

Summary
Aims
Following a traumatic brain injury (TBI), 5–50% of patients will develop posttraumatic epilepsy (PTE) with children being particularly susceptible. Currently, PTE cannot be prevented and there is limited understanding of the underlying epileptogenic mechanisms. We hypothesize that early after TBI the brain undergoes distinct cellular and synaptic reorganization that facilitates cortical excitability and promotes the development of epilepsy.
Methods
To examine the effect of pediatric TBI on cortical excitability, we performed controlled cortical impact (CCI) on juvenile rats (postnatal day 17). Following CCI, animals were monitored for the presence of epileptiform activity by continuous in vivo electroencephalography (EEG) and/or sacrificed for in vitro whole‐cell patch‐clamp recordings.
Results
Following a short latent period, all animals subjected to CCI developed spontaneous recurrent epileptiform activity within 14 days. Whole‐cell patch‐clamp recordings of layer V pyramidal neurons showed no changes in intrinsic excitability or spontaneous excitatory postsynaptic currents (sEPSCs) properties. However, the decay of spontaneous inhibitory postsynaptic currents (sIPSCs) was significantly increased. In addition, CCI induced over a 300% increase in excitatory and inhibitory synaptic bursting. Synaptic bursting was prevented by blockade of Na+‐dependent action potentials or select antagonism of glutamate or GABA‐A receptors, respectively.
Conclusion
Our results demonstrate that CCI in juvenile rats rapidly induces epileptiform activity and enhanced cortical synaptic bursting. Detection of epileptiform activity early after injury suggests it may be an important pathophysiological component and potential indicator of developing PTE.
Keywords: Cerebral cortex, Epilepsy, Neurophysiology, Synaptic plasticity, Traumatic brain injury
Introduction
Traumatic brain injury (TBI) is a leading cause of death and disability in children and often leads to the development of posttraumatic epilepsy (PTE) 1, 2, 3, 4. PTE develops in up to 20% of children and depends on several factors including the severity of injury, age of the patient, and injury site 5, 6. The underlying pathophysiology of PTE is poorly understood, but develops in the wake of injury and leads to spontaneous recurrent seizures. Over the long term, these posttraumatic seizures (PTS) may cause secondary brain damage through mechanisms including increased metabolic requirements, hypoxia, increased intracranial pressure, and/or excessive release of neurotransmitters 7. Exacerbating the clinical management of PTE is that acute and prophylactic use of antiepileptic drugs fail to prevent seizures 8 or reduce the risk of developing PTE 9, 10, 11. In humans, PTE most often develops slowly over months and even years. The slow development of PTE provides a unique temporal window to study and identify the epileptic changes as they occur “on the road” to PTE. Furthermore, evidence suggests that there may be a critical window following TBI for clinical intervention 12. Development of new therapeutic strategies in children requires an improved understanding of the processes and timing of events that occur early after injury resulting in the pathogenesis stages of PTE.
Injuries ranging from mild (concussion) to severe penetrating wounds and skull fractures may fall under the broad term of TBI. The incidence of PTE is significantly higher following severe TBI. Controlled cortical impact (CCI) has been used extensively as a model of head injury 13, 14, 15, 16 and in pediatric models 17 and more recently as an effective means to model severe TBI 13, 18, 19. Following CCI, studies have shown significant cavitation and neuronal cell loss at the site of injury 20, 21, 22, 23, 24, and hippocampal neuro‐ and synaptogenesis 25, 26. Direct injury‐induced seizures have been reported to occur within the first 48 h following CCI in adult animals 27. However, the development of spontaneous recurrent posttraumatic seizures (PTS) occurs in 12.5% to 36% of animals following a latent period of weeks to months 18, 28. In juvenile animals, we have previously shown that CCI induces necrotic loss of cortex, damage to the underlying corpus callosum and hippocampus, synaptic reorganization and deficits in spatial learning and memory 17, 29, 30. In this study, we examined the underlying mechanisms that may contribute to cortical hyperexcitability and epileptogenesis in juvenile animals following CCI.
Pyramidal (PYR) neurons are the major source of excitatory output from layer V, a lamina that has been implicated as the site of origin of interictal epileptiform discharge in both acute and chronic models of neocortical epileptogenesis 31, 32. A recent preliminary report by Yang et al. 19 has shown that CCI performed in juvenile rats rapidly induces spontaneous epileptiform activity in layer V cortical pyramidal neurons. In the present study, we examined the underlying mechanisms that may contribute to the development of this epileptiform activity. Utilizing electrophysiological approaches, we determine that CCI in juvenile rats induces the rapid development of in vivo epileptiform activity and the preferential enhancement of in vitro excitatory presynaptic burst discharges. These synaptic bursts occurred in the absence of significant changes in intrinsic excitability of layer V pyramidal neurons and are thought to be driven by altered afferent cortical synaptic input. Our findings suggest that juvenile animals undergo unique pathophysiological changes early after TBI that may be involved in the pathogenesis of PTE.
Materials and Methods
Protocols used for all experiments were approved by the University of Arizona Institutional Animal Care and Use Committee.
CCI Injury
To experimentally model TBI, a controlled cortical impact (CCI) was performed on 32 CCI and 14 control postnatal day 17 (P17) Sprague Dawley rats as previously described 17, 29, 30. In brief, male Sprague Dawley rats were sedated with isoflurane and injected interperitoneal (i.p.) with a mixture of ketamine (50 mg/kg) and xylazine (5 mg/kg) at 0.01 mL per 10 g of rat weight. Surgery site was shaved, and animals were fixed into a stereotaxic frame. A midline scalp incision was then performed over the right somatosensory region, lateral to the sagittal suture between bregma and lambda. The skull was exposed and a 6‐mm craniotomy was performed. Precaution was taken during the craniotomy to avoid damaging the underlying dura and inducing significant bleeding. A frontoparietal CCI (5 mm tip, 4 m/second, 2.0 mm depth) was performed using a pneumatic impactor (Amscien Instruments, Richmond, VA, USA). After the CCI, the bone flap that was removed during the craniotomy was placed over the injury site and secured with dental cement. During this time, electroencephalography (EEG) leads were mounted in some animals and secured with dental cement. The skin was then sutured closed and the incision area swabbed with betadine. Animal temperature was maintained with an electric heating pad and monitored postsurgery until ambulatory (<3 h). Following the initial recovery, animals were returned to standard housing and monitored daily. A portion of animals that received CCI were connected to EEG starting 24 h postinjury (n = 16). All animals were then allowed to recover until further experimentation began on postinjury day (PID) 14.
In Vivo Seizure Monitoring with Electroencephalography
Rats subjected to CCI or age‐matched controls were implanted with epidural recording electrodes. Experimental evidence indicates craniotomy may induce alterations to the cortex 33, 34. As such, we considered the craniotomy a component of the injury process and used appropriate naïve age‐matched control animals. Epidural recording electrodes were made from #0–80 × 1/8 inch stainless steel screws and up to four were implanted at the following stereotaxic co‐ordinates: AP: 2.0 mm, Lateral: ±3.0, Depth: 1 mm; AP: −4.0 mm, Lateral: −3.0 mm, Depth: 1 mm; AP: −8.0 mm, Lateral: +3.0, Depth: 1 mm (Figure 1). On PID 1, after recovery from surgery, animals were placed in acrylic cages where they could move freely and were connected through commutators to the recording system. Animals were singly housed during this period. EEG signals were recorded continuously for 13 days postinjury using an Xltek 128 channel Neurolink IP amplifier (1.0 Hz and 70 Hz cutoffs, 512 Hz sampling rate). Two independent, blinded, and trained personnel analyzed the digital EEG files and their results were compared for consistency and averaged. As previously described, epileptiform activity was defined by the presence of epileptiform discharges or seizure‐like events 35.
