal bacteria promotes resistance to pneumonia in newborns. To test this hypothesis, we exposed pregnant mouse dams to a combination of ampicillin, gentamicin, and vancomycin, three commonly used antibiotics in pregnant women and human newborns (22)(Fig. 1A), beginning 5 days before delivery. Antibiotics were discontinued immediately after birth and newborn mice were challenged intratracheally with Streptococcus pneumoniae (S. pneumoniae) serotype 19 A, a leading cause of pneumonia in human newborns (23).
Figure 1. Intestinal commensal bacteria promote resistance to S. pneumoniae in newborn mice via IL22.
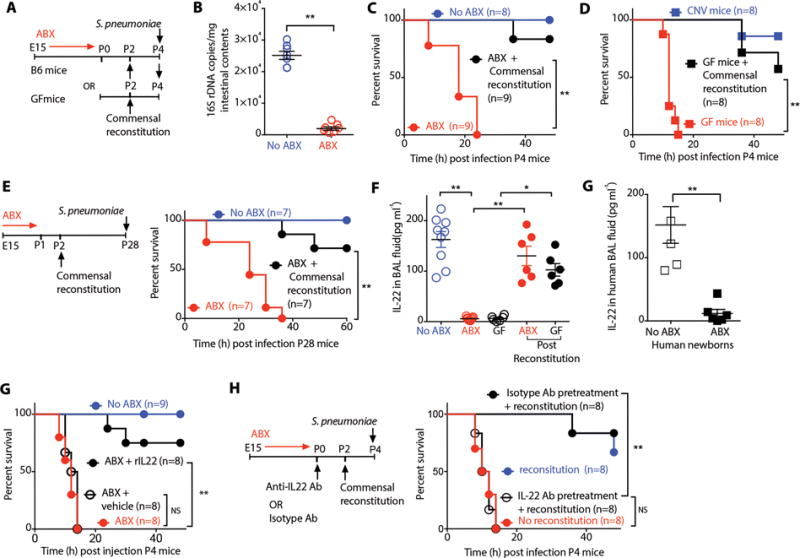
(A) Study design. (B) Intestinal commensal bacteria enumerated in postnatal day 4 (P4) newborn mice exposed to antibiotics (ABX) or no antibiotics (ABX-free) quantified using real-time PCR.. (C) Survival of ABX-free or ABX-exposed P4 mice or ABX-exposed newborn mice reconstituted with intestinal commensal bacteria and infected with S. pneumonia. (D) Survival of germ-free (GF) or conventionally housed (CNV) mice or age-matched GF mice reconstituted with intestinal commensal bacteria and infected with S. pneumoniae. (E) Survival of ABX-free or ABX-exposed P14 mice or ABX-exposed newborn mice reconstituted with intestinal commensal bacteria and infected with S. pneumoniae. (F) The amount of IL-22 in the bronchial lavage (BAL) fluid of P4 ABX-free or ABX-exposed mice or GF newborn mice or ABX-exposed or GF newborn mice reconstituted with intestinal commensal bacteria in early life. None of the newborn mice in this experimental group were inoculated with S. pneumoniae. (G) The amount of IL-22 in the BAL fluid of human newborns who were exposed to ABX or no ABX. (H) Survival of P4 ABX-free or ABX-exposed newborn mice treated with IL-22 intratracheally and infected with S. pneumoniae. (I) Survival of P4 ABX-free or ABX-exposed newborn mice treated with anti-IL22 antibody or isotype control antibody before reconstitution with intestinal commensal bacteria and infection with S. pneumoniae. Data are representative of three independent experiments. Results are shown as the mean ± s.e.m (Student’s t-test or ANOVA or Wilcoxon signed-rank test). *P ≤ 0.05; **P ≤ 0.01. Number of individual animals [n] are indicated.
This early life antibiotic exposure reduced not only the total number of commensal bacteria (Fig. 1B) but also disrupted the succession of bacterial species in the intestine of newborn mice (Fig. S1A,B, Table S1A). Six hours post infection, we observed an increased bacterial load in the lungs and the bronchial lavage (BAL) fluid (Fig. S1C) and increased susceptibility in newborn mice whose dams were exposed to antibiotics (ABX-exposed) as compared to age-matched mice whose dams were not exposed to antibiotics (ABX-free) (Fig. 1C). Germ-free (GF) mice, which lack commensal bacteria, similarly were more susceptible to challenge with S. pneumoniae as compared to the age-matched conventionally raised (CNV) mice (Fig. 1D). We paralleled these observations using Escherichia coli K1 or Candida albicans, other common causes of pneumonia in newborns (Fig. S1D,E) (24). Since disruption of commensal bacteria in infancy is associated with increased susceptibility to inflammatory disorders like allergen-induced airway hyperreactivity (25) and colitis (26) in later life, we ascertained whether disruption of postnatal commensal colonization led to durable changes in host resistance to infection. We found that increased susceptibility to pneumonia after early life ABX-exposure persisted beyond the neonatal period, until at least four weeks of age (Fig. 1E). This persistence in susceptibility contrasted with the transient susceptibility to infection that occurs in ABX-exposed adult mice (27, 28), highlighting the critical nature of commensal exposure in early life.
We reversed the commensal disruption in ABX-exposed newborns by transferring intestinal contents from a newborn mouse in the early postnatal period as done previously (Fig. 1A) (29). Reconstitution of intestinal commensal bacteria restored resistance to pneumonia in ABX-exposed and GF newborn mice (Fig. 1C,D). This protection against S. penumoniae persisted beyond the neonatal period. ABX-exposed mice that received intestinal contents in the early postnatal period likewise showed increased resistance to infection at least for as long as four weeks after birth compared to their littermates that did not receive intestinal bacterial reconstitution (Fig. 1E,F).
Whether lung-resident commensal bacteria educate the mucosal immune system, like the intestinal commensal bacteria, remains a source of controversy (30, 31) and the effect of early life antibiotics on lung commensal colonization in human newborns remains unexplored. We found no difference in the composition of lung commensal bacteria in ABX-free and ABX-exposed mice (Fig. S1F,G, Table S1B), perhaps related to our experimental strategy of limiting ABX-exposure to the pregnant dams and not the newborn mice. Lack of differences in lung-resident commensal bacteria in the ABX-exposed newborn mice as compared to ABX-free newborns suggest that intestinal commensal bacteria rather than lung commensals mediate the resistance to pneumonia, although this possibility cannot be completely excluded.
Postnatal colonization by commensal bacteria promotes interleukin (IL)-22 dependent mucosal defenses in newborn mice
We hypothesized that disruption of commensal colonization mediated changes in the expression of genes related to various aspects of innate lung defense. We carried out RNA sequencing analysis of lung mucosal RNA isolated from newborn mice on day 0-4. Unsupervised analysis revealed consistent transcriptional changes in ABX-exposed newborns as compared to ABX-free murine newborns (Fig. S1H, Table S2). Differentially expressed genes included interleukin (IL)-22, a cytokine critical in lung epithelial repair (Fig. S1H) (32, 33) and host defense against pathogens (32, 34, 35). We found decreased concentrations of IL-22 in the BAL fluid of ABX-exposed or GF newborn mice as compared to ABX-free newborn mice (Fig. 1F). We confirmed these observations in human newborns, finding reduced concentrations of IL-22 in the BAL fluid from human newborns exposed to prolonged duration of ABX (Fig. 1G, Table 1).
Table 1.
Demographic characteristics of human newborn infants.
| Patient Demographics |
Clinical History | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Race | GA (wks) |
Age at exam (days) |
Lobe lavage |
Asthma | Chronic Cough |
History of Mechanical Ventilation |
Ventilator Dependent |
History of Pneumonia |
Acute Respiratory Failure |
Interstitial Lung Disease |
Steroids | Antibiotic use |
Indications for study |
Study Findings |
| M | Caucasian | 34 | 56 | RLL | No | No | Yes | Yes | No | No | Yes | Yes | Yes | Surfactant Deficiency | Intestitial lung disease |
| M | Caucasian | 35 | 54 | RLL | No | No | Yes | Yes | No | No | No | Yes | Yes | airway obstruction | tracheal rings |
| M | Caucasian | 36 | 66 | RUL | No | No | No | No | No | No | No | No | No | airway obstruction | normal exam |
| M | Caucasian | 36 | 37 | LLL | No | No | Yes | No | No | No | No | No | No | ALTE | normal exam |
| F | African American | 36 | 68 | RUL | No | No | No | No | No | No | No | No | No | airway obstruction | laryngomalacia |
| F | Caucasian | 37 | 72 | RUL | No | No | Yes | Yes | No | No | No | Yes | No | subglottic stenosis | subglotiic stenosis |
| M | African American | 37 | 51 | RLL | No | No | No | No | No | No | No | No | No | airway obstruction | tracheal compression |
| M | Caucasian | 37 | 68 | RLL | No | No | No | No | No | No | No | No | Yes | airway obstruction | larynomalaia |
| F | African American | 37 | 48 | RUL | No | No | Yes | No | No | No | No | Yes | Yes | Stridor | laryngeal edema |
| M | African American | 37 | 54 | LLL | No | No | No | No | No | No | No | No | Yes | airway obstruction | normal exam |
| M | Caucasian | 38 | 80 | Lingula | No | No | No | No | No | No | No | No | No | tracheomalacia | mild tracheomalacia |
| M | Other | 38 | 76 | RUL | No | No | Yes | No | No | No | No | Yes | No | tracheomalacia | tracheomalacia |
| F | Caucasian | 39 | 54 | RLL | No | No | No | No | No | No | No | Yes | No | airway obstruction | laryngeal hemangioma |
| M | Caucasian | 39 | 42 | RUL | No | No | No | No | No | No | No | No | Yes | tracheomalacia | tracheomalacia |
| M | Caucasian | 40 | 61 | RLL | No | No | No | No | No | No | No | No | Yes | tracheomalacia | mild tracheomalacia |
Reconstitution with intestinal contents from age-matched neonatal mice restored IL-22 levels in BAL fluid of ABX-exposed or GF newborn mice (Fig. 1G). Similarly, treatment with recombinant IL-22 intratracheally restored host resistance to pneumonia in ABX-exposed newborn mice (Fig. 1H). To interrogate the importance of IL-22 in newborn’s resistance to pneumonia, we blocked IL-22 signaling with an IL-22 neutralizing antibody (36). Treatment of newborn mice with an anti-IL-22 antibody blocked the restoration of host resistance in ABX-exposed newborn mice after reconstitution of intestinal commensal bacteria (Fig. 1I). IL-22 acts via a transmembrane receptor complex that consists of IL-22R1, a receptor subunit that is shared by related cytokine IL-20 (37). We found no difference in concentrations of IL-20 in BAL fluid of ABX-exposed or GF newborn mice compared to ABX-free newborn mice (Fig. S1I). Blockade of IL-20 signaling by treatment with a neutralizing antibody directed against IL-20 (38) did not block restoration of host resistance in ABX-exposed newborn mice after reconstitution of intestinal commensal bacteria (Fig. S1J) (36). These findings demonstrate a central and non-redundant role for IL-22 in host defense against pneumonia (32, 33) and importantly, implicate IL-22 as a critical mediator by which commensal bacteria promote resistance to pneumonia in newborn mice . IL-22 bioactivity is negatively regulated by IL-22-binding protein (IL-22BP), a secreted receptor that binds to soluble IL-22 with higher affinity than IL-22R1 and functions as an antagonist (39). We did not evaluate the role of endogenous IL-22BP in our study. Several lung resident immune cells are known to secrete IL-22BP(40), but the role of endogenous IL-22BP in pulmonary host defense remains unclear and may represent an additional regulatory layer in the ontogeny of lung defense in the newborn.
