

Abstract
General regulatory factors (GRFs), such as Reb1, Abf1, Rap1, Mcm1, and Cbf1, positionally organize yeast chromatin through interactions with a core consensus DNA sequence. It is assumed that sequence recognition via direct base readout suffices for specificity and that spurious nonfunctional sites are rendered inaccessible by chromatin. We tested these assumptions through genome-wide mapping of GRFs in vivo and in purified biochemical systems at near–base pair (bp) resolution using several ChIP-exo–based assays. We find that computationally predicted DNA shape features (e.g., minor groove width, helix twist, base roll, and propeller twist) that are not defined by a unique consensus sequence are embedded in the nonunique portions of GRF motifs and contribute critically to sequence-specific binding. This dual source specificity occurs at GRF sites in promoter regions where chromatin organization starts. Outside of promoter regions, strong consensus sites lack the shape component and consequently lack an intrinsic ability to bind cognate GRFs, without regard to influences from chromatin. However, sites having a weak consensus and low intrinsic affinity do exist in these regions but are rendered inaccessible in a chromatin environment. Thus, GRF site-specificity is achieved through integration of favorable DNA sequence and shape readouts in promoter regions and by chromatin-based exclusion from fortuitous weak sites within gene bodies. This study further revealed a severe G/C nucleotide cross-linking selectivity inherent in all formaldehyde-based ChIP assays, which includes ChIP-seq. However, for most tested proteins, G/C selectivity did not appreciably affect binding site detection, although it does place limits on the quantitativeness of occupancy levels.
Specificity for gene regulation originates from features within DNA that are selectively recognized by DNA binding proteins. How selectivity is achieved, particularly in the context of an entire genome, is not fully understood. Recent work has suggested that some proteins recognize both the sequence and shape of a DNA binding site, with shape not uniquely specified by sequence (Rohs et al. 2009; Slattery et al. 2014; Yang et al. 2017). DNA shape, as a component of site-specific recognition but distinct from nucleotide base recognition, has thus far had limited experimental investigation within the physiological context of an entire genome.
Here we utilize a variety of “ChIP-seq” DNA binding assays to investigate DNA sequence and shape contributions to genome-wide DNA binding specificity. This includes ChIP-exo which improves positional resolution through use of lambda exonuclease. The exonuclease trims each strand of DNA molecules in the 5′-3′ direction until it is stopped by the formaldehyde-induced protein-DNA cross-link (Rhee and Pugh 2011). To examine genome-wide protein-DNA in a purified in vitro system at high positional resolution, we developed an exonuclease version of PB-seq (protein binding with deep sequencing) called PB-exo. PB-seq was developed to capture the binding affinities for a protein across an entire genome in the absence of chromatin (Guertin et al. 2012). The highly defined nature of the system also allowed us to uncover and precisely define sequence-specificity in formaldehyde cross-linking, which is widely used in epigenome mapping.
Beyond direct DNA sequence readout, a less appreciated aspect of site-specificity is DNA shape readout. Although intrinsically related to DNA sequence, a particular DNA shape can arise from multiple possible arrangements of nucleotide bases that create a distinct shape of the sugar-phosphate backbone (Rohs et al. 2009; Slattery et al. 2014). Standard motif discovery methods, such as MEME (Bailey et al. 2009) and HOMER (Heinz et al. 2010), rely on position weight matrices and do not report on DNA shape. However, by combining DNA shape analysis with traditional position weight matrices (Yang et al. 2014), improved predictions of in vivo binding are achieved (Gordan et al. 2013; Zhou et al. 2015; Mathelier et al. 2016). The role of DNA shape and sequence are so intertwined that it has not been feasible to define a general role for DNA shape that is separate from DNA sequence readout (Abe et al. 2015). One attempt to do so (Zentner et al. 2015) was subsequently invalidated (Zentner et al. 2015; Rossi et al. 2017).
As important as site specificity is, proteins also need to avoid binding similar, fortuitous, or nonbiological sites. Evolution may have purged nonbiologically important sites from the genome, or at least reduced them to a level where any residual binding has little impact on biological fitness (Dermitzakis and Clark 2002; Moses et al. 2006). Additionally, other DNA binding proteins, most notably histones, may compete for binding and render intrinsically high affinity sites inaccessible (Guertin and Lis 2013). How these mechanisms play out across a genome has not been fully worked out because of the complexity of site-specific DNA binding mechanisms, many of which involve interactions with other proteins. Consequently, specificity may be distributed across broad regions of DNA, where any individual nucleotide position may contribute relatively little and thus tolerate degeneracy (Rhee and Pugh 2011).
To circumvent these issues, we chose to study a class of site-specific DNA binding proteins that possess a relatively tight consensus and do not require binding partners. These factors (Reb1, Abf1, Mcm1, Rap1, and Cbf1) organize nucleosomes and are referred to as general regulatory factors (GRFs) (Yu and Morse 1999; Yarragudi et al. 2004; Raisner et al. 2005; Badis et al. 2008; Hartley and Madhani 2009; Hughes and de Boer 2013). By directing nucleosome organization, GRFs help maintain nucleosome-free promoter regions (NFRs) (Badis et al. 2008; Hartley and Madhani 2009), thereby giving the transcription machinery access to the DNA. While GRF binding and their cognate sites are enriched within promoter NFRs (Rhee and Pugh 2011), thousands of additional seemingly equivalent motifs are not bound. They reside both within NFR regions and in nucleosome-encased gene bodies. While, in principle, other proteins might prevent binding, this premise has not been experimentally verified on a genomic scale.
Here we interrogate factor binding on a genomic scale using a variety of assays that probe distinct aspects of DNA binding and provide the first detailed investigation of ChIP cross-linking bias. We examine site selection preferences defined in vivo with intrinsic preferences defined in vitro in a manner that identifies distinct shape and sequence readouts of the DNA. At the most fundamental level, this work addresses why site-specific DNA recognition in vivo may differ from predicted binding or from binding in an isolated in vitro system.
Results
Description and application of PB-exo to Reb1
In developing PB-exo as an in vitro version of ChIP-exo (Supplemental Fig. S1A), purified sheared genomic DNA was incubated with purified GRFs. Binding was trapped by formaldehyde cross-linking, then treated according to our standard ChIP-exo protocol (Rhee and Pugh 2012). We examined five GRFs (Reb1, Abf1, Mcm1, Rap1, and Cbf1) and one phosphate starvation-response transcription factor (Pho4).
We initially validated in vitro PB-exo by examining Reb1 at the previously-defined 975 primary TTACCCK Reb1 binding sites (Rhee and Pugh 2011) and comparing to in vivo ChIP-exo. Like ChIP-exo, lambda exonuclease digestion in PB-exo improved upon PB-seq by concentrating the signal at cognate binding sites (Fig. 1A,B). Exonuclease stop sites appeared as DNA strand-specific peak patterning when viewed in composite (Fig. 1B) and allowed for the near-bp identification of protein–DNA cross-linking points. The precise locations of exonuclease stop sites were essentially identical between ChIP-exo and PB-exo, although they were quantitatively different in relative tag counts. Four primary cross-linking points were identified at −18, −10, +5, and +9 bp relative to the Reb1 motif midpoint. We interpret differences in peak intensities between ChIP-exo and PB-exo (red frame in Fig. 1B) to reflect differences in formaldehyde-accessible contacts in vivo compared to in vitro binding with pure protein.
Figure 1.

