

Abstract
Well-differentiated (WD) liposarcoma is a low-grade mesenchymal tumor with features of mature adipocytes and high propensity for local recurrence. Often, WD patients present with or later progress to a higher-grade nonlipogenic form known as dedifferentiated (DD) liposarcoma. These DD tumors behave more aggressively and can metastasize. Both WD and DD liposarcomas harbor neochromosomes formed from amplifications and rearrangements of Chr 12q that encode oncogenes (MDM2, CDK4, and YEATS2) and adipocytic differentiation factors (HMGA2 and CPM). However, genomic changes associated with progression from WD to DD have not been well-defined. Therefore, we selected patients with matched WD and DD tumors for extensive genomic profiling in order to understand their clonal relationships and to delineate any defining alterations for each entity. Exome and transcriptomic sequencing was performed for 17 patients with both WD and DD diagnoses. Somatic point and copy-number alterations were integrated with transcriptional analyses to determine subtype-associated genomic features and pathways. The results were, on average, that only 8.3% of somatic mutations in WD liposarcoma were shared with their cognate DD component. DD tumors had higher numbers of somatic copy-number losses, amplifications involving Chr 12q, and fusion transcripts than WD tumors. HMGA2 and CPM rearrangements occur more frequently in DD components. The shared somatic mutations indicate a clonal origin for matched WD and DD tumors and show early divergence with ongoing genomic instability due to continual generation and selection of neochromosomes. Stochastic generation and subsequent expression of fusion transcripts from the neochromosome that involve adipogenesis genes such as HMGA2 and CPM may influence the differentiation state of the subsequent tumor.
Keywords: overgrowth
INTRODUCTION
Sarcomas are malignancies showing mesenchymal differentiation that have been classified into more than 70 histological subtypes (Taylor et al. 2011a; Fletcher 2013). They are generally grouped according to the tissue or cell type that they most resemble. The liposarcoma subtypes are tumors with varying degrees of adipocytic differentiation. These are categorized into four main groups: well-differentiated (WD), dedifferentiated (DD), myxoid, and pleomorphic (Taylor et al. 2011a; Fletcher 2013). Myxoid liposarcomas are characterized by a disease-defining translocation DDIT3 with FUS or EWSR1, whereas pleomorphic liposarcomas show complex karyotypes, often alongside TP53 mutations (Knight et al. 1995; Hosaka et al. 2002; Mariño-Enríquez et al. 2014). The most common forms are the WD and DD liposarcomas (Fletcher et al. 2002). These are intimately related in that DD liposarcoma arises in the context of WD liposarcoma (Lahat et al. 2008). The differences in overall histology and behavior of these tumors are striking (Fletcher et al. 2002; Bill et al. 2016). WD liposarcoma is a low-grade tumor expressing markers of mature adipocytes with a high propensity for local recurrence (Laurino et al. 2001). Some WD tumors exhibit, either de novo or later progress to, a higher-grade, more aggressive, typically nonlipogenic form that can metastasize (Henricks et al. 1997). This tumor is termed “dedifferentiated” liposarcoma. DD liposarcoma patients have poorer survival, have a higher mitotic index, and are more likely to experience metastasis (Dalal et al. 2006; Mussi et al. 2008).
Molecular characterizations using independent WD and DD tumors have shown similar profiles. There are no obvious differences in the somatic mutation landscapes between WD and DD liposarcomas as determined by targeted sequencing for which subtype-specific therapy can be specified (Kanojia et al. 2015). In addition, both of these tumors harbor amplified segments from Chr 12q that form the basis of ring or rod neochromosomes, the origins of which are unknown (Pedeutour et al. 1999; Garsed et al. 2014). Often, fragments from additional chromosomes become incorporated into these neochromosomes, resulting in multiple genomic rearrangements involving Chr 12q and other chromosomal regions. Breakage-fusion-bridge cycles mediate amplification of Chr 12q, which may generate the striking number of structural rearrangements and amplified oncogenes that were observed in two DD liposarcoma tumor samples using whole-genome sequencing (Taylor et al. 2011b) and the lack of continuity in the amplified Chr 12q regions in WD/DD liposarcomas as seen by FISH analysis (Italiano et al. 2008). The high copy oncogenes from Chr 12q include MDM2, CDK4, HMGA2, and YEATS2, which drive tumorigenesis (Papenfuss and Thomas 2015). However, current clinical trials of MDM2 and CDK4 inhibitors as single agents have not yielded significant responses (Ray-Coquard et al. 2012; Dickson et al. 2016; Wagner et al. 2017).