Disruption of commensal bacteria in the early postnatal period leads to durable changes in the repertoire of IL-22 producing immune cells in the lungs of newborn mice, contributing to increased susceptibility to pneumonia
The identity of the IL-22-producing cells in the newborn mouse lung is unknown. We found that neither neutrophils (CD45+Ly6G+) nor macrophages (CD45+F4/80+) nor T cells (CD45+CD4+) were a significant source of IL-22 in the lungs of newborn mice (Fig. 2A). The majority of IL-22 producing cells in the murine newborn lung were lineage negative (CD45+CD3−CD8−CD11b−CD19−MHCII−F4/80−CD161−Ly6G−) lymphocytes. We further characterized these lineage-negative lymphocytes based on expression of surface markers CD4, CD117, CD127, NkP46 or CCR6 and transcription factors RORγt, T-Bet or Eomes. More than 90% of IL-22 producing cells were lineage negative (CD3−CD8−CD11b−CD19−MHCII−F4/80−CD161−Ly6G−F4/80−) lymphocytes expressing surface markers NKp46, CCR6, CD117 and transcription factor RORγt identifying them as ILC3 lymphoid cells (Fig 2A, S2A).
Figure 2. Intestinal commensal bacteria direct postnatal trafficking of IL-22+ILC3 innate lymphoid cells to murine newborn lung.
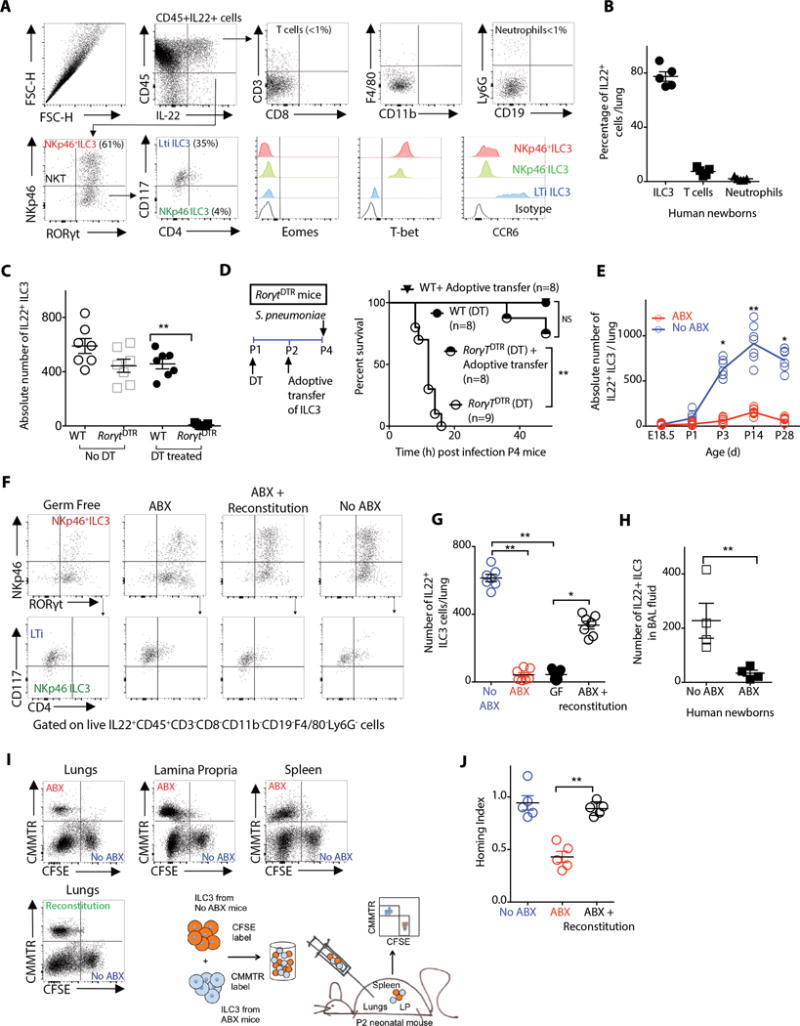
(A) Representative flow cytometry plots of distinct subsets of IL-22+ cells in the lungs of postnatal day 4 (P4) newborn mice. Shown are relative frequencies of IL22+ T cells (CD45+CD3+) or neutrophils (CD45+Ly6G+) or lineage negative (CD3−CD8−CD11b−CD19−F4/80−CD161−Ly6G−F4/80−) lymphocytes in the lungs of P4 newborn mice. (B) The relative frequencies of distinct subsets of IL-22+ cells in the bronchial lavage (BAL) fluid of human newborn infants.. (C) Absolute numbers of IL22+ILC3 in the lungs of P4 wild-type (WT) or RorγtiDTR newborn mice treated with diphtheria toxin (DT) or no DT. (D) Survival of P4 WT or RorγtiDTR newborn mice treated with DT (ILC3-depleted) that received adoptive transfer of ILC3 and then were infected with S. pneumoniae. (E) The absolute number of IL-22+ILC3 in the lungs of ABX-free or ABX-exposed newborn mice at different time points after birth. (F) Representative flow cytometry plots and (G) absolute numbers of IL-22+ILC3 in the lungs of P4 GF or ABX-free or ABX-exposed or ABX-exposed newborn mice reconstituted with intestinal commensal bacteria in early life. (H) The absolute numbers of IL-22+ILC3 in the BAL fluid of human newborns exposed to ABX or no ABX. (I) ILC3 from P4 ABX-free newborn mice were labeled with carboxyflourosceinsuccimidylester (CFSE). ILC3 from age-matched P4 ABX-exposed or ABX-exposed newborn mice reconstituted with commensal bacteria were labeled with chloromethylbenozylaminotetramethylrhodamine (CMMTR). An equal number of CFSE- or CMMTR-labeled ILC3 were adoptively transferred into ABX-exposed newborn mice. Representative flow cytometry plots and absolute numbers of CFSE+ or CMMTR+ ILC3 in lung, spleen or small intestine were determined 12 h following adoptive transfer. (J) Relative capability of ILC3 from ABX-free or ABX-exposed or ABX-exposed newborn mice reconstituted with intestinal commensal bacteria in early life to traffic to the lungs (Homing index). Data and plots are representative of three independent experiments. Results are shown as the mean ± s.e.m (Student’s t-test or ANOVA or Wilcoxon signed-rank test). *P ≤ 0.05; **P ≤ 0.01. Number of individual animals [n] are indicated.
We tested these observations in human newborns. ILC3 (CD45+CD3−CD8−CD14−CD19−CD69− RORγt+), but not neutrophils (CD45+CD3−CD8−CD19−CD69+), NK cells (CD45+CD3−CD8−CD19−CD56+), CD4+ T cells (CD45+CD3+CD4+) or CD8+ T cells (CD45+CD8+) were a primary source of IL-22 in the lungs of human newborns (Fig. 2B, S2B). These findings illustrate an important difference in the cellular sources of IL-22 in the lung of newborn humans compared to adult humans, as several groups have reported that NK cells (41), Th17 (42) and γδ T cells (43) are the principal sources of IL-22 in adult human lungs.
ILC3 lymphoid cells developmentally depend on RORγT and continuously express this transcription factor (44, 45). Therefore, to interrogate the importance of ILC3 in the resistance of newborn mice to pneumonia, we bred transgenic mice expressing cre recombinase under the control of the RORγt promoter (46) with transgenic mice expressing inducible diphtheria toxin receptor (iDTR) (47) to generate RorγtDTR mice. Treatment of newborn RorγtDTR mice with diphtheria toxin (DT) decreased the number of ILC3 in the lungs (Fig. 2C, S2C), reduced IL-22 in BAL fluid (Fig. S2D) and made the DT-treated RorγtDTR newborn mice more susceptible to pneumonia (Fig. 2D). Adoptive transfer of lung ILC3 restored host resistance to pneumonia in newborn RorγtDTR mice treated with DT (Fig. 2D). Together, these data confirm that IL-22+ILC3 are necessary and sufficient in promoting host resistance to pneumonia in newborn mice (48).
We sought to determine if disruption of commensal colonization alters the repertoire of IL-22 producing cells in the newborn mouse lung. We found significantly decreased (P<0.01) numbers of IL-22+ILC3 (Fig. 2E–G) but not neutrophils or T cells or NK cells (for all, P>0.05) (Fig. S2E) in the lungs of ABX-exposed or GF newborn mice as compared to ABX-free newborn mice. The decrease in the numbers of IL-22+ILC3 persisted beyond the newborn period till at least four weeks of life (Fig. 2E). We confirmed these observations in human newborns and found significantly decreased numbers of lung IL-22+ILC3 in the BAL fluid of human newborns exposed to prolonged duration of antibiotics (Fig. 2H). We then questioned if reversing the commensal disruption would correct the immune alterations in ABX-exposed newborn mice. We found that reconstitution with commensal bacteria restored the numbers of IL-22+ILC3 in the lungs of ABX-exposed or GF newborn mice (Fig. 2F,G), although individual IL-22 expression did not change (Fig. S2F). These data illustrate that disruption of commensal bacteria in early postnatal development alters the repertoire of IL-22-producing cells in the newborn lungs.
Commensal bacteria direct the postnatal trafficking of IL-22+ILC3 in the murine newborn lung
We tested whether reduced numbers of IL-22+ILC3 in the lungs of GF or ABX-exposed newborn mice could be explained by differences in proliferation or apoptosis of IL-22+ILC3. We assessed cell proliferation or apoptosis by quantifying the number of IL22+ILC3 lymphoid cells expressing Ki67 or annexin, respectively. We found that an increased number of ILC3 in the lungs was not due to changes in proliferation or apoptosis (Fig. S2G). We, therefore, hypothesized that a decrease in the absolute numbers of IL-22+ILC3 in ABX-exposed newborn mice was due to a reduced ability of ILC3 from ABX-exposed newborns to traffic preferentially to the lungs. To test this, we used a competitive trafficking assay (49) to determine the advantage of ILC3 isolated from ABX-free newborn mice to traffic to the lungs as compared to ILC3 from ABX-exposed newborn mice. We found that ILC3 from ABX-exposed newborn mice had decreased ability to traffic selectively into the lungs, but not the spleen or small intestine as compared to ILC3 isolated from ABX-free newborn mice (Fig. 2I,J).