Figure 1.
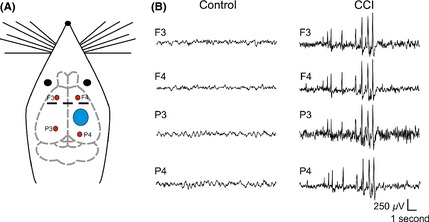
Controlled cortical impact (CCI) induces epileptiform activity. Left: Schematic representation of rat brain indicating site of CCI injury (blue circle) and EEG recording electrodes (red circles). Middle and Right: Epidural EEG recordings from rats made 14 days after CCI. Middle panel is from a control animal without observable epileptiform activity and right panel from a CCI animal that displayed spontaneous epileptiform discharges.
Epileptiform discharges (ED) were defined by rhythmic transients containing spikes and uniform sharp waves that lasted between 1 and 5 seconds. High‐amplitude rhythmic discharges that were clearly distinguishable from background and lasted for >5 seconds have been considered seizures 36. As simultaneous behavioral seizure activity was not monitored, this activity has been classified as seizure‐like.
Preparation and Maintenance of Brain Slices
On PID 14–19, the rats were deeply anaesthetized with inhalation of isoflurane and decapitated. The brain was rapidly removed and coronal slices (350 μm thick) prepared on a vibratome (VT 1200; Leica, Nussloch, Germany) as previously described 37, 38. Slices were obtained from the somatosensory cortex that contained the injury site in CCI animals or from corresponding control cortex in sham animals. The site of CCI was readily identifiable in slices as significant cavitation and tissue loss. Initial harvesting of brain slices was performed in an ice‐cooled (4°C) carboxygenated (95% O2, 5% CO2) high sucrose solution containing the following (in mM): 234 sucrose, 11 glucose, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4H2O, 10 MgS47H2O, 0.5 CaCl22H2O. Slices were then incubated for 1 h at 32°C in carboxygenated artificial CSF (aCSF) containing (in mM): 126 NaCl, 26 NaHCO3, 2.5 KCl, 10 Glucose, 1.25 Na2H2PO4H2O, 1 MgSO47H2O, 2 CaCl2H2O, pH 7.4. Slices were then returned to room temperature before being moved to the recording chamber for whole‐cell patch‐clamp recording.
In Vitro Electrophysiological Recording
Slices prepared from CCI (n = 32) or sham (n = 14) animals were submerged in flowing carboxygenated aCSF heated to 32°C. Submerged slices were first visualized under 4× brightfield for identification of layer V cortex. For slices from CCI rats, recordings were made in the peri‐injury zone within 2 mm of the injury‐induced cavitation. Recordings from control slices were made in the corresponding cortex to the peri‐injury zone of CCI animals. Whole‐cell recordings were obtained from regular spiking cortical pyramidal neurons using an upright microscope (Axioexaminer; Carl‐Zeiss, Thornwood, NY, USA) fitted with infrared differential interference contrast optics. Regular spiking (RS) pyramidal neurons were distinguished based on their current‐clamp firing behavior 39. The electrode capacitance and bridge circuit were appropriately adjusted. The series resistance (R s) of neurons chosen for analysis was <20% of membrane input resistance and monitored for stability. Membrane potential was not corrected for a calculated 10 mV liquid junction potential. A Multiclamp 700 A patch‐clamp amplifier (Axon Instruments, Union City, CA, USA) was used for both current‐ and voltage‐clamp mode. Recordings were obtained at 32°C using borosilicate glass microelectrodes (tip resistance, 2.5–3.5 MΩ). For excitatory recordings, electrodes were filled with an intracellular solution containing (in mM): 135 KGluconate, 4 KCl, 2 NaCl, 10 HEPES, 4 EGTA, 4 Mg ATP, 0.3 Na TRIS. For recording of inhibitory events, an intracellular solution containing the following was used (in mM): 70 KGluconate, 70 KCl, 2 NaCl, 10 HEPES, 4 EGTA, 4 Mg ATP, 0.3 GTP. This internal solution has been used previously 38, 40 and shown to facilitate detection of inhibitory events. The calculated E Cl was approximately −16 mV, resulting in inward GABAA currents at a holding potential of −70 mV. Inhibitory events were pharmacologically isolated by bath application of 2‐Amino‐5‐phosphonopentanoic acid (d‐APV; 50 μM) and 6,7‐dinitroquinoxaline‐2,3‐dione (DNQX, 20 μM). Sodium (Na+)‐dependent action potentials were blocked by bath application of tetrodotoxin (TTX, 1 μM).
Data Analysis
Data were analyzed using pCLAMP (Axon Instruments, Sunnyvale, CA, USA), Prism (GraphPad, La Jolla, CA, USA) and MiniAnalysis (Synaptosoft, Decatur, GA, USA) software and are presented as means ± SEM. For detection of spontaneous synaptic events automated threshold detection was employed through MiniAnalysis and detected events were subsequently manually verified. Synaptic burst events were detected based on previously published characteristics 32 and were defined by a minimum of three synaptic events occurring in 250 milliseconds that temporally summated. Input resistance was calculated from the voltage response to the input of a current step (1 second, 50 mV). Adaptation index was calculated as 100 × (1 − F Last/F 2), where F Last corresponds to the firing rate of the last interspike interval and F 2 the second interspike interval. Many of the pyramidal neurons had a high variability in the first interspike interval, so the second interspike interval was chosen for analysis. Statistical significance was tested using an unpaired t test, and differences were determined to be significant if P < 0.05.
Results
To model severe TBI in pediatric patients, we subjected 17‐day‐old rats to CCI and compared them to age‐matched controls. As previously described, the CCI procedure results in a significant cavity in the cortex at the site of the injury and extensive necrosis 17, 19.
Epileptiform Activity is Rapidly Induced In Vivo Following Traumatic Brain Injury
To monitor for the development of epileptiform activity, we performed electroencephalography (EEG) recordings from CCI animals (n = 16). Following recovery from the CCI surgery, EEG activity was continuously recorded for the first 2 weeks. Epileptiform activity was detected postrecording based on previously published characteristics 35 and as detailed in the methods. Two trained personnel where blinded to the animals’ experimental condition and the independent grading of the EEG recordings was averaged. Within the first 24 h of recording, 87.5% of CCI animals developed epileptiform activity. Following this initial “immediate” epileptic activity CCI animals displayed a quiescent period with no detectable epileptic activity. The length and timing of this latent period varied across animals but occurred between postinjury day 3 and 7. Following this latent period and by postinjury day 14, all CCI animals subsequently developed “late” recurrent epileptiform activity. This epileptiform activity synchronized across EEG leads (Figure 1) and is not observed in control animals 28, 41, 42, 43. On average, 16.4 ± 3 epileptiform events were detected in the second week of EEG recording following this latent period. This activity resembles epileptiform discharges and interictal spiking that has been previously shown in other epileptic animal models and in human TBI patients 35, 44, 45. In seven of 16 CCI animals, prolonged seizure‐like events were also detected that lasted >30 seconds (Figure 2). Seizure‐like events were detected on average 5.57 ± 1.4 times in the second week of recording after CCI and had an average duration of 134.2 ± 11 seconds. In humans, epileptiform activity that occurs immediately after injury (<24 h) is thought to be direct injury induced and a poor predictor of PTE 46, 47. However, even a single “late” seizure that occurs more than 1 week after injury has a 65–90% chance of progressing to recurrent seizures and PTE 48, 49. The development of “late” epileptiform activity within 14 days after CCI, and in advance of PTE, suggests the presence of emerging epileptogenic activity. As PID 14 was the earliest time point that all CCI animals reliably displayed this “late” stage in vivo epileptiform activity, it was subsequently chosen for further in vitro experimentation.
