

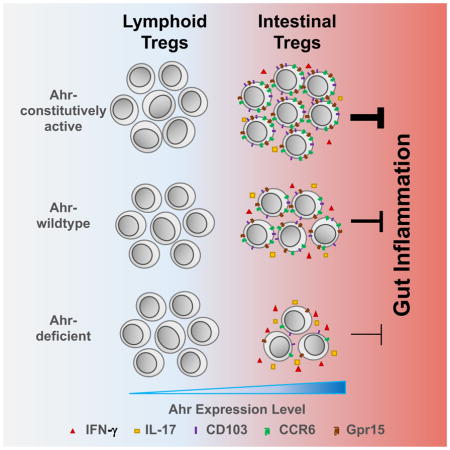
SUMMARY
Local environment may impact the development and function of tissue-resident T regulatory cells (Tregs) that are crucial for controlling inflammation. Although the aryl hydrocarbon receptor (Ahr), an environmental sensor, is expressed by Tregs, its role in Treg cell development and/or function remains elusive. Here, we generated mouse genetic models to ablate or activate Ahr expression specifically in Tregs. We showed that Ahr was expressed more abundantly by peripherally induced Treg (pTregs) in the gut, and its expression was independent of microbiota. Ahr was important for Treg gut homing and function. Ahr inhibited pro-inflammatory cytokines produced by Tregs but was dispensable for Treg stability. Furthermore, Ahr-expressing Tregs had enhanced in vivo suppressive activity compared to Tregs lacking Ahr expression in a T cell transfer model of colitis. Our data suggest that Ahr signaling in Tregs may be important for gut immune homeostasis.
eTOC Blurb
Ye et al. fnd that Ahr is most abundantly expressed by peripherally derived Tregs (pTreg) in the gut. Ahr expression and activation are important for Treg gut homing and function to suppress intestinal inflammation.
INTRODUCTION
A fundamental question is how environmental cues instruct regulatory T cell (Treg) development and function to maintain host immune homeostasis. Nutrients and metabolites that are produced by microbiome and diet, can act in the gut on Tregs to trigger tolerance. The aryl hydrocarbon receptor (Ahr) is an environmental sensor that detects not only xenobiotic ligands such as environmental pollutants (e.g., dioxin) but also physiological compounds generated by host cells, microbiota, and diet (e.g., amino acid tryptophan metabolites)(Zhou, 2016). Thus, deciphering the Ahr-mediated molecular pathways in Tregs offers the potential for developing novel therapies to treat immune dysregulation. The role of Ahr in immune cells has only been recently appreciated when Ahr was identified as a molecular link between the environment and the host immune system. It has been reported that Ahr is downregulated in the intestinal tissue of patients with inflammatory bowel disease (IBD), thus highlighting the clinical relevance of the Ahr pathway in human autoimmunity (Monteleone et al., 2011). Ahr has been relatively well studied in T helper (Th)17 cells and group 3 innate lymphoid cells (i.e., ILC3s) for its role in induction of effector cytokines (e.g., Interleukin (IL)-17 and IL-22) (Esser et al., 2009; Qiu and Zhou, 2013). However, its role in regulatory T cells (Tregs) specified by the Forkhead transcription factor Foxp3 remains controversial, with conflicting data showing Ahr expression in Tregs and either positive or negative regulation of Treg differentiation by Ahr. Of note, these data are largely derived from loss-of-function analysis using Ahr complete null mice or gain-of-function analysis by ligand administration (Nguyen et al., 2013; Stockinger et al., 2014; Zhou, 2016). These approaches may confound the interpretation of the results since the broad expression of Ahr in other cell types will likely influence Treg development and/or function. For example, Ahr-deficient macrophages produce more IL-6 (Kimura et al., 2009), a cytokine known to suppress Foxp3 expression (Bettelli et al., 2006). In addition, Ahr deficiency in ILC3s leads to aberrant outgrowth of gut commensal Segmented Filamentous Bacteria (SFB), causing elevated intestinal Th17 cells (Qiu et al., 2013) that have a reciprocal relationship with Tregs during differentiation (Bettelli et al., 2006; Zhou et al., 2008). Thus, it is essential to elucidate the Treg cell-autonomous role of Ahr in mucosal immunology by genetic approaches.
In spite of shared expression of Foxp3, tissue-resident Tregs may have different gene expression profiles or functions compared to their counterparts in lymphoid organs or TGF-β-induced Treg cells in vitro (i.e., iTregs), suggesting a complex mode of tissue-specific regulation of Tregs by Foxp3 and co-factors (Burzyn et al., 2013). It remains elusive how the surrounding environment contributes to the differentiation, maintenance, and function of these tissue-resident Tregs. Microbiota and dietary metabolites (e.g., retinoic acid and short-chain fatty acids) have been shown to influence the differentiation and function of gut Tregs, highlighting the crucial environmental impacts on Tregs (Atarashi and Honda, 2011; Bollrath and Powrie, 2013; Mucida et al., 2009).
Ahr belongs to the basic helix-loop-helix (bHLH)/Per-Arnt-Sim (PAS) family of proteins. The PAS domains consist of two regions, PAS-A and PAS-B, and are known to function as an interface for dimerization with the Ahr nuclear translocator (ARNT), and in ligand binding (Stevens et al., 2009). An Ahr deletion mutant lacking the PAS-B domain (ΔPAS-B) has been shown to constitutively dimerize with ARNT, bind to DNA, and activate transcription in a ligand-independent manner (i.e., constitutively active (CA)-Ahr) (McGuire et al., 2001). Ahr expression was thought to be ubiquitous in vertebrate cells; however, recent data suggest that its expression is regulated by certain environmental cues (e.g., cytokines) (Kimura et al., 2008; Quintana et al., 2008; Veldhoen et al., 2008; Zhou, 2016). Previous studies expressing the constitutively active Ahr ΔPAS-B (i.e. CA-Ahr) in transgenic mice have shed light on in vivo activation of Ahr (Andersson et al., 2002; Nohara et al., 2005; Stockinger et al., 2014; Tauchi et al., 2005; Zhou, 2016). However, in these studies, CA-Ahr was expressed in mice under the control of artificial genes and/or promoters out of the Ahr endogenous context (i.e., human CD2 minigene, SV40 and/or keratin-14 promoter), thus likely confounding the data interpretation in regard to the role of Ahr in Tregs (Stockinger et al., 2014). Here, we developed a conditional knock-in mouse model that expressed CA-Ahr and an IRES-GFP under the regulation of the Ahr endogenous locus (AhrCAIR mice) coupled with Foxp3Yfp-Cre approach to provide insights into in vivo expression and activation of Ahr in a Treg-specific manner. In addition, we utilized Ahrf/− Foxp3Yfp-Cre mice to probe cell-intrinsic role of Ahr in Tregs. Systemic analysis of Tregs in different anatomic locations revealed a gut-specific expression pattern of Ahr in Tregs and a role for Ahr in directing Treg gut homing and function.
RESULTS
Development of Ahr conditional knock-in mice expressing a constitutively active form of Ahr with GFP reporter
To investigate the expression pattern of Ahr and the consequence of Ahr activation in vivo, we used a Cre-loxP site-specific recombination approach to generate an Ahr knock-in mouse model (AhrCAIR mice), in which constitutively active Ahr (CA-Ahr) that lacks the PAS-B domain is expressed in a Cre-inducible manner and marked by GFP. Specifically, using homologous recombination, a Flag-CA-Ahr-IRES-GFP (abbreviated as CAIR) allele preceded by a loxP-flanked strong transcriptional termination sequence (STOP) was introduced into the endogenous Ahr locus and confirmed by Southern blot (Figures S1A and S1B). AhrCAIR/+ mice had one Ahr-null allele due to the transcriptional STOP and the other Ahr-wildtype allele. When crossed with EIIa-Cre transgenic mice to delete the STOP in the germline and subsequently removing the EIIa-Cre by breeding, AhrdCAIR/+ mice express CA-Ahr and GFP (Figure S1C and data not shown). GFP is under the control of the endogenous Ahr regulatory locus, thus reflecting Ahr transcription. No GFP expression or Ahr transcript can be detected in AhrCAIR/CAIR mice, indicating complete ablation of Ahr transcription before introduction of Cre-recombinase (data not shown). Accordingly, AhrCAIR/CAIR mice have a defect in innate lymphoid cells in the gut, thus phenocopying Ahr null mice (Figure S1D and (Kiss et al., 2011; Lee et al., 2011; Qiu et al., 2012)).