Genome-wide in vitro binding of Reb1. (A) Heat maps comparing ChIP-exo and PB-exo at 975 TTACCCK Reb1 primary sites (rows) (Rhee and Pugh 2011). Distances are from the underlined motif reference point. ChIP-exo of strain BY4741 shows background, with rows linked to the Reb1 ChIP-exo sort. In vitro PB-exo was sorted independently. Blue indicates tag 5′ ends located on the same strand as the motif, whereas red are located on the opposite strand. (B) Composite of tag 5′ ends for ChIP-exo (green) and PB-exo (purple) of Reb1 at 975 primary sites. Density above the x-axis represents tags on the motif strand, whereas opposite strand density is inverted below the x-axis. The orange hashtags represent prominent cross-linking points calculated by pairing adjacent peaks above and below the x-axis. Dashed black lines represent peaks that are common in ChIP-exo and PB-exo. Dashed green and purple lines represent the peaks that are enriched in ChIP-exo and PB-exo, respectively, and are highlighted by the red box. The blue brackets highlight the “shoulder” regions that contain higher cross-linking in the ChIP-exo samples. (C) Composite of tag 5′ ends for ChIP-exo (green) and WhIP-exo (blue) of Reb1 at Reb1 primary sites. Annotation descriptions and the ChIP-exo trace are the same as in B. (D) Composite plots of nucleosome midpoints generated by MNase H3 ChIP-seq at different groups of Reb1 motif occurrences located in promoters. (E) Relative occupancy at sites detected in both ChIP-exo and PB-exo assays (+/+) versus sites detected only by PB-exo (−/+) and the percentage of those sites located in ORFs for all proteins in this study (except Abf1). Abf1 was excluded because its G/C cross-linking bias made for a potentially misleading comparison. The 25th, 50th, and 75th percentiles are marked. The proteins are arranged, left to right, by their propensity to cause nucleosome depletion (Kaplan et al. 2009). (F) Composite plots of nucleosome midpoints generated by MNase H3 ChIP-seq at different groups of Reb1 motif occurrences located in ORFs.
To further investigate this, we replaced purified protein with a crude whole-cell extract (termed WhIP-exo), which provided a complex source of Reb1 and other factors in an in vitro context. Importantly, libraries were not formed when exogenous DNA was omitted, indicating chromatin was not contaminating the extract. The WhIP-exo pattern with Reb1 contained characteristics of both ChIP-exo and PB-exo (Fig. 1C), indicating that cellular factors (as opposed to technical aspects of the assay) modulate Reb1/DNA interactions. As such, we concluded that in moving from a factor-rich to a factor-depleted system (ChIP-exo → WhIP-exo → PB-exo), site-specific protein-DNA interactions were altered that are manifested in quantitative and qualitative changes in the exonuclease stop sites.
Promiscuous sites-specific binding on nucleosome-free DNA
Abf1, Mcm1, Rap1, Cbf1, and Pho4 were assayed by PB-exo and ChIP-exo and analyzed in the same manner as Reb1. As discussed throughout, for all six proteins, the vast majority of binding events identified in ChIP-exo were also detected by PB-exo. These events were concentrated in promoter regions at the center of NFRs (defined by MNase H3 ChIP-seq) (e.g., Fig. 1D and Supplemental Fig. S1B–F). Additionally, PB-exo identified hundreds of unique “in vitro-only” binding events not observed by ChIP-exo for all proteins except Abf1 (a technical reason discussed below explains this exception). The PB-exo tag counts at in vitro-only bound sites were significantly lower than sites bound in both ChIP-exo and PB-exo (Fig. 1E, +/+ vs. −/+ bars), indicating that they likely represent weak/promiscuous site-specific binding. Most in vitro-only sites were located in ORFs (Fig. 1E, % ORF sites), where they presumably are occluded in vivo by chromatin (Fig. 1F). Indeed, even those in vitro-only binding events within promoters were also nucleosomal in vivo (Fig. 1D). Thus, GRFs like Reb1 have an intrinsic ability to bind weak promiscuous sites when they are not normally occupied by chromatin.
G/C specificity of formaldehyde cross-linking in ChIP
For one protein, Abf1, we detected relatively fewer in vitro interactions using PB-exo, despite a multitude of sites being detected in vivo (Supplemental Fig. S2A). Abf1 is an essential S. cerevisiae chromatin organizer (Rhode et al. 1989), with a simple PB-exo cross-linking pattern. Major opposite-strand exonuclease stops were detected at +4 and +16 relative to the motif midpoint (Fig. 2A,B), reflecting a single major point of cross-linking. Among the 913 Abf1 motif occurrences located in promoters, only 130 (14%) were detectably bound by Abf1 in vitro, of which 121 (93%) were also identified by ChIP-exo (Supplemental Fig. S2A). An additional 102 sites were detected only in vivo by ChIP-exo (discussed below). Like Reb1, we observed that the distribution pattern of cross-links was not entirely identical between in vivo ChIP-exo and in vitro PB-exo (Fig. 2B), which further validated our findings with Reb1 that other factors affect binding interactions in vivo. Two additional minor cross-links were detected in vivo, upstream (more 5′) of the Abf1 motif, which may reflect additional in vivo interactions that are not occurring in the purified system.
Figure 2.

Genome-wide in vitro Abf1 binding reveals formaldehyde G/C specificity. (A) The left panel shows a four-color plot representation of 30-bp sequences centered on the motif midpoint. Each row represents a motif occurrence that passed our FIMO threshold. The black arrow points to the calculated cross-linking point. The remaining panels show, for each assay, tag 5′ ends distributed around motif occurrences located in promoters and sorted first by PB-exo, then ChIP-exo, and finally native PB-seq tag counts. Rows are linked across all data sets. The horizontal dashed line demarcates our threshold for binding in at least one assay. Sites with tags below the dashed line were not considered bound, because the tags generally did not form peak pairs or were not particularly enriched above background. (B) Tag counts for ChIP-exo (green) and PB-exo (purple) for Abf1 at Abf1 motif occurrences. (C) MEME logos obtained from the top 500 peak-pairs from each assay. The orange hashtag represents the calculated cross-linking point. (D) Left panel, frequency of G/C within 30-bp sequences centered on the Abf1 motif midpoint for the top 100 sites bound in PB-exo (purple), the top 100 remaining sites bound in ChIP-exo but not PB-exo (blue), and the top 100 remaining sites bound in native PB-seq but not the other two assays (orange). Blue asterisks highlight alternate cross-linking sites observed in ChIP-exo. Frequencies occurring within the Abf1 motif were not plotted. The dashed black line indicates the background G/C content. Right panel, four-color plot representation of A/T (red) or G/C (green) for sequences centered on the motif reference point. Colored boxes represent groups used in the left panel.
The number of expected Abf1 binding sites (913), based on motif occurrence, far exceeded the 130 bound sites identified in vitro. We therefore investigated whether a technical aspect of the assay was underreporting the number of Abf1-bound locations (false negatives). To this end, we performed “native PB-seq” (Guertin et al. 2012), which is a simplified genome-wide assay that forgoes formaldehyde cross-linking and exonuclease digestion. This less stringent immunoprecipitation resulted in higher background but nonetheless revealed hundreds of additional Abf1-bound promoter-enriched motifs that were not detected by PB-exo or ChIP-exo (Fig. 2A, right panel; Supplemental Fig. S2A,B). Thus, some technical bias in the Abf1 ChIP/PB-exo assays was producing substantial false negatives. This bias was likely formaldehyde, since a set of bound sites similar to native PB-seq were obtained for Abf1 using a formaldehyde-free assay (MNase-based ORGANIC vs. formaldehyde-base X-ChIP) (Kasinathan et al. 2014). The extremely high-resolution of PB-exo next allowed us to identify the precise nucleotide(s) responsible for the bias.
We focused on the nucleotides residing between the exonuclease stop points (+4 and +16 relative to the Abf1 motif midpoint). Analysis of the 500 most-occupied locations from each assay found a strong enrichment of G/C at +8 in the ChIP-exo and PB-exo data sets that was not present in native PB-seq (Fig. 2A,C) and was not in previously published protein-binding microarray (PBM) data (Gordan et al. 2011). When sorted by tag counts (occupancy) in the PB-exo assay, 98 of the top 100 bound locations had a G/C at +8 (Fig. 2D). The other two were algorithmically misassigned to a neighboring motif. Since the G/C enrichment at +8 was specific to formaldehyde, we conclude that formaldehyde cross-linking of Abf1 occurs selectively through G/C at +8.
