

Abstract
Information that regulates gene expression is encoded throughout each gene but if different regulatory regions can be understood in isolation, or if they interact, is unknown. Here we measure mRNA levels for 10,000 open reading frames (ORFs) transcribed from either an inducible or constitutive promoter. We find that the strength of cotranslational regulation on mRNA levels is determined by promoter architecture. By using a novel computational genetic screen of 6402 RNA-seq experiments, we identify the RNA helicase Dbp2 as the mechanism by which cotranslational regulation is reduced specifically for inducible promoters. Finally, we find that for constitutive genes, but not inducible genes, most of the information encoding regulation of mRNA levels in response to changes in growth rate is encoded in the ORF and not in the promoter. Thus, the ORF sequence is a major regulator of gene expression, and a nonlinear interaction between promoters and ORFs determines mRNA levels.
Precise control of gene expression is essential (Skotheim et al. 2008) and the way this regulatory information is encoded in enhancers, promoters, 5′ untranslated regions (UTRs), and other regulatory regions throughout the gene sequence has been well characterized (Kudla et al. 2009; Dvir et al. 2013; Keren et al. 2013; Shalem et al. 2013, 2015). Steady-state mRNA levels are determined by the combination of synthesis and decay rates. It was generally assumed that promoters mostly determine synthesis rates, while sequences in the 5′ UTR, open reading frame (ORF), and 3′ UTR determine mRNA translation and degradation rates. While there is now evidence that synthesis, decay, and translation are tightly coupled, there is no clear delineation of regulatory mechanisms within different parts of a gene, and the precise mechanisms that link the promoter to cytoplasmic processes remain unknown (Wolffe and Meric 1996; Ladomery 1997; Haimovich et al. 2013).
As the location that regulates the initiation of transcription, the promoter is a reasonable place to start thinking about mRNA expression. In yeast, genes can broadly be split into two classes based on promoter architecture: those with a consensus and evolutionarily conserved TATA box and those without one (TATA-less). TATA-binding protein (TBP) binds TATA+ promoters at the TATA box, but TBP also binds TATA-less promoters at a TATA-like sequence that is one or two mismatches away from the TATA consensus. TATA-less promoters are thus also referred to as TATA-like. While recent work suggests that this distinction is not quite binary, multiple lines of evidence have shown that, on many different levels, genes that lack a consensus evolutionarily conserved TATA box are fundamentally different from the TATA+ class (Kubik et al. 2017; Taatjes 2017). Genes lacking a TATA box are depleted of nucleosomes around the transcription start site (TSS) and exhibit lower transcriptional plasticity (Tirosh and Barkai 2008; Rhee and Pugh 2012). For simplicity, we will generally refer to the TATA+ class as constitutive and the TATA-like (TATA-less) class as inducible.
Features within the ORF itself, such as codon usage, correlate with steady-state mRNA levels (Akashi 2003; Neymotin et al. 2014, 2016). There are two proposed reasons for this correlation: natural selection for specific codon usage and an active role in codon usage in regulating mRNA levels. In first, codon usage is shaped by ribosome dynamics and tRNA pools, with selection for accurate and fast translation, especially among abundant proteins (Plotkin and Kudla 2011). In the second, codon usage directly affects mRNA levels (Chen et al. 2017), likely through translation-coupled decay. There is ample evidence for both models, suggesting a model in which both the act of translation and selection on codon usage are together responsible for the observed correlation between codon usage and gene expression among native genes.
In addition to effects from codon usage, the stability of transcripts is determined by multiple other processes, one of which is nonsense-mediated decay (NMD). Initially identified as a quality control mechanism for transcripts that contain an aberrant premature termination codon (PTC) within the ORF, there is now abundant evidence that NMD affects the expression of between 5% and 20% of native transcripts in yeast and mammalian cells (Behm-Ansmant et al. 2007; Kervestin and Jacobson 2012; Tani et al. 2012; He and Jacobson 2015). The precise mechanism is still unclear, but three models have been proposed: the Exon Junction Complex (EJC) model, the Upf1 3′-UTR sensing and potentiation model, and the faux 3′-UTR model (Le Hir et al. 2000; Amrani et al. 2004; Hogg and Goff 2010; He and Jacobson 2015). Previous observations suggest that the EJC model is the main source for NMD in mammals, but yeast mainly triggers NMD through the other two mechanisms (Lindeboom et al. 2016). The latter have a common denominator: long 3′ UTRs, which result from PTCs but can also be found in native mRNAs. Thus, 3′ UTR length encodes regulatory information in a way that depends on active translation.
To further complicate the relationship between sequence and expression, some transcription factors (TFs) influence mRNA stability and localization (Bregman et al. 2011; Zid and O'Shea 2014; Braun et al. 2015). Thus, in addition to affecting mRNA synthesis rates, promoters play a role in other parts in the life of an mRNA. However, three major questions remain: By which mechanisms do elements in the promoter influence the lifecycle of the mRNA, where in the gene are different types of regulatory information encoded, and are there genetic interactions between discrete regulatory regions?
Promoters influence mRNA levels. Coding sequences influence mRNA levels. To determine if there is a genetic interaction between these two spatially distinct regulatory regions, we generated a pooled library of over 10,000 yeast genomic DNA fragments cloned into plasmids containing either inducible TATA box containing GALL promoter or the constitutive TATA-less RPL4A promoter. This library exhibits over four orders of magnitude of expression, most of which can be predicted using a sequence-feature–based mathematical model. In both the library and in native transcripts, cotranslational features such as codon usage more strongly influence mRNA levels when expression is driven by a constitutive promoter. To identify the molecular mechanism for this difference in cotranslational regulation between promoter architectures, we performed a computational genetic screen across 6402 RNA-seq experiments and found that the RNA helicase Dbp2 specifically insulated transcripts of TATA+, but not TATA-less promoters, from the effects of cotranslational regulation on steady-state mRNA levels. Finally, we used RNA-seq and promoter-YFP expression data to show that, specifically for TATA-less promoters, the ORF contains more regulatory information than the promoter.