Peripherally induced gut Tregs express higher amounts of Ahr
The role of Ahr in Tregs has been controversial (Zhou, 2016). To examine Ahr expression specifically in Tregs, we utilized Foxp3Yfp-Cre mice (i.e., male Foxp3Yfp-Cre/Y or female Foxp3Yfp-Cre/Yfp-Cre mice), in which a YFP-Cre recombinase fusion protein was inserted into the 3′ UTR of the Foxp3 locus downstream of a viral IRES. Thus, both YFP and Cre-recombinase were expressed by Foxp3+ Tregs in Foxp3Yfp-Cre mice (note: Foxp3 expression was reported by YFP) (Rubtsov et al., 2008). To determine Ahr expression on a per cell basis, we generated AhrCAIR/+Foxp3Yfp-Cre mice in which Ahr-expressing Tregs were marked by GFP after Cre-recombinase-mediated deletion of the STOP cassette. Compared with other tissues, Ahr (as reported by GFP fluorescence) was most abundantly expressed in the YFP+ (i.e., Foxp3+) Tregs in both small and large intestines of AhrCAIR/+Foxp3Yfp-Cre mice (Figures 1A and 1B). Preferential expression of Ahr in gut Tregs was further confirmed at the endogenous Ahr mRNA level by realtime RT-PCR in sorted Tregs from Foxp3Yfp-Cre mice (Figure S1E). Ahr protein was also detected in gut Tregs (Figures S1F and S1G) by intracellular staining using Ahr antibody, consistent with its higher expression by gut Tregs revealed by GFP in the reporter mice (AhrCAIR/+Foxp3Yfp-Cre). Nrp1 has been suggested as a surface marker to distinguish thymus-derived Tregs (Nrp1+ tTregs) versus peripherally derived Tregs (Nrp1− pTregs) (Weiss et al., 2012; Yadav et al., 2012). Recent data further show that microbiota- and food antigen-induced pTregs can be distinguished by RORγt expression (Kim et al., 2016). Examination of Ahr reporter mice (AhrCAIR/+Foxp3Yfp-Cre) revealed specific expression pattern of Ahr in the different subsets of Tregs in various tissues. tTregs in the gut had higher expression of Ahr than their counter parts in the lymphoid organs (e.g., the spleen and mesenteric lymph nodes) (Figures 1C and 1D). Compared to tTregs, pTregs had even higher levels of Ahr expression (Figures 1C and 1D). The microbiota-induced Tregs (Nrp1−RORγt+ Tregs) expressed the highest amounts of Ahr in various organs (i.e., spleen, mLN, small and large intestines) (Figure 1E). An enrichment of RORγt expression was further observed in GFP+ Tregs as opposed to GFP− Tregs in the gut of Ahr reporter mice (AhrCAIR/+Foxp3Yfp-Cre), suggesting that Ahr preferentially marks RORγt+ Tregs (Figures S1H and S1I). However, in the gut, food antigen-induced pTregs (Nrp1−RORγt− Tregs) also expressed significant amounts of Ahr (Figure 1E). Further analysis of endogenous Ahr expression by intracellular staining showed that both tTregs and pTregs in the gut had higher expression of Ahr compared to those in other tissues (Figure S2A–S2C). Together, these data indicate differential expression of Ahr by Tregs at different anatomical locations, among which the gut pTregs express the highest amounts of Ahr.
Figure 1. Ahr is highly expressed by intestinal pTregs.

(A) Flow cytometry analysis of GFP by Tregs in the small intestine (SI), large intestine (LI), thymus (Thy), spleen (Sp), peripheral lymph node (pLN), mesenteric lymph node (mLN), lung, liver and fat of the indicated littermate mice. Data are representative of two independent experiments.
(B) Percentages of GFP+ Tregs in various tissues of AhrCAIR/+Foxp3Yfp-Cre mice. Data are shown as mean ± SD (n ≥ 3 mice per group). ANOVA followed by Bonferroni’s test for SI or LI versus other tissues.
(C) Flow cytometry analysis of GFP and Nrp1 expression by Tregs in various tissues of the indicated mice. Data are representative of three independent experiments.
(D) Percentages of GFP+ cells among Nrp1+ Tregs or Nrp1− Tregs from various tissues of AhrCAIR/+Foxp3Yfp-Cre mice. Each two symbols connected with a line represent data from the same individual mouse (n ≥ 4 mice per group).
(E) Percentages of Ahr+ cells measured by intracellular staining in Nrp1+RORγt−, Nrp1−RORγt+ or Nrp1−RORγt− Tregs in various tissues of Ahrf/−Foxp3Yfp-Cre mice. Data are shown as mean ± SD (n=3 mice per group).
See also Figure S1 and S2.
Treg-specific deletion of Ahr impairs pTregs in the gut
No apparent difference in intestinal Treg cell percentage or number was observed in Ahr null mice (i.e., Ahr−/−) or Ahrf/fCd4-cre mice (Figures S3A and S3B). Since Ahr plays a complex role in various immune cells that could potentially influence Tregs (e.g., antigen-presenting cells, innate lymphoid cells, Th17/Th22 cells, and Tr1 cells) (Apetoh et al., 2010; Basu et al., 2012; Gandhi et al., 2010; Kimura et al., 2009; Qiu et al., 2012; Quintana et al., 2008; Veldhoen et al., 2008), we next asked whether Ahr functions in a Treg-autonomous fashion to regulate Treg cell development in vivo. To this end, we deleted Ahr in Tregs by generation of Ahrf/−Foxp3Yfp-Cre mice. As expected, Ahr expression was ablated in Tregs but not in other cell populations in Ahrf/−Foxp3Yfp-Cre mice due to Foxp3-driven Cre-mediated deletion (Figure S3C). Thymic Treg development was unaffected by Ahr deficiency, consistent with the low expression of Ahr in the thymus (Figures S3D and 1A). We observed a marked decrease in Foxp3+ Tregs in the gut but not in other organs of Ahrf/−Foxp3Yfp-Cre mice (Figures 2A, S3E and S3F), despite no apparent defects in intestinal Treg cell proliferation or survival as revealed by Ki67 and Annexin V staining (data not shown). Together, these data indicate a tissue-specific and cell-intrinsic role of Ahr in regulating peripheral Treg cell compartment.
Figure 2. Ahr play essential roles in the intestinal pTreg.

(A and D) Percentages and absolute numbers of Tregs among CD4+ T cells in various tissues of the indicated mice. Data are shown as mean ± SD (n ≥ 5 mice per group).
(B and E) Flow cytometry analysis of RORγt and Nrp1 gated on Tregs in various tissues of the indicated littermate mice. Data are representative of four (B) or three (E) independent experiments.
(C and F) Absolute numbers of Nrp1−, Nrp1+, Nrp1−RORγt+, Nrp1−RORγt−, Nrp1+RORγt+, or Nrp1+RORγt− Tregs in various tissues of the indicated mice. Data are shown as mean ± SD (n ≥ 6 mice per group).
See also Figure S3.
