

Abstract
Brain tumors typically arise sporadically and do not affect several family members simultaneously. In the present study, we describe clinical and genetic data from two patients, a mother and her daughter, with familial brain tumors. Exome sequencing revealed a germline missense mutation in the TP53 and ATRX genes in both cases, and a somatic copy-neutral loss of heterozygosity (LOH) in TP53 in both atypical teratoid/rhabdoid tumor (AT/RT) and astrocytoma tumors. ATRX mutation was associated with the loss of ATRX protein expression. In the astrocytoma case, R132C missense mutation was found in the known hotspot site in isocitrate dehydrogenase 1 (IDH1) and LOH was detected in TP53. The mother carried few other somatic alterations, suggesting that the IDH1 mutation and LOH in TP53 were sufficient to drive tumor development. The genome in the AT/RT tumor was atypically aneuploid: Most chromosomes had experienced copy-neutral LOH or whole-chromosome gains. Only Chromosome 18 had normal diploid status. INI1/hSNF5/SMARCB1 was homozygously deleted in the AT/RT tumor. This report provides further information about tumor development in a predisposed genetic background and describes two special Li–Fraumeni cases with a familial brain tumor.
Keywords: neoplasm of the central nervous system
INTRODUCTION
An atypical teratoid/rhabdoid tumor (AT/RT) is a highly malignant embryonal tumor of the brain occurring most often in infants. Regardless of intensive therapy, the outcome of the patients remains poor: the 5-yr overall survival rate is ∼30% (Fischer-Valuck et al. 2017). A mutation or deletion in the INI1/hSNF5/SMARCB1 gene occurs in the majority of AT/RT tumors, and this genetic alteration is the key finding in the diagnostic procedure (Louis et al. 2016). Recently, Johann et al. (2016) reported the existence of three epigenetic AT/RT subgroups. In this study, whole-genome DNA and RNA sequencing did not reveal any recurrent genomic aberrations beyond biallelic deletion of INI1. Thus, different genetic abnormalities were not able to explain the differences between the AT/RT subgroups (Johann et al. 2016).
Diffusely infiltrating astrocytomas are the most common primary brain tumors in adult populations. They are subdivided into low-grade diffuse astrocytomas (Grade II), anaplastic astrocytomas (Grade III), and glioblastomas (Grade IV). Anaplastic astrocytomas are the rarest type of diffusely infiltrating astrocytoma (Louis et al. 2016). The standard treatment modality for diffusely infiltrating astrocytomas is surgery combined with radiation and/or chemotherapy, but tumors acquire resistance rapidly. Mutations in isocitrate dehydrogenase 1 (IDH1) and IDH2 are found in >95% of Grade II–III gliomas and are associated with better patient prognosis than in IDH wild-type tumors (Louis et al. 2016). However, the 5-yr survival rate of IDH-mutated anaplastic astrocytomas is ∼55%–60% (Yan et al. 2009; Haapasalo et al. 2014).
Most brain tumors are sporadic and occur in patients with no known predisposing germline variants. Several genetic variants are known to cause predisposition to brain tumors, but these variants also increase the risk of other tumor types, and are rare in the human population. Such variants include hemizygous alterations of the neurofibromatosis 1 (NF1) (Gutmann et al. 2003), TP53 (Bougeard et al. 2015), as well as alterations in multiple DNA repair genes, such as mismatch repair (MMR) genes (Wei et al. 1997), X-ray cross-complementary genes (XRCC) (Kiuru et al. 2008), and O6-methylguanine-DNA methyltransferase (MGMT) genes (Felini et al. 2007). Germline mutations of the TP53 tumor suppressor gene on Chromosome 17p13.1 cause Li–Fraumeni syndrome (Malkin et al. 1990). The TP53 gene encodes a transcription factor (p53) that normally regulates the cell cycle and prevents damaged cells from dividing and forming tumors (Malkin et al. 1990). The Li–Fraumeni mutations can be inherited, or they can be de novo mutations arising early during embryogenesis or the development of germ cells in the parent. This syndrome has been shown to be associated with malignancies in a variety of tissue types (Iavarone et al. 1992; Sidransky et al. 1992; Nichols et al. 2001; Malkin 2011; Sorrell et al. 2013). Brain tumors, such as astrocytic tumors are possible (Guha and Malkin 2017), but there are no previous reports on AT/RT with Li–Fraumeni syndrome. Here, we report the exome sequences and detailed clinical history from two patients from the same family that have Li–Fraumeni syndrome. An anaplastic astrocytoma was diagnosed from the mother, whereas the child suffered from a rare AT/RT with complex copy-number alterations, including homozygous loss of INI1/hSNF5/SMARCB1 locus.