Results
ORF-encoded sequence features play an active role in regulating gene expression in native transcripts
To determine the ability of coding sequences to regulate gene expression, we developed a method to decouple native genomic sequences from their 5′ and 3′ regulatory context (Fig. 1A; Supplemental Fig. 1). We digested the yeast genome with four restriction enzymes and inserted the resulting random genomic DNA fragments into a plasmid containing the inducible GALL promoter, a start codon, the ClaI restriction site, and three staggered stop codons (Supplemental Fig. 1A). This generated a pool of over 10,000 plasmids, each of which contains a single random fragment of the yeast genome. The cloned random gDNA fragments derive from the entire yeast genome and therefore contain parts of native coding sequences, as well as UTRs, promoters, transcription terminators, and other chromosomal features (Supplemental Fig. 1B; Supplemental Table 1). We transformed the plasmids into yeast and measured expression of each transcript as the ratio between RNA and DNA abundance (Fig. 1B). In spite of having the same promoter, 5′ UTR, and transcription terminator, the expression of the gDNA fragments varies of over four orders of magnitude (Fig. 1C; Supplemental Fig. 2). To determine the sources of this regulation and to quantify the contribution of individual sequence features to changes in expression, we computed over 4000 sequence features for each insert, such as dimer and trimer nucleotide counts, GC content, codon bias, and the presence of premature stop codons (see Methods). We fit a linear model with a minimal number of predictive features to the experimental data and found that a model with seven features can accurately predict expression (R2 = 0.64 for all inserts and R2 = 0.61 for inserts with no premature stop codon) (Figs. 1D, 2B; Supplemental Fig. 3), suggesting that a small number of ORF-encoded sequence features generate a large amount of the variation in gene expression. To determine if ORF-encoded sequence features regulate expression in native genes, we used the same model without taking into account 3′ UTR length to predict expression of native genes from their coding sequence (Fig. 1E).
Figure 1.
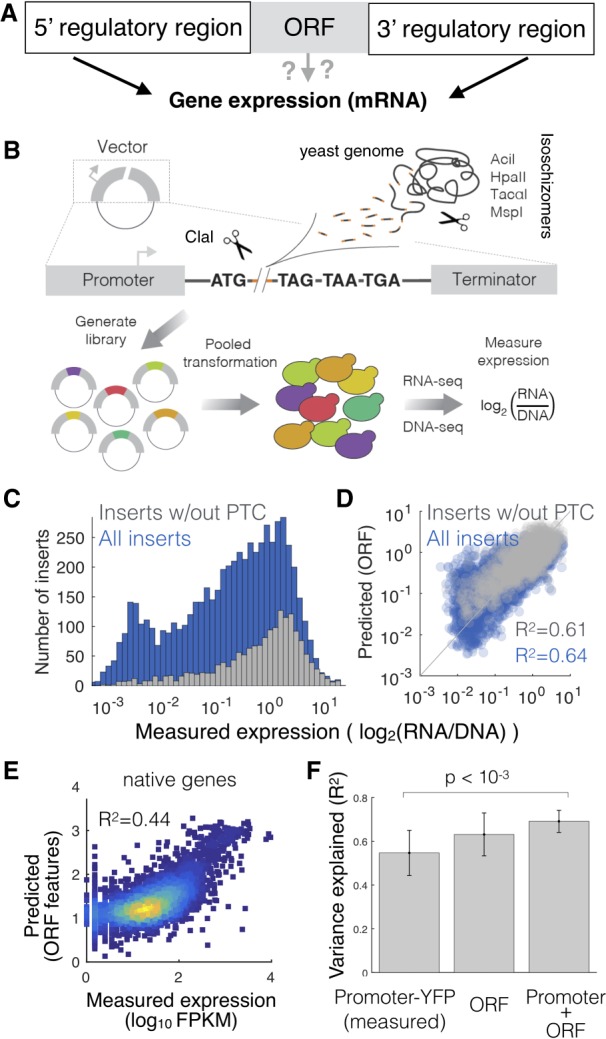
An expression library of genomic fragments to quantify the ability of ORF-encoded sequence features to regulate gene expression. (A) A schematic view of the following initial question: Does the ORF determine gene expression? (B) Scheme of the gDNA library preparation and expression measurements. (C) Measured expression distributions for all inserts (blue) and only those lacking a premature termination codon (PTC; gray). (D) Measured versus predicted expression levels in the gDNA library. Expression is predicted from the sequence of each gDNA insert using a 10-fold cross-validated linear model (R2 is calculated across all test data from all cross-validations). (E) Expression predicted from the sequence of each native yeast ORF using the same features as for the gDNA library. (F) Including ORF-encoded features in a model of expression increases the ability of promoter-YFP data to predict steady-state mRNA levels. Error bars, SD from 10-fold cross-validation.
Figure 2.

A gDNA library with different promoters identifies sequence features that interact with the promoter to determine gene expression. (A) The gDNA library was cloned under the control of either the TATA+ GALL promoter or the TATA-less ribosomal RPL4A promoter and the expression of both libraries measured in yeast growing on galactose as a carbon source. (B) Coefficients from the multiple linear model based on ORF sequence features from libraries with the two different promoters. Outlined are the features used for predicting expression in both the libraries or native genes. tAI is the tRNA adaptation index. Nucleotides followed by numbers refer to the position in a codon; e.g., A/G1 is the fraction of codons with an A or G at position 1. (C,D) Lines show the median expression for inserts binned by 3′ UTR length (C) or codon bias (D). Correlation values are for unbinned data, and the P-value is a test for a significant difference between the two correlation values using bootstrapping. (E) NMD effect, measured as the log2 ratio in mRNA (TPM) between upf1 and wild-type cells for native transcripts. Lines show the median NMD effect across transcripts binned by 3′ UTR length for TATA-containing (red) and TATA-less promoters (blue). The P-value is for a t-test for a difference in mean NMD strength for all unbinned data between TATA and TATA-less genes. (F) The makeup of two linear models, one that predicts mRNA levels from promoter-YFP data, and the other that includes codon bias (tAI) as an additional predictor. For both models, native genes are split into two classes, TATA and TATA-less, and tAI effect is the difference in R2 between the two models. (G) Difference in tAI effect for random samplings of equal numbers of genes from each class. (H,I) An ORF-encoded sequence feature model was trained to predict mRNA levels for TATA and TATA-less promoters (R2 = squared Pearson correlation coefficient).
The ability of the ORF-feature model to predict expression of native genes could be because coding sequences play an active regulatory role, or it could be due to coevolution and selection for certain sequence properties in genes whose expression levels are determined by more stereotypical regulatory regions such as the promoter. To differentiate correlation from causation, we took advantage of data in which the strength of 859 promoters driving YFP was measured (Keren et al. 2013). We find that ORF features are equally good as promoters in predicting steady-state mRNA levels and that a combined model including both promoter-YFP data and ORF features performs significantly better (Fig. 1F). Thus, ORF-encoded sequence features play an active role in regulating gene expression in native transcripts.