We further investigated the role of Ahr in regulation of gut Tregs. Gated on Foxp3, Treg were separated into different populations based on expression of Nrp1 and/or RORγt (Figure 2B). Both the absolute number and percentage of Nrp1− Tregs that represent pTregs were significantly reduced in the gut of Ahrf/−Foxp3Yfp-Cre mice, compared to Ahrf/+Foxp3Yfp-Cre littermate control mice (Figures 2B, 2C and S3G). Accordingly, the percentage but not absolute number of Nrp1+ Tregs was increased (Figures 2C and S3G). Further examination of pTregs revealed that percentages and absolute numbers of both RORγt+ pTregs and RORγt− pTregs were significantly decreased in the gut, but not in the spleen or mesenteric lymph nodes (Figures 2B, 2C and S3G). Intriguingly, although their identity remains to be determined, a population of Tregs that express both Nrp1 and RORγt was upregulated in the gut of Ahrf/−Foxp3Yfp-Cre mice (Figures 2B and 2C and S3G). Together, these data support a model that Ahr facilitates the differentiation and/or accumulation of pTregs in a tissue- and cell-specific manner.
Treg-specific activation of Ahr promotes pTregs in the gut
Using the AhrCAIR/+Foxp3Yfp-Cre mice, we next investigated the effect of in vivo Ahr activation on the accumulation of intestinal Foxp3+ Tregs. AhrCAIR/+Foxp3Yfp-Cre mice exhibited an increase in the percentage and number of Foxp3+ Tregs in the large intestine (Figures 2D and S3H). Consistently, activation of Ahr by in vivo administration of ligand FICZ led to enhancement of Foxp3+ Tregs in the gut (Figure S3I). In addition, genetic activation of Ahr in Tregs led to enhancement of both Nrp1+ Tregs and Nrp1− Tregs (Figure 2F). Of note, enhancement of Nrp1+ Tregs was only observed in the large intestine but not in other organs, despite the evidence that Nrp1+ Tregs are derived from the thymus (Weiss et al., 2012; Yadav et al., 2012). Further analyses of Nrp1− Tregs indicated that the number and percentage of RORγt− but not RORγt+ pTregs were increased in AhrCAIR/+Foxp3Yfp-Cre mice, suggesting that Ahr activation preferentially enhances RORγt− populations within pTreg compartment in the gut (Figures 2E, 2F and S3J).
Environmental factors impact on gut Tregs and Ahr expression
The intestinal microbiota, including Clostridium species, enhanced the number and function of Tregs (Atarashi et al., 2011; Geuking et al., 2011). To investigate whether microbiota regulate Ahr expression, and modulate Treg compartment in the gut, we treated Ahr+/+Foxp3Yfp-Cre mice with broad-spectrum antibiotics. Consistent with previous report (Atarashi et al., 2011), the percentages of Tregs, especially the Nrp1−RORγt+ Tregs, were markedly reduced upon antibiotics treatment (Figures 3A and 3B). The reduction of Nrp1−RORγt+ Tregs was confirmed in germ-free mice, demonstrating that their accumulation and differentiation are dependent on microbiota (Figures 3E and 3F). Intriguingly, Ahr expression in Tregs was unaffected in antibiotics-treated mice or germ-free mice (Figures 3C, 3D, 3G and 3H), suggesting that environmental factors other than microbiota drive the gut-specific expression of Ahr in Tregs. In contrast, AhrCAIR/+Foxp3Yfp-Cre mice were resistant to antibiotics treatment and showed no changes in percentages of Tregs or their expression of Ahr, consistent with the constitutively active nature of CA-Ahr (Figures 3I and 3J).
Figure 3. Microbiota have distinct impact on gut Tregs.

(A, B, C, D, I, and J) 3-week-old mice were treated with antibiotics (ABX) or PBS for 2 to 3 weeks. Mice were analyzed for the percentages of Tregs among CD4+ T cells (A), percentages of Nrp1−RORγt+ cells among Tregs (B), and percentages of Ahr+ cells among tTregs (C) and pTregs (D) in the gut of Ahr+/+Foxp3Yfp-Cre mice, or percentages of Tregs among CD4+ T cells (I) and Ahr-GFP+ cells among Tregs (J) in the gut of AhrCAIR/+Foxp3Yfp-Cre mice. Data are representative of four independent experiments (A–D) or complied from three independent experiments (I and J), and shown as mean ± SD (n ≥ 4 mice per group).
(E, F, G, and H) Germ-free wildtype mice were gavaged with or without feces collected from specific pathogen free (SPF) mice. Mice were analyzed after 3 weeks after transfaunation. Flow cytometry analysis of RORγt and Nrp1 gated on CD4+TCRβ+Foxp3+ cells (E), or of Ahr and Foxp3 gated on CD4+TCRβ+ cells (G). Percentages of Nrp1−RORγt+ cells among Tregs (F) and percentages of Ahr+ cells among Tregs (H). Data are representative of two independent experiments and shown as mean ± SD (n ≥ 3 mice per group).
Dietary components have been reported to play essential roles in the development of ILC3 and intraepithelial lymphocytes (IELs) via action of Ahr (Kiss et al., 2011; Li et al., 2011). Our efforts to determine the effect of dietary Ahr ligand on Tregs by feeding mice with a synthetic diet AIN-76A deficient in plant-derived Ahr ligands (Kiss et al., 2011) did not lead to conclusive results on Treg development and/or Ahr expression, although consistent reduction of gut ILC3 was observed by using the same diet (data not shown). These data suggest a complex interaction of endogenous and exogenous Ahr ligands with other factors (e.g., microbiota) to affect gut Tregs. Together, the data demonstrated that environmental cues of microbiota and/or dietary Ahr ligand have distinct impact on Ahr expression and Treg compartments in the gut.
Ahr deficiency leads to alteration of the transcriptional program in intestinal Tregs
Tregs may utilize multiple mechanisms to exert the suppressive functions that maintain immune homeostasis and prevent autoimmunity (Vignali, 2012). To investigate the role of Ahr in gut Treg transcriptional programming, we purified the large intestinal YFP+ (Foxp3+) Tregs from Ahrf/+ Foxp3Yfp-Cre or Ahrf/− Foxp3Yfp-Cre mice by FACS sorting and performed a genome-wide analysis of mRNA expression by high throughput sequencing (RNA-Seq). Gene expression profiling analysis indicated that the majority of genes in gut Tregs were not significantly affected by genetic ablation of Ahr, indicating no drastic perturbation of the Treg transcriptional program without Ahr under the steady state (Figure 4A). This was consistent with the observation that Ahrf/−Foxp3Yfp-Cre mice did not develop spontaneous autoimmunity (data not shown). However, a small cohort of genes (n = 59) displayed deregulated expression in steady state gut Tregs upon deletion of Ahr (≥ 1.5 fold; q ≤ 0.05, p-values adjusted for multiple test comparisons) (Figure S4). Specifically, the expression of certain genes that are important for Treg homing and/or function in the gut (e.g., Ccr6, Gpr15, Itgae, and Rgs9) was decreased in Ahr-deficient Tregs (Figure 4B). The expression of granzyme that are associated with Treg cytotoxic function (e.g., Gzma) was also downregulated in the absence of Ahr (Figure 4B). On the other hand, Th1-associated genes were found to be expressed at increased levels in Ahr-deficient Tregs. For example, the expression of the cytokine Ifng, chemokine Ccl5, and the Th1-associated key transcription factor Tbx21 was enhanced in Ahr-deficient Tregs (Figures 4B and 4C). The expression of other genes that are associated with Treg function (e.g., Ctla4, Il2ra, Il10, Tnfrsf18, Runx1, Runx3, Entpd1 or Nt5e) was not significantly altered in the absence of Ahr, indicating a selective requirement for Ahr in Foxp3+ Treg transcriptional regulation (data not shown). Additionally, gene set enrichment analysis using gene sets derived from the KEGG pathway database identified two pathways that were significantly enriched: Cytokine-cytokine receptor interaction and Chemokine signaling pathway (q-value ≤ 0.01). Differentially regulated genes related to the pathway analysis include: Ccl4, Ccl5, Ifng, Csf1, Ccl20 (data not shown)(Mootha et al., 2003; Subramanian et al., 2005). Together, these data suggest that Ahr may regulate intestinal Treg development, maintenance, homing/retention, and function by multiple mechanisms.
Figure 4. Regulation of intestinal Treg transcriptional program by Ahr.