Figure 2.
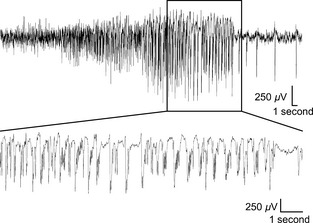
Controlled cortical impact (CCI) selectively induces electrographic seizure‐like activity. Representative recording from a single EEG electrode during a seizure‐like event. This example was recorded 9 days after CCI. These “late” seizures were infrequent but present in 7 of 16 CCI animals. (Top) Note the presence of a spontaneous electrographic seizure‐like event defined by a high‐amplitude (>2× baseline) rhythmic discharge that lasted for >5 seconds. (Bottom) An expanded timescale of the boxed area showing details of the epileptic activity.
Epileptiform Synaptic Bursting is Induced in vitro Following TBI
Epileptogenesis has been extensively studied in numerous animal models and is generally thought to occur as the result of disruption to intrinsic excitability, synaptic inhibition, and/or synaptic excitation 50. To investigate the contribution of these mechanisms to CCI‐induced epileptiform activity, we examined for intrinsic and synaptic electrophysiological changes in cortical brain slices. Epileptiform activity detected by EEG was widespread and synchronous within and across cortical hemispheres. Pyramidal neurons in layer V are the major output pathway of the cortex and have been implicated in network synchronization 51. As such, whole‐cell patch‐clamp recordings of physiologically identified layer V pyramidal neurons were made from animals 14 days after CCI or in age‐matched control. All recordings were performed in the peri‐injury zone (i.e., within 2 mm of injury site) or corresponding control cortex.
Intrinsic Excitability
The intrinsic membrane properties of a neuron have been repeatedly shown to be altered in various models of epilepsy 52, 53. Neurons that have pathological enhancements to intrinsic excitability may be spontaneous generators of epileptic activity. To examine this possibility, we first recorded for changes in the intrinsic membrane properties from CCI (n = 35) and control pyramidal neurons (n = 23). Recordings made under current clamp revealed no statistical difference in neuronal resting membrane potential between control (−67.5 ± 1.0 mV) and CCI (−67.7 ± 0.9 mV) (P < 0.92) or input resistance (control = 198.5 ± 16.1 MΩ; CCI = 192.3 ± 12.9 MΩ, P < 0.76). Next, we evaluated the firing–current (f‐I) relationship in control and CCI animals. A series of current steps (−150 pA to 300 pa, 50 pA steps, 1 second) were injected through the patch pipette, and the membrane voltage response was recorded (Figure 3A). We examined for changes in the firing frequency and adaptation index but found no statistical difference (Figure 3B). Finally, using a rheobase protocol (50 mseconds, 5 pA steps), we examined for changes in membrane excitability. We found no statistical difference in rheobase current or action potential properties (threshold, amplitude, and half‐width) (Figure 4). Overall, these results suggest that a change in the intrinsic membrane excitability of layer V pyramidal neurons does not significantly contribute to the development of epileptiform activity early following CCI.
Figure 3.
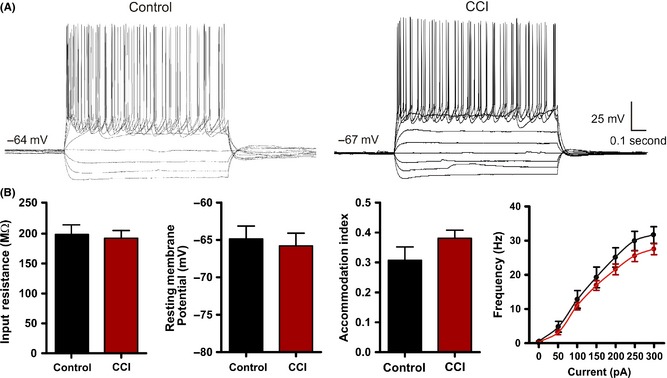
Intrinsic excitability is not altered by Controlled cortical impact (CCI). (A) Current‐clamp recordings in response to intracellular current steps (−150 pA to 250 pA, 1 second) in pyramidal neurons from control or CCI‐injured animals. Note the similarity in the intrinsic cellular response. (B) Bar charts of average response values of various intrinsic membrane properties from control (n = 23) and CCI (n = 35). No statistical difference was found for input resistance (P = 0.76), resting membrane potential (P = 0.92), accommodation index (P = 0.23) or firing frequency.
Figure 4.
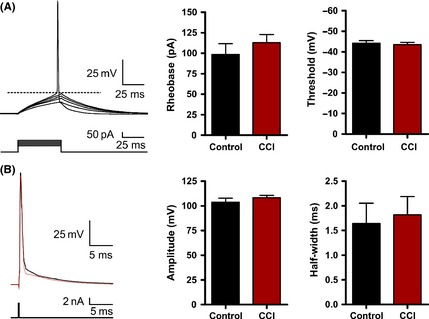
Action potential firing is not altered by Controlled cortical impact (CCI). (A) Representative whole‐cell current‐clamp recording in response to a series of 50 mseconds injection (5 pA steps). Bar charts of average values for control or CCI. Rheobase was calculated as the minimum current that produced an action potential. Threshold was measured at the greatest change in calculated slope. (B) Current‐clamp single action potential step (2 nA, 0.5 mseconds) was injected to measure action potential properties. No statistical difference was found for action potential threshold (P = 0.51), amplitude (P = 0.33), or half‐width (P = 0.80).
Spontaneous Synaptic Activity
The generation of epileptiform activity and seizures is thought to occur through altered network activity and increased neuronal recruitment that may involve changes to synaptic properties and efficacy 50. We tested this possibility by examining for changes in postsynaptic currents received by layer V pyramidal neurons.
Excitation
First, under voltage clamp (V hold = −70 mV), we examined for changes in spontaneous postsynaptic currents. For these experiments, pharmacological isolation of excitatory glutamatergic postsynaptic currents (EPSCs) was not possible as GABA antagonists are known to disinhibit the slice and may promote epileptiform activity, and thereby mask CCI‐induced changes. To minimize the detection of inhibitory events, neurons were held near and positive of the reversal potential of chloride (calculated ECl− = −80 mV). This allowed detection of only excitatory positive‐directed (inward current) events in isolation from any small inhibitory (outward current) events. At the end of some sPSC recordings, these events were confirmed to be glutamatergic as they were blocked by bath application of glutamate receptor antagonists (n = 5). Using this approach, we examined for changes in synaptic properties between neurons recorded from control (n = 20) and CCI (n = 24) animals. Specifically, the interevent interval (i.e., frequency) of excitatory sEPSCs was not statistically different between control (295.6 ± 42.9 mseconds) and CCI (243.2 ± 26.7) (P = 0.28). Similarly, we found no statistical difference between control and CCI in the amplitude of sEPSCs (control = 16.8 ± 1.2 pA; CCI = 17.4 ± 1.4 pA, P = 0.75), charge transfer (control = 69.5 ± 7.0 fC; CCI = 73.1 ± 5.4 fC, P = 0.70), or decay (control = 3.6 ± 0.2 mseconds; CCI = 3.5 ± 0.2 mseconds, P = 0.77) (Figure 5). The data suggest that CCI does not alter average excitatory synaptic activity.