To better examine the molecular bases for the differences in histopathology and clinical behavior between synchronous WD and DD liposarcomas as well as their relatedness, we collected a novel and unique data set of patients who developed both WD and DD tumors during the course of their disease (Table 1; Fig. 1A,B). With this cohort, we conducted extensive exome and transcriptome profiling of somatic alterations, differential expression, and fusions of each subtype. We then determined both intra- and interpatient differences between WD and DD tumors.
Table 1.
Clinical characteristics and outcomes of patients
| Patient ID | Type | Site of sample analyzed | Age at diagnosis | Sample analyzed: primary/recurrent (date) | Preceding therapya | Date of initial diagnosis | Primary: WD/DDb (treatment) | Recurrences (treatment, path if availableb) | Site(s) of recurrence | D/AW/AWO (Date of death or last F/U) |
|---|---|---|---|---|---|---|---|---|---|---|
| DWN-1 | DD | Abdomen | 59 | Recurrent (3/20/07) | No | 10/28/02 | DD (S) | 2004 (R → S, WD), 2005 (C → S, WD), 2007 (S, DD), 2007 (S, DD), 2009 (C → S, DD) | Retroperitoneum and peritoneal cavity | D (8/7/2009) |
| WD | Retroperitoneum | Recurrent (6/2/09) | Yes (C) | |||||||
| N | Fat | |||||||||
| DWN-2 | DD | Retroperitoneum | 53 | Recurrent (6/3/08) | No | 7/28/05 | Cellular WD (R → S) | 2006 (S, DD), 2007 (S, DD), 2008 (S, DD), 2008 (attempted S, DD) | Retroperitoneum and peritoneal cavity | D (3/16/2009) |
| WD | Retroperitoneum | Recurrent (6/3/08) | No | |||||||
| N | Fat | |||||||||
| DWN-3 | DD | Retroperitoneum | 52 | Primary (12/9/2014) | No | 11/18/14 | DD with cartilaginous metaplasia (S) | 2016 (C, DD) | Cervical spine, right iliac bone, right psoas | AW (6/29/2017) Receiving C |
| WD | Retroperitoneum | Primary (12/9/2014) | No | |||||||
| N | Kidney | |||||||||
| DWN-4 | DD | Retroperitoneum | 52 | Primary (4/28/2015) | Yes (C) | 12/23/14 | DD (C → S) | None | NA | AWO (3/10/2017) |
| WD | Retroperitoneum | Primary (4/28/2015) | Yes (C) | |||||||
| N | Kidney | |||||||||
| OWN-5 | DD | Retroperitoneum | 68 | Primary (5/5/2015) | No | 2/24/15 | DD (S) | None | NA | AWO (5/25/2017) |
| WD | Retroperitoneum | Primary (5/5/2015) | No | |||||||
| N | Kidney | |||||||||
| DWN-6 | DD | Left thigh | 61 | Recurrent (7/6/2015) | Yes (R) | 12/22/10 | Cellular WD (S) | 2015 (R → S, DD) | Left thigh | AWO (4/19/2017) |
| WD | Left thigh | Recurrent (7/6/2015) | Yes (R) | |||||||
| N | Skeletal muscle | |||||||||
| DWN-7 | DD | Retroperitoneum | 79 | Primary (7/20/2015) | No | 5/22/15 | DD (S) | None | NA | AWO (6/22/2017) |
| WD | Retroperitoneum | Primary (7/20/2015) | No | |||||||
| N | Kidney | |||||||||
| DWN-8 | DD | Retroperitoneum | 57 | Primary (9/3/2015) | Yes (C → R) | 11/10/14 | DD (C → R → S → C) | None | NA | AWO (6/30/2017) |
| WD | Retroperitoneum | Primary (9/3/2015) | Yes (C → R) | |||||||
| N | Skeletal muscle | |||||||||
| DWN-10 | DD | Pelvis/retroperitoneum | 41 | Recurrent (12/10/2015) | Yes (C) | 8/2/12 | DD (S) | 2013 (C → S, WD), 2014 (S, DD), 2015 (C → S, DD), 2016 (C → R) | Pelvis, retroperitoneum | AW (6/20/2017) Awaiting S |