(A) Large intestinal Tregs were sorted from Ahrf/+Foxp3Yfp-Cre (Control) or littermate Ahrf/−Foxp3Yfp-Cre mice by flow cytometry and subjected to genome-wide mRNA analysis (RNA-Seq). Dot plot shows comparison of gene expression between the two groups. Genes significantly changed (q-value ≤ 0.05) in Tregs are highlighted in red (up-regulated) and green (down-regulated). Dashed lines delineate two-fold change.
(B) Log2 ratio of indicated gene expression derived from RNA-Seq of (Ahrf/−Foxp3Yfp-Cre)/(Ahrf/+Foxp3Yfp-Cre) Tregs were depicted in bar graph. Data are derived from two independent experiments.
(C) Large intestinal Tregs were sorted from Ahrf/+Foxp3Yfp-Cre or littermate Ahrf/−Foxp3Yfp-Cre mice and mRNA expression of indicated genes was analyzed by realtime RT-PCR. Dots represent biological repeats. Data are shown as mean ± SD.
See also Figure S4.
Ahr promotes homing/retention molecules in Tregs and enhances their gut homing
The expression of CCR6, a chemokine receptor expressed by certain CD4+ T cells (e.g., Tregs and Th17 cells) (Ivanov et al., 2006; Kleinewietfeld et al., 2005; Yamazaki et al., 2008), was reduced in Ahr-deficient large intestinal Tregs at the mRNA and protein level (Figures 4B, 5A and S5A). In contrast, the expression of CCR6 by ILC3 (i.e., CD4+TCRβ− LTi-like cells) was unaffected in Ahr-deficient mice (Figure S5B). The expression of CCL20, the ligand for CCR6, was also reduced in Ahr-deficient large intestinal Tregs (Figure 4C), indicating the regulation of CCR6-CCL20 axis by Ahr in Tregs. GPR15, an orphan guanine nucleotide-binding protein (G protein)-coupled receptor, plays a key role in Treg homing specifically to the large intestine (Kim et al., 2013). We observed decreased expression of Gpr15 in Ahr-deficient Tregs (Figures 4B and 4C), supporting a hypothesis that Ahr regulates Treg gut homing/retention. The expression of CD103 is important for Treg activation and function and contributes to the retention of Tregs at inflammatory sites (Feuerer et al., 2009; Geuking et al., 2011; Huehn et al., 2004; Lehmann et al., 2002; Rausch et al., 2008; Stephens et al., 2007; Suffia et al., 2005). Consistent with the finding that Itgae, the gene encoding CD103, was downregulated in Ahr-deficient Tregs (Figure 4C), Tregs isolated from the gut of Ahrf/−Foxp3Yfp-Cre mice showed a defect in CD103 protein expression, compared to those from littermate controls (Figure 5B). In contrast, there was no apparent defect in the CD103 expression in Tregs found in the spleen or mesenteric lymph node (Figure 5B), suggesting that Ahr is important for promoting the accumulation of CD103+ Tregs in the gut. Furthermore, the enhancement of CD103+ Tregs was also observed in the gut of AhrCAIR/+Foxp3Yfp-Cre mice compared to littermate Ahr+/+Foxp3Yfp-Cre in which CA-Ahr was expressed in Foxp3+ cells (Figure 5C), and in vivo administration of FICZ led to increased CD103 expression by the intestinal Tregs (Figure 5D). Of note, upon examination of CD103 expression in the gut of AhrCAIR/+Foxp3Yfp-Cre mice, intestinal Foxp3+ Tregs with different levels of Ahr based on GFP intensity showed a positive correlation between Ahr and CD103 expression (Figure 5E), suggesting that Ahr expression and activation enhances CD103 expression in Tregs. Further determination of the differentially expressed genes in Nrp1+ tTregs and Nrp1− pTregs confirmed the requirement of Ahr in regulation of CD103, CCR6, Gpr15, and Ccl20 protein and/or mRNA expression (Figure S5C–S5H). However, the expression of other gut homing molecules (i.e., α4β7 and CCR9) (Berlin et al., 1995; Zabel et al., 1999) in Tregs was unaffected upon Ahr deletion (Figure S5I–S5L).
Figure 5. Ahr promotes gut homing of Tregs.

(A) Flow cytometry analysis of CCR6 and YFP in the large intestine of indicated littermate mice. Data are representative of 2 independent experiments.
(B–C) Percentages of CD103+ cells among Tregs in various tissues of the indicated mice. Data are compiled from three (B) and two (C) independent experiments and shown as mean ± SD (n ≥4 mice per group).
(D) 10-day-old mice were injected with FICZ or DMSO control for 7 days. Percentages of CD103+ cells gated on CD4+TCRβ+Foxp3+ cells were shown. Data are compiled from two independent experiments and shown as mean ± SD.
(E) The expression of GFP and YFP gated on CD4+TCRβ+ cells was analyzed by flow cytometry (the left column). The expression of CD103 by each individual Treg population with different level of Ahr expression depicted in color (i.e., red, blue, green, and grey–the right column) was analyzed by flow cytometry (the middle column). Data are representative of three independent experiments.
(F and G) Large intestinal LPLs from Ahr+/+Foxp3Yfp-Cre CD45.1/45.2 mice (Ctr) and Ahrf/−Foxp3Yfp-Cre CD45.2/45.2 (cKO) mice were mixed and around 1×107 cells were adoptively transferred into Rag1−/− mice. Migration of donor cells gated on CD4+Foxp3− non-Tregs or CD4+Foxp3+ Tregs was determined by flow cytometry. Cell migration in indicated organs is expressed in term of migration index using the formulation: Migration index = (ratio of cKO/Ctr cells in post-transfer)/(ratio of cKO/Ctr cells in input). Data are shown as mean ± SD (n = 4 mice per group).
See also Figure S5.
The effect of Ahr on the expression of multiple homing molecules prompted us to further examine if Ahr directs Treg homing to the gut. Large intestinal LPLs from congenically marked Ahr+/+Foxp3Yfp-Cre CD45.1/45.2 mice or Ahrf/−Foxp3Yfp-Cre CD45.2/45.2 mice were mixed at equal ratio and transferred to Rag1−/− mice. Twenty hours later, we examined Foxp3+ Tregs in various organs of recipient mice (Figure 5F). Ahr deficiency in Tregs had no impact on distribution of non-Tregs (i.e., Foxp3− cells) among various host tissues; however, it markedly reduced Treg homing to the large intestine (Figure 5G). To further determine this hypothesis, we generated Ahrf/− Foxp3Yfp-Cre/+ female mice in which Ahr expression was ablated in YFP+ Tregs but not YFP− Tregs due to random X-chromosome inactivation. Notably, ratio of YFP+ Tregs/YFP− Tregs was significantly reduced in Ahrf/−Foxp3Yfp-Cre/+ female mice compared to Ahr+/+Foxp3Yfp-Cre/+ female mice, suggesting a key role for Ahr in Treg compartment (Figures S5M and S5N).
Ahr controls Treg function but not Foxp3 stability
Our gene profiling analysis revealed that Ahr-deficient Tregs had increased Th1- associated gene expression (Figures 4A to 4C). Thus, we examined the IFN-γ production by Tregs from Ahrf/−Foxp3Yfp-Cre mice under the steady state. Compared to their littermate controls, Ahr-deficient Tregs in the large intestine produced more IFN-γ (Figures S6A and S6B).