RESULTS
Clinical Presentation and Family History
The first case is a 2-yr-old girl, born at a gestational age of 33 wk with a birth weight of 1.8 kg. She exhibited slow motor development and left side weakness after birth and received regular physiotherapy for the first months of her life. The first symptom was asymmetrical convulsion during a fever at the age of 2. Two weeks later, the girl suffered from right-sided weakness, irritation, and tiredness in the mornings. Central nervous system (CNS) magnetic resonance imaging (MRI) revealed a cerebral, cystic, large tumor, but there were no signs of obstruction of the cerebrospinal fluid (CSF) flow (Fig. 1A,B). A left-sided craniotomy was performed under general anesthesia, and a microneurosurgical radical resection was performed. A postoperative cranial MRI (1 wk later) showed that most of the tumor was resected, and there was a 2-cm wide residual tumor in the anterior border of the resection cavity, which was resected. There were no new neurological deficiencies after the procedure. Neuropathological diagnosis was highly malignant AT/RT. The cellular marker for proliferation MIB-1, an antibody against Ki-67 antigen, index was 20%, immunohistochemical INI1 staining was negative (i.e., pathological) (Fig. 1C). The patient was treated with induction chemotherapy (methotrexate, vincristine, etoposide, cyclophosphamide, and cisplatin) followed by three cycles of high-dose chemotherapy (thiotepa, carboplatin) with autologous stem cell transplantation. New cranial and spinal MRI was performed 3 mo after the surgeries, but they did not reveal any evident residual or new tumors.
Figure 1.

(A) Timeline of the clinical courses and patient treatments. Chemotherapy: induction therapy using HD-MTX (high-dose methotrexate), VCR (vincristine), cisplatin, cyclophosphamide, and etoposide. (B) MRI of the primary AT/RT of the child. (L) left side, (R) right side. (C) Histology of the primary tumor of the child (200× magnification). Scale bar, 20 µm. (D) MRI of the primary tumor of the mother. (L) left side, (R) right side. (E) Histology of the primary anaplastic astrocytoma of the mother (200× magnification). Scale bar, 20 µm.
Nine months after diagnosis, a control cranial MRI revealed three recurrences in the upper medial and parasagittal region of the resection cavity (Fig. 1A). A recraniotomy and microneurosurgical tumor resection was performed under general anesthesia (Supplemental Fig. 1). A postoperative MRI showed a small residual tumor with contrast enhancement, and an immediate third tumor resection was performed. Local radiotherapy was given 50.4/1.8 Gy after the third surgery. The patient suffered from headaches and nausea, and two new extra-axial occipital tumors appeared a month after radiation as seen in the MRI. The patient received palliative care and died a little more than 1 yr after the diagnosis.
The second case (mother) was ∼30 yr old and was previously considered a healthy woman. Her mother had been diagnosed with carcinoma of the uterus in her 40s. The case had previously suffered from an episode of seizures years earlier, and valproic acid was initiated as an anticonvulsant medication. Two years later, she had another episode of symmetrical convulsions. An MRI of the CNS revealed a left-sided, frontal, cystic tumor, which was edemic and compressed the left lateral ventricle, but there were no signs of obstruction of the CNS fluid flow (Fig. 1D). Craniotomy with microneurosurgical radical resection was performed. Histopathological diagnosis was diffusively infiltrating astrocytoma Grade III (i.e., anaplastic astrocytoma). The tumor was weakly positive in IDH1 R132H-immunostaining (Fig. 1E) and the MIB-1 proliferation index was 5%. The mother received postoperative radiotherapy to tumor bed 59.4/1.8 Gy. Six control cranial MRIs during the 3-yr follow-up revealed no residuals or recidives. Six years after diagnosis, the patient was asymptomatic. As the child was diagnosed with AT/RT, genetic analyses were done because of family history. The results showed a germline TP53 mutation in both cases, which is known to cause Li–Fraumeni syndrome.
Predisposing Germline Variants
Exome-sequencing data were generated from blood-derived DNA samples from both patients (mother and child) to characterize additional predisposing germline mutations. As already observed with Sanger sequencing, both patients had a germline nonsynonymous single-nucleotide variant (SNV) in the TP53 gene (Table 1; Fig. 2A; Supplemental Table 1). It was a G>A substitution in the coding strand, leading to glycine>serine amino acid substitution (exon7:c.G733A, p.G245S in the transcript NM_000546). This variant has been previously linked to Li–Fraumeni, at least in sarcoma patients (Toguchida et al. 1992), and it is one of the most frequently reported TP53 missense mutations (7.4% of all) in the COSMIC database (http://cancer.sanger.ac.uk/cosmic).
Table 1.
Variant information for ATRX and TP53 germline variants, which were present in both cases
| Gene | Chromosome | HGVS coding DNA reference | HGVS protein reference | Variant type | Predicted effect | dbSNP/dbVar ID | Genotype | ClinVar ID |
|---|---|---|---|---|---|---|---|---|
| ATRX | Chr X:76856021 | NM_000489:exon23: c.5579A>G | NM_000489:exon23: p.N1860S (p.Asn1860Ser) | Nonsynonymous SNV | Asn1860Ser | rs45439799 | Heterozygous | VCV000011724.1 |
| TP53 | Chr 17:7577548 | NM_000546:exon7:c.733G>A | NM_000546:exon7:p.G245S (p.Gly245Ser) | Nonsynonymous SNV | Gly245Ser | rs28934575 | Heterozygous | VCV000012365.2 |
HGVS, Human Genome Variation Society; dbSNP, the Single Nucleotide Polymorphism database; dbVar the Database of Genomic Structural Variation.
Figure 2.

Both the mother and the child carried a germline mutation in both TP53 and ATRX genes. (A) The sequencing coverage and TP53 allele fraction information in blood and tumor samples as visualized in integrative genomics viewer (IGV). The green color represents the variant allele. Copy-neutral LOH was observed in all the tumor samples. The sequencing coverage range is indicated in square brackets. The exon, transcript base, and amino acid information in the major transcript (NM_000546) are marked above the figure. (Bottom) TP53 mutation is located at the DNA-binding domain. (B) The sequencing coverage and ATRX allele fraction information in blood and tumor samples as visualized in IGV. The orange color represents the variant allele. Copy-neutral LOH was observed in both AT/RT tumor samples. The exon, transcript base, and amino acid information in the major transcript (NM_000489) are marked above the figure. (Bottom) ATRX mutation is located at the SNF2N domain.