| WD | Pelvis | Recurrent (12/10/2015) | Yes (C) | |||||||
| N | Smooth muscle | |||||||||
| FF-1 | DD | Retroperitoneum | 34 | Primary (12/18/1997) | No | 12/18/97 | DD (S) | 1998 (C) | Retroperitoneum | D (12/4/1998) |
| WD | Retroperitoneum | Primary (12/18/1997) | No | |||||||
| N | Appendix | |||||||||
| FF-3 | WD | Retroperitoneum | 25 | Recurrent (6/17/2005) | No | 8/20/89 | Cellular WD (S) | 1993 (S, ?), 1997 (S, ?), 1999 (S, DD), 1999 (C → S, ?), 2002 (C), 2005 (S, DD) | Retroperitoneum | D (1/17/2010) |
| DD | Retroperitoneum | Recurrent (6/17/2005) | No | |||||||
| N | Small bowel | |||||||||
| FF-4 | DD | Peritoneum | 65 | Recurrent (1/23/2006) | No | 2000 | ? (S) | 2004 (S, ?), 2005 (C) | Retroperitoneum | D (2/3/2008) |
| WD | Kidney | Recurrent (1/23/2006) | No | |||||||
| N | Kidney | |||||||||
| FF-5 | WD | Retroperitoneum | 67 | Primary (11/19/1997) | Yes (C/R) | 8/19/97 | DD (C/R → S) | 1999 (C) | Retroperitoneum, lung | D (10/12/2001) |
| DD | Retroperitoneum | Primary (11/19/1997) | Yes (C/R) | |||||||
| N | Colon | |||||||||
| FF-7 | WD | Diaphragm | 77 | Residual primary (4/5/2005) | No | 1/17/05 | DD (S) | 2006 (S, DD) | Retroperitoneum and peritoneal cavity | D (4/6/2006) |
| DD | Left upper quadrant | Residual primary (4/5/2005) | No | |||||||
| N | Fat | |||||||||
| FF-8 | WD | Retroperitoneum | 75 | Recurrent (10/30/1998) | No | 9/30/97 | DD (S) | 1998 (C → S, DD), 1998 (S, DD) | Retroperitoneum and peritoneal cavity | D (6/8/1999) |
| DD | Retroperitoneum | Recurrent (10/30/1998) | No | |||||||
| N | Skeletal muscle and fat | |||||||||
| FF-9 | DD | Retroperitoneum | 56 | Primary (5/1/2003) | No | 3/21/03 | DD (S) | 2004 (C → S, DD), 2005 (R → S, DD), 2005 (C, DD) | Retroperitoneum and peritoneal cavity | D (3/8/2006) |
| WD | Retroperitoneum | Primary (5/1/2003) | No | |||||||
| N | Kidney | |||||||||
| FF-10 | WD | Retroperitoneum | 43 | Recurrent (1/21/2005) | No | 4/11/94 | ? Scrotal mass (S) | 2002 (C/R → S, DD), 2005 (S, DD), 2005 | Retroperitoneum and peritoneal cavity | D (10/28/2007) |
| DD | Retroperitoneum | Recurrent (1/21/2005) | No | |||||||
| N | Spleen |
S, surgery; R, radiation; C, chemotherapy; C/R, chemoradiation; ?, data unavailable; D, died with disease; AW, alive with disease; AWO, alive without recurrence.
aAny therapy within 3 months prior to surgery.
bDD: Any presence of DD liposarcoma in the specimen.
Figure 1.

Whole-exome profiling of concurrent well-differentiated (WD) and dedifferentiated (DD) liposarcomas. (A,B) Representative hematoxylin and eosin stains of liposarcomas used in this study: WD and DD. (C) Boxplots of the total numbers of somatic mutations called by MuTect when comparing tumors with their matched normal samples. Only fresh frozen cases of WD (left) and DD (right) liposarcomas were included here. A paired t-test comparing the somatic mutation burdens between WD and DD liposarcomas was not significant (P = 0.36). (D) Venn diagram example showing the number of somatic mutations in patient 3 (DWN3) as called by MuTect.