To further investigate the role of Ahr in Treg cell maintenance and stability, we sorted splenic Tregs from Ahrf/+Foxp3Yfp-Cre or littermate Ahrf/−Foxp3Yfp-Cre mice, and adoptively transferred them into Rag1−/− mice (Figure 6A). Ahr-deficient Tregs maintained the Foxp3 expression at a comparable extent to wildtype Tregs (Figure 6B) but aberrantly produced more pro-inflammatory cytokines (i.e., IFN-γ and IL-17) in the gut (Figures 6C and S6C). Consistently, Ahr deficiency in Tregs did not alter CpG methylation or chromatin status at the Foxp3 locus, but a trend of more open chromatin conformation at the Ifng and Il17a loci was detected by ATAC-Seq (Figures 6D, 6E and S6D). To exclude extrinsic effects caused by Rag1−/− recipients and directly compare the fitness between Ahr–sufficient and –deficient Tregs in a competitive setting, we sorted splenic YFP+ Tregs from Ahr+/+Foxp3Yfp-Cre mice (congenically marked by CD45.1 and CD45.2) and Ahrf/−Foxp3Yfp-Cre mice (congenically marked by CD45.2), mixed them at a 1:1 ratio, and then transferred them into the same Rag1−/− host (Figure 6F). Ahr-deficient Tregs maintained Foxp3 expression but produced more IFN-γ and IL-17 (Figures 6G, S6E and S6F), consistent with a key role for Ahr in prevention of aberrant production of pro-inflammatory cytokines by Tregs.
Figure 6. Ahr suppresses pro-inflammatory cytokine production in intestinal Tregs.

(A–C) CD4+YFP+ Tregs were purified from the spleen of Ahrf/+Foxp3Yfp-Cre or littermate Ahrf/−Foxp3Yfp-Cre mice. 2×105 cells from each group were transferred to Rag1−/− mice for analysis (A). Percentages of Foxp3+ cells among CD4+TCRβ+ cells (B), and percentages of IFN-γ+ or IL-17+ cells among Tregs (C) were analyzed by intracellular staining followed by flow cytometry. Data are representative of two independent experiments and shown as mean ± SD (n = 5 mice per group).
(D) Heat maps show DNA methylation status of Treg-specific demethylated region (TSDR) at the Foxp3 CNS2 locus in YFP+ or YFP− cells sorted from large intestinal LPLs of Ahrf/+Foxp3Yfp-Cre or Ahrf/−Foxp3Yfp-Cre littermate mice. Data are representative of two independent experiments.
(E) Normalized ATAC-Seq signal (RPM) profiles of Ifng and Il17a gene loci in Tregs sorted from large intestinal LPLs of Ahrf/+Foxp3Yfp-Cre or Ahrf/−Foxp3Yfp-Cre littermate mice. Data are representative of two independent experiments.
(F and G) CD4+YFP+ Tregs were purified from the spleen of Ahr+/+Foxp3Yfp-Cre CD45.1/45.2 or Ahrf/−Foxp3Yfp-Cre CD45.2/45.2 mice and mixed at 1:1 ratio. 4×105 mixed cells were transferred to Rag1−/− mice for analysis (F). Percentages of IFN-γ and IL-17-producing cells among CD45.1/45.2 Tregs or CD45.2/45.2 Tregs were analyzed by intracellular staining followed by flow cytometry (G). Each two symbols connected with a line represent data from the same individual mouse (n = 6 mice per group). Data are representative of two independent experiments.
See also Figure S6.
Ahr activation in Tregs is protective against T cell-mediated colitis
To further investigate the potential role of Ahr in Treg function, we utilized a T cell transfer model of colitis (Powrie et al., 1994). Specifically, we transferred CD45RBhi naïve CD4+ T cells into Rag1−/− mice together with purified Tregs with (i.e., GFP+YFP+) or without the expression of Ahr (i.e., GFP−YFP+) from AhrCAIR/+Foxp3Yfp-Cre mice, in which these cells have developed and co-existed in the same microenvironment. Compared to GFP− Tregs, CA-Ahr-expressing GFP+ Tregs suppressed T cell-induced wasting disease and colitis more efficiently, as assessed by body weight loss (Figures 7A and 7B), colon length and histology, and neutrophil/lymphocyte infiltration (Figures 7C to 7G). In addition, less CD45.1− cells (i.e., CD45.2+ Tregs) were found in the gut, but not in the spleen, of Rag1−/− mice that were adoptively transferred with GFP− Tregs compared to GFP+ Tregs (Figures 7H and S7A), consistent with the defective homing of Ahr-deficient Tregs to the gut. Furthermore, less pro-inflammatory cytokine-producing T cells were detected in mice that received GFP+ Tregs than in those that received GFP− Tregs in the colitis model (Figure 7I). Co-transferred GFP− Tregs maintained Foxp3 expression compared to GFP+ Tregs, suggesting that Ahr is dispensable for Foxp3 expression and/or stability (Figure S7B).
Figure 7. Ahr is required for optimal Treg suppressive function in vivo.

(A–B) Weight change of Rag1−/− mice at various times after adoptive transfer of CD4+CD25+CD45RBhigh (CD45.1/45.1) cells without (Teff only) or with Ahr-expressing (CD4+YFP+GFP+) or Ahr-non-expressing (CD4+YFP+GFP−) Tregs isolated from AhrCAIR/+Foxp3Yfp-Cre mice (CD45.2/45.2). Asterisks denote the significance of the difference between the percent weight change of the “Teff + GFP+ Tregs” and “Teff + GFP− Tregs” cohorts. Data are representative of two independent experiments (n=3–7 per group). (B) Linear regression lines were generated for the weight loss curves from Rag1−/−cohorts that were co-injected with Teff and Tregs.
(C–L) Rag1−/− mice were scarified 2 weeks after adoptive transfer of Teff without or with GFP+ or GFP− Tregs as (A–B) for hematoxylin/eosin staining of representative cecum and colon, scale bar, 500 μm (C), colon length (D), clinical histology score (E), percentages of infiltrating Gr-1+CD11b+ neutrophils among CD45.2+ LPLs (F), absolute numbers of CD4+CD45.1+ T cells (G), percentages and absolute numbers of GFP+ or GFP−CD45.1− cells (H) and absolute numbers of the indicated cytokine-producing CD45.1+CD4+ T cells in the large intestine (I). Data are representative of two independent experiments and shown as mean ± SD (n= 4–5 mice per group).
See also Figure S7.
In contrast to Tregs from Ahrf/+Foxp3Yfp-Cre mice that suppressed T-cell transfer colitis, Tregs purified from Ahrf/−Foxp3Yfp-Cre mice had impaired suppressive function, as revealed by worsened gut histopathological changes and enhanced gut inflammation in Rag1−/− recipient mice, while maintaining Foxp3 expression (Figures S7C–S7H). Thus, the loss-of-function data further support a positive role of Ahr in promoting Treg suppressive function in vivo.
DISCUSSION
Our data revealed a distinct expression pattern of Ahr in different tissue-resident Tregs. Under the steady state, tissue-resident Tregs in the gut expressed the highest amounts of Ahr. High Ahr expression in gut-associated Tregs, especially in peripherally induced Nrp1− Tregs may represent a mechanism of tissue adaptation, rendering gut Tregs readily activated by environmental cues (e.g., ligands that are abundantly present in the gut) to exert their suppressive function locally for gut homeostasis. Consistent with the literature (Atarashi et al., 2011; Geuking et al., 2011), our data showed that lack of gut microbiota caused Treg reduction, especially Nrp1−RORγt+ Tregs. We cannot rule out a possibility that Ahr activity is regulated by microbiota through Ahr ligands. However, microbiota appeared to play a dispensable role for Ahr expression in gut Tregs, suggesting a model that microbiota regulate Tregs via mechanism(s) independent of Ahr expression (e.g., by regulating retinoic acid and short-chain fatty acids to affect Foxp3 expression) (Arpaia et al., 2013; Benson et al., 2009; Coombes et al., 2007; Furusawa et al., 2013; Mucida et al., 2007; Nolting et al., 2009; Schambach et al., 2007; Singh et al., 2014; Smith et al., 2013; Sun et al., 2007).
Our data suggest a complex role for Ahr in different subsets of Tregs. Of note, there was no apparent defect in iTreg differentiation when Ahr-deficient naïve CD4+ T cells were stimulated by TGF-β in vitro (data not shown). Likewise, comparable in vivo Treg differentiation by OVA treatment was observed in recipient mice that was adoptively transferred with naïve non-Treg cells from Ahrf/−Foxp3Yfp-Cre OT-II mice, compared to those from Ahrf/+Foxp3Yfp-Cre OT-II mice (data not shown). In addition, Tregs in the thymus were not affected by Ahr deficiency, indicating that Ahr is dispensable for thymic development of Tregs.