Quite surprisingly, both cases carried an ATRX SNV in their germline (Table 1; Fig. 2B; Supplemental Table 1). This was an A>G conversion in the coding strand, leading to asparagine>serine conversion in the amino acid sequence (exon23:c.A5579G, p.N1860S in the transcript NM_000489). The variant has been linked to X-linked α-thalassemia/mental retardation syndrome (Gibbons et al. 1995). The allele frequency of the A>G variant is 0.011 in the Finnish cohort in the ExAC database (Supplemental Table 1) (http://exac.broadinstitute.org/). Both tumors were ATRX-negative in the immunohistochemistry analysis (Fig. 1C,E).
Somatic Alterations
Tumor samples from both mother and child were analyzed with exome sequencing to reveal genetic alterations involved in tumor development. One freshly frozen sample was obtained from the mother during the first surgery and two tumor samples were obtained from the child: FFPE sample from the primary surgery and a freshly frozen sample from a relapse taken at 9 mo after initial diagnosis. Tumor purities were very high (>90%) in the child's tumor samples, whereas the predicted purity was <60% in the mother's tumor sample.
The mother had an R132C somatic IDH1 mutation, which is the second most common IDH mutation in diffuse gliomas (Supplemental Table 2; Hartmann et al. 2009). Allele fraction for this mutation was only 0.24, suggesting also rather low (∼50%) tumor purity. Additional missense mutations were detected in six other genes in the mother's tumor, but none of them have been shown to have any oncogenic role in diffuse gliomas. The proportion of the TP53 germline variant reads was increased in the mother's tumor sample (Fig. 2A; Supplemental Table 1), suggesting copy-neutral loss of the wild-type variant, which is commonly observed in Grade II–III diffuse gliomas (Hattori et al. 2016). This was indeed the case according to the copy-number analysis that utilized both sequence coverage and heterozygous allele frequencies in the germline (Fig. 3A; Supplemental Fig. 2). In addition to loss of heterozygosity (LOH) in TP53 and the surrounding region in 17p13, only a hemizygously deleted region was observed in the beginning of Chromosome X (Fig. 3A; Supplemental Fig. 2). No deletions in 1p or 19q were detected.
Figure 3.
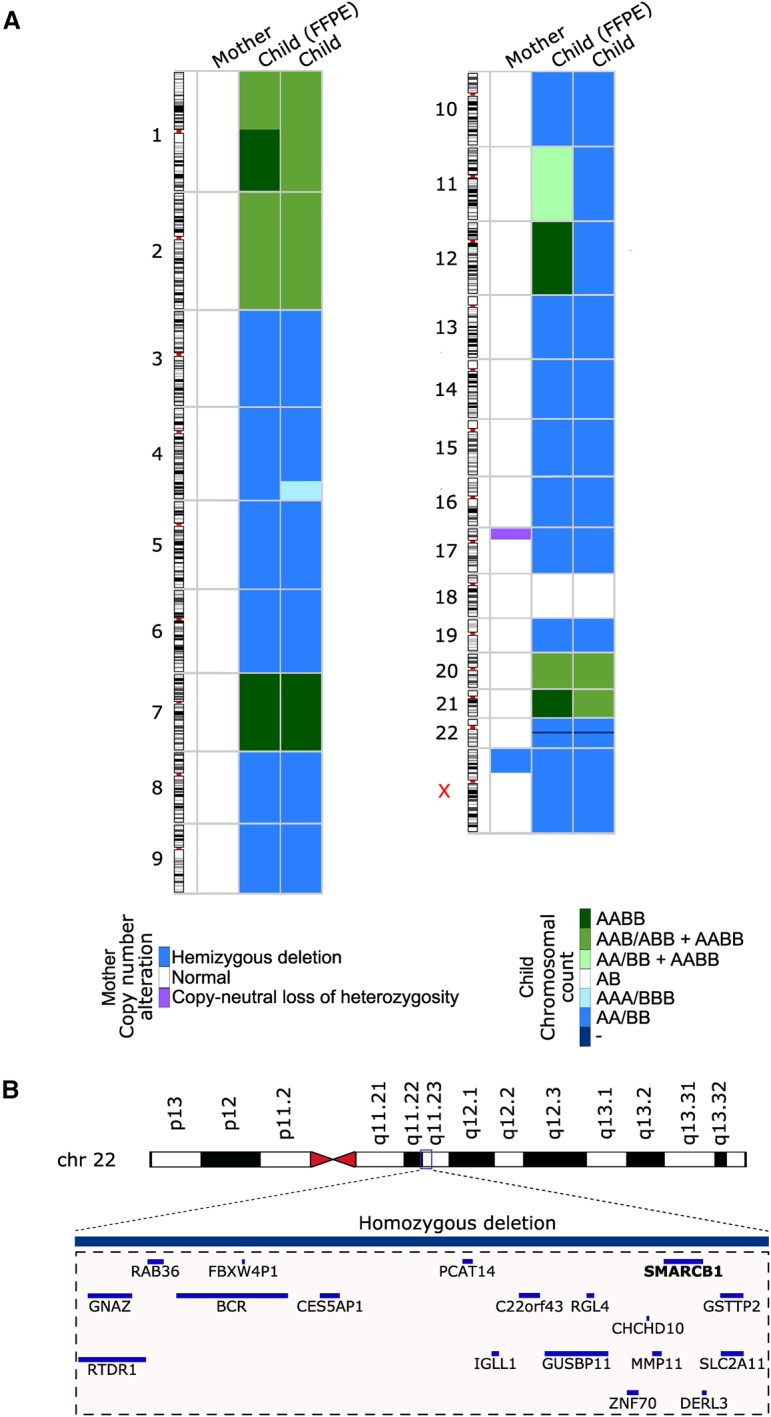
Somatic copy-number alterations in mother's and child's tumors. (A) Copy-neutral LOH was observed in most chromosomes in the child's tumor, but chromosome gains were also prevalent. In the mother, there was a copy-neutral LOH in Chr 17p.3 and a hemizygous deletion in Chr X. In the chromosomal counts figure for the child, A represents a genomic region inherited from the mother and B a genomic region inherited from the father. Normal chromosomal count is thus marked with AB and, for example, copy-neutral LOH in either paternal or maternal chromosomal region is marked with AA/BB. If a tumor contains two subclones with different counts for a genomic region, the chromosomal counts for each subclone are separated with a + sign. (B) A region in Chr 22q11.23 was homozygously deleted in both tumor samples obtained from the child. Genes falling within this region, including SMARCB1/hSNF5/INI1, are visualized in the figure.