The question arises as to how G/C cross-linking selectivity at +8 accounts for the 102 additional Abf1-bound sites detected only by ChIP-exo and not by PB-exo (Supplemental Fig. S2A). This was not due to low sequencing depth, because the average tag counts for sites detected in both assays were equivalent. The in vivo “ChIP-exo only” sites showed increased preference for G/C at +9 (alternative to cross-linking at +8) and −2 (minor cross-link) (“*” in Fig. 2D), which suggests that the in vivo conformation and/or interactions of Abf1 are sufficiently different from the in vitro setting to allow for a broader search space for G/C (by 1 bp) within individual sites and thus more detection.
The bound Abf1 sites that were captured only by the formaldehyde-independent native PB-seq assay and not by ChIP-exo were devoid of G/C at +8, as expected (Fig. 2D). Within the limits of the assay, Abf1 occupancy (tag counts) at these “native only” sites were on par with those detected by ChIP-exo, indicating that they were not intrinsically weak sites. This result, along with other in vitro studies of Abf1 (Beinoraviciute-Kellner et al. 2005), indicates that the nucleotide composition at the +8 (and +9) position does not significantly affect the affinity of Abf1 for DNA, but only the ability to capture the binding event by formaldehyde cross-linking.
Given this new insight, we reexamined our Reb1 data and assessed the extent to which formaldehyde G/C specificity had limited its detection. We compared the formaldehyde-based assays to native PB-seq and found that the penetrance of Reb1 binding across all sites was similar in vitro and in vivo irrespective of formaldehyde (Supplemental Fig. S2C). We attribute this to the presence of four potential cross-linking sites per binding site, resulting in four opportunities to obtain at least one cross-link. In this case, formaldehyde G/C specificity was not limiting our ability to detect binding of Reb1, as it was for Abf1. Sites that had a higher measured occupancy in PB-exo or ChIP-exo relative to native PB-seq also had higher G/C content at the calculated cross-linking points (Supplemental Fig. S2D, compare peaks at orange dashed lines across panels). For all proteins in this study other than Abf1, we found that formaldehyde cross-linking did not appreciably limit our ability to detect bound sites. However, it did shift the rank order of binding sites based on cross-linking efficiency.
Taken together, these results demonstrate that formaldehyde cross-linking has strong specificity for G/C. This can potentially result in the underrepresentation of binding events in all ChIP-based assays for those proteins possessing only one point of cross-linking to DNA. This underreporting is applicable to ChIP-seq, although the low resolution of the assay does not allow the number of cross-linking points to be determined. Proteins with multiple points of cross-linking, which exonucleases are well-suited to distinguish, will be less impacted due to the increased likelihood of at least one point having a G/C. Moreover, if the in vivo milieu provides increased flexibility to the protein, as seen for Abf1, then a neighboring G/C may react in place of a “head-on” A/T. Thus, binding site occupancy measured in formaldehyde-based assays, like ChIP, is semiquantitative and requires caution when comparing occupancy at individual sites. Ensemble comparisons are less affected due to effects of averaging.
TTACCCK does not fully delineate Reb1 intrinsic specificity
PB-exo allows us to consider all potential DNA binding events without the complexity imposed in vivo, and decipher site specificity determinants that are intrinsic to the protein. We started with the simplifying assumption that a purified protein like Reb1 would bind to all instances of its core motif located on otherwise protein-free genomic DNA. For Reb1, we first considered only exact matches to the core motif, TTACCCK, which we expected to be sufficient for DNA binding.
Remarkably, purified Reb1 bound only ∼20% of 749 exact TTACCCK occurrences in ORFs (PB-exo, Supplemental Fig. S3A,B). This compares with >60% of 780 occurrences at promoters. These binding sites were detectable across multiple assays and quantitatively reproducible within each assay (Supplemental Fig. S4A,B). Since the in vitro systems were devoid of nucleosomes, exclusion by nucleosomes could not explain why Reb1 did not bind “perfect” TTACCCK sequences in these experiments.
In striking contrast, we detected Reb1 binding to many locations in vitro that lack a “perfect” TTACCCK sequence (Supplemental Fig. S4C, diminished motif color uniformity), and so we reexamined the rules for Reb1 specificity. We examined 13,612 full and deeply degenerate motif occurrences, defined by a relatively low FIMO P-value threshold of <0.001 (an order of magnitude lower than the default) (Bailey et al. 2009). Of these, 4071 sites were bound by Reb1 in vitro when assayed by PB-exo, which is vastly more than the 1264 bound sites detected in vivo by ChIP-exo (Supplemental Fig. S4C). Thus, in contrast to Reb1 being intrinsically unable to bind certain “perfect” sites (which we examine below), Reb1 can bind a large number of degenerate sites in vitro but cannot bind them in vivo. They likely represent weaker nonbiological sites that occur randomly. If chromatin precludes Reb1 binding to these sites, there would be no apparent evolutionary pressure to purge them from the genome.
The ability of Reb1 to bind promiscuously to sites in vitro led us to reexamine data from a prior study (Kasinathan et al. 2014) reporting a broader in vivo binding of Reb1 than ChIP-exo (Rhee and Pugh 2011). That study essentially conducted native ChIP on extracted non-cross-linked chromatin, called ORGANIC, to identify in vivo binding. The approach involves extended in vitro incubation times, which raises the possibility of Reb1 redistribution to “in vitro only” sites. Indeed, we observed that 516 of our 2807 “in vitro only” sites were also identified in the ORGANIC assay (Supplemental Fig. S4C, middle panel). This represents nearly 30% of all Reb1 locations identified by ORGANIC. Those binding locations contained substantially fewer tags and had weaker motifs when compared to sites that were identified by ChIP-exo. They were also among the stronger “in vitro only” sites, which suggests that they occur primarily in vitro.
DNA shape immediately flanking TTACCCK is a major determinant of Reb1 binding
The inability of Reb1 to bind certain core TTACCCK motifs in vitro led us to consider whether additional specificity might be explained by the extended motif VTTACCCGNH (IUPAC nomenclature) (Rhee and Pugh 2011). The flanks (V…….NH) were originally ignored due to their high degeneracy. Previous NMR studies of S. cerevisiae Reb1 (Davis and Stillman 1997) and crystallography studies of S. pombe Reb1 (Jaiswal et al. 2016), which are 61% similar, show that both bind to the same consensus motif and adopt an abnormal DNA shape with a large minor groove and a large bend at the 5′ end of the motif (position −4). Presumably, this distortion is stabilized via local interactions with Reb1. Reb1 interactions with the distorted region are predominantly through the DNA backbone (Supplemental Fig. S5A, cyan space-fill), which invokes an indirect or shape readout of that portion of the binding site. In contrast, those contacts that occur over the core TTACCCK motif are indeed base contacts (Supplemental Fig. S5A, green space-fill). Conceivably, at the flanks of the motif, distortion of the DNA helix towards a shape that accommodates Reb1 may be attainable through a variety of sequences, and this might explain the deep degeneracy of the core motif extension (V……. NH). We therefore examined whether intrinsic DNA shape (Fig. 3A) might be involved in Reb1 site specificity (Zhou et al. 2013).
Figure 3.