RESULTS
Clonal Origin of Both WD and DD Tumor Components
Comparisons of somatic aberrations between matched tumors can be used to determine their degree of relatedness. For each of these patients, the total numbers of somatic point mutations did not differ significantly between the WD and DD components. DD components often had slightly higher mutation burdens than their matched WD samples, but this did not reach significance (Table 2; Fig. 1C, and paired t-test P = 0.36). No recurrent point mutation was found across samples. Few (two to 11) somatic mutations were detected in both matched WD and DD tumors in any given patient (Table 2; Fig. 1D; Supplemental Table 1A–C). The majority of these shared mutations were also at low allele frequencies (0.1–0.2, Supplemental Table 1A). Furthermore, only one known cancer gene, NAV3, with a nonsynonymous mutation was common in both WD and DD tumors from patient DWN3 (uc001syp.3, p.R2241Q, variant allele frequencies of 0.77 and 0.80 in WD and DD tumors, respectively). Together, these findings suggest that coincident WD and DD liposarcomas within the same patient are related, with clonal origin and early divergence. Because the numbers of somatic mutations were very few in these samples, we were limited in our ability to characterize the clonal population structure and mutation signature differences between WD and DD components (Supplemental Fig. 1E). From the data available, pairwise comparisons in mutation signatures did not reveal any differences between sample pairs. Whole-genome sequencing would improve the characterization of whether WD is truly the clonal origin of DD.
Table 2.
Total tallies of shared mutations between the WD and DD components, and the percentage of shared mutations in each WD and DD component, respectively
| Patient | Total WD mutations | Total DD mutations | Shared mutations | Total WD functional mutations | Total DD functional mutations | Shared functional mutations |
|---|---|---|---|---|---|---|
| DWN1 | 105 | 159 | 2 | 30 | 45 | 0 |
| DWN2 | 185 | 29 | 11 | 53 | 1 | 2 |
| DWN3 | 60 | 71 | 8 | 12 | 22 | 3 |
| DWN4 | 80 | 90 | 9 | 16 | 14 | 1 |
| DWN5 | 101 | 117 | 9 | 22 | 38 | 2 |
| DWN6 | 67 | 162 | 8 | 7 | 27 | 1 |
| DWN7 | 145 | 172 | 6 | 30 | 29 | 1 |
| DWN8 | 97 | 148 | 11 | 13 | 22 | 0 |
| DWN10 | 47 | 212 | 3 | 11 | 18 | 1 |
Functional mutations: nonsynonymous, stop-gains, stop-losses, and mutations affecting splice sites.
WD, well-differentiated; DD, dedifferentiated.
Recurrent WD and DD Copy-Number Alterations
We delineated somatic copy-number profiles from the exome data and identified focal regions of amplifications or losses (Fig. 2A–D) (see Methods). Canonical amplifications in CDK4, MDM2, and YEATS2 were observed in all samples (Supplemental Table 2). Recurrent copy-number amplifications were observed in Chromosomes 1q, 5p, 6q, and 12q that were recapitulated as focal amplifications in both tumor types (Fig. 2A,C; Supplemental Table 3). As a note, the reported recurrent Chr 1p32 amplification in c-JUN was present in 3 of 17 samples (DWN6-DD, DWN7-DD, and FF7-WD) (Coindre et al. 2010; Kanojia et al. 2015). In addition, loss of TP53 was found in one sample (DWN3-DD), and three patients had TERT amplification (DWN2-WD, DWN2-DD, DWN10-DD, and FF7-DD) in Chr 5p. By GISTIC, additional focal amplifications were seen in regions Chr 1p, Chr 10q, Chr 14q, Chr 17p, Chr 18q, and Chr 19p, with the signal for Chr 14q and Chr 5p being slightly stronger in DD liposarcoma (Fig. 2A,C; Supplemental Table 3A–C). Recurrent losses in Chr 13 occurred in five patients (DWN1-WD, DWN3-DD, DWN10-DD, FF5-DD, and FF9-WD), which includes known tumor suppressors DLEU1, FOXO1, GJB2, PDSSB, USP12, ARL11, RASL11A, IFT88, THSD1, SPRY2, DACH1, CDX2, TRIM13, KLF5, ING1, EDNRB, BRCA2, TPTE2, KL, STARD13, PCDH17, SOX1, RB1, TSC22D1, PCDH8, DLEU2, LATS2, GPC5, PDX1, KCNRG, FLT3, INTS6, PCDH9, and OLFM4, although this did not give any significant focal peak.