The daughter's tumor exhibited biallelic loss of INI1/hSNF5/SMARCB1 on Chromosome 22 (Fig. 3A,B), supported by negative INI1 staining (Fig. 1C). Biallelic loss of INI1 is the only known recurrent alteration in AT/RT and is used as major criteria for AT/RT diagnosis (Louis et al. 2016). Surprisingly, the child's tumor harbored several additional copy-number alterations. Most of her tumor genome was occupied by whole-chromosome losses and/or gains (Fig. 3A; Supplemental Fig. 2). Copy-neutral LOH was observed in Chromosomes 3–6, 8–17, 19, 22, and X with a few exceptions: In the sample taken at diagnosis, Chr 12 was tetraploid, and there was a subclonal population of cells with tetraploid Chr 11. Furthermore, there was a third copy gain in the end 4q in the sample taken upon relapse. Both samples showed focal biallelic loss of INI1 locus. In addition to LOH regions, Chromosomes 1, 2, 7, 20, and 21 were either tetraploid or there was a combination of subclonal tetraploid and triploid chromosome counts. In the FFPE sample taken at diagnosis, the proportion of tetraploid subclone was ∼30% for all the other chromosomes, except Chr 11, where the tetraploid proportion was ∼20% of the tumor content. In the relapse sample, the proportion of tetraploid subclone was significantly smaller: ∼3% of the tumor content. Surprisingly, Chr 18 was the only chromosome with a normal diploid chromosome count in the child's tumors, which clearly differs from the typical AT/RT tumors that lack copy-number alterations outside Chr 22 (Louis et al. 2016). In addition, 13 chromosomes (Chromosomes 3, 4, 6, 8, 9, 10, 12, 13, 15, 17, 19, 22, and X) of 17 chromosomes with LOH were maternal; only Chromosomes 5, 11, 14, and 16 with LOH were inherited from the father (Supplemental Table 1). Furthermore, the child had 28 nonsynonymous mutations in her tumors: nine mutations were present in both tumor specimens, one only in the primary tumor, and 18 only in the recurrent tumor (Supplemental Table 2). None of the mutations had existing COSMIC IDs.
Our literature search revealed two genes, namely DOK3 and SH3GLB2, which were mutated in both AT/RT tumors and have been previously linked to tumor suppression in lung and breast cancer, respectively (Berger et al. 2010; Zhang et al. 2011). Furthermore, two putative tumor suppressor genes, NEK4 and KLF14, were mutated only in the recurrent AT/RT sample. NEK4 has been reported to regulate the entry into replicative senescence and DNA-damage-induced cell cycle arrest in fibroblasts (Nguyen et al. 2012), whereas KLF14 inhibits KRAS signaling and has demonstrated tumor suppressor activity (Tetreault et al. 2013; Fan et al. 2015).
DISCUSSION
Our results show that the mother and the child had a germline nonsynonymous SNV in the gene TP53. In addition, they both carried an ATRX SNV in their germline. As suspected, the mother had a somatic IDH1 mutation R132C and the daughter's tumor exhibited biallelic loss of INI1/hSNF5/SMARCB1.