Our studies using loss-of-function and gain-of-function approaches demonstrate that Ahr promotes the expression of the gut homing and activation marker CD103, encoded by Itgae, in Tregs. Of note, deletion of Itgae in Tregs itself is not sufficient to affect Treg cell function (Annacker et al., 2005) in a T cell-transfer model of colitis, suggesting that other mechanism(s) may compensate for the loss of CD103 in Tregs. Consistent with this notion, we also observed a decrease of Gpr15, a recently identified orphan guanine nucleotide-binding protein (G protein)-coupled receptor that plays a key role in homing of Tregs to the large intestine (Kim et al., 2013). CCL20 can be expressed by Th17 cells that are abundantly present in the gut (Yamazaki et al., 2008), in agreement with the involvement of CCR6-CCL20 axis as an autocrine and/or paracrine recruitment/retention mechanism for Th17 cells and Tregs homing to mucosal tissues especially during inflammation (Lee et al., 2013). Thus, decreased CCR6/CCL20 expression in Ahr-deficient Tregs may affect their homing to the gut. In agreement, genetic ablation of Ahr in Tregs caused reduction of Tregs, especially Nrp1− Tregs, only in the large intestinal lamina propria but not in other tissues. In addition, competitive homing assay showed the most marked reduction of Ahr-deficient Tregs in the large intestine, consistent with the important role for Ahr in Treg gut homing.
Our mouse model (i.e., AhrCAIR) allows the expression of CA-Ahr under the regulation of the endogenous Ahr locus and simultaneously marks Ahr expression by a GFP reporter to track Ahr expression and function in a cell type-specific manner in vivo after crossing to Cre transgenic mice. However, several important considerations need to be kept in mind. First, the reporter GFP reflects Ahr transcription; Second, we are using a destabilized GFP protein, and its half-life is likely different from endogenous Ahr protein. Third, the Ahr locus drives not only GFP expression but also a constitutively active form of Ahr (CA-Ahr) in AhrdCAIR reporter mice, and CA-Ahr may feedback and influence the expression of endogenous Ahr. Thus, we have examined and compared Ahr expression both by GFP in AhrdCAIR reporter mice and by intracellular staining and realtime RT-PCR of endogenous Ahr protein/mRNA in wildtype mice. Our data suggest that Ahr mRNA and protein expression can be reported by GFP. It is important to point out that the protein expression of CA-Ahr allele was considerably lower compared to the wildtype allele in Tregs as shown in immunoblotting by a yet unknown mechanism. Nevertheless, in line with the phenotypes that we observed using Ahr ligands, our data favor a model in which CA-Ahr driven by the endogenous locus control regions in AhrCAIR/+Foxp3Yfp-Cre mice was physiologically active. This is further supported by the opposing phenotypes we observed in Ahr-deficient mice compared to AhrCAIR/+Foxp3Yfp-Cre mice.
In the model of T cell transfer-mediated colitis, we were unable to transfer gut Tregs due to limited cell number and therefore used splenic Tregs. It is important to note that the Ahr-expressing Tregs (GFP+YFP+ cells) in AhrCAIR/+Foxp3Yfp-Cre mice co-expressed RORγt. In addition, Ahr-expressing Tregs in the spleen of AhrCAIR/+Foxp3Yfp-Cre mice showed more activated phenotypes as revealed by increased percentages of CD44hiCD62Llow Tregs (data not shown). Despite no apparent Treg phenotype in the spleen under the steady-state conditions, defective functions of the splenic Ahr-deficient Tregs in suppressing T cell transfer-mediated colitis were observed, consistent with the more suppressive function of RORγt+ Tregs and/or activated Tregs in various animal models of disease (Kim et al., 2016; Luo et al., 2016; Ohnmacht et al., 2015; Sefik et al., 2015). In addition, upon adoptive transfer to a lymphopenic Rag-deficient environment, Ahr-deficient splenic Tregs showed enhanced proinflammatory cytokine production (i.e., IFN-γ and IL-17) in the gut, consistent with their impaired suppressive functions.
Although Ahr was co-expressed with RORγt in Tregs, overall RORγt+ Tregs did not show a reduction in Ahr-deficient mice. In addition, no alteration of RORγt transcripts in Ahr-deficient Tregs was detected by RNA-Seq analysis. These data suggest that Ahr is unlikely to regulate RORγt transcription in Tregs. Ahr was reported to regulate in vitro Foxp3 expression induced by TGF-β (Quintana et al., 2008). However, our data suggest that Ahr did not regulate Foxp3 expression in vivo, as evidenced by lack of detectable changes in transcription of Foxp3 gene by RNA-Seq analysis, TSDR methylation status at the Foxp3 locus by bisulfite sequencing, or chromatin conformation by ATAC-Seq. In addition, we did not observe Foxp3 stability changes upon deletion of Ahr in Tregs. Together, these data suggest a model that Ahr preferentially marks gut Tregs, and multiple mechanisms of action of Ahr may work in concert to mediate Treg gut adaptation. Perturbation of Ahr pathway may lead to impaired Treg cell homing and function, and thus diminish its capacity to control inflammation in the gut.
EXPERIMENTAL PROCEDURES
Mice
All mice were maintained in SPF or Germ-free facilities at Northwestern University (NU) and the University of Florida (UF). The mice were littermate controlled and were 6–10 week old unless otherwise indicated in the text. C57BL/6-SJL (CD45.1), Ahrf/f, Cd4-cre mice were purchased from Taconic Farms and EIIa-cre mice were purchased from Jackson Laboratory. Foxp3Yfp-Cre and Ahr−/− mice were described previously (Fernandez-Salguero et al., 1995; Rubtsov et al., 2008). All studies were approved by the Animal Care and Use Committees of NU and UF.
Isolation of Intestinal LPLs and Flow Cytometry
Isolation of intestinal LPLs and flow cytometry were done as previously described (Qiu et al., 2012). CD4+ T cells from spleen and peripheral lymph nodes were purified with CD4+ T cell isolation kit (Stemcell). Antibodies were purchased from eBioscience, BD Pharmingen, BioLegend or TONBO. CD16/32 antibody was used to block the nonspecific binding to Fc receptors before all surface staining. For transcription factor staining, cells were fixed and permeabilized with Foxp3 staining buffer Kit (eBioscience). For cytokine staining, cells were stimulated with 50 ng/ml PMA, 500 ng/ml ionomycin for 4 hours and Brefeldin A (2 μg/ml) was added 2 hours before cell harvest. Dead cells were discriminated by Live and Dead violet viability kit (Invitrogen). Sample acquisition was performed on FACSCantoII or LSRFortessa (BD Biosciences) and analyzed with FlowJo (version 10.2; Tree Star).
Antibiotics Treatment
Mice were gavaged daily with 200 μl/day of broad-spectrum antibiotics (“ABX” including 1 g/l ampicillin, 1 g/l neomycin, 1 g/l metronidazole, 0.5 g/l vancomycin, and 1 g/l gentamycin) for 2 to 3 weeks.
RNA-Seq
CD4+YFP+ Tregs were sorted by flow cytometry from the large intestinal LPLs of Ahrf/+Foxp3Yfp-Cre or littermate Ahrf/−Foxp3Yfp-Cre mice. About 8×105 sorted Tregs pooled from 4–5 mice per group were lysed in Trizol (Invitrogen), and RNA was extracted with RNAeasy Mini Kit (Qiagen). Total RNA was treated with Ribo-zero kit and RNAseq libraries were generated using kits from Illumina. Detailed data processing is described in the supplemental experimental procedures.
Realtime RT-PCR
Treg RNA was isolated with Trizol (Invitrogen). cDNA was synthesized using GoScript™ Reverse Transcription kit (Promega). Realtime RT-PCR was performed using SYBR Green (Biorad) and various primer sets (Table S1). Reactions were run using CFX Connect™ (Biorad). The results were displayed as relative expression values normalized to β-actin.