Figure 2.

Somatic copy-number alterations in concurrent WD and DD liposarcomas. (A) Focal amplifications and (B) deletions in WD samples as determined by GISTIC 2.0. (C) Focal amplifications and (D) deletions in DD samples as determined by GISTIC 2.0. (E) Gene-level burdens of gains and (F) losses in WD (left) and DD (right) samples, both fresh frozen and FFPE.
Higher Number of Losses in DD Tumors Than in WD Tumors
In general, deletions were more abundant in DD than in WD tumors (P = 0.025 for deleted genes; P = 0.14 for amplified genes) (Fig. 2E,F). No gene was included in a segment of copy-number alteration that was enriched in either WD or DD tumors by Fisher's exact test. There were a significantly higher number of copy-number alterations in Chromosome 12 (P = 0.016) and in Chr 12q (P = 0.022) in DD than for WD samples (Supplemental Fig. 2A,B). This implies that neochromosomes of DD tumors go through more breakage-fusion- bridge cycles than do WD tumors. No gene within Chr 12q was amplified or deleted significantly in either WD or DD tumors. Next, we used a pathway-level approach to identify enrichments of somatic copy-number changes in WD or DD tumors. To do this, we compared the copy-number alteration enrichment scores for each pathway between WD and DD samples (see Methods). There, we found that immune-related pathways scored higher in copy-number losses in DD than in WD samples that are also differentially expressed between these two subtypes in our data (Fig. 3B,C).
Figure 3.

Differentially expressed genes between WD and DD liposarcomas. (A) Volcano plot denoting the genes that are significantly higher in expression in WD liposarcomas (red; 357 genes) and in DD liposarcomas (blue; 395 genes). Gene Set Enrichment Analysis (GSEA) plots of pathways that are significantly more highly expressed in (B) DD and (C) WD tumors based on GSEA analysis.
Differentially Expressed Genes Support Pathological Differences
When comparing the overall gene expression differences between WD and DD samples, 357 genes were more highly expressed in WD compared to DD tumors (Fig. 3A). The top-ranking genes, including FABP4, ADIPOQ, LPL, LEP, and PTGER3, have roles in lipid metabolism and differentiation. Of the known adipocytic differentiation markers, the levels of PPPARγ, CEBPB, CEBPD, FOXO1, FOXO3, FOS, JUN, MYC, and CDKN1A were also higher in WD when compared to DD samples. The data supports the more mature adipocytic nature of WD tumors. Apart from the adipocytic pathways (Fig. 3B), the immune system and bone morphogenesis pathways were also enriched in WD samples. The 395 genes with lower expression in WD tumors when compared to DD tumors are assumed to be mostly genes that are up-regulated in DD liposarcomas. For these, the highest-ranking genes were involved in DNA replication, cell cycle, cell division, and DNA repair (Fig. 3C). Notably, many of the high-ranking genes in DNA repair belonged to DNA homologous recombination (HR) repair genes such as GINS4, BRCA2, XRCC2, RAD51AP1, RAD51, XRCC1, POLQ, FEN1, and RAD54B.
Few Differentially Expressed Genes Are Caused by Copy-Number Alterations
This unique data set provided the opportunity to study whether copy-number alterations in each paired set of matched tumors would impact gene expression differences between WD and DD tumor groups. These tumor-specific aberrations could provide insight into their pathological and clinical differences. First, we approached this at the gene level, based on the exome copy-number segments (Supplemental Fig. 2C,D). For instance, if there were amplifications within the WD component of a particular patient that were also amplified in DD tumors from other patients or appearing in both WD and DD components in other patients, then these would be filtered out because they could occur in either tumor type and did not drive intrinsic differences between WD and DD tumor types. Thus, we identified WD- and DD-specific amplifications and losses for further study. Although there were numerous subtype-specific amplifications and losses, very few gene expression differences could be explained by them. Of 907 WD-specific gains, only 11 were overexpressed in their cognate samples when compared to all DD samples, including transcription factor FOS, antigen-processing factor LONRF1, and PAX3/7 binding protein PAXBP1. Similarly, from the 1177 DD-specific gains, only 22 were lower in expression in WD when compared to DD samples (including genes related to DNA repair RMI2 and BABM1, mesenchymal stem cell factor TUFT1, heterochromatin factor CBX1, and DGK1, which regulates intracellular concentrations of diacylglycerol). Similar strategies were used to identify DD-specific losses. Of these 7888, 55 were recurrent in at least four patients, including the Golgi-related genes BOK, GAL3ST2, SLC30A1, and STK25. Three of these genes, BOK, GAL3ST2, and STK25, are on Chr 2q. The most recurrently deleted genes were RNU6-19P and SRGAP2D, which were seen in seven and eight patients, respectively. Out of these losses, 63 are more highly expressed in DD liposarcoma (including PPP2R5A, GOS2, and NR4A3). None of the WD-specific losses were down-regulated in our WD cohort. PTEN is a DD-specific loss in three DD samples (DWN3-DD, DWN5-DD, and DWN7-DD) and its expression is lower in these samples when compared to their matched WD tumor (data not shown).