Distinct DNA shape features help define Reb1 and Abf1 binding. (A) DNA shape parameters. The angle or distance reported for each parameter is indicated by red lines. (B) Line plots of variations in roll for top versus bottom 100 promoter Reb1 motif occurrences, defined by the sort in Supplemental Figure S3A. Dashed black line denotes genome-wide median. Blue and yellow stars represent positions with significant positive or negative roll (|Z| > 2, Mann-Whitney U test), respectively, for the top versus bottom 100. Shaded boxes highlight the nucleotides outside the core motif with significant shape differential. (C) Heat map representation of four DNA shape parameters for Reb1 from B. Z-scores are based on the Mann-Whitney U test. Orange hashtags indicate the location of Reb1 cross-linking points. The red box highlights the region of the motif with the greatest concentration of significant positions across all four DNA shape parameters. Helical twist and roll are inter-bp values. (D) List of specific sequences considered as exact Abf1 motif occurrences. (E) Four-color plot of sequences (left) centered on the motif midpoint for all instances of CGTnnnnnACGAC in promoters, representing one of the eight specific sequence configurations. (Right panel) Heat map of tags sorted by native PB-seq. The blue and red boxes indicate the top versus bottom 50 occupied sites. (F) Line plots of variations in minor groove width for Abf1 motif occurrences. Blue/bound or red/unbound thick lines represent the average of the thin lines, which reflect shape profiles for the eight individual Abf1 motif configurations. The dashed black line indicates the genome-wide median. Blue and yellow stars represent positions with significant larger or smaller minor groove width (|Z| > 3, Mann-Whitney U test), respectively, for the combined top versus bottom 400 sites. The position of the consensus Abf1 motif is labeled along the x-axis.
Averaged shape profiles of the top and bottom 100 Reb1-bound TTACCCK sites in promoters were compared (Fig. 3B). Where sequences are constant (e.g., TTACCC), we expected and observed large shape parameters that are invariant between the top and bottom groups. Since a unique sequence provides a unique shape, the two contributions are not resolvable. However, degenerate sequences largely eliminate sequence readout and therefore allow shape contributions to be assessed. Across all four computationally predicted DNA shape features, the top 100 bound sites possessed statistically significant shape deviations from the least-bound sites, which occurred at the 5′ end of the motif (Fig. 3B,C, red box). Statistical significance was determined using the Mann-Whitney U test, which reports the probability that the distributions of DNA shape parameters at each nucleotide are equal (Gordan et al. 2013). For visualization purposes, we display the average of these distributions. These deviations indicated that Reb1 prefers to bind motifs which start with (1) less positive helical twist, (2) more positive roll, (3) larger minor groove width, and (4) less negative propeller twist than otherwise equivalent lowly bound motifs (all designated as negative Z-scores).
The nucleotide content at the −4 position according to the MEME output was 52% G, 33 % A, 13% C, and 2% T. When we modeled the nucleotide content at this −4 position and computed its DNA shape, we observed that the most positive roll at the 5′ end of the Reb1 motif occurred when G or A is present, whereas the most negative roll occurs with T (Supplemental Fig. S5B). When motifs found in ORFs were examined in the same manner, essentially similar rules were found (Supplemental Fig. S5C). Thus, a specific shape aspect that is intrinsic to the ends of Reb1 motifs, and not strongly evident in the base sequence, appears to be a key factor in Reb1 binding. This need not reflect an actual shape; rather it might represent a propensity for distortion towards the final bound shape observed in the crystal structure. For Reb1 motifs, having a “T” at −4 reduces this propensity such that it precludes Reb1 binding. We further confirmed these findings using a motif defined by a MEME position weight matrix, rather than a fixed sequence (Supplemental Fig. S5D–F).
A confounding aspect of natural promoter regions is that they represent a potentially complex evolutionary integration of a variety of DNA properties that not only produce Reb1 binding sites, but also NFRs and transcriptional regulation. These other events might place constraints on Reb1 binding site composition. To address this concern, we examined the binding of yeast Reb1 to human DNA, which serves as high complexity DNA that has essentially no evolutionary ties to yeast Reb1. Reb1 bound to 21,443 sites in the human genome in vitro (Supplemental Fig. S6A). MEME analysis of the top 1000 locations confirmed Reb1's cognate motif (Supplemental Fig. S6B) and produced a qualitatively similar composite plot of cross-linking points (Supplemental Fig. S6C) as with yeast DNA. DNA shape analysis of the bound motifs produced the same correlation for positive roll at the −4 position with binding affinity as was observed in the yeast genome (Supplemental Fig. S6D). We therefore conclude that evolutionary constraints within the yeast genome were not biasing our results, and that an orthogonal approach towards site determination further demonstrates the dual role of DNA shape and sequence in Reb1 site recognition.
Distinct DNA shape features describe determinants of site recognition for multiple GRFs in regions of low DNA sequence readout
We next looked for DNA shape features in Abf1 binding, since ∼30% of the binding events (measured by native PB-seq) displayed normal binding to weak motifs (Fig. 2A, black motif P-value indicators). To avoid sequence effects, we analyzed only those sites having an exact match to the core motif (Fig. 3D; Cho et al. 1995). Of these, only about half were bound (Fig. 3E). The DNA shape properties of the top 50 bound sites across each of the eight sequence variants (400 in total) were averaged and compared to the bottom 50 sites (also 400 unbound sites in total) (Supplemental Fig. S6E). Bound sites had a consistently distinct shape pattern (e.g., minor groove width) compared to the unbound sites (Fig. 3F, −4 to +1: transition from negative to positive Z-scores) in the region of the motif where DNA sequence readout was lowest (Fig. 2C). Bound sites displayed a minor groove wedge between the motif left side and the motif center, whereas unbound sites were comparatively constant. Thus, beyond sequence, DNA shape also contributes to Abf1 site specificity.
We employed a similar strategy to study two additional GRFs, Mcm1 and Rap1 (Fig. 4; Supplemental Figs. S7, S8), which contain low DNA sequence readout in the center of their motifs (Fig. 4A,D). As with the other GRFs, Mcm1 and Rap1 were bound in promoters in vivo (Supplemental Figs. S7A,B, S8A,B). PB-exo identified the in vivo sites, along with hundreds of in vitro-only, of which most were located in ORFs (Supplemental Figs. S7C,D, S8C,D). To study the DNA shape aspects of Mcm1 binding, we analyzed a subset of bound and unbound motif occurrences from Supplemental Figure S7A that contained all of the most highly conserved nucleotides of the 16-bp pseudosymmetric motif (TTnCCnnnTnnGGnAA) (Fig. 4A,B; Shore and Sharrocks 1995; Hughes and de Boer 2013). DNA shape analysis of these motifs revealed an intrinsically negative roll at the motif midpoint (Fig. 4C, shaded areas), which aligns with the nucleotides that are bent by Mcm1 in the crystal structure (Supplemental Fig. S9A, top panel; Tan and Richmond 1998). However, Mcm1 makes no base-specific interactions in this area (Supplemental Fig. S9A, bottom panel), despite an enrichment of “T” at position 0. Nucleotide substitution modeling suggests this T may instead promote negative roll and the preferred DNA bend (Supplemental Fig. S9B). The unbound instances of the consensus sequence possess large spikes in positive roll that would inhibit proper bending of DNA upon Mcm1 binding (Fig. 4C, yellow starred nucleotides in the motif center). Thus, modest differences in DNA shape parameters appear to be a sufficient distinguishing factor for bound versus unbound sites.
Figure 4.

Influence of DNA shape on Mcm1/DNA and Rap1/DNA complex formation. (A) MEME logo obtained from the top 500 peak-pairs from Mcm1 PB-exo. (B, left panel) Four-color plot of sequences centered on the motif midpoint for all combined instances of TTnCCnnnTnnGGnAA in promoters or ORFs. (Right panel) Heat map of tags sorted by PB-exo. Sites above the black dashed line contain a peak pair. (C) Line plots of variations in roll for the top (green) versus bottom 20 (orange) motif occurrences. The dashed black line indicates the median roll of all DNA sequences. Blue and yellow stars represent positions with significant positive and negative roll (|Z| > 2, Mann-Whitney U test), respectively, for the top compared to the bottom sites. The position of the consensus Mcm1 motif is labeled along the x-axis. The tan shaded area indicates the nucleotides in the motif center that are bent in the structure presented in Supplemental Fig. S9A. (D) MEME logo obtained from the top 500 peak-pairs from Rap1 PB-exo. (E, left panel) Four-color plot of sequences centered on the motif midpoint for all combined instances of ACCCRnRCA in promoters or ORFs. (Right panel) Heat map of tags sorted by PB-exo. The dashed lines represent groups of 100 sites. (F) Line plots of variations in minor groove width for groups of 100 motif occurrences. Colored lines correspond to groups in E. The dashed black line indicates the genome-wide median. The position of the consensus Rap1 motif is labeled along the x-axis. Relevant shape effects are highlighted by shaded area. Blue and yellow stars represent positions with significant large or small minor groove width (|Z| > 2, Mann-Whitney U test), respectively, for the top 100 sites compared to the set of sites ranked 301–400 (light blue).