We then asked if reversing the commensal disruption would restore the ability of ILC3 to traffic to the lungs. We similarly determined the advantage of ILC3 isolated from ABX-exposed newborn mice that had received transfer of commensal bacteria to traffic to the lungs as compared to ILC3 from ABX-exposed newborn mice that had received no such transfer. Reconstitution of commensal bacteria restored the ability of ILC3 from ABX-exposed newborns to traffic selectively to the lungs (Fig. 2I,J). Tissue-selective ILC3 trafficking has been described for the small intestine and secondary lymphoid tissues (50, 51). Our data unveils a role for intestinal commensal bacteria in selective trafficking of ILC3 into the lungs.
Commensal bacteria modulate expression of CCR4 on ILC3 lymphoid cells and direct their postnatal trafficking into lung
Chemokines control the trafficking and positioning of immune cells and are critical for development and recruitment of immune cells in disease (52). We first identified the repertoire of chemokine receptors on IL22+ILC3 from the lungs or small intestine (SI) of newborn mice. We found that C-C chemokine receptor (CCR) 4 was highly expressed by a majority of IL-22+ILC3 from the newborn murine lung but not from the newborn murine SI (Fig. S3A). We found no difference in expression of CCR6, 7, 9 or C-C chemokine ligand (CCL) 20 nor C-X-C chemokine receptor (CXCR) 3 or 5 on IL-22+ILC3 from the newborn lung or SI (Fig. S3A). Tissue-selective ILC3 trafficking has been described for the intestine (50, 51), but not for the lungs. Like the intestine, the lung has a large mucosal surface, which is in continuous contact with the environment and therefore could potentially benefit from lung-selective ILC3 trafficking.
We hypothesized that exposure to commensal bacteria in early life may modulate the expression of lung-specific homing receptors on IL-22+ILC3 lymphoid cells and thus increase their ability to traffic to the lungs. We examined the numbers and frequencies of CCR4 expressing IL22+ILC3 in the lungs of ABX-exposed or ABX-free newborn mice. We found that the majority of IL22+ILC3 from the lungs of ABX-free newborn mice were CCR4high as compared to the lung ILC3 from ABX-exposed mice or GF mice, which were CCR4low (Fig. 3A). We found no difference in expression of CCR6, 7, 9 CCL20, CXCR3 or CXCR5 on IL22+ILC3 from the lungs of ABX-free mice or ABX-exposed mice (Fig. 3A). CCR4 is critical for homeostatic trafficking of T lymphocytes (53) and Tregs (54) into the lungs. CCL17, one of the ligands for CCR4 is expressed by the lung epithelium (55). We therefore hypothesized that commensal bacteria use a similar mechanism to direct trafficking of ILC3 into the newborn lungs. To test this hypothesis we determined the capability of ILC3 isolated from newborn mice lacking CCR4 (Ccr4−/−) to traffic to the lungs as compared to ILC3 from age-matched wild-type littermates. We found that ILC3 from newborn Ccr4−/− mice had decreased ability to traffic into the lungs as compared to ILC3 isolated from wildtype littermates (Fig. 3B). Furthermore, the newborn Ccr4−/−mice were more susceptible to pneumonia compared to wildtype littermates (Fig. 3C). We then asked if an adoptive transfer of wildtype ILC3, which express CCR4 and therefore traffic into the lungs, could improve host resistance in newborn Ccr4−/−mice. We found that adoptive transfer of ILC3 from wildtype newborn mice to age-matched Ccr4−/−mice restored host resistance to pneumonia (Fig. 3C). These data illuminate a role for CCR4 in trafficking of ILC3 into the lungs and promoting newborn’s resistance to pneumonia. Nevertheless, the origin of ILC3 that traffic to the lungs is unknown. ILC3 are concentrated within the SI (51), and the spatial proximity of ILC3 with commensal bacteria in the small intestine supports the notion that ILC3 in intestinal mucosa may be directed by the commensal bacteria to traffic to the lung.
Figure 3. Intestinal dendritic cells mediate cross talk between commensal bacteria and IL22+ILC3 innate lymphoid cells and induce ILC3 to traffic to the murine newborn lung.
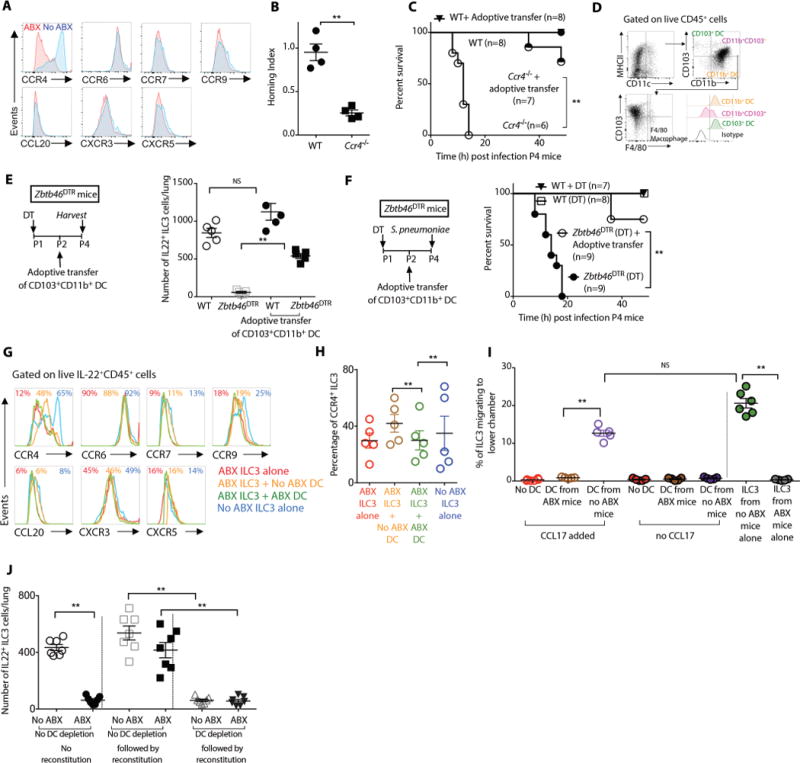
(A) Representative flow cytometry histograms showing expression of C-C chemokine receptor (CCR) 4, 6, 7 and 9, C-X-C chemokine receptor (CXCR) 3 and 5 and C-C chemokine ligand (CCL) 20 by IL22+ILC3 innate lymphoid cells in the lung of postnatal day 4 (P4) ABX-free or ABX-exposed newborn mice. (B) An equal number of ILC3 from P4 WT or Ccr4−/− newborn mice were adoptively transferred into age-matched ABX-exposed newborn mice, and the ability of ILC3 to traffic to the lungs (homing index) was determined. (C) Survival of P4 WT or Ccr4−/− newborn mice that received adoptive transfer of WT ILC3 after infection with S. pneumoniae. (D) Representative flow cytometry plots and relative frequencies of distinct subsets of mononuclear phagocytes in the small intestine of P4 newborn mice. (E) The absolute numbers of IL22+ILC3 in the lungs of P4 WT or Zbtb46DTR newborn mice treated with DT (CD11b+CD103+ DC-depleted) that received adoptive transfer of CD11b+CD103+ DCs. (F) Survival of P4 WT or Zbtb46DTR newborn mice treated with DT (CD11b+CD103+ DC-depleted) that then received adoptive transfer of CD11b+CD103+ DCs, after infection with S. pneumoniae . (G) ILC3 isolated from lungs of P4 ABX-exposed mice were co-cultured with CD11b+CD103+ DCs isolated from age-matched ABX-exposed or ABX-free mice and examined for surface expression of various chemokine receptors. A representative flow cytometry plot is shown and (H) relative frequencies of IL-22+ILC3 cells expressing CCR4. (I) ILC3 isolated from lungs of P4 ABX-exposed mice were co-cultured either alone or with CD11b+CD103+ DCs isolated from age-matched ABX-exposed or ABX-free newborn mice. The ability of these ILC3 to migrate in vitro in response to a gradient of the chemokine ligand (CCL) 17 is shown. (J) The absolute numbers of IL22+ILC3 in the lungs of P4 Zbtb46DTR newborn mice either exposed to ABX or no ABX that were then treated with DT (CD11b+CD103+ DC-depleted) or no DT (no DC depletion) before they were reconstituted with commensal bacteria. Data and plots are representative of three independent experiments. Results are shown as the mean ± s.e.m (Student’s t-test or ANOVA or Wilcoxon signed-rank test,). *P ≤ 0.05; **P ≤ 0.01. Number of individual animals [n] are indicated.
CD103+CD11b+ DC capture antigen from commensal bacteria and induce expression of CCR4 on ILC3
We sought to identify the mechanisms by which intestinal commensal bacteria induce the expression of CCR4 on ILC3. Murine ILC3 do not express pattern recognition receptors and therefore are unlikely to directly sense the commensal bacteria (56). Mononuclear phagocytes like DC and macrophages, not only detect a range of microbial signals (57), but can also cross talk with ILC3 in the intestine (58). We found that the majority of mononuclear phagocytes in the intestine of newborn mice were CD45+CD11b+CD103+F4/80− cells (CD103+CD11b+ DC) (Fig. 3D), consistent with previous findings (59). Transcription factor ZBTB46 is selectively expressed by CD103+ CD11b+ DCs and their committed progenitors but is not expressed by monocytes, macrophages, or other lymphoid or myeloid lineages (60, 61). We used mice that express diptheria toxin receptor (DTR) under the control of Zbtb46, (Zbtb46DTR), which allows for the depletion of CD103+ CD11b+ DC (60) after treatment with DT. Twenty-four hours later, there was a decreased number of intestinal CD11b+CD103+ DCs (Fig. S3C). Depletion of CD103+CD11b+ DCs was associated with a decrease in IL-22 in the BAL (Fig. S3D), reduced numbers of ILC3 in the lungs and increased susceptibility to pneumonia of DT-treated Zbtb46DTR newborn mice (Fig. 3E,F). Adoptive transfer of CD103+ CD11b+ DC into age-matched newborn Zbtb46DTR mice treated with DT (CD103+CD11b+ DC-depleted newborn mice) restored the number of lung ILC3 and improved the resistance of newborn mice to pneumonia (Fig. 3E,F).
There is increasing evidence that intestinal DCs act as conductors of ILC traffic to the intestine and secondary lymphoid tissue (50). We tested whether CD103+CD11b+ DCs induced CCR4 expression on IL-22+ILC3. We co-cultured CD103+CD11b+ DCs isolated from the small intestine of ABX-free or ABX-exposed newborn mice with ILC3 isolated from the lungs of ABX-free or ABX-exposed newborn mice. CD103+CD11b+ DCs isolated from the small intestine of ABX-free newborn mice were more efficient than CD103+CD11b+ DCs isolated from the small intestine of ABX-exposed newborn mice at inducing CCR4 expression on IL-22+ILC3 (Fig. 3G,H) and restoring the ability of lung ILC3 from ABX-exposed animals to migrate in response to the CCR4 ligand, CCL17 in vitro (Fig. 3I). We found no change in expression of CCR6, 7, 9 CCL20, CXCR3 or CXCR5 (Fig. 3G). Conversely, co-culture of CD11b+CD103− DCs isolated from small intestine of either ABX-free or ABX-exposed newborn mice did not change the CCR4 expression on IL-22+ILC3 (Fig. S3E), suggesting CD11b+CD103− DCs were not as efficient as CD11b+CD103+ DCs at modulating CCR4 expression on IL-22+ILC3. These observations are consistent with other reports (62–64), which suggest that distinct DC subsets respond differentially to similar environmental cues.