Second, we used an integrative pathway analysis to identify those pathways in which the genes were enriched in both copy-number alterations and gene expression changes in the same direction (see Methods). Immune-related pathways are both enriched by copy-number losses specifically in DD samples and are significantly down-regulated in DD samples compared to WD samples (Supplemental Table 4A,B). Interestingly, an immune signature analysis (see Methods) also indicated that DD samples have lower immune infiltration scores of 28 different immune cell types than do WD samples (Bindea et al. 2013). As a note, the two pathway-level enrichment analyses aforementioned did not show any differences in pathways related to adipocyte differentiation.
DD Samples Had a Higher Number of Fusions
Nine-hundred and thirty-three putative gene fusion transcripts were identified in the nine pairs of frozen WD and DD samples, with a median of 39 fusions per sample (see Methods, Supplemental Table 5A–D). DD samples had a significantly higher number of fusions than their paired WD samples (Fig. 4A,B; P = 0.0115 by paired t-test). A significant proportion of these fusions involve Chromosome 12 (median = 0.44) (Fig. 4C). In addition, DD samples shared lower proportion (average = 17%) of their fusions with their cognate WD samples than did the WD samples (average = 6.9%) (P = 0.0167 by paired t-test). These again imply that neochromosomes of DD tumors may go through more breakage-fusion-bridge cycles than WD tumors after divergence from their clonal origin. Closer examination revealed that the majority of these fusions were not in the correct orientation (head-to-tail), representing collateral damage without any important functional roles in tumor growth. However, there were intact fusions involving HMGA2 and CPM of Chr 12q that were more recurrent in DD samples (6/9, P = 0.049 and 5/9, P = 0.13, respectively) than in WD tumors (1/9 and 1/9, respectively) (Supplemental Table 5C,D, Supplemental Methods). The protein domains in which identified breakpoints were predicted to occur showed that the HMGA2 fusion transcripts found in DD samples all contained intact AT-hook DNA binding motifs, whereas the fusion in the DWN1–WD sample had a truncated AT-hook DNA binding motif that may result in a nonfunctional chimeric protein. In addition, HMGA2 was also significantly overexpressed in DD samples when compared to WD samples (Supplemental Table 5C, P = 0.04; log2 of fold change = 1.97). Fusions involving carboxypeptidase M (CPM) on Chr 12q were also more prevalent in DD tumors than in WD tumors. These rearrangements would cause truncated transcripts of CPM to be depressed, which follows the significantly lower levels of CPM found in DD tumors when compared to WD tumors (P = 0.01; log2 of fold change = 1.77).
Figure 4.