Rap1 is another GRF that organizes chromatin, binds promoters of genes that encode ribosomal and glycolytic proteins, and binds telomeres (Shore 1994; Ganapathi et al. 2011; Hughes and de Boer 2013). When the core DNA sequence of the Rap1 motif (Fig. 4D) was held constant (ACCCRnRCA), less than half of the sites were detectably bound (Fig. 4E). Most of the in vitro bound sites were weak Rap1 motifs (as defined by MEME) located in ORFs that were not bound in vivo (Supplemental Fig. S10A,B). DNA shape analysis revealed that Rap1 motifs possess an intrinsically wide minor groove spanning the central degenerate region of the motif that was wider at binding-competent sites (Fig. 4F). A clear trend was observed between increased width of the minor groove in the central degenerate region of the motif and increased Rap1 binding in vitro. Like Mcm1, Rap1 does not make base-specific contacts with this central region (Supplemental Fig. S10C; Le Bihan et al. 2013) but may take advantage of the wider minor groove to splay the DNA. For all the GRFs tested here, we conclude that although sites may possess all the conserved nucleotides needed to make proper base-specific hydrogen bonding, favorable DNA shape features are also required for high affinity binding.
Two E-box proteins use different mechanisms to achieve site specificity
To compare the importance of DNA shape in determining binding of GRFs versus a transcription factor, we next examined Cbf1 and Pho4, both of which recognize the same core sequence. Cbf1 is a GRF that binds the palindromic E-box motif (CACGTG) and utilizes DNA shape to discriminate between potential binding sites (Gordan et al. 2013). The same motif is bound by the transcription factor Pho4, yet the two possess distinct specificities despite having the same class of basic helix-loop-helix DNA binding domain and core recognition sequence (Zhou and O'Shea 2011). We therefore compared the DNA sequence/shape specificities of the two proteins. We considered all instances of the E-box motif (CACGTG) in promoters and ORFs, sorted by in vitro Cbf1 occupancy (Fig. 5A). Like the other GRFs, almost all (88% of 113) in vivo Cbf1-bound promoter sites were also detected in vitro (Supplemental Fig. S11A). There were also a substantial number of low occupancy “in vitro-only sites,” typically in ORFs (Fig. 5A). Pho4 displayed a highly similar, symmetrical PB-exo pattern with identical points of cross-linking to Cbf1 relative to the E-box midpoint (Supplemental Fig. S11B). However, unlike Cbf1, Pho4 bound to virtually all E-boxes in vitro (96%) (Fig. 5A; Supplemental Fig. S11C). That was not the case in vivo, where only 5% were bound by Pho4, under activating conditions as determined by ChIP-seq (Supplemental Fig. S11D; Zhou and O'Shea 2011).
Figure 5.

Genome-wide in vitro Cbf1 and Pho4 binding locations. (A) Annotation descriptions are the same as in Figure 2A, except for Cbf1 and Pho4. Data are sorted by Cbf1 PB-exo tag counts ±30 bp from the motif center, and rows across all data sets are linked. The Pho4 ChIP-seq data under phosphate starvation is from Zhou and O'Shea (2011). (B) Line plots of variations in roll for the top (red) versus top (blue) 100 Pho4 PB-exo-bound E-box motif occurrences. The bottom (brown) 100 Cbf1 PB-exo motif occurrences are also shown but were not included in the statistical analysis. The dashed black line indicates the genome-wide median. Blue and yellow stars represent positions with significant large or small roll (|Z| > 2, Mann-Whitney U test), respectively. The shaded area designates the positions just outside the core motif that possessed significant differences in DNA shape. The position of the consensus E-box motif is labeled along the x-axis. (C) Composite plots of nucleosome dyads generated by MNase H3 ChIP-seq for different groups of E-box motif occurrences. The data were collected from cells grown in YPD, but the Pho4-in vivo bound sites were defined by data collected under phosphate starvation conditions.
Previous studies have shown that Cbf1 prefers to bind E-boxes with a “T” at the 5′ end of the E-box (Zhou and O'Shea 2011). This manifests as a specific DNA shape flanking both sides of the palindromic core motif (Gordan et al. 2013). Our PB-exo experiments confirmed these preferences for Cbf1 in DNA sequence (Supplemental Fig. S11E) and DNA shape readout (Fig. 5B). Importantly, the DNA shape features of the top sites bound by Pho4 in vitro were not significantly different from random E-box sites (Supplemental Fig. S11F). However, Pho4-bound E-boxes were significantly different from Cbf1-bound E-boxes (Fig. 5B). While the discriminatory DNA shape (and DNA sequence) information at one end of the motif is sufficient to support binding, at the strongly bound Cbf1 motifs it is enriched at both ends (Fig. 5A, sequence plot in top panel with a “T” on the 5′ and “A” on the 3′ end). Where both sides of the E-box lacked the preferred negative roll, Cbf1 was not bound (Fig. 5B). Pho4 discriminates far less on the flanks, allowing it to bind all E-boxes in vitro. The widespread in vitro binding of Pho4 raises the question as to how this is prevented in vivo.
To understand the observed difference in sites bound by Pho4 in vivo and in vitro, we looked at nucleosome occupancy at E-boxes via MNase H3 ChIP-seq under normal growth conditions. We divided the promoter E-boxes into three groups: in vivo Cbf1-bound in normal growth; in vivo Pho4-bound under phosphate starvation; and promoter sites (<500 bp upstream of an ATG codon) that are not bound by either Cbf1 or Pho4 in vivo. Consistent with its role as a GRF, Cbf1-bound E-boxes existed in the center of NFRs surrounded by well-positioned nucleosomes (Fig. 5C, red trace). Under normal growth conditions (where Pho4 is not present in the nucleus), the Pho4 targets were enriched in NFRs (blue trace), although partially occluded by nucleosomes. In contrast, other E-boxes in promoters were generally occluded by nucleosomes (green trace) in a manner that was not significantly different from random sites (black trace). This is consistent with the idea that Pho4 possesses the intrinsic ability to bind every E-box, but in vivo is prevented from binding by chromatin unless assisted by chromatin remodelers (Svaren et al. 1994) that are targeted at promoter regions.
Discussion
We have used a genome-wide approach to examine site-specific DNA binding in vivo and in vitro. Like traditional in vitro assays, PB-exo provides information about transcription factor binding across a genome in the absence of cellular influences such as chromatin structure or other binding partners. In contrast, PBMs use custom oligonucleotides as a DNA substrate, which has been very successful in finding the preferred DNA recognition sequence of transcription factors. However, when these predictions are extrapolated to the genome, the overlap between in vivo and in vitro binding sites is limited since sites with mismatches to the core sequence are commonly missed (Mukherjee et al. 2004; Grau et al. 2013; Orenstein and Shamir 2014; Zhou et al. 2015). PB-exo uses an entire genome as both substrate and a nonspecific DNA competitor. With this setup, a large collection of potential binding sites is sampled within a single reaction. The system is highly adaptable, as purified protein may be swapped for whole-cell extract (WhIP-exo) to study DNA assembly of complex mixtures of proteins, as demonstrated here. To avoid complications associated with any co-evolved constraints within the DNA, the source DNA may be from an evolutionarily distant organism. Thus, PB-exo (and its variations) is well-suited to compare in vitro versus in vivo binding when using other genome-wide assays such as ChIP-exo, and for understanding complex mechanisms of site-specific DNA interactions.