T Cell-mediated Transfer Model of Colitis
Naïve 4×105 CD4+CD25−CD45RBhigh cells from the spleen and lymph nodes of CD45.1+ congenic mice were intravenously injected into Rag1−/− mice with or without 4×104 CD4+YFP+ splenic Tregs sorted from Ahrf/+Foxp3Yfp-Cre or littermate Ahrf/−Foxp3Yfp-Cre mice or CD4+YFP+GFP+ or CD4+YFP+GFP− splenic Tregs sorted from AhrCAIR/+Foxp3Yfp-Cre mice. The weight change of recipient Rag1−/− mice were monitored for 8 weeks or the mice were sacrificed for analysis 2 weeks after cell transfer.
Methylation Analysis
Genomic DNA of FACS-sorted large intestinal CD4+TCRβ+YFP+ Tregs from Ahrf/+Foxp3Yfp-Cre or littermate Ahrf/−Foxp3Yfp-Cre mice was isolated with DNeasy tissue kit (Qiagen). Bisulfate conversion was performed with EZ DNA methylation Kit (Zymo Research). The PCR product was cloned using pGEM-T easy vector system (Promega) for sanger sequencing using T7 primer.
Transposase-Accessible Chromatin Sequencing (ATAC-Seq)
5×104 sorted large intestinal CD4+TCRβ+YFP+ Tregs were subjected to ATAC-Seq according to a published protocol (Buenrostro et al., 2013) with a modification in the library purification step. Specifically, the library was cleaned up with 1.2x SPRIselect beads (Beckman Coulter), to exclude the small fragments, before sequencing with Illumia HiSeq 2500. Detailed data processing is described in the supplemental experimental procedures.
Statistical Methods
Unless otherwise noted, statistical analysis was performed with the unpaired Student’s t test on individual biological samples. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001
Supplementary Material
HIGHLIGHTS.
Ahr is preferentially expressed by intestinal pTregs independent of the microbiota.
Ahr regulates Treg homing to the gut.
Ahr is important for in vivo Treg function but not Foxp3 expression.
Ahr-expressing Tregs have enhanced suppressive activity in a model of colitis.
Acknowledgments
We thank the entire L.Z. lab and Dr. Dorina Avram for their help and suggestions. We thank Dr. L. Yang at the University of Florida for cell sorting support. We thank Genomics Facility (the University of Chicago) for their services and assistance. Transgenic and Targeted Mutagenesis Laboratory (TTML) (Northwestern University) (Dr. Lynn Doglio) for generation of AhrCAIR mice. This work was in part made possible from an NIH instrumentation grant (1S10 OD021676-01), and University of Florida Department of Medicine Gatorade Fund (CJ) The work was supported by the National Institutes of Health (AI089954 and DK105562 to L.Z.), and by a Cancer Research Institute Investigator Award (LZ). Liang Zhou is a Pew Scholar in Biomedical Sciences, supported by the Pew Charitable Trusts, and an Investigator in the Pathogenesis of Infectious Disease, supported by Burroughs Wellcome Fund. H.S. is an ANRF Scholar and Hellman Fellow. J.W.B. was funded by NIH Diversity Supplement 3R01DK105562-08S1. The authors have no financial conflict of interest.
Footnotes
AUTHOR CONTRIBUTIONS
J.Y., J.Q., A.U., H.S. and S.L. designed the study and performed experiments. J.W.B. contributed to the data analysis. C.J. provided reagents and suggestions. Z.E.C. performed histological analysis. J.Y., J.Q. and L.Z. wrote the paper with input from all authors. L.Z. conceived, designed, and coordinated the project.
ACCESSION NUMBERS
The RNA-Seq data and ATAC-Seq data reported in this paper have been deposited in the Gene Expression Omnibus under the accession number GSE106083.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersson P, McGuire J, Rubio C, Gradin K, Whitelaw ML, Pettersson S, Hanberg A, Poellinger L. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:9990–9995. doi: 10.1073/pnas.152706299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annacker O, Coombes JL, Malmstrom V, Uhlig HH, Bourne T, Johansson-Lindbom B, Agace WW, Parker CM, Powrie F. Essential role for CD103 in the T cell-mediated regulation of experimental colitis. The Journal of experimental medicine. 2005;202:1051–1061. doi: 10.1084/jem.20040662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D, Burns EJ, Sherr DH, Weiner HL, Kuchroo VK. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nature immunology. 2010;11:854–861. doi: 10.1038/ni.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, Rudensky AY. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Honda K. Microbiota in autoimmunity and tolerance. Current opinion in immunology. 2011;23:761–768. doi: 10.1016/j.coi.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu R, O’Quinn DB, Silberger DJ, Schoeb TR, Fouser L, Ouyang W, Hatton RD, Weaver CT. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity. 2012;37:1061–1075. doi: 10.1016/j.immuni.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson MJ, Elgueta R, Schpero W, Molloy M, Zhang W, Usherwood E, Noelle RJ. Distinction of the memory B cell response to cognate antigen versus bystander inflammatory signals. The Journal of experimental medicine. 2009;206:2013–2025. doi: 10.1084/jem.20090667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin C, Bargatze RF, Campbell JJ, von Andrian UH, Szabo MC, Hasslen SR, Nelson RD, Berg EL, Erlandsen SL, Butcher EC. alpha 4 integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell. 1995;80:413–422. doi: 10.1016/0092-8674(95)90491-3. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Bollrath J, Powrie FM. Controlling the frontier: regulatory T-cells and intestinal homeostasis. Seminars in immunology. 2013;25:352–357. doi: 10.1016/j.smim.2013.09.002. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nature immunology. 2013;14:1007–1013. doi: 10.1038/ni.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. The Journal of experimental medicine. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser C, Rannug A, Stockinger B. The aryl hydrocarbon receptor in immunity. Trends in immunology. 2009;30:447–454. doi: 10.1016/j.it.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Feuerer M, Hill JA, Mathis D, Benoist C. Foxp3+ regulatory T cells: differentiation, specification, subphenotypes. Nature immunology. 2009;10:689–695. doi: 10.1038/ni.1760. [DOI] [PubMed] [Google Scholar]
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, Kozoriz D, Weiner HL, Quintana FJ. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nature immunology. 2010;11:846–853. doi: 10.1038/ni.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuking MB, Cahenzli J, Lawson MA, Ng DC, Slack E, Hapfelmeier S, McCoy KD, Macpherson AJ. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity. 2011;34:794–806. doi: 10.1016/j.immuni.2011.03.021. [DOI] [PubMed] [Google Scholar]
- Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, Debes GF, Lauber J, Frey O, Przybylski GK, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. The Journal of experimental medicine. 2004;199:303–313. doi: 10.1084/jem.20031562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Kim KS, Hong SW, Han D, Yi J, Jung J, Yang BG, Lee JY, Lee M, Surh CD. Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science. 2016;351:858–863. doi: 10.1126/science.aac5560. [DOI] [PubMed] [Google Scholar]
- Kim SV, Xiang WV, Kwak C, Yang Y, Lin XW, Ota M, Sarpel U, Rifkin DB, Xu R, Littman DR. GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science. 2013;340:1456–1459. doi: 10.1126/science.1237013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Naka T, Nakahama T, Chinen I, Masuda K, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. The Journal of experimental medicine. 2009;206:2027–2035. doi: 10.1084/jem.20090560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A. 2008;105:9721–9726. doi: 10.1073/pnas.0804231105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, Diefenbach A. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. 2011;334:1561–1565. doi: 10.1126/science.1214914. [DOI] [PubMed] [Google Scholar]
- Kleinewietfeld M, Puentes F, Borsellino G, Battistini L, Rotzschke O, Falk K. CCR6 expression defines regulatory effector/memory-like cells within the CD25(+)CD4+ T-cell subset. Blood. 2005;105:2877–2886. doi: 10.1182/blood-2004-07-2505. [DOI] [PubMed] [Google Scholar]