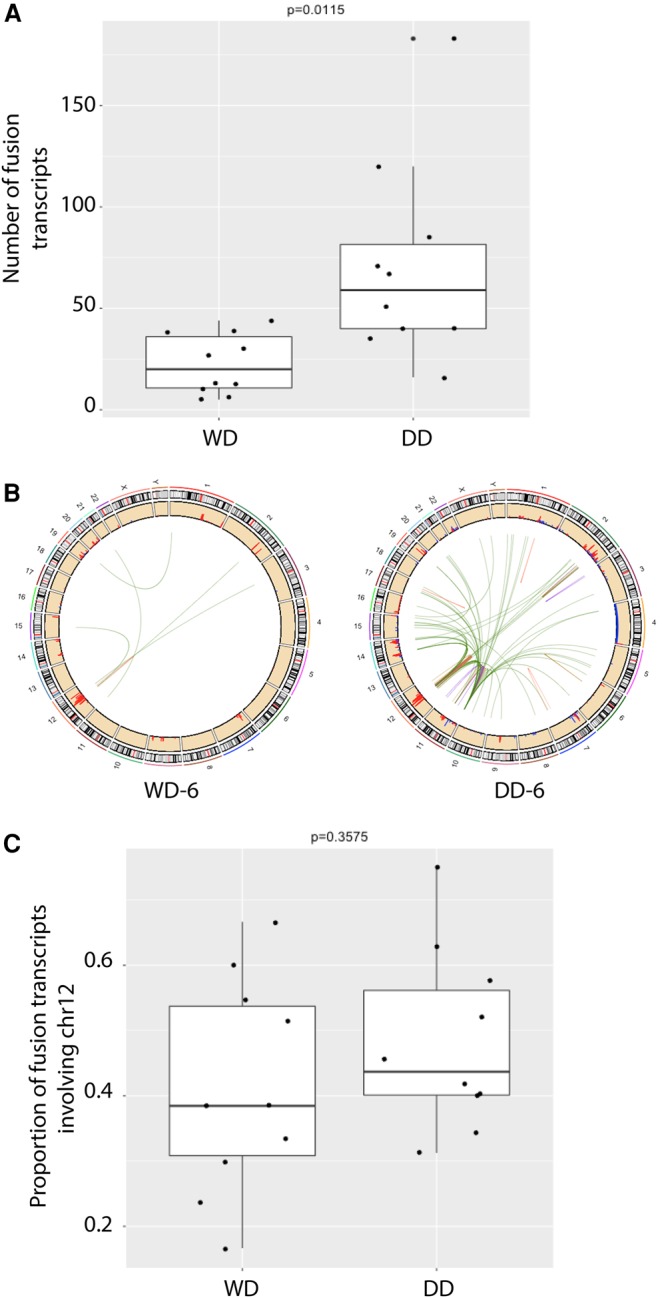
Number of fusion transcripts in concurrent WD and DD liposarcomas. (A) Boxplots of the total number of fusion transcripts called by at least two callers. (B) Circos plots of these consensus fusion calls in the WD and DD tumors from patient DWN-6. (C) The proportion of fusion transcripts that occur on Chromosome 12.
DISCUSSION
The molecular basis of WD liposarcoma progression to DD liposarcoma has not been well studied. The patients in this study presented with both tumor types, some as recurrent tumors from an initial pretreated WD diagnosis and others as a concurrent progression of DD adjacent to WD liposarcoma. With exome and transcriptome profiling of these tumors, we have several lines of evidence to suggest that these coincident tumors arise from a common origin and diverged early. First, the existence of shared point mutations between WD and DD components indicate that they were derivatives from a common ancestral clone as these shared events are rare in independent tumors from two individuals. Second, the total numbers of shared point mutations per patient were few despite having comparable overall mutation burdens in both tumor types. This suggests that although the point mutations accumulated at a similar rate in each tumor type, the large proportion of tumor-specific point mutations indicates an early and continual divergence. Therefore, the propensity for dedifferentiation is determined early on in the disease rather than as a result of slow progressive accumulation of mutations. Copy-number alterations (especially amplified Chr 12q) may be responsible for determining the initiation of malignancy because there were no clear drivers in the somatic point mutations.
Many of the copy-number alterations observed are likely due to the breakage-fusion-bridge events that continue to generate and alter the neochromosomes at each cell division. Our data support the early existence and amplifications of Chr 12-based neochromosomes. First, gains from Chr 12 are highest in magnitude in matched WD and DD liposarcomas, are 100% recurrent across all patients, and are accompanied by coamplifications of Chr 6q23 and Chr 1p32, both of which harbor known oncogenes. Next, we observed that most of the shared fusion transcripts between WD and DD tumors (mean = 71.9%) involve genes in Chr 12. Because the number of copy-number losses and fusions was higher in DD than in WD tumors, we infer that DD tumors have undergone more cycles of breakage-fusion-bridge than their matched WD tumors. This would be consistent with the high number of amplifications and overexpression of CDK4 and MDM2 from Chr 12q in DD tumors when compared to WD tumors that would contribute to the rapid cell division and thus increased breakage-fusion-bridge events experienced by DD tumors.