G/C specificity of formaldehyde cross-linking
PB-exo serves as a strong in vitro counterpart to in vivo ChIP experiments because it uses the same method as ChIP to capture protein-DNA interactions (i.e., formaldehyde cross-linking), and thus has the same limitations. The Abf1 PB-exo data set provides a clear example of formaldehyde cross-linking specificity. The G/C requirement on either strand at the preferred cross-linking point appears to be essentially absolute. This is consistent with in vitro studies showing that formaldehyde predominantly cross-links guanine to the side-chains of lysine or cysteine (Lu et al. 2010). Which strand G/C resides on appears to matter little because the cross-linked N2 of G resides in essentially the same helical location on either strand (Lu et al. 2010). G/C requirements at individual cross-linking points can be found in all other formaldehyde-dependent data sets presented here. However, these other proteins display multiple cross-links to DNA. As such, there are correspondingly more opportunities for cross-links and thus more tolerance for an absence of G/C at any particular cross-linking site (Fig. 6A).
Figure 6.
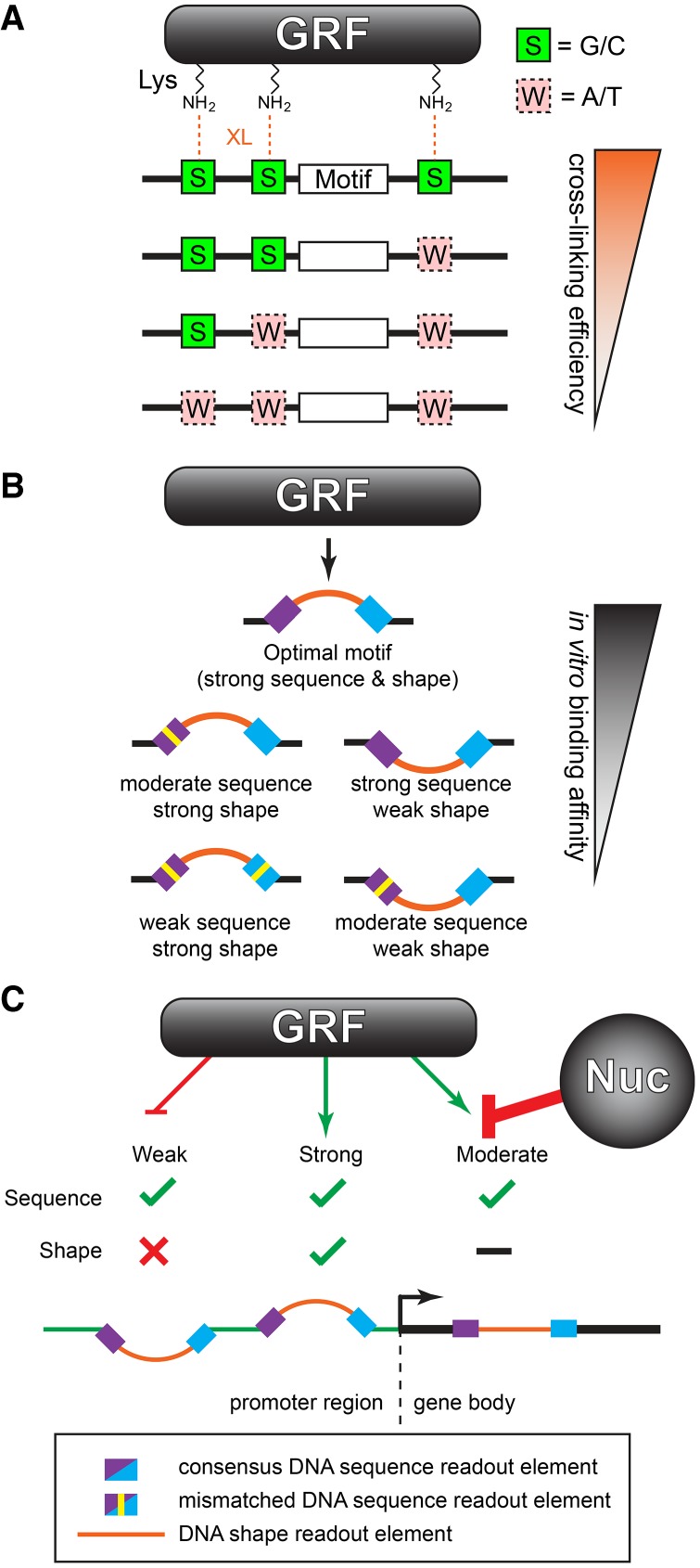
Genome-wide determinants of sequence-specific DNA binding. (A) Formaldehyde cross-linking (XL) efficiency is influenced by the fortuitous occurrence of G/C in the vicinity of lysine (Lys) side chains of the protein that interact with DNA. (B) GRF binding is specified by a combination of DNA sequence and shape readout. (C) Functional GRF binding sites having proper sequence and shape typically reside in promoters. Weaker sites may exist outside of promoters but are rendered inaccessible by chromatin. Strong consensus motifs that lack proper shape features do not bind GRFs and so may arise anywhere in the genome without consequence.
Indeed, this was quite evident for Reb1, which has at least three cross-linking points. More than 90% of the Reb1 binding events lacked evidence of G/C bias. Only in the top ∼10% of tag-enriched sites in ChIP-exo or in PB-exo was there a modest bias towards G/C at cross-linking points. The fortuitous presence of at least one G/C at any of the possible cross-linking points (including our observed ±1-bp tolerance) caused few binding events to be missed (as verified by native PB-seq). Consequently, ChIP assays are approximately quantitative, meaning that there may be less physiological site-to-site variability in occupancy than formaldehyde-based assays would suggest. ChIP-based assays can be quite definitive, with robust reproducibility, for detecting binding events. However, it may be less definitive about which binding sites are more highly occupied than others, except in more extreme comparisons. Thus, more tag counts (occupancy) at a location, even after normalization, may not always reflect relatively more binding. A general exception to this notion is where binding events are considered in aggregate, such that cross-linking biases become averaged out between comparison groups.
How DNA sequence and shape contribute to binding specificity
DNA binding specificity in genome regulation arises where proteins recognize nonuniform properties of DNA. Nonuniformity originates from the distinct physical and chemical properties of bases and their sequential order. While nonuniformity is clearly manifested through direct base readout (i.e., hydrogen bonding of protein amino acid side chains with an ordered arrangement of DNA bases), it is now appreciated that nonuniformity is further manifested indirectly via effects that base stacking and composition impart on the shape of a chemically uniform sugar-phosphate backbone. Base stacking and backbone conformation may be manifested through a wide range of parameters such as roll, propeller twist, helical twist, minor groove width, and sugar pucker, etc., of which some may be computationally predicted through DNA base pentamer sequences (Zhou et al. 2013; Li et al. 2017).
While a specific DNA sequence is expected to generate a single predominant intrinsic shape, the reverse is not necessarily true. Rather, a particular DNA shape (within parameter limits) may arise from many different sequence combinations. Additionally, neighboring sequences contribute to DNA shape. Neighboring effects are not well-captured in MEME because each nucleotide position is compiled independently of the identity of neighboring nucleotides. Consequently, MEME logos do not clearly capture shape information. Our findings suggest how DNA shape is contained within motif regions having low sequence definition. Shape specificity is likely also important within regions of direct sequence readout, where the motif base sequence is well-defined. However, at this time, separating direct from indirect readout within those regions is not feasible using genomic data alone, but these distinctions are illuminated when combined with atomic-level structural information of protein-DNA interactions. When bound by a GRF, the DNA is forced to adopt a unique and specific conformation that deviates from the dimensions of averaged B-form DNA. These twists, bends, and duplex deformations occur at specific base pairs and in a specific direction (as represented by the sign of the Z-scores) within the motif and correspond to positions with the greatest differences in DNA shape between bound and unbound motif occurrences. The genomic data show that true GRF binding sites are comprised of a combination of DNA sequence and DNA shape elements and that deviations in either will modulate the affinity of the GRF for DNA (Fig. 6B). No evidence currently exists that shape alone can drive site-specific GRF binding (Zentner et al. 2015; Rossi et al. 2017).