The transcriptional regulator ATRX (α-thalassemia/mental retardation syndrome X-linked) protein is encoded by the ATRX gene, located in Xq21.1. ATRX contains an ATPase/helicase domain, and it belongs to the SWI/SNF family of chromatin remodeling proteins (Gibbons et al. 2000). ATRX directly interacts with DNA as well as with several functional partners, presumably by assembling with different chromatin remodeling or transcriptional regulatory complexes. In addition, reduced ATRX levels induce chromosome segregation defects (Mayes et al. 2014). Similarly, INI1 is a member of the SWI/SNF family and plays a role as a tumor suppressor in malignant rhabdoid tumors (Versteege et al. 1998). Furthermore, alternative lengthening of telomeres (ALT) is a mechanism by which ∼10%–15% of cancer cells can extend their telomeres without the enzyme telomerase. Loss of ATRX/DAXX function appears to be required for ALT (Amorim et al. 2016). Approximately 10%–15% of human cancers are suspected to use the ALT pathway, including a subset of nervous system tumors (e.g., medulloblastoma, low-grade diffuse gliomas, and ganglioneuroblastoma) (Lovejoy et al. 2012). By lengthening the telomere, ALT contributes to the unrestricted cell division that is a hallmark of cancer and likely makes cancer cells less vulnerable to chemotherapy or radiation therapy. These ALT-positive tumors use recombination to sustain immortality (Dilley and Greenberg 2015). Recent findings suggest that hampering ALT might be a potential new cancer treatment modality. ALT is disrupted by the inhibition of protein kinase ATR leading to cell apoptosis (Lovejoy et al. 2012). ATRX is located in Chromosome X, and thus one of its alleles is epigenetically silenced. Both our patients had ATRX variants in their germline. There are discordant reports about the pathogenicity of the detected ATRX variant in ClinVar, and the variant frequency was 1.1% in a Finnish cohort, which also suggests a lower pathogenicity. However, this variant was associated with the loss of ATRX protein expression in both tumors. In addition, ATRX staining was negative in the endovascular cells in the mother and in all the cells surrounding the blood vessels in the child, suggesting that the normal ATRX allele has been imprinted in these cells. Other germline mutations that are not covered by exome sequencing might also be responsible for the ATRX loss in the variant-containing locus. Nevertheless, our data show that ATRX loss or defect can be present also in nonmalignant cells, which might lead to undesired outcome after ATR inhibitor based treatments. Germline status and ATRX protein detection will be informative for ATRX-mutated cases if these treatment modalities become part of the clinical practice in the future.
The IDH mutation is frequently observed with TP53 and ATRX mutations in astrocytic tumors (Cai et al. 2014). IDH mutation is widely used as a clinical tool to sort glial tumors (Louis et al. 2016), and patients with IDH mutation have better outcomes than IDH wild-type diffuse glioma patients (Ichimura 2012). The mother carried a somatic IDH R132C mutation in her tumor. Antibodies raised against IDH1 R132H mutation gave a weakly positive signal in immunohistochemistry, which suggests cross-reactivity against R132C mutation. The mother's outcome remains stable after 6 yr, which is better than the average prognosis. The lack of other oncogenic alterations is somewhat surprising, but as both TP53 and ATRX harbored a pathogenic variant already in the germline, the IDH1 hotspot mutation might have been enough to trigger the development of a diffuse glioma. These three genes are the most commonly altered genes in 1p19q nondeleted IDH-mutated diffuse gliomas, such as anaplastic astrocytoma (Cancer Genome Atlas Research Network et al. 2015).
The complexity of copy-number changes observed in the child's tumor cells is striking. How could this kind of genome have arisen? Most likely there was a loss of one pair for most chromosomes, followed by genome-wide duplication of chromosome counts. The loss of one chromosome to generate triploid counts could have happened soon after in Chr 2 and Chr 20, and possibly in Chr 1p. The fact that there was a combination of both tetraploid and hemizygously deleted diploid Chr 11 chromosome counts in the child's first sample (child FFPE) suggests that the observed gains and losses took place sequentially, rather than simultaneously in all the chromosomes. The hemizygous loss of Chr 17 has generated a homozygous missense mutation in TP53, which has likely damaged the p53-dependent cell cycle checkpoint mechanisms (Vaziri et al. 2003). The fact that homozygous loss in Chr 22 has similar boundaries in both chromosomes suggests that the INI1 locus was fully deleted before the suggested genome-wide gain in chromosome counts. It is difficult to determine whether Chr 18 has remained diploid throughout all these genome rearrangements or whether one copy of both maternal and paternal chromosomes has been deleted after a tetraploid stage.
Li–Fraumeni is known to predispose humans to cancer but the question remains as to why both the mother and the child developed a brain tumor. It seems that the diffuse gliomas need other genetic alterations, such as IDH and ATRX mutations, for their development. The mother obtained somatic IDH1 mutation, and she carried an ATRX mutation in her germline. In AT/RT, the literature has revealed only one recurrent genetic mutation—namely, homozygous loss of INI1, which was also observed in the tumor of the child.
To our knowledge, this is the first study to show that Li–Fraumeni syndrome with accompanying somatic LOH in a patient with AT/RT. This highlights the fact that even in rare brain tumors, the anamnesis of family history is essential, and modern genetic analyses of tumors and heredity should be performed.