Cotranslational regulation is stronger for mRNAs regulated by TATA-less promoters
While the majority of experiments that investigated transcriptional, cotranslational, and post-translational regulation used inducible promoters (Meaux et al. 2008; Shah et al. 2013; Shalem et al. 2013; Puchta et al. 2016; Radhakrishnan et al. 2016), most of these promoters are in fact not condition specific. Promoters can be roughly divided into two broad categories: those with a TATA box (SAGA-dominated and inducible or regulated) and those lacking a conserved TATA box (TFIID-dominated and constitutive) (Struhl 1986; Basehoar et al. 2004; Huisinga and Pugh 2004; Tirosh and Barkai 2008; de Jonge et al. 2017). To determine if ORF-encoded sequence features are regulatory for constitutive promoters, we built the random gDNA fragment library in a second plasmid in which the regulated TATA box containing GALL promoter was replaced with the equally strong constitutive ribosomal RPL4A promoter (Fig. 2A; Supplemental Fig. 4). We found that codon bias (tRNA adaptation index [tAI]) (dos Reis et al. 2003) and 3′ UTR length, both of which require active translation to be interpreted by the cell (Kervestin and Jacobson 2012; Gardin et al. 2014; Presnyak et al. 2015), have more importance in a model trained on RPL4Apr data compared with GALLpr data (Fig. 2B). Both 3′ UTR length and codon bias have a stronger effect on expression when transcription is driven by the constitutive RPL4A promoter (Fig. 2C,D; Supplemental Fig. 5).
To determine if this promoter-specific effect of cotranslational regulation affects native genes, we divided promoters into two categories: those with an evolutionarily conserved TATA box (inducible) and those without (TATA-less, or constitutive) (Basehoar et al. 2004). To measure the effect of NMD separately for each class of transcript, we used RNA-seq measurements from a Δupf1 strain, which is defective in NMD (Smith et al. 2014). Consistent with the gDNA library, native transcripts from TATA-less promoters are affected by NMD in a manner that increases with 3′ UTR length, while transcripts from promoters with an evolutionarily conserved TATA box are unaffected by NMD (Fig. 2E; Supplemental Fig. 6). To determine if the effect of codon bias on native genes is stronger for TATA-less promoters, we used a linear model to predict mRNA levels either from promoter-YFP expression data alone (Keren et al. 2013) or from a model that includes both promoter-YFP data and codon bias (tAI) (dos Reis et al. 2003) and measured the increase in R2 when including codon bias as a model feature (Fig. 2F). Consistent with results from the gDNA library, codon bias is more strongly predictive of mRNA levels for transcripts driven by TATA-less promoters (Fig. 2G). These differences are not due to differences in expression, mRNA stability, or codon bias since the distribution of mRNA expression, degradation rates, and tAI are similar between the two classes of genes (Supplemental Fig. 4). To determine if ORF-encoded sequence features are more predictive of native expression levels for TATA-less genes, we predicted expression from sequence separately for the two classes of genes. ORF features are more predictive of expression in TATA-less genes (Fig. 2H,I). In both, for the gDNA library and native genes, cotranslational regulation affects mRNA levels more strongly when expression is driven by TATA-less promoters, suggesting the existence of some mechanism that carries information from the promoter to the ribosome.
A genetic screen of 6402 RNA-seq experiments identifies the RNA helicase Dbp2 as TATA-containing promoter-specific insulator damper of cotranslational regulation
To identify the molecular mechanism underlying the difference in cotranslational regulation between TATA and TATA-less promoters, we performed a computational genetic screen for mutants that alter our ability to predict mRNA levels from promoter-YFP data. We screened 6402 RNA-seq experiments, all RNA-seq experiments that have been done in Saccharomyces cerevisiae (Ziemann et al. 2015), for mutants that affect the ability of codon bias to predict expression in a promoter class–specific manner (Fig. 3A). For each of the 6402 experiments, we calculated the increase in R2 upon including codon bias as a predictive feature. Codon bias has more predictive power for TATA-less genes across thousands of RNA-seq experiments (Fig. 3B). We identified a single mutant that significantly reduces this difference across multiple biological replicates. Deletion of DBP2 (Beck et al. 2014) increases the effect of codon bias on gene expression specifically for TATA-containing genes (Fig. 3C; Supplemental Fig. 7). Furthermore, DBP2 expression varies across the data set and this variation correlates with the effect of tAI. Conditions and mutants with low DBP2 expression have a higher tAI effect on expression of TATA+ but not TATA-less genes (Supplemental Fig. 7A). This suggests that Dbp2, a cotranscriptionally loaded RNA helicase (Cloutier et al. 2012; Ma et al. 2016), specifically reduces the effect of cotranslational regulation on mRNA levels in a promoter-specific manner. Interestingly, Dbp2 physically interacts with nucleosomes, Pol II–associated general transcription factors (GTFs), and the ribosome (Supplemental Fig. 8; Stark et al. 2006).
Figure 3.

A computational genetic screen for mutants that alter promoter-specific cotranslational regulation. (A) We analyzed expression data for 6402 RNA-seq experiments in S. cerevisiae and, for each experiment, predicted mRNA levels from promoter-YFP data alone or promoter-YFP and codon bias (tAI). (B) For each for the 6402 experiments, we calculated the ability of tAI to improve R2 for TATA-less and TATA-containing genes. Highlighted is a single experiment (Beck et al. 2014) in which the R2 changes in a promoter class–specific manner between wild-type and mutant cells. (C) Shown are the changes in R2 for wild-type and Δdbp2 cells for TATA-containing and TATA-less transcripts. Each point corresponds to the effect of tAI on expression from equal-sized random samples of TATA (red) and TATA-less (blue) genes. Boxplots show the median and interquartile range. P-values are from t-tests. (D) Synthetic system for measuring the effect of NMD on gene expression. NMD is the ratio in mCherry expression between plasmids with and without a PTC. (E) In each genotype, we measured mCherry expression 16 times (biological replicates) for each of the four plasmids. As NMD effect is the ratio between two plasmids, for each promoter and genotype we calculated 82 ratios. Boxplots show the median and interquartile range for each set of ratios.
To confirm the results of the screen and to determine if Dbp2 plays a role in other forms of cotranslational regulation of mRNA levels, such as NMD, we built a set of four synthetic NMD reporters (Fig. 3D). Each plasmid contains two fluorescent proteins, mCherry and YFP, with or without a PTC in the intervening linker. In the presence of a stop codon in the linker, YFP becomes a long 3′ UTR, and this targets the transcript for NMD (Muhlrad and Parker 1999). Each of the two constructs is driven by either a constitutive (RPL4A) or a regulated (GALL) promoter. We transformed the plasmids into wild-type, upf1Δ, and dbp2Δ yeast and measured mCherry and YFP expression (Supplemental Fig. 4). The NMD effect is the ratio of the mCherry signal in the noPTC and PTC constructs. In wild-type cells, the effect of NMD is stronger in the TATA-less RPL4Apr plasmids, consistent with the gDNA library and native genes. Consistent with the computational RNA-seq screen, transcripts from the inducible GALL promoter are more strongly affected by NMD in a Δdbp2 strain (Fig. 3E). This suggests a model in which Dbp2 mutes the effect of codon bias and NMD on mRNA levels specifically for TATA-containing promoters.