Finally, to test the hypothesis that CD103+CD11b+ DCs capture antigen from commensal bacteria and direct the trafficking of ILC3 into the lungs, we treated newborn Zbtb46DTR mice with DT before reconstitution with commensal bacteria. Depletion of CD103+CD11b+ DCs abrogated the increase in ILC3 number in ABX-exposed newborn Zbtb46DTR mice after reconstitution with intestinal commensal bacteria (Fig. 3J). Treatment of Zbtb46DTR newborn mice with DT depleted the CD103+CD11b+ DCs in extra-intestinal sites including the lungs. CD103+CD11b+ DCs are exceedingly rare in the newborn murine lungs (65). Therefore intestinal CD103+CD11b+ DCs, but not lung resident CD103+CD11b+ DCs are likely to play a major role in selective trafficking of ILC3 to the lung. Since previous reports (66, 67) implicated IL-1β in crosstalk between intestinal mononuclear phagocytes and small intestine ILC3, we measured IL-1β in the supernatant from co-cultures of DCs and ILC3. We found no difference in the IL-1β levels in co-culture supernatants of CD103+ CD11b+ DCs isolated from the small intestine of ABX-free or ABX-exposed newborn mice and co-cultured with ILC3, (Fig. S3F), suggesting that IL-1β may not be involved in cross-talk between DCs and ILC3.
Discussion
Distinct immune responses specifically adapted for fetal and early postnatal life render newborns more vulnerable to infection (1). Lack of understanding of immune development in early life contributes to our inability to reduce neonatal morbidity due to pneumonia, which unfortunately kills more newborns than any other cause. While a series of coordinated events control the development of a newborn’s immune system (3), few are as important as the interaction with successive waves of commensal bacteria, which colonize the newborn’s intestine immediately after birth (4, 5). Disruption of commensal colonization in the critical window of the early postnatal period has enduring consequences for the developing immune system, exemplified by increased risk of inflammatory disorders like asthma and increased risk of respiratory infections beyond infancy in those infants exposed to prolonged antibiotic treatment or delivered by caesarean section (10–14). To develop therapeutic interventions to decrease morbidity in the newborn period and beyond, we need to better understand the role of commensal colonization in the development of the newborn immune system.
Prior studies investigating commensal bacteria-driven immune maturation have prioritized the use of GF mice (68). It is now accepted that the immune system while not immature in early life, differs fundamentally from immune responses in adults (1). Importantly, the newborn’s intestine is colonized by successive waves of diverse commensal bacteria, and newborn’s intestinal microbiota is fundamentally distinct from that of adult mice (29). Thus, commensal reconstitution studies in adult GF mice do not fully capture the complex interaction between the developing host and evolving microbiota. We exposed pregnant mouse dams to a combination of ABX, used commonly to treat mother and infants. This not only disrupted the sequential colonization of the newborn’s intestine by different commensal bacteria but also made the newborn mice more susceptible to pneumonia, replicating the key observations from epidemiological studies (10–14). Perhaps more importantly, the susceptibility to pneumonia in ABX-exposed newborn mice persisted beyond the newborn period, in contrast to previous reports (28) (27), showing that ABX exposure in adult mice induced only transient susceptibility to infection. Reversing the commensal disruption restored the resistance to pneumonia in the ABX-exposed newborn mice and, importantly, this protection persisted beyond the newborn period. These results coupled with other recent reports (25, 26) highlight the importance of therapeutic interventions addressing commensal dybiosis in early life that have lasting consequences.
T cells (32) and NK cells (41) are primary mediators of innate mucosal defense against respiratory pathogens in adults. We found that at birth, the mouse lung is populated by a few IL-22+ T cells or NK cells, but is home to a significant number of IL-22+ ILC3 innate lymphoid cells. While IL22+ILC3 maintain tissue homeostasis in the small intestine, the role of IL22+ILC3 in lung mucosal defense remains a source of controversy (69). ILC3 were dispensable for protection against Influenza A pneumonia (70). Reports ascribing an important role for ILC3 in lung immune homeostasis have used adult mice deficient in recombination activating gene (Rag) 2 or interleukin 2 receptor, gamma (Il2rg) (48, 71), which are profoundly immunodeficient, or treated with antiCD90.2 antibodies (35) resulting in nonspecific depletion of several immune cell types thus confounding interpretation of results from these animals. We, therefore, generated RorγtiDTR mice, a genetic tool to selectively deplete ILC3. By first showing that ILC3 depletion in early life rendered the newborn mice more susceptible to pneumonia, we were able to demonstrate that IL-22+ ILC3 were necessary to promote the resistance of newborn mice to pneumonia. Second, by restoring the resistance to pneumonia in ILC3 depleted-newborn mice after adoptive transfer of ILC3, we established that ILC3 were sufficient to promote newborn’s resistance to pneumonia. One limitation of RorγtiDTR mice is potential depletion of the RORγt+ T cells after DT treatment. However, both human newborns (72) as well as murine newborns (73) are relatively lymphopenic compared to adults. Absolute numbers of IL22+ T cells were low in the newborn mice (Fig. 2B), suggesting that depletion of RORγt+ T cells in our RorγtiDTR mice may have had only a marginal effect on the newborn’s resistance to infections.
Having shown that ILC3 played a critical role in promoting the resistance of newborn mice to pneumonia, we investigated the role of commensal bacteria in the postnatal development of lung IL22+ILC3. Several groups have demonstrated that colonization with commensal bacteria increased the number of IL22+ILC3 in the small intestine of adult GF mice (74, 75). Similarly, an increase in the number of IL-22+ILC3 in the postnatal period has been described for the small intestine (76, 77) but not for the lung, raising the possibility that this phenomenon is restricted to the newborn’s intestine. Our data challenge this assumption and illuminate a role for intestinal commensal bacteria in the postnatal accumulation of IL-22+ILC3 in the lungs. We noted an abrupt increase in the number of IL-22+ILC3 in the lungs immediately after birth and observed an age-dependent increase in the number of lung IL22+ILC3, peaking at two weeks of age. This postnatal increase in lung IL22+ILC3 was absent in GF or ABX-exposed newborn mice and was reversed by transfer of commensal bacteria in the early postnatal period. Previous studies have used transcriptional activation and cytokine production to delineate a role for commensal bacteria in the ontogeny of ILC3 (75, 76). But tissue selective-trafficking as a mechanism through which commensal bacteria direct the ontogeny of ILC3 in postnatal lungs has not been investigated. ILC3 are thought to establish residency in the developing intestine by tissue-specific migration (50, 51). We show that exposure to commensal bacteria in early life directs the lung-specific IL-22+ ILC3 trafficking and thus contributes to the postnatal accumulation of IL22+ILC3 in the newborn lung. This study did not address the potential role of commensal bacteria in long-term maintenance of lung IL-22+ILC3, which needs to be clarified in the context of recent reports that ILC homeostasis at peripheral sites is dependent primarily on self-renewal (78).
Small intestine-specific trafficking of ILC3 depends on expression of chemokine receptors CCR9 and α4β7 by ILC3 (50). In contrast, we found that lung-specific trafficking of IL-22+ILC3 was mediated by CCR4, a homing receptor also used by other immune cells (79) for homeostatic trafficking into the lung. CCR4 is activated by chemokine CCL17 or CCL20. CCL17 is expressed by the lung epithelium (55). There is evidence that intestinal DCs act as conductors of ILC3 traffic to the small intestine and secondary lymphoid tissue but not to the lungs (50, 51). We, therefore, evaluated the role of intestinal DCs in instructing IL-22+ILC3 to traffic to the lung and found that intestinal CD103+CD11b+ DCs increased expression of the lung homing receptor CCR4 by IL-22+ILC3. The ability to up-regulate CCR4 expression was dependent on the exposure of intestinal CD103+CD11b+ DCs to commensal bacteria as CD103+CD11b+ DCs from ABX-exposed newborn mice failed to increase the expression of CCR4 on IL22+ILC3, suggesting that the ability to induce CCR4 expression and thus direct the tissue-specific migration of IL22+ILC3 is a cell extrinsic property. Many factors such as components of bacterial cell walls, bacterial metabolites, and intestinal epithelial cell-derived cytokines are known to condition the functional properties of CD103+CD11b+ DCs (80). Identification of this signal remains an area of active investigation. The role of lung CD103+CD11b+ DCs in lung-specific trafficking of IL-22+ILC3 remains unclear. The rarity of CD103+CD11b+ DCs in the newborn murine lung (65) precluded us from evaluating whether these cells could induce IL22+ILC3 to traffic to the lungs as efficiently as intestinal CD103+CD11b+ DCs, a potential limitation of our study.
In conclusion, our data demonstrate the importance of commensal exposure in a defined developmental window during the newborn period in the development of pulmonary mucosal immunity in mice. We illuminate a critical role for intestinal commensal bacteria in lung-selective trafficking of IL22+ILC3. This was mediated by intestinal CD103+CD11b+ DCs, which induced expression of the lung homing signal CCR4 on the IL22+ILC3. Lung-selective trafficking contributed to the postnatal accumulation of IL22+ILC3, promoting the newborn’s resistance to pneumonia. These data also potentially explain the association between caesarean delivery or widespread use of antibiotics and an increased risk of infections in newborn infants. Finally, similar mechanisms could influence the development of other pulmonary inflammatory disorders like asthma, which is also associated with caesarean delivery and antibiotic use in early life (81) and lead to new therapeutic agents to mitigate the risk associated with early-life antibiotic exposure in children.
Materials and Methods
Study design
Institutional Animal Care and Use Committee at Cincinnati Children’s Hospital Medical Center (CCHMC) approved all the animal studies (IACUC2014-0055), which were carried out in accordance with NIH’s Guide for the Care and Use of Laboratory Animals. We bred Rorγt-Cre mice with Rosa26-iDTR mice to generate RorγtiDTR mice. We maintained C57/BL6, Rosa26-iDTR mice, Rorγt-Cre or Zbtb46DTR mice at CCHMC animal facility. We maintained the germ-free (GF) C57/BL6 neonatal mice in plastic isolator cages with autoclaved feed and water at CCHMC Germ-Free Core facility. After birth, neonatal mice from multiple litters were pooled and redistributed to control for the founder effect and to minimize in-cage variations. We used neonatal C57/BL6, Zbtb46DTR, RorγtDTR or GF C57/BL6 mice between postnatal ages P1 and P14 and appropriate, age-, sex- and genetic strain-matched controls to account for any variations in data. We treated pregnant female mice (C57/BL6 or Zbtb46DTR) with sterile drinking water mixed with three different antibiotics (ampicillin, gentamicin, and vancomycin; all at 1 mg ml−1) starting from embryonic day 15 until the delivery of neonatal mice. We determined group sizes necessary for adequate statistical power analysis using preliminary data sets. There was no randomization designed in the experiments, and we did not exclude any samples. The investigators were not blinded to group allocation during collection and analysis of the data.