METHODS
Immunohistochemical Analysis and Pathological Evaluation
The tumor samples were fixed in 4% phosphate-buffered formaldehyde and processed into paraffin blocks. The sections were stained with hematoxylin and eosin (H&E). An experienced neuropathologist evaluated the tumor samples and determined the histopathological type and grade according to the criteria presented by the World Health Organization (WHO) (Louis et al. 2007). The tumor immunostaining for IDH1 was performed using R132H point mutation–specific mouse monoclonal antibody (Dianova GmbH) as previously described (Mäkelä et al. 2014). ATRX and INI1 immunostainings were performed using Ventana Benchmark GX immunoautomate (Ventana Medical Systems). DISCOVERY EZ Prep solution (# 950-102, Ventana Medical Systems) was used for deparaffinization (72°C). Epitope retrieval was performed at 95°C in Cell Conditioning Solution CC1 (# 950-124), for 92 min for ATRX detection and 32 min for INI1 detection. Antibody incubation duration was 60 min with a rabbit polyclonal anti-ATRX antibody (dilution 1:500, HPA0001906, Atlas antibodies) and 28 min with a mouse monoclonal antibody anti-INI-1 (dilution 1:200, Clone: 25/BAF47, 612110, B&D Laboratories), both at 36°C. Both immunostainings were detected and visualized using OptiView DAB IHC Detection Kit (# 760-7000, Ventana Medical Systems) together with Optiview Amplification kit (# 760-099, Ventana Medical Systems). All the washes were performed using Tris-based reaction buffer solution (# 950-300, Ventana Medical Systems). Nuclei were visualized with Hematoxylin II (#790-2208, Ventana Medical Systems).
Exome Sequencing
DNA was isolated from the blood samples using a QIAGEN isolation kit, from the frozen samples with a QIAamp DNA Mini kit (QIAGEN) with RNAse treatment, and from the FFPE sample with a turXTRAC FFPE DNA kit (Covaris). Library construction and exome sequencing were performed at Beijing Genomics Institute (BGI), Hong Kong. A SureSelect Human All Exon V5 (target size 50 M) kit (Agilent) was used for library preparation, and samples were sequenced with Illumina HiSeq2000 technology. The pursued sequencing depth was 50× for blood-derived samples and 100× for tumor samples (Supplemental Table 3).
Genome Alignment
The raw reads from the whole-exome sequencing experiment for each sample were aligned to the human reference genome (hg19) using Bowtie2 (version 2.2.5) (Langmead and Salzberg 2012) and converted to BAM format using SAMtools (version 0.1.19) (Li et al. 2009). Duplicate reads were marked and removed from the resulting BAM files using SAMBLASTER (version 0.1.21) (Faust and Hall 2014).
Somatic Copy-Number Aberration Analysis and Tumor Purity Estimation
Aligned reads were counted in 1000-bp windows across the entire genome using BEDTools-2.25.0 (Quinlan and Hall 2010). Coverage log ratios were calculated relative to a median reference derived from the two normal blood samples. GC fractions were calculated for all 1000-bp windows, and Loess regression was applied to coverage log ratios to correct for GC content bias. After GC bias correction, samples were corrected for differences in overall coverage using median-of-ratios normalization.
Heterozygous single-nucleotide polymorphisms (SNPs) were identified for each patient by searching their normal blood sample for genomic sites that displayed an allele fraction close to 50%. The alternate allele read count r was required to satisfy 0.05 ≤ BinCDF(r; n, 0.5) ≤ 0.95, and the total read count had to be at least 50. The list of heterozygous SNPs was further filtered to only include genomic sites annotated in the KAVIAR (Glusman et al. 2011) or ExAC databases (Lek et al. 2016). Polymorphic indels and SNPs within 10 bp of indels (both somatic and germline) were discarded from the list of heterozygous SNPs to mitigate alignment artifacts.
Tumor purities were estimated based on the observed allele fractions for the detected heterozygous germline SNPs located in regions with copy-neutral LOH in each tumor sample.
Germline and Somatic Variant Calling
Variants were called using an inhouse variant calling pipeline based on SAMtools mpileup output (Li et al. 2009). Variants were called heterozygous if, at a given locus, at least 20% of all reads showed the alternate allele or homozygous alternate if, at a given locus, at least 80% of all reads showed the alternate allele. Annotation for both germline and somatic variants was performed using ANNOVAR (Wang et al. 2010) in combination with custom scripts. Variants were further annotated with impact on protein coding, Cosmic ID (Bamford et al. 2004), and the SNP database (dbSNP) ID (Sherry et al. 2001). Furthermore, annotations included allele frequencies reported in 1000 Genomes (The 1000 Genomes Project Consortium 2010), the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP6500, unpublished; http://evs.gs.washington.edu/EVS/), SISu, and ExAC (Lek et al. 2016). To select somatic variants, we discarded the variants that were also detected in the respective germline sample. The rest of the somatic variants were manually inspected to remove sequencing artifacts.
We discarded germline variants if the population frequency of alternate alleles was >0.01 in either of the three following cohorts: all five populations of 1000 Genomes (The 1000 Genomes Project Consortium 2010), the Sequencing Initiative Suomi cohort (SISu, unpublished; http://www.sisuproject.fi/) that consists of exomes of 10,000 Finnish individuals, and all seven populations of the ExAC cohort (Lek et al. 2016). In addition, we devised a Bayesian scoring scheme that assigns each variant locus a score using the total, reference, and alternate allele read count information. The score represents the confidence that at least 1 (Bayes_score_1; Supplemental Table 1) or 2 (Bayes_score_2; Supplemental Table 1) of the samples have variation (i.e., heterozygous or homozygous alternate) in a given locus. All the germline variants with a Bayesian score of <0.9999999 or lack of allele frequency information in all the above reference cohorts were discarded.
ADDITIONAL INFORMATION
Data Deposition and Access
Genome data have been deposited at the European Genome-phenome Archive (EGA), which is hosted at the European Bioinformatics Institute (EBI) and the Centre for Genomic Regulation (CRG), under accession number EGAS00001002716. The variants were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession numbers SCV000693709–SCV000693712.