For TATA-less promoters, growth rate–mediated regulation of expression is implemented in the coding sequence, not the promoter
Why implement regulation in coding sequences? In response to changes in growth rate, cells alter the expression of thousands of genes (Brauer et al. 2008; García-Martínez et al. 2016). In yeast, expression of translation-related genes is positively correlated with growth rate, while genes associated with environmental stress response, respiration, and oxidative phosphorylation are negatively correlated with growth rate (Fig. 4A; Supplemental Table 2; Brauer et al. 2008). TATA-less genes tend to be positively correlated with growth rate, while TATA-containing genes are negatively correlated (Fisher's exact test P = 10−27, odds ratio = 4.0). To determine if the information encoding regulation of expression in response to changes in growth rate is in the promoter, we took advantage of a data set in which promoter-YFP expression data for 859 genes were measured in 10 different environmental conditions (Keren et al. 2013). For each gene we calculated the slope, how promoter-YFP expression changes with growth rate (Fig. 4A), and compared it to the change in steady-state mRNA levels upon changes in growth. We find that changes in the mRNA levels of constitutive genes (TATA-less) are less accurately predicted by promoter-YFP data (Fig. 4B). Therefore, for TATA-less genes, information encoding growth rate–mediated regulation of expression lies outside of the promoter. To determine if the information that regulates expression with growth rate is encoded in the ORF, we asked if ORF-encoded sequence features can predict how mRNA levels change as a function of growth rate. We find that ORF features predict growth-mediated changes poorly for TATA genes and better for TATA-less genes (Fig. 4C). For constitutive genes, there is more regulatory information in the ORF than in the promoter (Fig. 4D,E).
Figure 4.

ORF features regulate expression in response to changes in growth rate. (A) Brauer et al. (2008) grew yeast at six different growth rates in each of six different environments and measured how the expression of each gene changed as a function of growth rate (slope GR-mRNA). TATA-less genes (blue; N = 1088) increase in expression, while those with TATA boxes (red; N = 401) more often decrease. The P-value is from a Fisher's exact test. (B) Keren et al. (2013) measured promoter-YFP expression at different growth rates. Each point shows the slope (expression change as a function of growth rate) for promoter-YFP (y-axis) and steady-state mRNA (x-axis) for TATA (red) or TATA-less (blue) genes. (C) GR-mRNA was predicted from ORF features allowing us to calculate the slope between growth rate and the contribution of ORF to expression (GR-ORF). Shown is the relationship between GR-ORF and GR-mRNA (see A), for each type of genes. (D) GR-mRNA was predicted from both GR-promoter and ORF features (GR-(ORF + promoter)). Shown is the relationship between this and GR-mRNA, for each type of genes (E) R2 values for a model that predicts GR-mRNA from GR-promoter, ORF features, or ORF + promoter for each type of genes. Error bars are standard deviation, calculated with bootstrapping.
Discussion
We used a pooled library of random mRNAs under the control of either the TATA+ inducible GALL promoter or the TATA-less constitutive RPL4A promoter to identify features inside of transcripts that exhibit quantitatively different effects on steady-state mRNA levels as a function of promoter architecture. Our observation that codon usage affects mRNA levels when we use fragments of the yeast genome shows that naturally existing variation in codon bias among native yeast genes directly affects mRNA expression. The fact that our predictions of mRNA levels are not perfect (R2 < 1) means that there is still much to be understood regarding the exact molecular mechanisms by which this occurs. The storage of public data and metadata in a standardized format allowed us to identify the mechanism by which the effect of sequences in the ORF are modulated by promoter architecture. Overall, the better and more quantitative data become, the clearer it is how little in gene regulation we truly understand (Cohen 2017).
Genetic screens involving targeted and random mutation in both cis and trans regulatory elements have played a major role in defining the machinery that controls gene expression (Struhl 1995; Sharon et al. 2012). However, the global output of all biomedical research is exponentially greater than the ability of any single laboratory. In the genomic era, naturally existing genetic variation has been utilized to understand gene regulation (Rockman and Kruglyak 2006; Lindeboom et al. 2016). In this era of publically available data, we are rapidly approaching a point at which functional genomic data will be available for almost all mutants in a wide variety of conditions. Here we show that publically available expression data can be used to discover molecular mechanisms underlying entirely novel types of gene regulation.
How gene expression is regulated was recognized as a problem prior to the identification of DNA as the genetic material (Avery et al. 1944; Monod 1947). Understanding how expression levels are encoded in the genome and how genetic variation affects expression has been one of the biggest challenges in molecular biology for the past 50 years (Beer and Tavazoie 2004; Rockman and Kruglyak 2006). Much of this work has focused on noncoding regions, specifically on the ability of TFs to bind distinct regulatory motifs in promoters and enhancers. The limited work understanding the regulatory potential of coding sequences has been done in an isolated manner, without considering the other regulatory regions of the gene (Kudla et al. 2009; Puchta et al. 2016; Radhakrishnan et al. 2016). Our results here show that coding sequences contain a large amount of regulatory information that affects gene expression and that this information interacts with information in the promoter architecture. Experimental systems and computational analyses that treat regulatory units in isolation miss molecular mechanisms that affect the expression of thousands of genes.
Why do organisms implement regulation at the level of the ORF? Measurements of steady-state mRNA levels from cells proliferating at different rates suggested that thousands of genes are regulated in response to changes in growth rate in both yeast (Brauer et al. 2008) and mammalian cells (M Badia, S Generoso, B Payer, LB Carey, in prep.). However, promoter activity, as measured by promoter-YFP signal, scales with growth rate similarly for most genes, suggesting little or no promoter-specific regulation with growth rate (Keren et al. 2013). Here we show that information encoded in the ORF and interpreted in a manner that depends on the promoter architecture and on the RNA helicase Dbp2 resolves this conflict. Constitutive genes, which are the majority of genes in yeast, are regulated mostly by sequences encoded in the ORF. Why? Translation-mediated regulation is less noisy than that mediated by TFs (Carey et al. 2013). Furthermore, it may be mechanistically easier to make coordinated quantitative changes in the mRNA levels of thousands of genes in a post-transcriptional manner than through the use of TFs. Therefore, implementing global regulation at the level of the ORF may be a more reliable and efficient means to coordinate changes in expression across thousands of genes.
To implement biologically useful regulation via coding sequences, the regulation needs to be targeted to some genes and not others. One way is to have large sequence composition differences between groups of genes. A second way is to somehow insulate some genes but not others from this mechanism of regulation. Dbp2 is well positioned to do the latter. As an RNA helicase, it is likely to be relatively sequence-neutral compared with many RNA binding proteins. It remains to be identified if Dbp2's helicase property enables regulation based on differences in secondary structure, as was recently observed for Dhh1 (Jungfleisch et al. 2017). In addition, Dbp2 associates with chromatin in a nascent RNA–dependent manner and is cotranscriptionally loaded onto mRNAs (Cloutier et al. 2012; Ma et al. 2016). Dbp2 is required for multiple stages of cotranscriptional mRNP assembly. This raises the possibility that Dbp2 specificity is encoded in the promoter via selective loading of Dbp2 onto certain classes of transcripts, such as those with TATA boxes. This would in turn regulate mRNP assembly and nuclear export. Dbp2 co-IPs with RPL2A (ribosomal 60S subunit protein L2A), TIF4631 (the translation factor eIF4G), and the SEC13 (nuclear pore complex) (Gavin et al. 2002), suggesting that Dbp2 may remain bound to the mRNA throughout its lifecycle, directly linking transcription initiation with translation.