As with most biological processes, the regulation of protein binding to DNA is a continuum. On one end of that spectrum, typical transcription factors like Pho4 do not appear to compete with nucleosomes and instead predominantly sample motifs that already exist in the NFRs generated by other factors. In vitro (PB-exo), Pho4 bound nearly every instance of an E-box motif across the yeast genome. However, in vivo, Pho4 is a low-abundance protein that is recruited to the nucleus upon phosphate starvation by other factors, to act at a few dozen genes (Komeili and O'Shea 1999; Zhou and O'Shea 2011). Since Pho4 appears unable to compete with nucleosomes, competent sites that are occluded by nucleosomes are invisible to Pho4. On the other end of the continuum, GRFs can compete with nucleosomes to promote the formation of NFRs (Lascaris et al. 2000; Raisner et al. 2005; Bai et al. 2011; Levo et al. 2017). Thus, nucleosomes alone would be less successful in masking competent binding sites for GRFs. GRFs rely on both DNA sequence and shape to define in vivo binding sites. We hypothesize that evolution has further shaped specificity by preventing the accumulation of strong GRF binding sites in nonregulatory regions. Below a certain threshold, weak sites across the genome need not be evolutionarily purged since they lack sufficient affinity to allow GRFs to outcompete nucleosomes (Fig. 6C). These possibilities remain to be tested.
In this study, we have reconciled in vivo site selection preference with intrinsic preferences defined in vitro using pure proteins and DNA. Importantly, beyond confirming and characterizing known specificity determinants (sequence and accessibility), we have identified determinants of site recognition that are best described by DNA shape. Our work provides a conceptual advance in relating sequence/shape recognition to experimentally measured genome binding, wherein other physiological constraints come into play. At its most fundamental level, our work shows why certain DNA binding motif occurrences are not true binding sites. The experimental evidence demonstrates that genome binding specificity is achieved through the interplay of at least three factors: DNA sequence; DNA shape; and occlusion by chromatin.
Methods
Cell growth
TAP-tagged Reb1, Mcm1, Rap1, Cbf1, and Abf1 Saccharomyces cerevisiae strains in a BY4741 background were obtained from Open Biosystems. For ChIP-exo and whole-cell extract preparation, cells were grown in 500 mL of yeast peptone dextrose (YPD) media at 25°C to an OD600 = 0.8. Formaldehyde was added to a final concentration of 1% for 15 min, then quenched with 125 mM glycine. For protein purification and genomic DNA preparation, cells were grown to an OD600 = 2.0.
Proteins
TAP-tagged Reb1, Mcm1, Rap1, Cbf1, and Abf1 were purified as previously described (Krogan et al. 2002). HA-Pho4 was a generous gift of N. Krietenstein and P. Korber (Universitat Munchen, Germany).
PB-exo
PB-exo was performed on purified proteins and purified genomic DNA that was sonicated to an average of 200 bp. A detailed description for the purification of genomic S. cerevisiae DNA and the procedure is provided in the Supplemental Material. In brief, purified protein and DNA were incubated in a binding buffer and then formaldehyde cross-linked. Following quenching, the sample was passed through a spin column to remove formaldehyde byproducts. The sample was then used as the starting material for the standard ChIP-exo protocol (Rhee and Pugh 2012).
WhIP-exo
WhIP-exo was performed as PB-exo, except the purified protein in the binding reaction was replaced with 150–350 µg (total protein) of whole-cell extract. TAP-tagged Reb1 whole-cell extract was prepared as previously described (Schultz 1999) using the “Breaking Cells in Coffee Mill” option to lyse yeast cells.
Native PB-seq
A detailed description of the procedure is provided in the Supplemental Material. In brief, the binding reaction was identical to PB-exo. Then the sample was cleaned up and used as the starting material for immunoprecipitation. This protocol was adapted from Guertin and Lis (2013) and library prep was adapted from Quail et al. (2008).
ChIP-exo
ChIP-exo was performed as previously described (Rhee and Pugh 2012). In brief, 250 mL of TAP-tagged S. cerevisiae cultures grown at 30°C to an OD600 of 0.8 were treated with 1% formaldehyde for 15 min, then quenched. Cells were disrupted by bead beating, and chromatin pellets were washed. Chromatin was solubilized by sonication and subjected to standard ChIP using IgG-sepharose. The first adaptor was ligated to the ChIP DNA while immobilized on beads, then subjected to exonuclease digestion. Next, the material was eluted, and the second adaptor ligated to the exonuclease treated end. The resulting libraries were subjected to Illumina sequencing.
Bioinformatic analyses
Peak calling was performed using the Genetrack (Albert et al. 2008) peak caller to call strand-independent peaks and then pairing those stranded peaks. Called peaks were then determined by the presence of the known DNA sequence motifs within ±30 bp of the midpoint of the paired peaks, as called using the MEME algorithm (Bailey et al. 2009). Unbound sites were defined as possessing the correct DNA motif but not peak-paired within the previous distance. Composite plots and heat maps were then oriented around these motifs unless specified otherwise. DNA shape analysis at bound and unbound locations was performed using the DNAshape webserver (Zhou et al. 2013). Extended details on the above procedures are available in the Supplemental Material.
Data access
All sequencing files and peak files from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE93662. Coordinate files, script parameters, and custom code used to generate the figures for this paper can be found in the Supplemental Material (Supplemental_Materials.zip) or downloaded from: https://github.com/CEGRcode/2018-Rossi_GenomeResearch.
Competing interest statement
B.F.P. has a financial interest in Peconic, LLC, which utilizes the ChIP-exo technology implemented in this study and could potentially benefit from the outcomes of this research.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants ES013768 to B.F.P. and CA168104 to M.J.R. We thank members of the Pugh laboratory and Center for Eukaryotic Gene Regulation, especially Bede Portz, for insightful discussion and critical comments on the manuscript. We also thank N. Krietenstein and P. Korber for their generous gift of HA-Pho4 protein.
Author contributions: M.J.R. performed the experiments; M.J.R., W.K.M.L., and B.F.P. conceived the experiments and analyses; M.J.R. and W.K.M.L. performed the analyses; M.J.R. and B.F.P. cowrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.229518.117.
Freely available online through the Genome Research Open Access option.