Ethics Statement
Patient consent was obtained in writing. The study was approved by the Finnish Committee of Ethics of Tampere University Hospital and by the National Supervisory Authority for Welfare and Health.
Acknowledgments
We thank Mr. Erkki Karttunen, Mrs. Satu Ranta, Mrs. Marita Nieminen, PhD, Juha Kesseli, and Ms. Paula Kosonen for their skillful technical assistance and Joanna Ilvesaro, PhD, for her expertise in immunohistochemical staining protocols. We acknowledge the personnel at Tampere University Hospital and Fimlab Laboratories for their contribution to sample collection and handling. The authors would also like to acknowledge the CSC–IT Center for Science, Ltd. (https://www.csc.fi/csc) for providing the computational resources.
Author Contributions
K.N. created all the consent forms and gathered the clinical data along with J.H., O.L., and P.H. H.H. has done the neuropathological assessments. E.A., M.A., and S.H. performed the computational analyses. K.J.G., J.T., J.H., and K.N. manually confirmed and interpreted the obtained results. K.N. and M.N. have provided financial support for the study. K.J.G., O.L., and M.N. supervised the project. All contributors took part in the writing and approved the manuscript.
Funding
This study was financially supported by grants from the Nona and Kullervo Väre Foundation (K.N.), Lea and Arvo Ylppö Foundation (K.N.), the Finnish Pediatric Research Foundation (K.N.), Tellervo Edessalo Foundation (O.L.), the Academy of Finland (grant 259038 to K.J.G., 269474 to M.N.), the Emil Aaltonen Foundation (M.A.), Päivikki ja Sakari Sohlberg Foundation (K.J.G.), Competitive State Research Financing of the Expert Responsibility area of Tampere University Hospital (J.H., H.H.; grants 9T042 and 9U041 to M.N.), and the Pirkanmaa Cancer Society.
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Amorim JP, Santos G, Vinagre J, Soares P. 2016. The role of ATRX in the alternative lengthening of telomeres (ALT) phenotype. Genes (Basel) 7: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR, et al. 2004. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer 91: 355–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AH, Niki M, Morotti A, Taylor BS, Socci ND, Viale A, Brennan C, Szoke J, Motoi N, Rothman PB, et al. 2010. Identification of DOK genes as lung tumor suppressors. Nat Genet 42: 216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougeard G, Renaux-Petel M, Flaman JM, Charbonnier C, Fermey P, Belotti M, Gauthier-Villars M, Stoppa-Lyonnet D, Consolino E, Brugières L, et al. 2015. Revisiting Li–Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol 33: 2345–2352. [DOI] [PubMed] [Google Scholar]
- Cai J, Yang P, Zhang C, Zhang W, Liu Y, Bao Z, Liu X, Du W, Wang H, Jiang T, et al. 2014. ATRX mRNA expression combined with IDH1/2 mutational status and Ki-67 expression refines the molecular classification of astrocytic tumors: evidence from the whole transcriptome sequencing of 169 samples. Oncotarget 5: 2551–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network, Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, et al. 2015. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 372: 2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilley RL, Greenberg RA. 2015. ALTernative telomere maintenance and cancer. Trends Cancer Res 1: 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G, Sun L, Shan P, Zhang X, Huan J, Zhang X, Li D, Wang T, Wei T, Zhang X, et al. 2015. Loss of KLF14 triggers centrosome amplification and tumorigenesis. Nat Commun 6: 8450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust GG, Hall IM. 2014. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30: 2503–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felini MJ, Olshan AF, Schroeder JC, North KE, Carozza SE, Kelsey KT, Liu M, Rice T, Wiencke JK, Wrensch MR. 2007. DNA repair polymorphisms XRCC1 and MGMT and risk of adult gliomas. Neuroepidemiology 29: 55–58. [DOI] [PubMed] [Google Scholar]
- Fischer-Valuck BW, Chen I, Srivastava AJ, Floberg JM, Rao YJ, King AA, Shinohara ET, Perkins SM. 2017. Assessment of the treatment approach and survival outcomes in a modern cohort of patients with atypical teratoid rhabdoid tumors using the National Cancer Database. Cancer 123: 682–687. [DOI] [PubMed] [Google Scholar]
- Gibbons RJ, Picketts DJ, Villard L, Higgs DR. 1995. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with α-thalassemia (ATR-X syndrome). Cell 80: 837–845. [DOI] [PubMed] [Google Scholar]
- Gibbons RJ, McDowell TL, Raman S, O'Rourke DM, Garrick D, Ayyub H, Higgs DR. 2000. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat Genet 24: 368–371. [DOI] [PubMed] [Google Scholar]
- Glusman G, Caballero J, Mauldin DE, Hood L, Roach JC. 2011. Kaviar: an accessible system for testing SNV novelty. Bioinformatics 27: 3216–3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha T, Malkin D. 2017. Inherited TP53 mutations and the LiFraumeni syndrome. Cold Spring Harb Perspect Biol 3: a026187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutmann DH, James CD, Poyhonen M, Louis DN, Ferner R, Guha A, Hariharan S, Viskochil D, Perry A. 2003. Molecular analysis of astrocytomas presenting after age 10 in individuals with NF1. Neurology 61: 1397–1400. [DOI] [PubMed] [Google Scholar]