Methods
Library plasmids construction
All plasmids are named and described in Supplemental Table 3. To generate the random gDNA fragment library, we used as a vector backbone the plasmid pRS416 carrying the URA3 selectable marker. We cloned into pRS416 the GALL inducible promoter and CYC1 transcriptional terminator, generating P47. The GALL promoter is a version of the GAL1 promoter truncated to remove the two most distal Gal4 binding sites and has ∼10% of the promoter activity as the fully intact GAL1 promoter (Mumberg et al. 1994). We next used Gibson assembly (NEB, E2611S) to insert the ATG-ClaI-Stop fragment (Supplemental Fig. 1A). This fragment was comprised of two annealed oligonucleotides, 174 and 175 (see Supplemental Table 4) in 10× oligo annealing buffer (400 µL 1M Tris at pH 8, 80 µL 0.5 M EDTA at pH 8, 800 µL 2.5 M NaCl, 2720 µL DNase/RNase-free water) in a reaction containing top- and bottom-strand DNA oligos (200 µM each), 2 µL of 10× oligo annealing buffer, and DNase/RNase-free water for a total volume of 20 µL. After incubation for 4 min at 94°C, we allowed the reaction to cool to room temperature for 10 min. To obtain the RPL4Apr plasmid (P86), we amplified using Phusion polymerase (Thermo Fisher Scientific, F530S) in GC buffer the RPL4A promoter from genomic DNA using primers 222 and 223 (Supplemental Table 4) and cloned the amplicon via Gibson assembly (NEB, E2611S) into the SacI and XbaI restriction sites of plasmid P47.
Library construction
The random yeast genomic sequences were obtained by cutting 40 µg of isolated genomic DNA (MasterPure yeast DNA purification kit, Epicentre, MPY80200) from yeast strain FY4, a prototrophic S288C strain, for 4 h at 37°C with a combination of four restriction enzymes (TaqαI, HpaII, MspI, and AciI). All restriction enzymes used in this study are from New England Biolabs (NEB). Size selection was performed using a 2.5% agarose gel and purified DNA was ligated into ClaI cut and dephosphorylated (rSAP, NEB, M0371S) vector (P47). Ligated products were desalted by drop dialysis using 13-mm-diameter Type-VS Millipore membrane (Merck Millipore, VSWP01300), and 3 µL was transformed by electroporation using the UltraClone kit with 10G elite DUO (Lucigen, 60117-1). Escherichia coli transformants were selected on LB medium (0.5% yeast extract, 1% NaCl, 15% bactotryptone) supplemented with 100 µg/mL ampicillin as a selection marker.
To obtain the RPL4Apr plasmid library with the same inserts that are present in the GALLpr library, we amplified with Q5 DNA polymerase reaction mix (NEB, M0491S) the library previously cloned into P47 using primers 345 and 198 (see Supplemental Table 4). After cutting pRS416-RPL4Apr (P86 in Supplemental Table 3) with XbaI and XhoI restriction enzymes, we cloned the PCR library fragments into pRS416-RPL4Apr via Gibson assembly (NEB, E2611S), obtaining the new promoter library that was transformed in E. coli electrocompetent cells (UltraClone Kit with 10G elite DUO, Lucigen, 60117-1) and plated on LB medium (0.5% yeast extract, 1% NaCl, 15% bactotryptone) supplemented with 100 µg/mL ampicillin as a selection marker. The constitutive and inducible library plasmids were purified using the NucleoBond XtraMidi plus EF (Machery-Nagel, 740422.50).
Yeast transformation
The gDNA libraries cloned into P47 (GALL) and P86 (RPL4A) were transformed into BY4741 (Y49) (Supplemental Table 3) via the lithium acetate method (Gietz and Woods 2006) and plated in glucose synthetic complete dropout plates lacking uracil for plasmid selection. In the same way, we transformed the GALL inducible library of the yeast native ORF in the mutant strains Y194 and Y195 (Supplemental Table 3). After 2 d of growth at 30°C, we collected all the transformants, and we resuspended them to OD of 0.5 in synthetic complete dropout plates lacking uracil supplemented with 2% galactose as a carbon source in order to get the GALL promoter induction. After growing the cells overnight, we scraped off the plates and inoculated four 500-mL flasks of the same medium to OD of 0.025 as biological replicates. The cultures were grown ∼10 h at 30°C to reach an exponential phase with an OD of 0.5, where we took 1.5-mL samples in order to extract plasmid DNA (MasterPure yeast DNA purification kit, Epicentre) and transcribed RNA (MasterPure yeast RNA purification kit, Epicentre) from all the biological replicas. We pelleted at 4°C and froze every sample at −80°C until processing.
NGS sample preparation
We used a gene-specific primer to generate the cDNA from each RNA preparation (Supplemental Fig. 1, primer 198 depicted in yellow; see Supplemental Table 4) using the ThermoScript RT-PCR System (Life Technologies, 11146-024). With barcoded primers, we amplified the second DNA strand and the library plasmids DNA in order to differentiate between replicas and sample origin, in this case, DNA and RNA (for details of barcoded primers combination for the libraries, see Supplemental Tables 4, 5). We note that because we made cDNA using a primer in the CYC1 terminator, any insert that results in transcription termination would give little or no mRNA expression in our assay. The PCR was set up as a 50-µL reaction of Q5 DNA polymerase (NEB, M0491S) in the following cycling conditions: for 30 sec at 98°C, for 10 sec at 98°C, for 30 sec at 55–64°C (depending on primer combination for demultiplexing on technical variability and biological replicates), and for 30 sec at 72°C (20 cycles) and 2 min at 72°C. PCR products were purified using MinElute PCR purification kit (Qiagen, 28004). NGS libraries were prepared from 100 ng of the purified DNA amplicons using an Ovation rapid DR system (Nugen, 0319-32) according to the manufacturer's instructions. Each library was visualized on a Bioanalyzer (Agilent Technologies) and quantified by qPCR with a Kapa Library quantification kit (Kapa Biosystems, KK4835). The libraries were sequenced through Illumina HiSeq 2500 125-bp paired-end reads method.