Murine neonatal pneumonia studies
We grew S. pneumoniae serotype 19A (ATCC 700674) or E. coli serotype K1 (82) or C. albicans (37°C, 200 rpm) in tryptic soy (TS) broth to log-phase growth. To mimic S. pneumoniae or E. coli or C. albicans pneumonia, we inoculated neonatal mice (postnatal day 4 or 14) with either S. pneumoniae serotype 19A (105 CFU g−1) or E. coli (104 CFU g−1) or C. albicans (105 CFU g−1) respectively via i.t. route. The animals were examined every 6 h for signs of distress and were euthanized 72 h later or earlier if moribund. To assess bacterial burden we homogenized the lung in sterile PBS. We plated serial dilutions of lung homogenates or BAL fluid in TSB agar plates and incubated (37°C, overnight) to count the number of CFU.
We pooled intestinal contents from no ABX-exposed P2 newborn mice. We transferred intestinal contents (200 μg in 50 μl PBS) or vehicle (50 μl PBS) to antibiotic-exposed neonatal P2 mice by a single oral gavage via fine polyethylene tubing as described before (83).
Isolation and characterization of IL-22+ cells in the murine neonatal lung
We pooled and cut the freshly lungs from 3-4 newborn mice and incubated (37 °C, 30 min) the cut tissues with shaking (150 r.p.m.) in digestion buffer (RPMI 1640 with 10% FBS, 15 mM HEPES, 1% penicillin/streptomycin (wt/vol) and 300 U ml−1 collagenase VIII) and pressed through a 100-µm nylon strainer to obtain single-cell suspension. The pooled preparation constituted a single data point in our analysis. We then incubated (37 °C, 5 h, 5% CO2) the cells (1 × 107) in culture medium containing RPMI 1640 with 10% FCS, 1× nonessential amino acids, 10 mM HEPES, 2 mM L-glutamine (all from Invitrogen) and 1% penicillin/streptomycin with 1:1,000 Golgi Stop (554724, BD Biosciences), 10 ng/ml phorbol 12-myristate 13-acteate (PMA) and 500 ng/ml calcium ionophore A23187 (both from Sigma-Aldrich). We washed and incubated (4 °C, 10 min) the cells (1 × 107) with anti-mouse CD16/CD32 and then re-incubated (4 °C, 30 min) with anti-mouse CD3 antibody (145-2C11), anti–mouse CD4 antibody (GK1.5), anti–mouse CD8 antibody (53–5.8), anti–mouse CD11b antibody (M1/70), anti–mouse CD19 antibody (6D5), anti-mouse Ly6G antibody (1A8), anti-mouse F4/80 antibody (BM8), anti-mouse CD117 antibody (2B8), anti–mouse NKp46 antibody (29A1.4), anti-mouse CCR4 antibody (2G12), anti-mouse CCR6 antibody (29-2L17), anti-mouse CCR7 antibody (4B12), anti-mouse CCR9 antibody (9B1), anti-mouse CCL20 antibody (114906), anti-mouse CXCR3 antibody (173), anti-mouse CXCR5 antibody (L138D7), anti-mouse Ki67 antibody (16A8) (all diluted 1:100, Biolegend). For intracellular staining, we washed and fixed (4 °C, 60 min) the surface-stained cells in 1× Cytofix/Cytoperm buffer (BD Biosciences) and permeabilized them (4 °C, overnight) using 1× Permeabilization Buffer (BD Biosciences) according to manufacturer instructions. We stained the cells intracellularly with anti–mouse IL-22 antibody (5164) or anti–mouse RORγt antibody (Q31-378) or anti-mouse T-bet antibody (4B10) or anti-mouse Eomes antibody (WD1928) (all diluted 1:50, Biolegend) and then washed (2×) and resuspended them in flow cytometry buffer. We collected the data with LSRII (BD Biosciences) and analyzed the data with FlowJo (Treestar).
Isolation and characterization of antigen presenting cells in the murine newborn intestine
We pooled and cut the freshly resected terminal ilea from 3-4 neonatal mice into 2- to 5-mm pieces and incubated (37°C,15 min) them in extraction buffer (HBSS, 15 mM HEPES and 1 mM EDTA) to remove the epithelial cells. We then incubated (37°C, 30 min) the cut tissues with shaking (150 rpm) in digestion buffer (RPMI 1640 with 10% FBS, 15 mM HEPES, 1% penicillin/streptomycin (wt/vol), and 300 U ml-1 collagenase VIII) and pressed through a 100-µm nylon strainer to obtain single-cell suspension. The pooled preparation constituted a single data point in our analysis. We then incubated (4 °C, 10 min) the cells (1 × 107) with anti-mouse CD16/CD32 and then re-incubated (4 °C, 30 min) with anti–mouse CD45 antibody (30-F11), anti–mouse CD103 antibody (2E7), anti-mouse CD11b antibody (M1/70), anti-mouse CD11c antibody (N418), anti-mouse MHCII antibody (M5/114.15.2), anti–mouse F4/80 antibody (BM8) and anti-mouse CX3CR1(SA011F11) (all diluted 1:100, Biolegend) and then washed (2×) and resuspended them in flow cytometry buffer. We collected the data with LSRII (BD Biosciences) and analyzed the data with FlowJo (Treestar).
Treatment of neonatal mice with neutralizing antibodies, recombinant IL-22 or diphtheria toxin
We injected neonatal B6 mice with an anti-IL22 antibody (8E11, a gift from Wenjun Ouyang) or anti-IL20 antibody (Clone PA5-47092, Invitrogen) or anti-IgG2A (54447, R&D) (all 5 μg per g body weight) via i.p. route on P1. For specific cell depletion, we treated neonatal RorγtiDTR or Zbtb46DTR mice with diphtheria toxin (1.5 ng, R&D) or vehicle via i.p. route on P1. We assessed ablation efficiency by flow cytometry 24 h later. For gain of function studies, we treated ABX-exposed neonatal mice with recombinant IL-22 (10 μg per g body weight) (cat. 414-CS, R&D) or vehicle via i.t. route on P2.
Adoptive transfer of ILC3 or CD103+CD11b+ DCs
For adoptive transfer, we pooled lungs or SI from 8-10 P2 newborn mice. We harvested approximately 1 × 106 Lin−CCR6+ from the pooled specimens by positive and negative selection as done previously by other groups (71). This resulting cell population was > 97% enriched for ILC3 (71). We harvested 1 × 107 intestinal CD103+CD11b+ DC from the pooled SI specimens by positive and negative selection as done previously (84). We adoptively transferred Lin−CCR6+ cells (0.5 × 106 cells/animal) or CD103+CD11b+ DC (0.5 × 106 cells/animal) via i.p. route on P2.
In vivo ILC3 migration assay
We first isolated lung Lin−CCR6+ cells (>97% ILC3) by positive and negative selection as done previously (71). We then incubated (37 °C, 20 min) ILC3 (1 × 107) from P3 GF or ABX-exposed newborns with carboxyfluorescein succinimidyl ester (CFSE) (5 mM). We incubated ILC3 from control (ABX-free) neonatal mice with chloromethyl-benzoyl-amino-tetramethylrhodamine (CMMTR) (10 mM). We quenched with an equal volume of 10% FBS, diluted 10 × with PBS and resuspended the cells in RPMI1640, supplemented with 2% FBS and 2 mM glutamine. We co-injected 106 cells of each population into ABX-exposed neonatal mice via i.p. route. We euthanized host mice 24 h. later and determined the numbers of injected ILC3 migrating into the lungs, spleen and SI by flow cytometry as done previously (50). We calculated the relative homing index as follows. [CFSE+ ILC3 in organ A]/[CMMTR+ ILC3 in organ A] ÷ [CFSE+ ILC3 in injected cells]/[CMMTR+ ILC3 in injected cells] as described before (49). We performed similar experiments after reconstitution of commensal bacteria and with lung Lin−CCR6+ cells from Ccr4−/− or WT mice.
Cell co-culture and chemotaxis assay
We isolated Lin−CCR6+ cells (>97%ILC3) from lungs (71) or CD103+CD11b+CD11c+ cells from the intestine of ABX-exposed or ABX-free newborn mice (P4). We then co-cultured lung ILC3 and intestinal CD103+CD11b+ DC or CD103−CD11b+ (105 cells each) in following combinations (ILC3 from ABX mice + DC from no-ABX mice, ILC3 from ABX-mice + DC from ABX mice or ILC3 from ABX-free mice + DC from ABX-free mice or ILC3 from ABX-free mice + DC from ABX mice, ILC3 from ABX or ABX-free mice alone or DC from ABX and ABX-free mice alone) in round-bottom plates in RPMI1640 supplemented with 2% FBS, 2 mM glutamine, and 50 µM β-mercaptoethanol. We harvested the supernatants and incubated the remaining cells were incubated with Golgi Plug and subsequently analyzed by flow cytometry. For chemotaxis assay, we loaded ILC3 (106 cells in 100 μl RPMI 2% FBS) into transwell inserts with pore size of 5 μm (Corning Transwell) and placed in wells containing ±20 nM CCL17 in RPMI1640, supplemented with 2% FBS, 2 mM glutamine, and 50 µM β-mercaptoethanol. We incubated (2 h, 37 °C) the plates and then analyzed the migrated and input cells by flow cytometry. We expressed results as percentages of ILC3 in migrated wells as compared to input wells as described before (50).
Sample collection and analysis of commensal bacteria in the lungs and small intestine of newborn mice
Given the technical challenges in collecting adequate bio specimens for 16s sequencing from newborn mice, we opted to pool bio-specimens from 8-10 newborn mice from 3 separate litters per treatment group. An unexpected benefit of this approach is the control of variations in the commensal bacteria attributed to founder and cage effect (85). We collected the entire left lobe of the lung using one heat treated sterile scissor per animal as described previously by other groups (86). We then cut open the terminal ileum using sterile scissors and removed the intestinal contents using sterile plastic loops as done previously (29). We snap froze (−80°C) the specimens for subsequent analysis (−80°C). We extracted the bacterial DNA from the whole lung or the intestinal contents using QIAamp DNA stool Mini Kit (Qiagen) using a previously described protocol (86) and quantified 16S ribosomal (r) DNA by RT-PCR using degenerate primers as described before (87). To analyze the commensal bacteria, we amplified the V2 region of microbial 16S rRNA by high fidelity PCR with barcoded 8F and 338R universal primers with A and B sequencing adaptors respectively and bifido primers (Roche) and sequenced them with Genome Sequencer GS-FLX Titanium system (Roche) at University of North Carolina Microbiome core facility (Chapel Hill, NC). The reagents used for DNA extraction, polymerase chain reaction and sequencing reaction are a common source of contamination in microbiome sequencing studies (88). To control for variation in the reagents, we processed all the samples using a common batch of DNA extraction reagents and PCR reagents. We included appropriate controls including a negative control (no template) and positive community control (intestinal contents from age-matched mice) from each batch of harvested tissues when performing 16 rRNA PCR or 16S rRNA sequencing. We decoded and processed the sequences using the QIIME software package (Version 1.7) and custom R package code (89). Analysis of the sequences from negative control indicated presence of several bacterial species suggesting potential contamination from the DNA extraction or sequencing reagents. The dominant bacterial species in the negative control were Rhodocyclae (17%), Rhizobiales (15%) Argobacterium (14%), Micrococcus (10%), Hydrogenophilus (9%), Neiserria (5%), Lysinibacillus (4%), Micrococcus (4%) and Tenericutes (4%). The relative abundance of these contaminants in experimental samples was less than 3% (Supplemental Table 1) and thus unlikely to alter the conclusions. We used phylogenetic diversity (PD) to compute and visualize α diversity and unweighted and weighted Unifrac for β diversity. We tested the observed differences in Unifrac distances between antibiotic treated groups and across different ages for significance using a t-test, and we corrected the reported P values for multiple comparisons using a Monte Carlo permutation procedure with 10,000 iterations. We deposited all data sets in a publicly available database (Figshare) and can be accessed at https://figshare.com/s/52f4aa2f8035fd505cf1.