- Haapasalo J, Hyartt A, Salmi M, Nordfors K, Lahtela SL, Kähkönen M, Helén P, Haapasalo H. 2014. Glioomien diagnoosi ja ennuste-molekyylidiagnostiikan mahdollisuudet. [Diagnosis and prognosis of gliomas—current prospects of molecular diagnostics]. http://uta32-kk.lib.helsinki.fi/bitstream/handle/10024/97277/glioomien_diagnoosi_2014.pdf?sequence=1. [PubMed] [Google Scholar]
- Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, et al. 2009. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 118: 469–474. [DOI] [PubMed] [Google Scholar]
- Hattori N, Hirose Y, Sasaki H, Nakae S, Hayashi S, Ohba S, Adachi K, Hayashi T, Nishiyama Y, Hasegawa M, et al. 2016. World Health Organization grade II-III astrocytomas consist of genetically distinct tumor lineages. Cancer Sci 107: 1159–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iavarone A, Matthay KK, Steinkirchner TM, Israel MA. 1992. Germ-line and somatic p53 gene mutations in multifocal osteogenic sarcoma. Proc Natl Acad Sci 89: 4207–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura K. 2012. Molecular pathogenesis of IDH mutations in gliomas. Brain Tumor Pathol 29: 131–139. [DOI] [PubMed] [Google Scholar]
- Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, Jones DTW, Sturm D, Hermann C, Segura Wang M, et al. 2016. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell 29: 379–393. [DOI] [PubMed] [Google Scholar]
- Kiuru A, Lindholm C, Heinävaara S, Ilus T, Jokinen P, Haapasalo H, Salminen T, Christensen HC, Feychting M, Johansen C, et al. 2008. XRCC1 and XRCC3 variants and risk of glioma and meningioma. J Neurooncol 88: 135–142. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. 2007. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114: 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, ed. 2016. WHO classification of tumours of the central nervous system WHO/IARC classification of tumours, 4th Ed. Rev. World Health Organization, Geneva, Switzerland. [Google Scholar]
- Lovejoy CA, Li W, Reisenweber S, Thongthip S, Bruno J, de Lange T, De S, Petrini JH, Sung PA, Jasin M, et al. 2012. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet 8: e1002772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkelä K, Nordfors K, Finne J, Jokilammi A, Paavonen T, Haapasalo H, Korja M, Haapasalo J. 2014. Polysialic acid is associated with better prognosis and IDH1-mutation in diffusely infiltrating astrocytomas. BMC Cancer 14: 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkin D. 2011. Li–Fraumeni syndrome. Genes Cancer 2: 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkin D, Li FP, Strong LC, Fraumeni JF Jr, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA. 1990. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250: 1233–1238. [DOI] [PubMed] [Google Scholar]
- Mayes K, Qiu Z, Alhazmi A, Landry J. 2014. ATP-dependent chromatin remodeling complexes as novel targets for cancer therapy. Adv Cancer Res 121: 183–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols KE, Malkin D, Garber JE, Fraumeni JF Jr, Li FP. 2001. Germ-line p53 mutations predispose to a wide spectrum of early-onset cancers. Cancer Epidemiol Biomarkers Prev 10: 83–87. [PubMed] [Google Scholar]
- Nguyen CL, Possemato R, Bauerlein EL, Xie A, Scully R, Hahn WC. 2012. Nek4 regulates entry into replicative senescence and the response to DNA damage in human fibroblasts. Mol Cell Biol 32: 3963–3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29: 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky D, Tokino T, Helzlsouer K, Zehnbauer B, Rausch G, Shelton B, Prestigiacomo L, Vogelstein B, Davidson N. 1992. Inherited p53 gene mutations in breast cancer. Cancer Res 52: 2984–2986. [PubMed] [Google Scholar]
- Sorrell AD, Espenschied CR, Culver JO, Weitzel JN. 2013. Tumor protein p53 (TP53) testing and Li–Fraumeni syndrome: current status of clinical applications and future directions. Mol Diagn Ther 17: 31–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetreault MP, Yang Y, Katz JP. 2013. Krüppel-like factors in cancer. Nat Rev Cancer 13: 701–713. [DOI] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toguchida J, Yamaguchi T, Dayton SH, Beauchamp RL, Herrera GE, Ishizaki K, Yamamuro T, Meyers PA, Little JB, Sasaki MS. 1992. Prevalence and spectrum of germline mutations of the p53 gene among patients with sarcoma. N Engl J Med 326: 1301–1308. [DOI] [PubMed] [Google Scholar]
- Vaziri C, Saxena S, Jeon Y, Lee C, Murata K, Machida Y, Wagle N, Hwang DS, Dutta A. 2003. A p53-dependent checkpoint pathway prevents rereplication. Mol Cell 11: 997–1008. [DOI] [PubMed] [Google Scholar]
- Versteege I, Sévenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O. 1998. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394: 203–206. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Bondy ML, Mao L, Gaun Y, Cheng L, Cunningham J, Fan Y, Bruner JM, Yung WK, Levin VA, et al. 1997. Reduced expression of mismatch repair genes measured by multiplex reverse transcription-polymerase chain reaction in human gliomas. Cancer Res 57: 1673–1677. [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. 2009. IDH1 and IDH2 mutations in gliomas. N Engl J Med 360: 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Brewster A, Guan B, Fan Z, Brown PH, Xu XC. 2011. Tumor-suppressor activity of RRIG1 in breast cancer. BMC Cancer 11: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.