Figure 5.
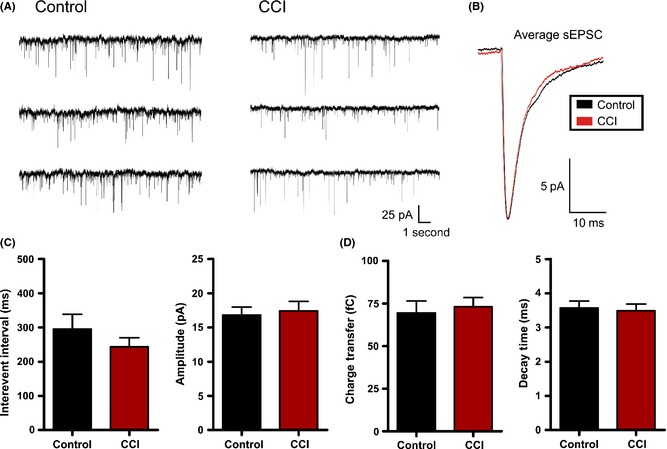
Controlled cortical impact (CCI) fails to alter excitatory postsynaptic currents. (A) Voltage‐clamp recordings of spontaneous postsynaptic current (sEPSC) in control or CCI‐injured animals. (B) Overlayed and amplitude‐scaled average sEPSC recorded from control (black) and CCI (red) animals. (C) and (D) Average sEPSC properties are plotted for control (n = 20) and CCI (n = 24). No statistical difference was found for sEPSC properties including interevent interval (P = 0.28), amplitude (P = 0.75), charge transfer (P = 0.70) or decay (P = 0.77). Vhold = −70 mV.
Inhibition
To directly examine for changes in inhibition, we recorded spontaneous inhibitory postsynaptic currents (sIPSCs) that were pharmacologically isolated by bath application of the glutamate receptor antagonists d‐AP‐V (50 μM) and DNQX (20 μM). We also utilized a modified internal patch solution with an elevated chloride concentration. This internal has been extensively used 38, 54 and increases the signal‐to‐noise ratio and detection fidelity of inhibitory synaptic events. Voltage‐clamp recordings were made at −70 mV from control (n = 9) or CCI (n = 16) neurons (Figure 6). We found no significant change in amplitude (control = 24.6 ± 2.4 pA; CCI 24.9 ± 3.3 pA, P = 0.95) or interevent interval (control = 385.1 ± 85.9 mseconds; CCI 328.2 ± 50.4 mseconds, P = 0.55). However, in contrast to excitatory synaptic activity, a significant increase in the decay time of inhibitory sIPSCs was observed (control = 5.2 ± 0.56 mseconds; CCI = 7.5 ± 0.70 mseconds, P < 0.03). A similar trend was observed in the charge transfer but it failed to reach statistical significance (control = 126.1 ± 17.6 fC; CCI = 185.1 ± 28.2 fC, P = 0.15) (Figure 6B). The net effect of these changes would be to increase the efficacy of inhibition following CCI by increasing the temporal window over which inhibition acts.
Figure 6.
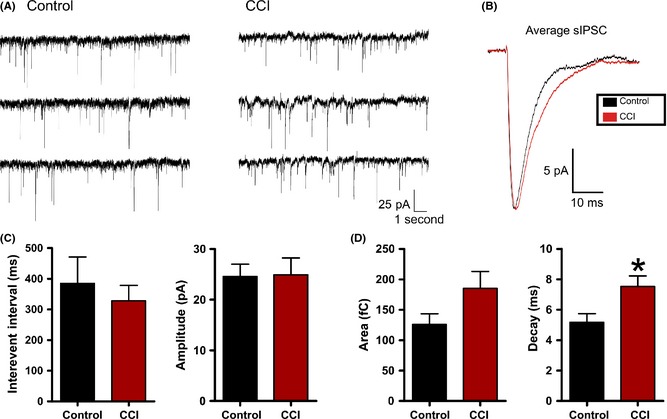
Controlled cortical impact (CCI) increases inhibitory synaptic decay. (A) Voltage‐clamp recordings of spontaneous inhibitory postsynaptic current (sIPSC) in control or CCI‐injured animals. For inhibitory recordings, glutamate receptor antagonists (APV/DNQX or kynurenate) were applied. (B) Overlayed and amplitude‐scaled average sIPSC recorded from control (black) and CCI (red) animals. (C) and (D) Average sIPSC properties for control (n = 9) or CCI (n = 16). No statistical difference was found for sIPSC interevent interval (P = 0.55), amplitude (P = 0.95) or charge transfer (P = 0.15). Vhold = −70 mV. *P < 0.05.
Synaptic Burst Discharges
Excitatory burst discharges are thought to increase synaptic efficacy by increasing the probability of inducing the postsynaptic cell to fire an action potential. In our initial experiments, we determined that the average sEPSC properties were not altered by CCI. However, during recording, we observed distinct spontaneous synaptic burst discharges that resembled the epileptiform activity observed in vivo. Based on previous reports 55, detection of synaptic bursts was determined by the presence of a minimum of three simultaneous sEPSCs within 250 mseconds that did not return to baseline. Synaptic bursts were then detected over a 5‐min period of sEPSC recording from control or CCI animals. These detection parameters were sensitive and allowed for detection of even a small number of synaptic bursts in a few control animals. At a single cell level, pyramidal neurons from CCI were 344% more likely to exhibit bursting (control = 23.1%; CCI = 79.5%). The average excitatory burst frequency in a neuron was also dramatically increased following CCI (1.575 ± 0.37 bursts/min) compared with control (0.050 ± 0.03) (P < 0.01). The average excitatory sEPSC burst in CCI animals consisted of 5.9 ± 1 synaptic events and lasted on average for 858.0 ± 240 milliseconds (Figure 7). Bath application of the Na+ channel blocker tetrodotoxin (TTX, 1 μM, n = 4) or the glutamate receptor antagonist kynurenate (2 mM, n = 5) eliminated excitatory burst discharges. Overall, these excitatory synaptic bursts represented <3.76% of all detected synaptic events (Figure 7B) but are thought to have a profound impact on synaptic coupling. On the inhibitory side, layer V pyramidal neurons were 340% more likely to exhibit inhibitory synaptic bursting following CCI (control = 22%; CCI = 75%). The average inhibitory neuronal burst frequency was also significantly increased following CCI (control = 0.111 ± 0.08 bursts/min; CCI = 0.700 ± 0.21 bursts/min, P < 0.05). The average inhibitory sIPSC burst in CCI animals consisted of 11.4 ± 2.6 synaptic events with a duration of 431.0 ± 71 milliseconds (Figure 8A). Again bath application of TTX eliminated inhibitory burst discharges (n = 5). On average, inhibitory synaptic bursting accounted for <2.71% of all detected synaptic events (Figure 8B). In comparison, CCI induced greater excitatory bursting than inhibitory bursting by both frequency of total neurons that displayed bursting and the average number of bursts per neuron. This suggests CCI may be preferentially increasing excitatory bursting, and thereby promote synaptic coupling.