Transcriptomic analysis
We sequenced high-quality RNA from the whole lung at CCHMC Sequencing Core Facility with an Illumina HiSeq 2500. We performed data alignment with TopHat, followed by gene quantification (FPKM) using Cufflinks. We carried out differential expression analysis with both FPKM and read count-based methods. We performed pathway and network analyses with Altanalyze as described before (90). We deposited all data sets in a publicly available database (Figshare) and can be accessed at https://figshare.com/s/52f4aa2f8035fd505cf1.
Human newborn studies
The Institutional review board at CCHMC approved all the human studies. The biological samples were initially collected from infants who underwent clinical evaluation of either upper airway obstruction or tracheaobronchomalacia, common anomalies of the airways in infants, after obtaining informed consent from the parents (IRB approval #2013-3309). The bronchial lavage (BAL) fluid samples were centrifuged (4°C, 10 min, 400 g). The resultant supernatant was frozen (−80 °C) and the cells were cryopreserved (−150 °C) in 90% FBS and 10% DMSO. We used the frozen supernatant and cryopreserved cells in our analysis (IRB approval #2015-7983). Since pneumonia (48) and asthma (71) are associated with ILC3 activation in the lungs, we excluded infants with a history of pneumonia (defined as worsening of respiratory status, increase or change in the quality of respiratory secretions, temperature instability with radiographic changes) or wheezing. We then selected infants who received antibiotics (ABX-exposed group) or no antibiotics (No ABX-group) and matched the respective treatment groups for gestational age, age at the time of procedure and history of mechanical ventilation. Characteristics of the subjects are provided in Table 1. After thawing, we incubated (37°C, 5 h, 5% CO2) the cells (0.5 × 106) in culture medium containing RPMI 1640 with 10% FCS, 1× nonessential amino acids, 10 mM HEPES, 2 mM L-glutamine (all from Invitrogen) and 1% penicillin/streptomycin with 1:1,000 Golgi Stop (554724, BD Biosciences), 10 ng/ml phorbol 12-myristate 13-acteate (PMA) and 500 ng/ml calcium ionophore A23187. We immunophenotyped the cells in BAL fluid as described before (91). We stained cells with anti-human CD3 antibody (SP34-2), anti-human CD8 antibody (B9.11), anti-human CD14 antibody (RMO52), anti-human CD19 antibody (J3-119), anti-human CD45 antibody (J.33), anti-human CD56 antibody (NK901), anti-human CD69 antibody (TP1.55.3) and anti-human NKp46 antibody (9E2) (all diluted 1:1000, Biolegend). For intracellular staining, we washed and fixed (4 °C, 60 min) the surface-stained cells in 1× Cytofix/Cytoperm buffer (BD Biosciences) and permeabilized them (4 °C, overnight) using 1× Permeabilization Buffer (BD Biosciences) according to manufacturer’s instructions. We stained the cells intracellularly with anti-human RORγt antibody (AFKJS-9), anti-human T-bet antibody (4B10) and anti-human IL-22 antibody (BG/IL22) (all diluted 1:50, Biolegend) and then washed (2×) and resuspended them in flow cytometry buffer. We collected the data with LSRII (BD Biosciences) and analyzed the data with FlowJo (Treestar).
ELISA
We measured IL-22 or IL-20 in the murine (69) or human BAL fluid (92) using commercially available kits (eBioscience) as described before. We measured IL-1β in the cell culture supernatant using commercially available kits (eBioscience) as per manufacturer’s instructions.
Statistical analyses
Each data point represents a pool of 3-4 newborn mice that were pooled before the isolation of leukocytes from the indicated tissue. All data met the assumptions of the statistical tests used. We compared differences between groups by either unpaired two-tailed Student’s t-test or ANOVA or Wilcoxon signed-rank test. We used the Kaplan-Meier log-rank test to compare survival between groups. (All in GraphPad Prism 6.0). P-values are indicated as follows: * p ≤ 0.05 or ** p ≤ 0.01.
Supplementary Material
Figure S1 (Related to Figure 1). Intestinal commensal bacteria promote resistance to S. pneumoniae in newborn mice via IL-22.
Figure S2 (Related to Figure 2). Intestinal commensal bacteria direct postnatal trafficking of IL-22+ILC3 innate lymphoid cells to murine newborn lung promoting resistance to pneumonia.
Figure S3 (Related to Figure 3). Intestinal dendritic cells mediate cross talk between commensal bacteria and ILC3 .
Table S1A. Relative abundance of different commensal bacteria in the intestine of ABX-free or ABX-exposed newborn mice.
Table S1B. Relative abundance of different commensal bacteria in the lung of ABX-free or ABX-exposed newborn mice.
Table S2. Differentially expressed genes in the lungs of postnatal day 4 ABX-free or ABX-exposed newborn mice.
Table S3. Tabulated raw data.
One sentence summary.
Postnatal colonization by intestinal commensal bacteria directs migration of innate lymphoid cells into the lungs and promotes newborn resistance to pneumonia.
Accessible Summary.
Interactions between host and intestinal commensals shape the development of immune cells in intestine. We report that host-commensal interactions extend beyond the local environment and shape the repertoires of immune cells at extra-intestinal sites like the lungs. Exposure to commensals in the developmental window of newborn period directs lung-selective trafficking of innate lymphoid cells (ILC3), a group of sentinel cells that maintain mucosal homeostasis. This was mediated by intestinal DCs, which induced expression of the lung homing signal CCR4 on the ILC3. Lung-selective ILC3 trafficking promoted the newborn’s resistance to pneumonia. These data explain the association between widespread use of antibiotics and an increased risk of pneumonia in newborn infants.
Acknowledgments
We thank W. Ouyang (Genentech) for providing anti-IL22 neutralizing antibody. We thank N. Butz for her assistance with microbial DNA isolation and the Children’s Hospital Research Foundation’s Flow Cytometry and Cell Sorting Core Laboratory for technical advice and support. We thank S. Way, C. Chougnet and H. Singh for their helpful comments. We thank the physicians, nurses and staff of CCHMC Pulmonary Medicine Division and all of the participating patients and their families.
Funding: H.D. is supported by 1K08HD084686 and Francis Family Foundation. T.A. is supported by 5K08DK093784, Burroughs Wellcome Fund, Crohn’s and Colitis Foundation of America, and Pew Trust. G.W is supported by 5R01AI099479 and 5R01HL105834 and J.W. is supported by 5R01HL095580, 4U01HL110964 and 5U01HL122642.
Footnotes
Author contributions: H.D. designed the in vivo experimental studies for commensal disruption and reconstitution, innate immune cell depletion and adoptive transfer of innate immune cells. J.G. designed in vitro co-culture and migration studies. T.A., G.W. and J.W. provided reagents. H.D., J.G. and K.O. carried out experiments. H.D. and J.G. analyzed the data and wrote the manuscript with input from T.A., G.W. and J.W.
Competing interests: Authors declare no conflicts of interest.
Data and Materials availability: All the data sets are deposited in a publicly available database (Figshare) and can be accessed at https://figshare.com/s/52f4aa2f8035fd505cf1.