NMD reporter plasmids construction and measurement
The four NMD reporter constructs GALLpr-mCherry-PTC-YFP, GALLpr-mCherry-linker-YFP, RPL4Apr-mCherry-PTC-YFP, and RPL4Apr-mCherry-linker-YFP (P71, 68, 74, and P78, respectively) (for details, see Supplemental Table 3) were generated by fusion PCR reaction with overlapping oligos amplifying mCherry with primers 538 and 539 (in case of stop codon presence in the linker between mCherry and YFP) or primers 538 and 540 (in case of stop codon absence in the linker between mCherry and YFP). YFP was amplified with primers 541 and 542 (for primers details, see Supplemental Table 4). For all PCR reactions, Phusion polymerase (Thermo Fisher Scientific, F530S) in GC buffer was used. After fusion of both fragments, the final product was purified using MinElute PCR purification kit (Qiagen, 28004) and cloned in the ClaI cut and dephosphorylated with rSAP (New England Biolabs, M0371S) pRS416-GALLpr and pRS416-RPL4Apr vectors (P47 and P86 in Supplemental Table 3). The four NMD reporter plasmids were transformed into Y49, and multiple transformants of each construct were inoculated into 96-well plates containing SCGal-URA media. Cells were grown overnight, diluted 1:50, and measured by flow cytometry after 15 h of growth, when the OD600 was around 0.5. Wells in which the cell density was too high or too low were discarded. All flow cytometry was performed on a BD LSRFortessa (BD Biosciences) with 488-nm and 561-nm lasers with 530/28 and 610/20 filters for YFP or mCherry, respectively. Analysis of flow-cytometry data was performed using a previously described custom MATLAB pipeline (Carey et al. 2013). These two promoters have approximately equal expression levels (Supplemental Fig. 4). The same procedure was followed for the constructs in Y194 and Y401 strains.
Sequencing data processing
Samples and replicates for the library were all compact in FASTQ files with 125-bp paired-end reads. The first step in processing the reads was removing 3′ Illumina adapters of those reads where the inserted ORF was <125 bp. Next, we took advantage of the oligos used during the PCR for the sequencing library preparation to demultiplex each sample and replicate (split reads by type of sample origin—DNA or RNA—and replicates 1 or 2). Then the oligos were trimmed from the read using Cutadapt (Martin 2011), leaving part of the unique genomic DNA sequence that was inserted in the plasmid as an ORF. To obtain the entire gDNA sequence inserted, we mapped all trimmed reads to the yeast genome using Bowtie 2 (Langmead and Salzberg 2012). Hits where the forward and reverse reads did not map uniquely, mapped to different chromosomes, the same strand, or the distance between them was >1 kb, were directly discarded. The rest of the hits were used to obtain the complete gDNA fragment used in the library and the number of reads mapped used as a quantification measure of the number of DNA or RNA molecules. For downstream analysis, we attached the 5′ and 3′ constant flanking sequence of the plasmid (which contained the designed ATG and stop codons) for later quantifying other features, such as 3′ UTR length (measured as the length from the first stop codon in the ORF to the start of the CYC1 transcription terminator) (Supplemental Table 6).
Expression values and data analysis
After obtaining the number of reads per unique sequence for each sample and replica, we first normalized read counts by the total amount of reads per sample and replica using the formula
where aij represents the read count for a unique sequence variant per replica and sample. S is the number of samples (RNA, DNA, and technical replicas) and R the total number of unique transcripts. In order to normalize by technical and biological bias we used as values of expression the log2 ratio between the averaged replicas DNA reads and RNA reads:
where ε represents a pseudocount (ε = 0.5). In order to normalize the measurements across all libraries, we calculated expression as the z-score value of this last log2 ratio.
It is possible that the effect of codon bias may be partially cotranscriptional; there is good experimental evidence that GC content affects the rate of mRNA synthesis, as well as limited evidence that codon bias itself does so (Newman et al. 2016; Zhou et al. 2016). To avoid possibly confounding effects, we regressed out the contribution of insert length and GC content (see Methods). This allowed us to measure the GC and length-independent role of 3′ UTR and tAI to expression.
Model to predict expression in the ORF library using inserts features
To build a multiple linear regression model to predict expression values based on the composition of the ORF (Supplemental Fig. 3 illustrates all steps followed), we used already described transcript features (tAI, 3′ UTR length, GC content, and transcript length) and others of unknown mechanism. From the subset of fragments without PTCs that are fully contained within native yeast ORFs, the directionality of the inserted fragment had a significant impact on expression (fragments oriented in the same direction as the native gene had higher expression that reverse oriented ones, t-test P < 10−11). Since the genomic orientation of the inserted ORF affects expression but the mechanism is unknown, we used a machine learning approach to identify the ORF features that better discriminate between forward- or reverse-oriented sequences to later include them in the final model to predict expression. We built three different classifier models (logistic regression, naïve Bayes, and TreeBagger) based on more than 4000 sequence features (bases counts, amino acid counts, codon counts, frequency of nucleotides in different position of codons, hexamers…). All three classifiers were trained on one-third of the data, and the number of features of each model was reduced to dozens by sequential feature reduction (“sequentialfs” MATLAB function). The three classifiers performed correctly (median AUC = 0.80) when tested in the remaining two-thirds of the data set. To determine what features could predict expression, we took all the selected features from the classifiers and used them in a linear model to predict expression. By using LASSO (Tibshirani 1996) to further reduce the number of predictors, we obtained five sequence features that could explain 11% of the variance in expression: frequency of A or G in the first position of the codon (AG1), A or C in the second position of the codon (AC2), frequency of A in the second position of the codon (A2), and the frequency of appearance of two the hexamers GAAAGA and ACGTTA. In the final model to predict expression of all variants in our library, we included the three features AG1, AC2, and A2 as these are likely to be functionally interpreted by the translating ribosome inside the cell. For all model predictions, we used a 10-fold cross-validation scheme to assess the accuracy of all models and to test for overfitting.
Predicting mRNA level of native genes based on ORF features
To measure the contribution of the coding region to changes in expression, we used a linear regression model that included exclusively ORF features selected previously using the gDNA library. 3′ UTR length and PTC features were not used for models for native genes. We used 10-fold cross-validation to test the accuracy of the model. mRNA expression levels are from van Dijk et al. (2015). All mRNA-seq data sets give essentially the same result; this can be seen in Figure 3B, which shows results from a more limited ORF sequence feature model that includes just a single predictor. The same model was used to predict the expression of genes for which the promoter contribution to expression was previously measured (Keren et al. 2013).
Yeast NMD strength analysis
To measure NMD strength for native yeast genes wild-type (SRR1258470, SRR1258471) and upf1 (SRR1258533,SRR1258534) RNA-seq reads were downloaded from NCBI SRA (PRJNA245106) (Smith et al. 2014). Expression was quantified using kallisto (Bray et al. 2016) (--single -l 180 -s 20) and orf_coding_all.fasta from SGD (yeast genome R64-2-1) (Cherry et al. 2012). For each gene, the NMD effect was defined as log2[mean(expression upf1)/mean(expression WT)]. To work in a parameter regime comparable to that of the gDNA library, only genes with 3′ UTR lengths (Nagalakshmi et al. 2008) from 200–400 nucleotides were analyzed.