Figure 7.
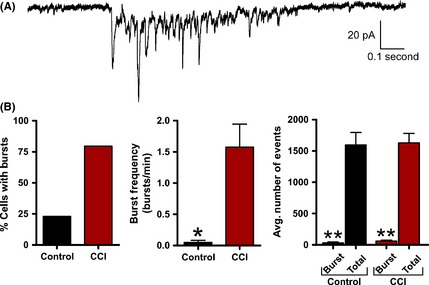
Excitatory synaptic bursts are induced by controlled cortical impact (CCI). (A) Voltage‐clamp recording of spontaneous excitatory burst discharge observed in a CCI animal with an epileptiform EEG. Note the burst is comprised of compound sEPSCs and resembles paroxysmal discharges observed in epileptic animals. (B) Bar charts of average values of various burst properties for control (n = 20) or CCI (n = 32). *P < 0.05, **P < 0.01.
Figure 8.
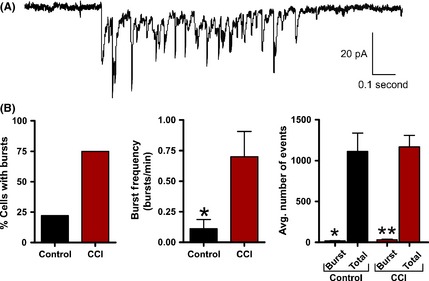
Inhibitory synaptic bursts are induced by controlled cortical impact (CCI). (A) Voltage‐clamp recording of spontaneous inhibitory burst discharge observed in a CCI animal with an epileptiform EEG. For inhibitory recording glutamate receptor antagonists (APV/DNQX or kynurenate) were applied. Note the burst is comprised of compound sIPSCs and resembles epileptiform discharges observed in epileptic animals. (B) Bar charts of average values of various burst properties for control (n = 9) or CCI (n = 16). *P < 0.05, **P < 0.01.
Discussion
In the weeks and months that follow after CCI, up to 36% of adult animals will develop spontaneous behavioral seizures 18 and over 85% have been shown to develop epileptiform activity 28. In humans, PTE develops following a latent period that can last from months to years 56. At the point in which seizures are clinically observable, the underlying neural activity and networks have undergone significant change. We believe this activity begins early after the injury and leads to hyperexcitability and subclinical electrographic changes well in advance of PTE. CCI in rodents has been effectively used to model TBI 13, 18, 57. However, these studies have primarily focused on CCI performed in adult animals. The outcome, incidence, and clinical management of TBI in children differ significantly from adults. We examined the development of epileptiform activity and the underlying pathophysiology that occurs in juvenile (P17) rats following CCI. The results of this study indicate that within 14 days of CCI injury epileptiform activity is induced that can be detected in vivo by EEG as synchronous discharges across multiple cortical regions. At a cellular and synaptic level, this epileptiform activity was accompanied by a lack of change in intrinsic membrane properties but a 44% increase in the decay of inhibitory synaptic input onto layer V pyramidal neurons. In addition, spontaneous synaptic bursting was significantly increased in both excitatory and inhibitory recordings following CCI. These mechanisms have not been reported following CCI in adult animals and may be unique to the response of juvenile animals to TBI.
Development of Epileptiform Activity Following CCI in Juvenile Rats
The hallmark of PTE is the development of spontaneous recurrent seizures. In humans, these seizures develop months to years following the initial injury 56. The progressive development of PTE suggests an evolving process that may begin early after injury. The seizures seen during the first 24 h of postinjury EEG recordings are thought to be injury induced and separate from the underlying epileptogenic processes that lead to PTE 27, 58, 59. Following a variable latent period, all CCI animals proceeded to develop spontaneous recurrent epileptiform activity by 14 days postinjury. Epileptiform activity and late seizures (i.e., that develop after the first week of injury) are known to be positive predictors for PTE, disease severity and outcome 60. Further, work recording EEG continuously for several months will be required to determine the prognostic value of the observed immediate and late epileptiform activity detected in this study. The study of PTE is complicated by the presence of multiple injury, repair, and adaptive processes initiated by the TBI—only a portion of which are presumed to be epileptogenic. Examination of animals early after injury at a time point when “late” recurrent epileptic activity has just begun has a potential reductionist advantage. Determining early pathophysiological changes within this period may help define a critical window or novel targets for therapeutic intervention. We have now validated that 14 days after injury is the earliest time point after CCI where juvenile animals reliably display in vivo and in vitro epileptiform activity.
Epileptogenesis has been extensively studied in numerous animal models and may result from a variety of mechanisms. While no common epileptogenic mechanism has been found, a combination of disruption to intrinsic cellular properties, synaptic inhibition, and/or synaptic excitation has been frequently reported 50. It has yet to be determined the impact of CCI in juvenile animals on intrinsic and synaptic changes that are thought to be epileptogenic. Overall, our results indicate that CCI fails to alter intrinsic membrane properties, neuronal firing or average excitatory synaptic activity while promoting burst discharges and enhanced inhibitory synaptic decay. Specifically, the intrinsic excitability of a neuron is determined in large part by its membrane properties and ion channels and when intrinsic excitability is enhanced it is commonly epileptogenic. However, following CCI in juvenile animals layer V pyramidal neurons displayed no change in intrinsic excitability. This included resting membrane potential, input resistance, action potential threshold, and rheobase. Similarly, there was no change in the firing properties (frequency or accommodation), input–output relationship (f‐I curve) or single action potential waveform. Together, these results suggest that alterations to intrinsic excitability in layer V pyramidal neurons do not significantly contribute to the observed development of epileptiform activity following CCI.
At a synaptic level, our results indicate that the average excitatory synaptic input onto layer V pyramidal neurons was not altered. There was no change in the amplitude, interevent interval or kinetics of excitatory spontaneous postsynaptic currents. The lack of change in sEPSCs after CCI suggests that altered properties of excitatory synapses onto layer V pyramidal neurons are not driving the development of epileptiform activity. In examining inhibitory drive onto excitatory neurons in the cortex, we similarly found no change in the amplitude or interevent interval of spontaneous inhibitory postsynaptic currents. Our finding of an increase in sIPSC decay is consistent with similar increases observed in other models of epilepsy 61. In general, an increase in synaptic decay is predicted to counteract the observed hyperexcitability by increasing the temporal integration window but may also be impacted by neural trauma‐induced changes in the chloride reversal potential 62. Altered intracellular chloride may also impact the kinetics of chloride‐dependent GABAergic inhibition 63 and would be in line with the observed CCI‐induced changes. Determining the relative role of increased synaptic decay in promoting or resisting epileptic changes following CCI remains an open question.
Development of Synaptic Bursting Following CCI in Juvenile Rats
The development of epilepsy is commonly associated with the synchronous discharge of neurons. In this study, we have identified unique epileptiform burst discharges following CCI. Excitatory burst firing is known to increase the fidelity of synaptic information transfer 64, 65 and may help to promote epilepsy by facilitating the propagation of local areas of hyperexcitability and synchrony. As no changes were observed in average excitatory synaptic IEI, it suggests the synaptic bursting is not due to altered presynaptic probability of release. The significant increase of synaptic bursts following CCI may initially appear at odds with a lack of change to synaptic properties (i.e., interevent interval). However, even after CCI, synaptic bursts represented <4% of total synaptic events. Therefore, the impact of synaptic burst events on synaptic properties may be diluted in standard analysis methods that average across all synaptic events. The data from this study highlight the need for discrete detection and analysis of synaptic bursting. Outside of effects on average synaptic properties it remains possible that CCI induces more targeted alterations to specific cell‐types or synapses.