- Lee AY, Eri R, Lyons AB, Grimm MC, Korner H. CC Chemokine Ligand 20 and Its Cognate Receptor CCR6 in Mucosal T Cell Immunology and Inflammatory Bowel Disease: Odd Couple or Axis of Evil? Frontiers in immunology. 2013;4:194. doi: 10.3389/fimmu.2013.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, Mantovani A, Kopan R, Bradfield CA, Newberry RD, Colonna M. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nature immunology. 2011;13:144–151. doi: 10.1038/ni.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann J, Huehn J, de la Rosa M, Maszyna F, Kretschmer U, Krenn V, Brunner M, Scheffold A, Hamann A. Expression of the integrin alpha Ebeta 7 identifies unique subsets of CD25+ as well as CD25- regulatory T cells. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:13031–13036. doi: 10.1073/pnas.192162899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, Wilhelm C, Veldhoen M. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell. 2011;147:629–640. doi: 10.1016/j.cell.2011.09.025. [DOI] [PubMed] [Google Scholar]
- Luo CT, Liao W, Dadi S, Toure A, Li MO. Graded Foxo1 activity in Treg cells differentiates tumour immunity from spontaneous autoimmunity. Nature. 2016;529:532–536. doi: 10.1038/nature16486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire J, Okamoto K, Whitelaw ML, Tanaka H, Poellinger L. Definition of a dioxin receptor mutant that is a constitutive activator of transcription: delineation of overlapping repression and ligand binding functions within the PAS domain. The Journal of biological chemistry. 2001;276:41841–41849. doi: 10.1074/jbc.M105607200. [DOI] [PubMed] [Google Scholar]
- Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, MacDonald TT, Pallone F, Monteleone G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology. 2011;141:237–248. 248 e231. doi: 10.1053/j.gastro.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature genetics. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Mucida D, Park Y, Cheroutre H. From the diet to the nucleus: vitamin A and TGF-beta join efforts at the mucosal interface of the intestine. Seminars in immunology. 2009;21:14–21. doi: 10.1016/j.smim.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- Nguyen NT, Hanieh H, Nakahama T, Kishimoto T. The roles of aryl hydrocarbon receptor in immune responses. International immunology. 2013;25:335–343. doi: 10.1093/intimm/dxt011. [DOI] [PubMed] [Google Scholar]
- Nohara K, Pan X, Tsukumo S, Hida A, Ito T, Nagai H, Inouye K, Motohashi H, Yamamoto M, Fujii-Kuriyama Y, Tohyama C. Constitutively active aryl hydrocarbon receptor expressed specifically in T-lineage cells causes thymus involution and suppresses the immunization-induced increase in splenocytes. Journal of immunology. 2005;174:2770–2777. doi: 10.4049/jimmunol.174.5.2770. [DOI] [PubMed] [Google Scholar]
- Nolting J, Daniel C, Reuter S, Stuelten C, Li P, Sucov H, Kim BG, Letterio JJ, Kretschmer K, Kim HJ, von Boehmer H. Retinoic acid can enhance conversion of naive into regulatory T cells independently of secreted cytokines. The Journal of experimental medicine. 2009;206:2131–2139. doi: 10.1084/jem.20090639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, Gaboriau-Routhiau V, Marques R, Dulauroy S, Fedoseeva M, et al. The microbiota regulates type 2 immunity through RORgammat(+) T cells. Science. 2015;349:989–993. doi: 10.1126/science.aac4263. [DOI] [PubMed] [Google Scholar]
- Powrie F, Leach MW, Mauze S, Menon S, Caddle LB, Coffman RL. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1994;1:553–562. doi: 10.1016/1074-7613(94)90045-0. [DOI] [PubMed] [Google Scholar]
- Qiu J, Guo X, Chen ZM, He L, Sonnenberg GF, Artis D, Fu YX, Zhou L. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity. 2013;39:386–399. doi: 10.1016/j.immuni.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, Zhou L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity. 2012;36:92–104. doi: 10.1016/j.immuni.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Zhou L. Aryl hydrocarbon receptor promotes RORgammat(+) group 3 ILCs and controls intestinal immunity and inflammation. Seminars in immunopathology. 2013;35:657–670. doi: 10.1007/s00281-013-0393-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- Rausch S, Huehn J, Kirchhoff D, Rzepecka J, Schnoeller C, Pillai S, Loddenkemper C, Scheffold A, Hamann A, Lucius R, Hartmann S. Functional analysis of effector and regulatory T cells in a parasitic nematode infection. Infect Immun. 2008;76:1908–1919. doi: 10.1128/IAI.01233-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR, Jr, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- Schambach F, Schupp M, Lazar MA, Reiner SL. Activation of retinoic acid receptor-alpha favours regulatory T cell induction at the expense of IL-17-secreting T helper cell differentiation. European journal of immunology. 2007;37:2396–2399. doi: 10.1002/eji.200737621. [DOI] [PubMed] [Google Scholar]
- Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, Burzyn D, Ortiz-Lopez A, Lobera M, Yang J, et al. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science. 2015;349:993–997. doi: 10.1126/science.aaa9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40:128–139. doi: 10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens GL, Andersson J, Shevach EM. Distinct subsets of FoxP3+ regulatory T cells participate in the control of immune responses. Journal of immunology. 2007;178:6901–6911. doi: 10.4049/jimmunol.178.11.6901. [DOI] [PubMed] [Google Scholar]
- Stevens EA, Mezrich JD, Bradfield CA. The aryl hydrocarbon receptor: a perspective on potential roles in the immune system. Immunology. 2009;127:299–311. doi: 10.1111/j.1365-2567.2009.03054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger B, Di Meglio P, Gialitakis M, Duarte JH. The aryl hydrocarbon receptor: multitasking in the immune system. Annual review of immunology. 2014;32:403–432. doi: 10.1146/annurev-immunol-032713-120245. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suffia I, Reckling SK, Salay G, Belkaid Y. A role for CD103 in the retention of CD4+CD25+ Treg and control of Leishmania major infection. Journal of immunology. 2005;174:5444–5455. doi: 10.4049/jimmunol.174.9.5444. [DOI] [PubMed] [Google Scholar]
- Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. The Journal of experimental medicine. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauchi M, Hida A, Negishi T, Katsuoka F, Noda S, Mimura J, Hosoya T, Yanaka A, Aburatani H, Fujii-Kuriyama Y, et al. Constitutive expression of aryl hydrocarbon receptor in keratinocytes causes inflammatory skin lesions. Mol Cell Biol. 2005;25:9360–9368. doi: 10.1128/MCB.25.21.9360-9368.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- Vignali DA. Mechanisms of T(reg) Suppression: Still a Long Way to Go. Frontiers in immunology. 2012;3:191. doi: 10.3389/fimmu.2012.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, Xiong H, Dolpady J, Frey AB, Ruocco MG, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. The Journal of experimental medicine. 2012;209:1723–1742. S1721. doi: 10.1084/jem.20120914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, Anthony BA, Sverdrup FM, Head R, Kuster DJ, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. The Journal of experimental medicine. 2012;209:1713–1722. S1711–1719. doi: 10.1084/jem.20120822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Yang XO, Chung Y, Fukunaga A, Nurieva R, Pappu B, Martin-Orozco N, Kang HS, Ma L, Panopoulos AD, et al. CCR6 regulates the migration of inflammatory and regulatory T cells. Journal of immunology. 2008;181:8391–8401. doi: 10.4049/jimmunol.181.12.8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel BA, Agace WW, Campbell JJ, Heath HM, Parent D, Roberts AI, Ebert EC, Kassam N, Qin S, Zovko M, et al. Human G protein-coupled receptor GPR-9–6/CC chemokine receptor 9 is selectively expressed on intestinal homing T lymphocytes, mucosal lymphocytes, and thymocytes and is required for thymus-expressed chemokine-mediated chemotaxis. The Journal of experimental medicine. 1999;190:1241–1256. doi: 10.1084/jem.190.9.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L. AHR Function in Lymphocytes: Emerging Concepts. Trends in immunology. 2016;37:17–31. doi: 10.1016/j.it.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.