Digital expression explorer analysis
We obtained the processed data of 6402 RNA-seq data set from the Digital Expression Explorer (DEE) database (Ziemann et al. 2015). Expression was calculated normalizing sequence read counts by transcript length and sum of reads in each experiment. The effect of codon bias on expression was inferred for each experiment as described in the Results (Fig. 2F). To ensure a meaningful calculation of the effect of codon bias (that assumes promoter-YFP data (Keren et al. 2013) to be correlated with mRNA levels), we removed experiments with a low correlation between expression and promoter-YFP data (correlation < 0.4). We next searched for experiments in which the promoter-dependent effect changes from the “wild-type” behavior. We grouped the expression data sets by experiment (BioProject; each setup corresponds to a different work) and graphed each of them separately in the same space (TATA/TATA-less versus “effect of codon bias on expression”). We manually looked for BioProjects in which a subset of similar samples (e.g., biological replicates of a mutant strain) differs consistently from another subset (e.g., wild-type strains).
Data access
DNA-seq and RNA-seq data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE100452. All code for figures and analysis are on GitHub (https://github.com/MikiSchikora/Promoter_architecture_Espinar17/) and available as Supplemental_Code.zip.
Supplementary Material
Acknowledgments
L.B.C. was supported by Ministerio de Economía y Competitividad (MINECO) (BFU2015-68351-P), AGAUR (2014SGR0974), and the Unidad de Excelencia María de Maeztu, funded by the MINECO (MDM-2014-0370). We thank Wenfeng Qian, Shaohuan Wu, Aaron New, Francesc Posas, and members of the Barcelona Yeast Group and the CRG/UPF Computational Genomics and Systems Biology groups for helpful comments on the results and manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.230458.117.
References
- Akashi H. 2003. Translational selection and yeast proteome evolution. Genetics 164: 1291–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A. 2004. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 432: 112–118. [DOI] [PubMed] [Google Scholar]
- Avery OT, Macleod CM, McCarty M. 1944. Studies on the chemical nature of the substance inducing transformation of pneumococcal types: induction of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type III. J Exp Med 79: 137–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basehoar AD, Zanton SJ, Pugh BF. 2004. Identification and distinct regulation of yeast TATA box-containing genes. Cell 116: 699–709. [DOI] [PubMed] [Google Scholar]
- Beck ZT, Cloutier SC, Schipma MJ, Petell CJ, Ma WK, Tran EJ. 2014. Regulation of glucose-dependent gene expression by the RNA helicase Dbp2 in Saccharomyces cerevisiae. Genetics 198: 1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer MA, Tavazoie S. 2004. Predicting gene expression from sequence. Cell 117: 185–198. [DOI] [PubMed] [Google Scholar]
- Behm-Ansmant I, Kashima I, Rehwinkel J, Saulière J, Wittkopp N, Izaurralde E. 2007. mRNA quality control: an ancient machinery recognizes and degrades mRNAs with nonsense codons. FEBS Lett 581: 2845–2853. [DOI] [PubMed] [Google Scholar]
- Brauer MJ, Huttenhower C, Airoldi EM, Rosenstein R, Matese JC, Gresham D, Boer VM, Troyanskaya OG, Botstein D. 2008. Coordination of growth rate, cell cycle, stress response, and metabolic activity in yeast. Mol Biol Cell 19: 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun KA, Dombek KM, Young ET. 2015. Snf1-dependent transcription confers glucose-induced decay upon the mRNA product. Mol Cell Biol 36: 628–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L. 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34: 525–527. [DOI] [PubMed] [Google Scholar]
- Bregman A, Avraham-Kelbert M, Barkai O, Duek L, Guterman A, Choder M. 2011. Promoter elements regulate cytoplasmic mRNA decay. Cell 147: 1473–1483. [DOI] [PubMed] [Google Scholar]
- Carey LB, van Dijk D, Sloot PMA, Kaandorp JA, Segal E. 2013. Promoter sequence determines the relationship between expression level and noise. PLoS Biol 11: e1001528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Li K, Cao W, Wang J, Zhao T, Huan Q, Yang YF, Wu S, Qian W. 2017. Codon-resolution analysis reveals a direct and context-dependent impact of individual synonymous mutations on mRNA level. Mol Biol Evol 34: 2944–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, Christie KR, Costanzo MC, Dwight SS, Engel SR, et al. 2012. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res 40: D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier SC, Ma WK, Nguyen LT, Tran EJ. 2012. The DEAD-box RNA helicase Dbp2 connects RNA quality control with repression of aberrant transcription. J Biol Chem 287: 26155–26166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen BA. 2017. How should novelty be valued in science? eLife 6: e28699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge WJ, O'Duibhir E, Lijnzaad P, van Leenen D, Groot Koerkamp MJ, Kemmeren P, Holstege FC. 2017. Molecular mechanisms that distinguish TFIID housekeeping from regulatable SAGA promoters. EMBO J 36: 274–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Reis M, Wernisch L, Savva R. 2003. Unexpected correlations between gene expression and codon usage bias from microarray data for the whole Escherichia coli K-12 genome. Nucleic Acids Res 31: 6976–6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvir S, Velten L, Sharon E, Zeevi D, Carey LB, Weinberger A, Segal E. 2013. Deciphering the rules by which 5′-UTR sequences affect protein expression in yeast. Proc Natl Acad Sci 110: E2792–E2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Martínez J, Troulé K, Chávez S, Pérez-Ortín JE. 2016. Growth rate controls mRNA turnover in steady and non-steady states. RNA Biol 13: 1175–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardin J, Yeasmin R, Yurovsky A, Cai Y, Skiena S, Futcher B. 2014. Measurement of average decoding rates of the 61 sense codons in vivo. eLife 3: e03735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin A-C, Bösche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon A-M, Cruciat C-M, et al. 2002. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415: 141–147. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Woods RA. 2006. Yeast transformation by the LiAc/SS carrier DNA/PEG method. Methods Mol Biol 313: 107–120. [DOI] [PubMed] [Google Scholar]
- Haimovich G, Choder M, Singer RH, Trcek T. 2013. The fate of the messenger is pre-determined: a new model for regulation of gene expression. Biochim Biophys Acta 1829: 643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Jacobson A. 2015. Nonsense-mediated mRNA decay: degradation of defective transcripts is only part of the story. Annu Rev Genet 49: 339–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg JR, Goff SP. 2010. Upf1 senses 3′ UTR length to potentiate mRNA decay. Cell 143: 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisinga KL, Pugh BF. 2004. A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae. Mol Cell 13: 573–585. [DOI] [PubMed] [Google Scholar]