In this study, it was not possible to isolate the source of the driver of synaptic bursts. As the bursts were sensitive to blockade with TTX, it suggests they are being driven by alterations to action potential‐dependent afferent input. Layer V pyramidal neurons predominantly receive input from all other cortical layers as well as from thalamus and are implicated in synchronization of cortical activity 51, 66, 67. Controlled cortical impact (CCI) induces a severe TBI that has been shown to alter cortex as well as other subcortical structures 17, 29, 68. Therefore, the data suggest that excitatory neurons projecting to layer V cortex have been altered by CCI to drive spontaneous burst discharges. The synchronous spontaneous epileptiform activity that was observed on EEG across cortical regions and hemispheres (Figure 1) suggests network recruitment and widespread propagation of the epileptic activity. The increase in excitatory burst discharges may promote epileptiform activity by increasing synaptic coupling onto layer V pyramidal neurons that are perfectly situated to increase cortical output and network synchrony. Determining if the synaptic bursting is specific to layer V pyramidal neurons, the location of the afferent driver of the epileptiform activity, and the specific contribution of layer V changes to in vivo epileptiform activity are areas of current investigation. In addition to excitatory bursting, distinct inhibitory bursting was similarly observed. Inhibitory bursting persisted in the presence of glutamatergic blockade (AP‐V and DNQX) suggesting inhibitory bursting is most likely intrinsically generated within inhibitory interneurons. This is in contrast to our findings on excitatory bursting and may reflect unique TBI‐induced changes to inhibitory interneurons.
Pediatric Traumatic Brain Injury
Traumatic brain injury that occurs in children differs from adults with a reduced mortality rate 69, increased incidence of skull fractures and epidural hematomas 70 and greater deficits in cognitive and behavioral functioning 71, 72. In this study, we have begun examining if the pathophysiology of TBI in children is unique. The pediatric brain is in the midst of a significant period of neurodevelopment and a host of age‐dependent physiological and neurobiological changes including cortical hypertrophy, synaptogenesis, use‐dependent pruning, enhanced glucose metabolism 73, 74, 75, increased neurotrophic factors 76, and altered excitatory amino acid receptors 77, 78. These changes may confer unique advantages and disadvantages to the outcome of a pediatric TBI event and shape the development of PTE. Injury in the brain of pediatric patients has been generally thought to result in an improved outcome yet evidence suggests that children may be particularly and uniquely sensitive to the effects of TBI 79, 80, 81. Experimentally, recent reports on TBI in adult animals have indicated a loss of inhibition and an increase in glutamatergic synaptic activity 13. These findings suggest a direct increase in cortical synaptic excitability that was not observed in our study of juvenile animals. The potential for unique mechanisms in adults versus children suffering from TBI have important implications to developing therapeutic strategies and directing the clinical management of patients.
In conclusion, CCI induced epileptiform activity and distinct synaptic bursting within 14 days of injury without altering the intrinsic properties of layer V pyramidal neurons. The development of epileptiform activity early after injury may be the first step “on the road” to PTE. Understanding how TBI alters cortical excitability early after injury may help define therapeutic targets and a critical window of intervention.
Conflict of Interest
The authors declare no conflicts of interest.
References
- 1. Annegers JF, Hauser WA, Coan SP, Rocca WA. A population‐based study of seizures after traumatic brain injuries. N Engl J Med 1998;338:20–24. [DOI] [PubMed] [Google Scholar]
- 2. Caveness WF, Meirowsky AM, Rish BL, et al. The nature of posttraumatic epilepsy. J Neurosurg 1979;50:545–553. [DOI] [PubMed] [Google Scholar]
- 3. Englander J, Bushnik T, Duong TT, et al. Analyzing risk factors for late posttraumatic seizures: A prospective, multicenter investigation. Arch Phys Med Rehabil 2003;84:365–373. [DOI] [PubMed] [Google Scholar]
- 4. Iudice A, Murri L. Pharmacological prophylaxis of post‐traumatic epilepsy. Drugs 2000;59:1091–1099. [DOI] [PubMed] [Google Scholar]
- 5. Appleton RE, Demellweek C. Post‐traumatic epilepsy in children requiring inpatient rehabilitation following head injury. J Neurol Neurosurg Psychiatry 2002;72:669–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barlow KM, Spowart JJ, Minns RA. Early posttraumatic seizures in non‐accidental head injury: Relation to outcome. Dev Med Child Neurol 2000;42:591–594. [DOI] [PubMed] [Google Scholar]
- 7. Evans RW. Neurology and Trauma, 2nd edn New York: Oxford University Press, 2006. [Google Scholar]
- 8. Bauer J, Burr W. Course of chronic focal epilepsy resistant to anticonvulsant treatment. Seizure 2001;10:239–246. [DOI] [PubMed] [Google Scholar]
- 9. Adelson PD, Bratton SL, Carney NA, et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents. Chapter 19. The role of anti‐seizure prophylaxis following severe pediatric traumatic brain injury. Pediatr Crit Care Med 2003;4:S72–S75. [PubMed] [Google Scholar]
- 10. Arango JI, Deibert CP, Brown D, Bell M, Dvorchik I, Adelson PD. Posttraumatic seizures in children with severe traumatic brain injury. Childs Nerv Syst 2012;28:1925–1929. [DOI] [PubMed] [Google Scholar]
- 11. Kochanek PM, Carney N, Adelson PD, et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents–second edition. Pediatr Crit Care Med 2012;13(Suppl 1):S1–S82. [DOI] [PubMed] [Google Scholar]
- 12. Graber KD, Prince DA. A critical period for prevention of posttraumatic neocortical hyperexcitability in rats. Ann Neurol 2004;55:860–870. [DOI] [PubMed] [Google Scholar]
- 13. Cantu D, Walker K, Andresen L, et al. Traumatic brain injury increases cortical glutamate network activity by compromising GABAergic control. Cereb Cortex 2014. doi: 10.1093/cercor/bhu041. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lighthall JW, Dixon CE, Anderson TE. Experimental models of brain injury. J Neurotrauma 1989;6:83–97. [DOI] [PubMed] [Google Scholar]
- 15. Liu N‐K, Zhang Y‐P, O'Connor J, et al. A bilateral head injury that shows graded brain damage and behavioral deficits in adultmice. Brain Res 2013;1499:121–128. [DOI] [PubMed] [Google Scholar]