References and Notes
- 1.Dowling DJ, Levy O. Ontogeny of early life immunity. Trends Immunol. 2014;35:299–310. doi: 10.1016/j.it.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marchant A, Kollmann TR. Understanding the ontogeny of the immune system to promote immune-mediated health for life. Front Immunol. 2015;6:77. doi: 10.3389/fimmu.2015.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacGillivray DM, Kollmann TR. The role of environmental factors in modulating immune responses in early life. Front Immunol. 2014;5:434. doi: 10.3389/fimmu.2014.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 2006;313:1126–1130. doi: 10.1126/science.1127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elinav E, Strowig T, Henao-Mejia J, Flavell RA. Regulation of the antimicrobial response by NLR proteins. Immunity. 2011;34:665–679. doi: 10.1016/j.immuni.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 6.Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, Peterson DA, Stappenbeck TS, Hsieh CS. Peripheral education of the immune system by colonic commensal microbiota. Nature. 2011;478:250–254. doi: 10.1038/nature10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gomez de Aguero M, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H, Steinert A, Heikenwalder M, Hapfelmeier S, Sauer U, McCoy KD, Macpherson AJ. The maternal microbiota drives early postnatal innate immune development. Science. 2016;351:1296–1302. doi: 10.1126/science.aad2571. [DOI] [PubMed] [Google Scholar]
- 8.Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol. 2007;5:e177. doi: 10.1371/journal.pbio.0050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, van den Brandt PA, Stobberingh EE. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics. 2006;118:511–521. doi: 10.1542/peds.2005-2824. [DOI] [PubMed] [Google Scholar]
- 10.Arboleya S, Sanchez B, Milani C, Duranti S, Solis G, Fernandez N, de los Reyes-Gavilan CG, Ventura M, Margolles A, Gueimonde M. Intestinal microbiota development in preterm neonates and effect of perinatal antibiotics. The Journal of pediatrics. 2015;166:538–544. doi: 10.1016/j.jpeds.2014.09.041. [DOI] [PubMed] [Google Scholar]
- 11.Kuppala VS, Meinzen-Derr J, Morrow AL, Schibler KR. Prolonged initial empirical antibiotic treatment is associated with adverse outcomes in premature infants. The Journal of pediatrics. 2011;159:720–725. doi: 10.1016/j.jpeds.2011.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark RH, Bloom BT, Spitzer AR, Gerstmann DR. Empiric use of ampicillin and cefotaxime, compared with ampicillin and gentamicin, for neonates at risk for sepsis is associated with an increased risk of neonatal death. Pediatrics. 2006;117:67–74. doi: 10.1542/peds.2005-0179. [DOI] [PubMed] [Google Scholar]
- 13.Cotten CM, Taylor S, Stoll B, Goldberg RN, Hansen NI, Sanchez PJ, Ambalavanan N, Benjamin DK, Jr, N. N. R. Network Prolonged duration of initial empirical antibiotic treatment is associated with increased rates of necrotizing enterocolitis and death for extremely low birth weight infants. Pediatrics. 2009;123:58–66. doi: 10.1542/peds.2007-3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mai V, Torrazza RM, Ukhanova M, Wang X, Sun Y, Li N, Shuster J, Sharma R, Hudak ML, Neu J. Distortions in development of intestinal microbiota associated with late onset sepsis in preterm infants. PloS one. 2013;8:e52876. doi: 10.1371/journal.pone.0052876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, Iwasaki A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:5354–5359. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fagundes CT, Amaral FA, Vieira AT, Soares AC, Pinho V, Nicoli JR, Vieira LQ, Teixeira MM, Souza DG. Transient TLR activation restores inflammatory response and ability to control pulmonary bacterial infection in germfree mice. Journal of immunology. 2012;188:1411–1420. doi: 10.4049/jimmunol.1101682. [DOI] [PubMed] [Google Scholar]
- 17.Dickson RP, Erb-Downward JR, Huffnagle GB. The role of the bacterial microbiome in lung disease. Expert Rev Respir Med. 2013;7:245–257. doi: 10.1586/ers.13.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Azad MB, Konya T, Persaud RR, Guttman DS, Chari RS, Field CJ, Sears MR, Mandhane PJ, Turvey SE, Subbarao P, Becker AB, Scott JA, Kozyrskyj AL, C.S. Investigators Impact of maternal intrapartum antibiotics, method of birth and breastfeeding on gut microbiota during the first year of life: a prospective cohort study. BJOG : an international journal of obstetrics and gynaecology. 2015 doi: 10.1111/1471-0528.13601. [DOI] [PubMed] [Google Scholar]
- 20.Glasgow TS, Young PC, Wallin J, Kwok C, Stoddard G, Firth S, Samore M, Byington CL. Association of intrapartum antibiotic exposure and late-onset serious bacterial infections in infants. Pediatrics. 2005;116:696–702. doi: 10.1542/peds.2004-2421. [DOI] [PubMed] [Google Scholar]
- 21.Tita AT, Landon MB, Spong CY, Lai Y, Leveno KJ, Varner MW, Moawad AH, Caritis SN, Meis PJ, Wapner RJ, Sorokin Y, Miodovnik M, Carpenter M, Peaceman AM, O’Sullivan MJ, Sibai BM, Langer O, Thorp JM, Ramin SM, Mercer BM, N. M.-F. M. U. N. Eunice Kennedy Shriver Timing of elective repeat cesarean delivery at term and neonatal outcomes. The New England journal of medicine. 2009;360:111–120. doi: 10.1056/NEJMoa0803267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tripathi N, Cotten CM, Smith PB. Antibiotic use and misuse in the neonatal intensive care unit. Clinics in perinatology. 2012;39:61–68. doi: 10.1016/j.clp.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaplan SL, Barson WJ, Lin PL, Stovall SH, Bradley JS, Tan TQ, Hoffman JA, Givner LB, Mason EO., Jr Serotype 19A Is the most common serotype causing invasive pneumococcal infections in children. Pediatrics. 2010;125:429–436. doi: 10.1542/peds.2008-1702. [DOI] [PubMed] [Google Scholar]
- 24.Stoll BJ, Hansen N, Fanaroff AA, Wright LL, Carlo WA, Ehrenkranz RA, Lemons JA, Donovan EF, Stark AR, Tyson JE, Oh W, Bauer CR, Korones SB, Shankaran S, Laptook AR, Stevenson DK, Papile LA, Poole WK. Changes in pathogens causing early-onset sepsis in very-low-birth-weight infants. The New England journal of medicine. 2002;347:240–247. doi: 10.1056/NEJMoa012657. [DOI] [PubMed] [Google Scholar]
- 25.Russell SL, Gold MJ, Hartmann M, Willing BP, Thorson L, Wlodarska M, Gill N, Blanchet MR, Mohn WW, McNagny KM, Finlay BB. Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep. 2012;13:440–447. doi: 10.1038/embor.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, Glickman JN, Siebert R, Baron RM, Kasper DL, Blumberg RS. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–493. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuijt TJ, Lankelma JM, Scicluna BP, de Sousa EMF, Roelofs JJ, de Boer JD, Hoogendijk AJ, de Beer R, de Vos A, Belzer C, de Vos WM, van der Poll T, Wiersinga WJ. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut. 2016;65:575–583. doi: 10.1136/gutjnl-2015-309728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mei J, Liu Y, Dai N, Hoffmann C, Hudock KM, Zhang P, Guttentag SH, Kolls JK, Oliver PM, Bushman FD, Worthen GS. Cxcr2 and Cxcl5 regulate the IL-17/G-CSF axis and neutrophil homeostasis in mice. The Journal of clinical investigation. 2012;122:974–986. doi: 10.1172/JCI60588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deshmukh HS, Liu Y, Menkiti OR, Mei J, Dai N, O’Leary CE, Oliver PM, Kolls JK, Weiser JN, Worthen GS. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nature medicine. 2014 doi: 10.1038/nm.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang YJ, Charlson ES, Collman RG, Colombini-Hatch S, Martinez FD, Senior RM. The role of the lung microbiome in health and disease. A National Heart, Lung, and Blood Institute workshop report. American journal of respiratory and critical care medicine. 2013;187:1382–1387. doi: 10.1164/rccm.201303-0488WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dickson RP, Huffnagle GB. The Lung Microbiome: New Principles for Respiratory Bacteriology in Health and Disease. PLoS Pathog. 2015;11:e1004923. doi: 10.1371/journal.ppat.1004923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA, Iwakura Y, Kolls JK. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nature medicine. 2008;14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nature medicine. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 34.Gessner MA, Werner JL, Lilly LM, Nelson MP, Metz AE, Dunaway CW, Chan YR, Ouyang W, Brown GD, Weaver CT, Steele C. Dectin-1-dependent interleukin-22 contributes to early innate lung defense against Aspergillus fumigatus. Infection and immunity. 2012;80:410–417. doi: 10.1128/IAI.05939-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mear JB, Gosset P, Kipnis E, Faure E, Dessein R, Jawhara S, Fradin C, Faure K, Poulain D, Sendid B, Guery B. Candida albicans airway exposure primes the lung innate immune response against Pseudomonas aeruginosa infection through innate lymphoid cell recruitment and interleukin-22-associated mucosal response. Infection and immunity. 2014;82:306–315. doi: 10.1128/IAI.01085-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang C, Wu L, Bulek K, Martin BN, Zepp JA, Kang Z, Liu C, Herjan T, Misra S, Carman JA, Gao J, Dongre A, Han S, Bunting KD, Ko JS, Xiao H, Kuchroo VK, Ouyang W, Li X. The psoriasis-associated D10N variant of the adaptor Act1 with impaired regulation by the molecular chaperone hsp90. Nature immunology. 2013;14:72–81. doi: 10.1038/ni.2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie MH, Aggarwal S, Ho WH, Foster J, Zhang Z, Stinson J, Wood WI, Goddard AD, Gurney AL. Interleukin (IL)-22, a novel human cytokine that signals through the interferon receptor-related proteins CRF2-4 and IL-22R. The Journal of biological chemistry. 2000;275:31335–31339. doi: 10.1074/jbc.M005304200. [DOI] [PubMed] [Google Scholar]
- 38.Wang M, Jeng KC, Ping LI. Exogenous cytokine modulation or neutralization of interleukin-10 enhance survival in lipopolysaccharide-hyporesponsive C3H/HeJ mice with Klebsiella infection. Immunology. 1999;98:90–97. doi: 10.1046/j.1365-2567.1999.00838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pociask DA, Scheller EV, Mandalapu S, McHugh KJ, Enelow RI, Fattman CL, Kolls JK, Alcorn JF. IL-22 is essential for lung epithelial repair following influenza infection. The American journal of pathology. 2013;182:1286–1296. doi: 10.1016/j.ajpath.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin JC, Beriou G, Heslan M, Chauvin C, Utriainen L, Aumeunier A, Scott CL, Mowat A, Cerovic V, Houston SA, Leboeuf M, Hubert FX, Hemont C, Merad M, Milling S, Josien R. Interleukin-22 binding protein (IL-22BP) is constitutively expressed by a subset of conventional dendritic cells and is strongly induced by retinoic acid. Mucosal immunology. 2014;7:101–113. doi: 10.1038/mi.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu X, Weiss ID, Zhang HH, Singh SP, Wynn TA, Wilson MS, Farber JM. Conventional NK cells can produce IL-22 and promote host defense in Klebsiella pneumoniae pneumonia. Journal of immunology. 2014;192:1778–1786. doi: 10.4049/jimmunol.1300039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sonnenberg GF, Nair MG, Kirn TJ, Zaph C, Fouser LA, Artis D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. The Journal of experimental medicine. 2010;207:1293–1305. doi: 10.1084/jem.20092054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simonian PL, Wehrmann F, Roark CL, Born WK, O’Brien RL, Fontenot AP. gammadelta T cells protect against lung fibrosis via IL-220. The Journal of experimental medicine. 2010;207:2239–2253. doi: 10.1084/jem.20100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nature immunology. 2004;5:64–73. doi: 10.1038/ni1022. [DOI] [PubMed] [Google Scholar]
- 45.Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, Mention JJ, Thiam K, Cerf-Bensussan N, Mandelboim O, Eberl G, Di Santo JP. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958–970. doi: 10.1016/j.immuni.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 46.Eberl G, Littman DR. Thymic origin of intestinal alphabeta T cells revealed by fate mapping of RORgammat+ cells. Science. 2004;305:248–251. doi: 10.1126/science.1096472. [DOI] [PubMed] [Google Scholar]