- Jungfleisch J, Nedialkova DD, Dotu I, Sloan KE, Martinez-Bosch N, Brüning L, Raineri E, Navarro P, Bohnsack MT, Leidel SA, et al. 2017. A novel translational control mechanism involving RNA structures within coding sequences. Genome Res 27: 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren L, Zackay O, Lotan-Pompan M, Barenholz U, Dekel E, Sasson V, Aidelberg G, Bren A, Zeevi D, Weinberger A, et al. 2013. Promoters maintain their relative activity levels under different growth conditions. Mol Syst Biol 9: 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kervestin S, Jacobson A. 2012. NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol 13: 700–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubik S, Bruzzone MJ, Shore D. 2017. TFIID or not TFIID, a continuing transcriptional SAGA. EMBO J 36: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla G, Murray AW, Tollervey D, Plotkin JB. 2009. Coding-sequence determinants of gene expression in Escherichia coli. Science 324: 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladomery M. 1997. Multifunctional proteins suggest connections between transcriptional and post-transcriptional processes. Bioessays 19: 903–909. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Hir H, Izaurralde E, Maquat LE, Moore MJ. 2000. The spliceosome deposits multiple proteins 20–24 nucleotides upstream of mRNA exon–exon junctions. EMBO J 19: 6860–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeboom RGH, Supek F, Lehner B. 2016. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat Genet 48: 1112–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma WK, Paudel BP, Xing Z, Sabath IG, Rueda D, Tran EJ. 2016. Recruitment, duplex unwinding and protein-mediated inhibition of the dead-box RNA helicase Dbp2 at actively transcribed chromatin. J Mol Biol 428: 1091–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J 17: 10–12. [Google Scholar]
- Meaux S, van Hoof A, Baker KE. 2008. Nonsense-mediated mRNA decay in yeast does not require PAB1 or a poly(A) tail. Mol Cell 29: 134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J. 1947. The phenomenon of enzymatic adaptation and its bearings on problems of genetics and cellular differentiation. Growth 11: 223–289. [Google Scholar]
- Muhlrad D, Parker R. 1999. Aberrant mRNAs with extended 3′ UTRs are substrates for rapid degradation by mRNA surveillance. RNA 5: 1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Müller R, Funk M. 1994. Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res 22: 5767–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman ZR, Young JM, Ingolia NT, Barton GM. 2016. Differences in codon bias and GC content contribute to the balanced expression of TLR7 and TLR9. Proc Natl Acad Sci 113: E1362–E1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neymotin B, Athanasiadou R, Gresham D. 2014. Determination of in vivo RNA kinetics using RATE-seq. RNA 20: 1645–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neymotin B, Ettore V, Gresham D. 2016. Multiple transcript properties related to translation affect mRNA degradation rates in Saccharomyces cerevisiae. G3 (Bethesda) 6: 3475–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JB, Kudla G. 2011. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet 12: 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presnyak V, Alhusaini N, Chen Y-H, Martin S, Morris N, Kline N, Olson S, Weinberg D, Baker KE, Graveley BR, et al. 2015. Codon optimality is a major determinant of mRNA stability. Cell 160: 1111–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchta O, Cseke B, Czaja H, Tollervey D, Sanguinetti G, Kudla G. 2016. Network of epistatic interactions within a yeast snoRNA. Science 352: 840–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Chen Y-H, Martin S, Alhusaini N, Green R, Coller J. 2016. The DEAD-Box protein Dhh1p couples mRNA decay and translation by monitoring codon optimality. Cell 167: 122–132.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF. 2012. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 483: 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman MV, Kruglyak L. 2006. Genetics of global gene expression. Nat Rev Genet 7: 862–872. [DOI] [PubMed] [Google Scholar]
- Shah P, Ding Y, Niemczyk M, Kudla G, Plotkin JB. 2013. Rate-limiting steps in yeast protein translation. Cell 153: 1589–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Carey L, Zeevi D, Sharon E, Keren L, Weinberger A, Dahan O, Pilpel Y, Segal E. 2013. Measurements of the impact of 3′ end sequences on gene expression reveal wide range and sequence dependent effects. PLoS Comput Biol 9: e1002934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sharon E, Lubliner S, Regev I, Lotan-Pompan M, Yakhini Z, Segal E. 2015. Systematic dissection of the sequence determinants of gene 3′ end mediated expression control. PLoS Genet 11: e1005147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon E, Kalma Y, Sharp A, Raveh-Sadka T, Levo M, Zeevi D, Keren L, Yakhini Z, Weinberger A, Segal E. 2012. Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat Biotechnol 30: 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skotheim JM, Di Talia S, Siggia ED, Cross FR. 2008. Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature 454: 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JE, Alvarez-Dominguez JR, Kline N, Huynh NJ, Geisler S, Hu W, Coller J, Baker KE. 2014. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Rep 7: 1858–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C, Breitkreutz B-J, Reguly T, Boucher L, Breitkreutz A, Tyers M. 2006. BioGRID: a general repository for interaction datasets. Nucleic Acids Res 34: D535–D539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K. 1986. Constitutive and inducible Saccharomyces cerevisiae promoters: evidence for two distinct molecular mechanisms. Mol Cell Biol 6: 3847–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K. 1995. Yeast transcriptional regulatory mechanisms. Annu Rev Genet 29: 651–674. [DOI] [PubMed] [Google Scholar]
- Taatjes DJ. 2017. The continuing SAGA of TFIID and RNA polymerase II transcription. Mol Cell 68: 1–2. [DOI] [PubMed] [Google Scholar]
- Tani H, Imamachi N, Salam KA, Mizutani R, Ijiri K, Irie T, Yada T, Suzuki Y, Akimitsu N. 2012. Identification of hundreds of novel UPF1 target transcripts by direct determination of whole transcriptome stability. RNA Biol 9: 1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R. 1996. Regression shrinkage and selection via the lasso. J R Stat Soc Series B Stat Methodol 58: 267–288. [Google Scholar]
- Tirosh I, Barkai N. 2008. Two strategies for gene regulation by promoter nucleosomes. Genome Res 18: 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk D, Dhar R, Missarova AM, Espinar L, Blevins WR, Lehner B, Carey LB. 2015. Slow-growing cells within isogenic populations have increased RNA polymerase error rates and DNA damage. Nat Commun 6: 7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffe AP, Meric F. 1996. Coupling transcription to translation: a novel site for the regulation of eukaryotic gene expression. Int J Biochem Cell Biol 28: 247–257. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Dang Y, Zhou M, Li L, Yu C-H, Fu J, Chen S, Liu Y. 2016. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc Natl Acad Sci 113: E6117–E6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zid BM, O'Shea EK. 2014. Promoter sequences direct cytoplasmic localization and translation of mRNAs during starvation in yeast. Nature 514: 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemann M, Kaspi A, Lazarus R, El-Osta A. 2015. Digital Expression Explorer: a user-friendly repository of uniformly processed RNA-seq data. ComBio 10.13140/RG.2.1.1707.5926 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.