- 16. Mannix RC, Zhang J, Park J, Lee C, Whalen MJ. Detrimental effect of genetic inhibition of B‐site APP‐cleaving enzyme 1 on functional outcome after controlled cortical impact in young adult mice. J Neurotrauma 2011;28:1855–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adelson PD, Fellows‐Mayle W, Kochanek PM, Dixon CE. Morris water maze function and histologic characterization of two age‐at‐injury experimental models of controlled cortical impact in the immature rat. Childs Nerv Syst 2013;29:43–53. [DOI] [PubMed] [Google Scholar]
- 18. Hunt RF, Scheff SW, Smith BN. Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp Neurol 2009;215:243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang L, Afroz S, Michelson HB, Goodman JH, Valsamis HA, Ling DSF. Spontaneous epileptiform activity in rat neocortex after controlled cortical impact injury. J Neurotrauma 2010;27:1541–1548. [DOI] [PubMed] [Google Scholar]
- 20. Anderson KJ, Miller KM, Fugaccia I, Scheff SW. Regional distribution of Fluoro‐Jade B staining in the hippocampus following traumatic brain injury. Exp Neurol 2005;193:125–130. [DOI] [PubMed] [Google Scholar]
- 21. Fox G, Fan L, Levasseur RA, Faden AI. Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J Neurotrauma 1998;15:599–614. [DOI] [PubMed] [Google Scholar]
- 22. Goodman JC, Cherian L, Bryan RM, Robertson CS. Lateral cortical impact injury in rats: Pathologic effects of varying cortical compression and impact velocity. J Neurotrauma 1994;11:587–597. [DOI] [PubMed] [Google Scholar]
- 23. Hall ED, Sullivan PG, Gibson TR, Pavel KM, Thompson BM, Scheff SW. Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: More than a focal brain injury. J Neurotrauma 2005;22:252–265. [DOI] [PubMed] [Google Scholar]
- 24. Smith DH, Soares HD, Pierce JS, et al. A model of parasagittal controlled cortical impact in the mouse: Cognitive and histopathologic effects. J Neurotrauma 1995;12:169–178. [DOI] [PubMed] [Google Scholar]
- 25. Rola R, Mizumatsu S, Otsuka S, et al. Alterations in hippocampal neurogenesis following traumatic brain injury in mice. Exp Neurol 2006;202:189–199. [DOI] [PubMed] [Google Scholar]
- 26. Scheff SW, Price DA, Hicks RR, Baldwin SA, Robinson S, Brackney C. Synaptogenesis in the Hippocampal CA1 field following traumatic brain injury. J Neurotrauma 2005;22:719–732. [DOI] [PubMed] [Google Scholar]
- 27. Nilsson P, Ronne‐Engström E, Flink R, Ungerstedt U, Carlson H, Hillered L. Epileptic seizure activity in the acute phase following cortical impact trauma in rat. Brain Res 1994;637:227–232. [DOI] [PubMed] [Google Scholar]
- 28. Statler KD, Scheerlinck P, Pouliot W, Hamilton M, White HS, Dudek FE. A potential model of pediatric posttraumatic epilepsy. Epilepsy Res 2009;86:221–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Card JP, Santone DJ, Gluhovsky MY, Adelson PD. Plastic reorganization of hippocampal and neocortical circuitry in experimental traumatic brain injury in the immature rat. J Neurotrauma 2005;22:989–1002. [DOI] [PubMed] [Google Scholar]
- 30. Jenkins LW, Peters GW, Dixon CE, et al. Conventional and functional proteomics using large format two‐dimensional gel electrophoresis 24 hours after controlled cortical impact in postnatal day 17 rats. J Neurotrauma 2002;19:715–740. [DOI] [PubMed] [Google Scholar]
- 31. Hoffman SN, Salin PA, Prince DA. Chronic neocortical epileptogenesis in vitro. J Neurophysiol 1994;71:1762–1773. [DOI] [PubMed] [Google Scholar]
- 32. Prince DA, Tseng GF. Epileptogenesis in chronically injured cortex: In vitro studies. J Neurophysiol 1993;69:1276–1291. [DOI] [PubMed] [Google Scholar]
- 33. Cole JT, Yarnell A, Kean WS, et al. Craniotomy: True sham for traumatic brain injury, or a sham of a sham? J Neurotrauma 2011;28:359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Olesen SP. Leakiness of rat brain microvessels to fluorescent probes following craniotomy. Acta Physiol Scand 1987;130:63–68. [DOI] [PubMed] [Google Scholar]
- 35. Ziyatdinova S, Gurevicius K, Kutchiashvili N, et al. Spontaneous epileptiform discharges in a mouse model of Alzheimer's disease are suppressed by antiepileptic drugs that block sodium channels. Epilepsy Res 2011;94:75–85. [DOI] [PubMed] [Google Scholar]
- 36. Horita H, Uchida E, Maekawa K. Circadian rhythm of regular spike‐wave discharges in childhood absence epilepsy. Brain Dev 1991;13:200–202. [DOI] [PubMed] [Google Scholar]
- 37. Anderson TR, Jarvis CR, Biedermann AJ, Molnar C, Andrew RD. Blocking the anoxic depolarization protects without functional compromise following simulated stroke in cortical brain slices. J Neurophysiol 2005;93:963–979. [DOI] [PubMed] [Google Scholar]
- 38. Anderson TR, Huguenard JR, Prince DA. Differential effects of Na+‐K+ ATPase blockade on cortical layer V neurons. J Physiol 2010;588:4401–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guatteo E, Bacci A, Franceschetti S, Avanzini G, Wanke E. Neurons dissociated from neocortex fire with “burst” and “regular” trains of spikes. Neurosci Lett 1994;175:117–120. [DOI] [PubMed] [Google Scholar]
- 40. Sun Q‐Q, Huguenard JR, Prince DA. Barrel cortex microcircuits: Thalamocortical feedforward inhibition in spiny stellate cells is mediated by a small number of fast‐spiking interneurons. J Neurosci 2006;26:1219–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gajda Z, Hermesz E, Gyengési E, Szupera Z, Szente M. The functional significance of gap junction channels in the epileptogenicity and seizure susceptibility of juvenile rats. Epilepsia 2006;47:1009–1022. [DOI] [PubMed] [Google Scholar]
- 42. Sato H, Sakamoto Y, Kamei C, Shimizu M. EEG activity changes in juvenile rats chronically treated with phenobarbital. Folia Psychiatr Neurol Jpn 1979;33:323–327. [DOI] [PubMed] [Google Scholar]
- 43. Zayachkivsky A, Lehmkuhle MJ, Fisher JH, Ekstrand JJ, Dudek FE. Recording EEG in immature rats with a novel miniature telemetry system. J Neurophysiol 2013;109:900–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Arndt DH, Lerner JT, Matsumoto JH, et al. Subclinical early posttraumatic seizures detected by continuous EEG monitoring in a consecutive pediatric cohort. Epilepsia 2013;54:1780–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vespa PM, Miller C, McArthur D, Eliseo M, Etchepare M, Hirt D, et al. Nonconvulsive electrographic seizures after traumatic brain injury result in a delayed, prolonged increase in intracranial pressure and metabolic crisis. Crit Care Med 2007;35:2830–2836. [PMC free article] [PubMed] [Google Scholar]
- 46. Dias MS, Carnevale F, Li V. Immediate posttraumatic seizures: Is routine hospitalization necessary? Pediatr Neurosurg 1999;30:232–238. [DOI] [PubMed] [Google Scholar]
- 47. Holmes JF, Palchak MJ, Conklin MJ, Kuppermann N. Do children require hospitalization after immediate posttraumatic seizures? Ann Emerg Med 2004;43:706–710. [DOI] [PubMed] [Google Scholar]