- 47.Buch T, Heppner FL, Tertilt C, Heinen TJ, Kremer M, Wunderlich FT, Jung S, Waisman A. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat Methods. 2005;2:419–426. doi: 10.1038/nmeth762. [DOI] [PubMed] [Google Scholar]
- 48.Van Maele L, Carnoy C, Cayet D, Ivanov S, Porte R, Deruy E, Chabalgoity JA, Renauld JC, Eberl G, Benecke AG, Trottein F, Faveeuw C, Sirard JC. Activation of Type 3 innate lymphoid cells and interleukin 22 secretion in the lungs during Streptococcus pneumoniae infection. J Infect Dis. 2014;210:493–503. doi: 10.1093/infdis/jiu106. [DOI] [PubMed] [Google Scholar]
- 49.Villablanca EJ, Mora JR. Competitive homing assays to study gut-tropic t cell migration. Journal of visualized experiments : JoVE. 2011 doi: 10.3791/2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim MH, Taparowsky EJ, Kim CH. Retinoic Acid Differentially Regulates the Migration of Innate Lymphoid Cell Subsets to the Gut. Immunity. 2015;43:107–119. doi: 10.1016/j.immuni.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mackley EC, Houston S, Marriott CL, Halford EE, Lucas B, Cerovic V, Filbey KJ, Maizels RM, Hepworth MR, Sonnenberg GF, Milling S, Withers DR. CCR7-dependent trafficking of RORgamma(+) ILCs creates a unique microenvironment within mucosal draining lymph nodes. Nat Commun. 2015;6:5862. doi: 10.1038/ncomms6862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annual review of immunology. 2014;32:659–702. doi: 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- 53.Mikhak Z, Strassner JP, Luster AD. Lung dendritic cells imprint T cell lung homing and promote lung immunity through the chemokine receptor CCR4. The Journal of experimental medicine. 2013;210:1855–1869. doi: 10.1084/jem.20130091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sather BD, Treuting P, Perdue N, Miazgowicz M, Fontenot JD, Rudensky AY, Campbell DJ. Altering the distribution of Foxp3(+) regulatory T cells results in tissue-specific inflammatory disease. The Journal of experimental medicine. 2007;204:1335–1347. doi: 10.1084/jem.20070081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sekiya T, Miyamasu M, Imanishi M, Yamada H, Nakajima T, Yamaguchi M, Fujisawa T, Pawankar R, Sano Y, Ohta K, Ishii A, Morita Y, Yamamoto K, Matsushima K, Yoshie O, Hirai K. Inducible expression of a Th2-type CC chemokine thymus- and activation-regulated chemokine by human bronchial epithelial cells. Journal of immunology. 2000;165:2205–2213. doi: 10.4049/jimmunol.165.4.2205. [DOI] [PubMed] [Google Scholar]
- 56.Magri G, Miyajima M, Bascones S, Mortha A, Puga I, Cassis L, Barra CM, Comerma L, Chudnovskiy A, Gentile M, Llige D, Cols M, Serrano S, Arostegui JI, Juan M, Yague J, Merad M, Fagarasan S, Cerutti A. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nature immunology. 2014;15:354–364. doi: 10.1038/ni.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annual review of immunology. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ermann J, Staton T, Glickman JN, de Waal Malefyt R, Glimcher LH. Nod/Ripk2 signaling in dendritic cells activates IL-17A-secreting innate lymphoid cells and drives colitis in T-bet-/-.Rag2-/- (TRUC) mice. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E2559–2566. doi: 10.1073/pnas.1408540111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, Flavell RA, Littman DR, Pamer EG. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity. 2012;36:276–287. doi: 10.1016/j.immuni.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, Idoyaga J, Cheong C, Yao KH, Niec RE, Nussenzweig MC. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. The Journal of experimental medicine. 2012;209:1153–1165. doi: 10.1084/jem.20112675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, Murphy KM. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. The Journal of experimental medicine. 2012;209:1135–1152. doi: 10.1084/jem.20120030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Igyarto BZ, Haley K, Ortner D, Bobr A, Gerami-Nejad M, Edelson BT, Zurawski SM, Malissen B, Zurawski G, Berman J, Kaplan DH. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity. 2011;35:260–272. doi: 10.1016/j.immuni.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muzaki AR, Tetlak P, Sheng J, Loh SC, Setiagani YA, Poidinger M, Zolezzi F, Karjalainen K, Ruedl C. Intestinal CD103(+)CD11b(-) dendritic cells restrain colitis via IFN-gamma-induced anti-inflammatory response in epithelial cells. Mucosal immunology. 2016;9:336–351. doi: 10.1038/mi.2015.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hao X, Kim TS, Braciale TJ. Differential response of respiratory dendritic cell subsets to influenza virus infection. Journal of virology. 2008;82:4908–4919. doi: 10.1128/JVI.02367-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roux X, Remot A, Petit-Camurdan A, Nahori MA, Kiefer-Biasizzo H, Marchal G, Lagranderie M, Riffault S. Neonatal lung immune responses show a shift of cytokines and transcription factors toward Th2 and a deficit in conventional and plasmacytoid dendritic cells. European journal of immunology. 2011;41:2852–2861. doi: 10.1002/eji.201041224. [DOI] [PubMed] [Google Scholar]
- 66.Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, Merad M. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. 2014;343:1249288. doi: 10.1126/science.1249288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, Swaminath A, Bonneau R, Scherl EJ, Littman DR. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. The Journal of experimental medicine. 2014;211:1571–1583. doi: 10.1084/jem.20140678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Taube C, Tertilt C, Gyulveszi G, Dehzad N, Kreymborg K, Schneeweiss K, Michel E, Reuter S, Renauld JC, Arnold-Schild D, Schild H, Buhl R, Becher B. IL-22 is produced by innate lymphoid cells and limits inflammation in allergic airway disease. PloS one. 2011;6:e21799. doi: 10.1371/journal.pone.0021799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T, Kubota M, Turner D, Diamond JM, Goldrath AW, Farber DL, Collman RG, Wherry EJ, Artis D. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nature immunology. 2011;12:1045–1054. doi: 10.1031/ni.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, Iwakura Y, Israel E, Bolger K, Faul J, DeKruyff RH, Umetsu DT. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nature medicine. 2014;20:54–61. doi: 10.1038/nm.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Collier FM, Tang ML, Martino D, Saffery R, Carlin J, Jachno K, Ranganathan S, Burgner D, Allen KJ, Vuillermin P, Ponsonby AL. The ontogeny of naive and regulatory CD4(+) T-cell subsets during the first postnatal year: a cohort study. Clin Transl Immunology. 2015;4:e34. doi: 10.1038/cti.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ridge JP, Fuchs EJ, Matzinger P. Neonatal tolerance revisited: turning on newborn T cells with dendritic cells. Science. 1996;271:1723–1726. doi: 10.1126/science.271.5256.1723. [DOI] [PubMed] [Google Scholar]
- 74.Satoh-Takayama N, Lesjean-Pottier S, Vieira P, Sawa S, Eberl G, Vosshenrich CA, Di Santo JP. IL-7 and IL-15 independently program the differentiation of intestinal CD3-NKp46+ cell subsets from Id2-dependent precursors. The Journal of experimental medicine. 2010;207:273–280. doi: 10.1084/jem.20092029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sanos SL, Bui VL, Mortha A, Oberle K, Heners C, Johner C, Diefenbach A. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nature immunology. 2009;10:83–91. doi: 10.1038/ni.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bouskra D, Brezillon C, Berard M, Werts C, Varona R, Boneca IG, Eberl G. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. 2008;456:507–510. doi: 10.1038/nature07450. [DOI] [PubMed] [Google Scholar]
- 77.Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, Mantovani A, Kopan R, Bradfield CA, Newberry RD, Colonna M. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nature immunology. 2012;13:144–151. doi: 10.1038/ni.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science. 2015;350:981–985. doi: 10.1126/science.aac9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nie Y, Waite J, Brewer F, Sunshine MJ, Littman DR, Zou YR. The role of CXCR4 in maintaining peripheral B cell compartments and humoral immunity. The Journal of experimental medicine. 2004;200:1145–1156. doi: 10.1084/jem.20041185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Scott CL, Aumeunier AM, Mowat AM. Intestinal CD103+ dendritic cells: master regulators of tolerance? Trends Immunol. 2011;32:412–419. doi: 10.1016/j.it.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 81.Stensballe LG, Simonsen J, Jensen SM, Bonnelykke K, Bisgaard H. Use of antibiotics during pregnancy increases the risk of asthma in early childhood. The Journal of pediatrics. 2013;162:832–838 e833. doi: 10.1016/j.jpeds.2012.09.049. [DOI] [PubMed] [Google Scholar]
- 82.Pluschke G, Pelkonen S. Host factors in the resistance of newborn mice to K1 Escherichia coli infection. Microb Pathog. 1988;4:93–102. doi: 10.1016/0882-4010(88)90051-4. [DOI] [PubMed] [Google Scholar]
- 83.Deshmukh HS, Liu Y, Menkiti OR, Mei J, Dai N, O’Leary CE, Oliver PM, Kolls JK, Weiser JN, Worthen GS. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nature medicine. 2014;20:524–530. doi: 10.1038/nm.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Atif SM, Uematsu S, Akira S, McSorley SJ. CD103-CD11b+ dendritic cells regulate the sensitivity of CD4 T-cell responses to bacterial flagellin. Mucosal immunology. 2014;7:68–77. doi: 10.1038/mi.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Campbell JH, Foster CM, Vishnivetskaya T, Campbell AG, Yang ZK, Wymore A, Palumbo AV, Chesler EJ, Podar M. Host genetic and environmental effects on mouse intestinal microbiota. ISME J. 2012;6:2033–2044. doi: 10.1038/ismej.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barfod KK, Roggenbuck M, Hansen LH, Schjorring S, Larsen ST, Sorensen SJ, Krogfelt KA. The murine lung microbiome in relation to the intestinal and vaginal bacterial communities. BMC Microbiol. 2013;13:303. doi: 10.1186/1471-2180-13-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hill DA, Hoffmann C, Abt MC, Du Y, Kobuley D, Kirn TJ, Bushman FD, Artis D. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal immunology. 2010;3:148–158. doi: 10.1038/mi.2009.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, Turner P, Parkhill J, Loman NJ, Walker AW. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:87. doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Salomonis N, Schlieve CR, Pereira L, Wahlquist C, Colas A, Zambon AC, Vranizan K, Spindler MJ, Pico AR, Cline MS, Clark TA, Williams A, Blume JE, Samal E, Mercola M, Merrill BJ, Conklin BR. Alternative splicing regulates mouse embryonic stem cell pluripotency and differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:10514–10519. doi: 10.1073/pnas.0912260107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bocsi J, Melzer S, Dahnert I, Tarnok A. OMIP-023: 10-color, 13 antibody panel for in-depth phenotyping of human peripheral blood leukocytes. Cytometry A. 2014;85:781–784. doi: 10.1002/cyto.a.22505. [DOI] [PubMed] [Google Scholar]
- 92.Scriba TJ, Kalsdorf B, Abrahams DA, Isaacs F, Hofmeister J, Black G, Hassan HY, Wilkinson RJ, Walzl G, Gelderbloem SJ, Mahomed H, Hussey GD, Hanekom WA. Distinct, specific IL-17- and IL-22-producing CD4+ T cell subsets contribute to the human anti-mycobacterial immune response. Journal of immunology. 2008;180:1962–1970. doi: 10.4049/jimmunol.180.3.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (Related to Figure 1). Intestinal commensal bacteria promote resistance to S. pneumoniae in newborn mice via IL-22.
Figure S2 (Related to Figure 2). Intestinal commensal bacteria direct postnatal trafficking of IL-22+ILC3 innate lymphoid cells to murine newborn lung promoting resistance to pneumonia.
Figure S3 (Related to Figure 3). Intestinal dendritic cells mediate cross talk between commensal bacteria and ILC3 .
Table S1A. Relative abundance of different commensal bacteria in the intestine of ABX-free or ABX-exposed newborn mice.
Table S1B. Relative abundance of different commensal bacteria in the lung of ABX-free or ABX-exposed newborn mice.
Table S2. Differentially expressed genes in the lungs of postnatal day 4 ABX-free or ABX-exposed newborn mice.
Table S3. Tabulated raw data.