References
- Abe N, Dror I, Yang L, Slattery M, Zhou T, Bussemaker HJ, Rohs R, Mann RS. 2015. Deconvolving the recognition of DNA shape from sequence. Cell 161: 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert I, Wachi S, Jiang C, Pugh BF. 2008. GeneTrack—a genomic data processing and visualization framework. Bioinformatics 24: 1305–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badis G, Chan ET, van Bakel H, Pena-Castillo L, Tillo D, Tsui K, Carlson CD, Gossett AJ, Hasinoff MJ, Warren CL, et al. 2008. A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Mol Cell 32: 878–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L, Ondracka A, Cross FR. 2011. Multiple sequence-specific factors generate the nucleosome-depleted region on CLN2 promoter. Mol Cell 42: 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beinoraviciute-Kellner R, Lipps G, Krauss G. 2005. In vitro selection of DNA binding sites for ABF1 protein from Saccharomyces cerevisiae. FEBS Lett 579: 4535–4540. [DOI] [PubMed] [Google Scholar]
- Cho G, Kim J, Rho HM, Jung G. 1995. Structure-function analysis of the DNA binding domain of Saccharomyces cerevisiae ABF1. Nucleic Acids Res 23: 2980–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis DR, Stillman DJ. 1997. Altered structure of the DNA duplex recognized by yeast transcription factor Reb1p. Nucleic Acids Res 25: 668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis ET, Clark AG. 2002. Evolution of transcription factor binding sites in Mammalian gene regulatory regions: conservation and turnover. Mol Biol Evol 19: 1114–1121. [DOI] [PubMed] [Google Scholar]
- Ganapathi M, Palumbo MJ, Ansari SA, He Q, Tsui K, Nislow C, Morse RH. 2011. Extensive role of the general regulatory factors, Abf1 and Rap1, in determining genome-wide chromatin structure in budding yeast. Nucleic Acids Res 39: 2032–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordan R, Murphy KF, McCord RP, Zhu C, Vedenko A, Bulyk ML. 2011. Curated collection of yeast transcription factor DNA binding specificity data reveals novel structural and gene regulatory insights. Genome Biol 12: R125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordan R, Shen N, Dror I, Zhou T, Horton J, Rohs R, Bulyk ML. 2013. Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep 3: 1093–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau J, Posch S, Grosse I, Keilwagen J. 2013. A general approach for discriminative de novo motif discovery from high-throughput data. Nucleic Acids Res 41: e197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin MJ, Lis JT. 2013. Mechanisms by which transcription factors gain access to target sequence elements in chromatin. Curr Opin Genet Dev 23: 116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin MJ, Martins AL, Siepel A, Lis JT. 2012. Accurate prediction of inducible transcription factor binding intensities in vivo. PLoS Genet 8: e1002610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley PD, Madhani HD. 2009. Mechanisms that specify promoter nucleosome location and identity. Cell 137: 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR, de Boer CG. 2013. Mapping yeast transcriptional networks. Genetics 195: 9–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal R, Choudhury M, Zaman S, Singh S, Santosh V, Bastia D, Escalante CR. 2016. Functional architecture of the Reb1-Ter complex of Schizosaccharomyces pombe. Proc Natl Acad Sci 113: E2267–E2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, et al. 2009. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458: 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasinathan S, Orsi GA, Zentner GE, Ahmad K, Henikoff S. 2014. High-resolution mapping of transcription factor binding sites on native chromatin. Nat Methods 11: 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komeili A, O'Shea EK. 1999. Roles of phosphorylation sites in regulating activity of the transcription factor Pho4. Science 284: 977–980. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Kim M, Ahn SH, Zhong G, Kobor MS, Cagney G, Emili A, Shilatifard A, Buratowski S, Greenblatt JF. 2002. RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomics approach. Mol Cell Biol 22: 6979–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lascaris RF, Groot E, Hoen PB, Mager WH, Planta RJ. 2000. Different roles for abf1p and a T-rich promoter element in nucleosome organization of the yeast RPS28A gene. Nucleic Acids Res 28: 1390–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bihan YV, Matot B, Pietrement O, Giraud-Panis MJ, Gasparini S, Le Cam E, Gilson E, Sclavi B, Miron S, Le Du MH. 2013. Effect of Rap1 binding on DNA distortion and potassium permanganate hypersensitivity. Acta Crystallogr D Biol Crystallogr 69: 409–419. [DOI] [PubMed] [Google Scholar]
- Levo M, Avnit-Sagi T, Lotan-Pompan M, Kalma Y, Weinberger A, Yakhini Z, Segal E. 2017. Systematic investigation of transcription factor activity in the context of chromatin using massively parallel binding and expression assays. Mol Cell 65: 604–617.e6. [DOI] [PubMed] [Google Scholar]
- Li J, Sagendorf JM, Chiu TP, Pasi M, Perez A, Rohs R. 2017. Expanding the repertoire of DNA shape features for genome-scale studies of transcription factor binding. Nucleic Acids Res 45: 12877–12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Ye W, Zhou L, Collins LB, Chen X, Gold A, Ball LM, Swenberg JA. 2010. Structural characterization of formaldehyde-induced cross-links between amino acids and deoxynucleosides and their oligomers. J Am Chem Soc 132: 3388–3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, Xin B, Chiu TP, Yang L, Rohs R, Wasserman WW. 2016. DNA shape features improve transcription factor binding site predictions in vivo. Cell Syst 3: 278–286.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses AM, Pollard DA, Nix DA, Iyer VN, Li XY, Biggin MD, Eisen MB. 2006. Large-scale turnover of functional transcription factor binding sites in Drosophila. PLoS Comput Biol 2: e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Berger MF, Jona G, Wang XS, Muzzey D, Snyder M, Young RA, Bulyk ML. 2004. Rapid analysis of the DNA-binding specificities of transcription factors with DNA microarrays. Nat Genet 36: 1331–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orenstein Y, Shamir R. 2014. A comparative analysis of transcription factor binding models learned from PBM, HT-SELEX and ChIP data. Nucleic Acids Res 42: e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail MA, Kozarewa I, Smith F, Scally A, Stephens PJ, Durbin R, Swerdlow H, Turner DJ. 2008. A large genome center's improvements to the Illumina sequencing system. Nat Methods 5: 1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raisner RM, Hartley PD, Meneghini MD, Bao MZ, Liu CL, Schreiber SL, Rando OJ, Madhani HD. 2005. Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell 123: 233–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF. 2011. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147: 1408–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF. 2012. ChIP-exo method for identifying genomic location of DNA-binding proteins with near-single-nucleotide accuracy. Curr Protoc Mol Biol Chapter 21: Unit 21.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhode PR, Sweder KS, Oegema KF, Campbell JL. 1989. The gene encoding ARS-binding factor I is essential for the viability of yeast. Genes Dev 3: 1926–1939. [DOI] [PubMed] [Google Scholar]
- Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B. 2009. The role of DNA shape in protein–DNA recognition. Nature 461: 1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi MJ, Lai WK, Pugh BF. 2017. Correspondence: DNA shape is insufficient to explain binding. Nat Commun 8: 15643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz MC. 1999. Chromatin assembly in yeast cell-free extracts. Methods 17: 161–172. [DOI] [PubMed] [Google Scholar]
- Shore D. 1994. RAP1: a protean regulator in yeast. Trends Genet 10: 408–412. [DOI] [PubMed] [Google Scholar]
- Shore P, Sharrocks AD. 1995. The MADS-box family of transcription factors. Eur J Biochem 229: 1–13. [DOI] [PubMed] [Google Scholar]
- Slattery M, Zhou T, Yang L, Dantas Machado AC, Gordan R, Rohs R. 2014. Absence of a simple code: how transcription factors read the genome. Trends Biochem Sci 39: 381–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svaren J, Schmitz J, Horz W. 1994. The transactivation domain of Pho4 is required for nucleosome disruption at the PHO5 promoter. EMBO J 13: 4856–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Richmond TJ. 1998. Crystal structure of the yeast MATα2/MCM1/DNA ternary complex. Nature 391: 660–666. [DOI] [PubMed] [Google Scholar]
- Yang L, Zhou T, Dror I, Mathelier A, Wasserman WW, Gordan R, Rohs R. 2014. TFBSshape: a motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Res 42: D148–D155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Orenstein Y, Jolma A, Yin Y, Taipale J, Shamir R, Rohs R. 2017. Transcription factor family-specific DNA shape readout revealed by quantitative specificity models. Mol Syst Biol 13: 910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarragudi A, Miyake T, Li R, Morse RH. 2004. Comparison of ABF1 and RAP1 in chromatin opening and transactivator potentiation in the budding yeast Saccharomyces cerevisiae. Mol Cell Biol 24: 9152–9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Morse RH. 1999. Chromatin opening and transactivator potentiation by RAP1 in Saccharomyces cerevisiae. Mol Cell Biol 19: 5279–5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Kasinathan S, Xin B, Rohs R, Henikoff S. 2015. ChEC-seq kinetics discriminates transcription factor binding sites by DNA sequence and shape in vivo. Nat Commun 6: 8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, O'Shea EK. 2011. Integrated approaches reveal determinants of genome-wide binding and function of the transcription factor Pho4. Mol Cell 42: 826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Yang L, Lu Y, Dror I, Dantas Machado AC, Ghane T, Di Felice R, Rohs R. 2013. DNAshape: a method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res 41: W56–W62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Shen N, Yang L, Abe N, Horton J, Mann RS, Bussemaker HJ, Gordan R, Rohs R. 2015. Quantitative modeling of transcription factor binding specificities using DNA shape. Proc Natl Acad Sci 112: 4654–4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.