- 48. Cuccurullo S. Physical Medicine and Rehabilitation Board Review, 2nd edn New York: Demos Medical Publishing, 2004. [Google Scholar]
- 49. Garga N, Lowenstein DH. Posttraumatic epilepsy: A major problem in desperate need of major advances. Epilepsy Curr 2006;6:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Prince DA, Connors BW. Mechanisms of interictal epileptogenesis. Adv Neurol 1986;44:275–299. [PubMed] [Google Scholar]
- 51. Telfeian AE, Connors BW. Layer‐specific pathways for the horizontal propagation of epileptiform discharges in neocortex. Epilepsia 1998;39:700–708. [DOI] [PubMed] [Google Scholar]
- 52. Willmore LJ. Post‐traumatic epilepsy: Cellular mechanisms and implications for treatment. Epilepsia 1990;31(Suppl 3):S67–S73. [DOI] [PubMed] [Google Scholar]
- 53. Yang L, Benardo LS, Valsamis H, Ling DSF. Acute injury to superficial cortex leads to a decrease in synaptic inhibition and increase in excitation in neocortical layer V pyramidal cells. J Neurophysiol 2007;97:178–187. [DOI] [PubMed] [Google Scholar]
- 54. Bacci A, Huguenard JR. Enhancement of spike‐timing precision by autaptic transmission in neocortical inhibitory interneurons. Neuron 2006;49:119–130. [DOI] [PubMed] [Google Scholar]
- 55. Gill R, Chang PK‐Y, Prenosil GA, Deane EC, McKinney RA. Blocking brain‐derived neurotrophic factor inhibits injury‐induced hyperexcitability of hippocampal CA3 neurons. Eur J Neurosci 2013;38:3554–3566. [DOI] [PubMed] [Google Scholar]
- 56. Agrawal A, Timothy J, Pandit L, Manju M. Post‐traumatic epilepsy: An overview. Clin Neurol Neurosurg 2006;108:433–439. [DOI] [PubMed] [Google Scholar]
- 57. Bolkvadze T, Pitkänen A. Development of post‐traumatic epilepsy after controlled cortical impact and lateral fluid‐percussion‐induced brain injury in the mouse. J Neurotrauma 2012;29:789–812. [DOI] [PubMed] [Google Scholar]
- 58. Chiaretti A, De Benedictis R, Polidori G, Piastra M, Iannelli A, Di Rocco C. Early post‐traumatic seizures in children with head injury. Childs Nerv Syst 2000;16:862–866. [DOI] [PubMed] [Google Scholar]
- 59. Yablon SA. Posttraumatic seizures. Arch Phys Med Rehabil 1993;74:983–1001. [PubMed] [Google Scholar]
- 60. Ramantani G. Neonatal epilepsy and underlying aetiology: To what extent do seizures and EEG abnormalities influence outcome? Epileptic Disord 2013;15:365–375. [DOI] [PubMed] [Google Scholar]
- 61. Calcagnotto ME, Paredes MF, Tihan T, Barbaro NM, Baraban SC. Dysfunction of synaptic inhibition in epilepsy associated with focal cortical dysplasia. J Neurosci 2005;25:9649–9657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Van den Pol AN, Obrietan K, Chen G. Excitatory actions of GABA after neuronal trauma. J Neurosci 1996;16:4283–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Houston CM, Bright DP, Sivilotti LG, Beato M, Smart TG. Intracellular chloride ions regulate the time course of GABA‐mediated inhibitory synaptic transmission. J Neurosci 2009;29:10416–10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Izhikevich EM, Desai NS, Walcott EC, Hoppensteadt FC. Bursts as a unit of neural information: Selective communication via resonance. Trends Neurosci 2003;26:161–167. [DOI] [PubMed] [Google Scholar]
- 65. Lisman JE. Bursts as a unit of neural information: Making unreliable synapses reliable. Trends Neurosci 1997;20:38–43. [DOI] [PubMed] [Google Scholar]
- 66. Peters A, Jones EG. Cerebral Cortex: Volume 1: Cellular Components of the Cerebral Cortex, 1st edn New York: Springer; 1984. [Google Scholar]
- 67. Wise SP. The laminar organization of certain afferent and efferent fiber systems in the rat somatosensory cortex. Brain Res 1975;90:139–142. [DOI] [PubMed] [Google Scholar]
- 68. Casella EM, Thomas TC, Vanino DL, et al. Traumatic brain injury alters long‐term hippocampal neuron morphology in juvenile, but not immature, rats. Childs Nerv Syst 2014;30:1333–1342. [DOI] [PubMed] [Google Scholar]
- 69. Luerssen TG, Klauber MR, Marshall LF. Outcome from head injury related to patient's age. A longitudinal prospective study of adult and pediatric head injury. J Neurosurg 1988;68:409–416. [DOI] [PubMed] [Google Scholar]
- 70. Sarkar K, Keachie K, Nguyen U, Muizelaar JP, Zwienenberg‐Lee M, Shahlaie K. Computed tomography characteristics in pediatric versus adult traumatic brain injury. J Neurosurg Pediatr 2014;13:307–314. [DOI] [PubMed] [Google Scholar]
- 71. Anderson V, Catroppa C, Morse S, Haritou F, Rosenfeld J. Functional plasticity or vulnerability after early brain injury? Pediatrics 2005;116:1374–1382. [DOI] [PubMed] [Google Scholar]
- 72. McKinlay A, Dalrymple‐Alford JC, Horwood LJ, Fergusson DM. Long term psychosocial outcomes after mild head injury in early childhood. J Neurol Neurosurg Psychiatry 2002;73:281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Adelson PD. Animal models of traumatic brain injury in the immature: A review. Exp Toxicol Pathol 1999;51:130–136. [DOI] [PubMed] [Google Scholar]
- 74. Chugani HT, Phelps ME, Mazziotta JC. Positron emission tomography study of human brain functional development. Ann Neurol 1987;22:487–497. [DOI] [PubMed] [Google Scholar]
- 75. Prins ML, Hales A, Reger M, Giza CC, Hovda DA. Repeat traumatic brain injury in the juvenile rat is associated with increased axonal injury and cognitive impairments. Dev Neurosci 2010;32:510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Friedman WJ, Olson L, Persson H. cells that express brain‐derived neurotrophic factor mRNA in the developing postnatal rat brain. Eur J Neurosci 1991;3:688–697. [DOI] [PubMed] [Google Scholar]
- 77. Giza CC, Maria NSS, Hovda DA. N‐methyl‐D‐aspartate receptor subunit changes after traumatic injury to the developing brain. J Neurotrauma 2006;23:950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Insel TR, Miller LP, Gelhard RE. The ontogeny of excitatory amino acid receptors in rat forebrain–I. N‐methyl‐D‐aspartate and quisqualate receptors. Neuroscience 1990;35:31–43. [DOI] [PubMed] [Google Scholar]
- 79. Anderson V, Moore C. Age at injury as a predictor of outcome following pediatric head injury: A longitudinal perspective. Child Neuropsychol 1995;1:187–202. [Google Scholar]
- 80. Ewing‐Cobbs L, Miner ME, Fletcher JM, Levin HS. Intellectual, motor, and language sequelae following closed head injury in infants and preschoolers. J Pediatr Psychol 1989;14:531–547. [DOI] [PubMed] [Google Scholar]
- 81. Taylor HG, Swartwout M, Yeates KO, Walz NC, Stancin T, Wade SL. Traumatic brain injury in young children: Post‐acute effects on cognitive and school readiness skills. J Int Neuropsychol Soc 2008;14:734–745. [DOI] [PMC free article] [PubMed] [Google Scholar]