The ongoing breakage-fusion-bridge cycles of the 12q region on neochromosomes may generate rearrangements that are involved in determining the adipocytic differentiation state of daughter cells. For instance, two genes on Chr 12q affect adipocyte differentiation: high-mobility group protein (HMGA2) and carboxypeptidase M (CPM). HMGA2 acts as a transcriptional regulating factor to promote adipogenesis and mesenchymal differentiation (Ozturk et al. 2014), whereas CPM activates extracellular proteins such as EGF and FGF (McGwire and Skidgel 1995) that are up-regulated during mesenchymal cell differentiation in various lineages, including adipocytes. Rearrangements disrupting the coding regions of both HMGA2 and CPM occurred significantly more frequently in DD than in their matched WD samples. The Cancer Genome Atlas sarcoma (TCGA SARC) set also revealed that out of all the sarcoma subtypes characterized, only DD liposarcomas have fusions involving HMGA2 (there are no WD liposarcomas in this data set) (Hu et al. 2018). Although a fusion transcript involving HMGA2 was found in one WD sample, it was predicted to truncate one of the AT-hook DNA binding motifs and may result in a nonfunctional chimeric protein. Several studies also showed that in DD liposarcoma, rearrangements of HMGA2 to distant or extrachromosomal regions result in the loss of Let-7 miRNA binding sites within the 3′-untranslated regions (3′ UTRs) (Meza-Zepeda et al. 2001; Taylor et al. 2011b). This leads to the overexpression of a truncated protein that activates the sarcoma protooncogene SSX1 in mesenchymal stem-like cells (Henriksen et al. 2010). It now appears that the balance between HMGA2 and Let-7 determines whether a cell will be stem-like, invasive and self-renewing, or differentiating into mature and less malignant cells in multiple types of cancer (Copley et al. 2013; Yu et al. 2015). The fact that we found HMGA2 to be more highly expressed in DD than in WD tumors and CPM with comparatively little expression in DD than in WD favors the idea that both rearrangement and expression control of genes on Chr 12q would determine the ultimate levels of wild-type and mutant transcripts of such genes. The differentiation state that is observed pathologically would then be the combined effects of both the timing and levels of these various products. The exact conditions for developing DD liposarcoma must be rarer and rate-limiting because WD tumors often recur as WD before reappearing as DD and also that a DD tumor mass is often smaller than its synchronous WD tumor. But once developed, these DD malignancies then grow rapidly, show extensive local infiltration, and sometimes metastasize. Therefore, we hypothesize that ongoing breakage-fusion-bridge cycles of the neochromosomes in the clone origin of WD and DD liposarcomas generate a heterogeneous population of daughter cells. At some point in time, stochastic development of daughter cells with certain, still-undefined, levels of the HMGA2 fusion transcripts without the 3′ UTR and disrupted CPM transcripts then become a DD mass (Mayr et al. 2007).
Epigenetic mechanisms and noncoding RNAs may explain the expression differences between the two subtypes that we cannot account for by point mutations and copy-number alterations as seen in the recent pan-sarcoma analysis (The Cancer Genome Atlas Research Network 2017). Methylation of CEBPA and KLF14 promoters was reported to alter their gene expression in DD more than WD liposarcoma, but we did not observe these expression differences in our samples (Supplemental Fig. 3; Taylor et al. 2011b). In addition, enriched H3K9me3 methylation of the transcription factor, KLF6, appears to result in lower expression in DD liposarcomas when compared to WD liposarcomas (Keung et al. 2015; Keung and Rai 2016). When overexpressed, KLF6 decreases proliferation of DD cells and increase latency periods of DD xenografts (Keung et al. 2015; Keung and Rai 2016). However, this differential effect was trending in our data and was not significantly different, although KLF2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, and 15 were higher in WD than in DD samples (Supplemental Fig. 3). These alternative driving mechanisms may elucidate the unexpected links we found to bone morphogenesis, telomere maintenance, and other pathways that were differentially expressed in our data. The concordance of our expression data with an independent expression microarray analysis of the dysregulated genes between independent WD and DD tumors (CIDEA, LMNB2, MFAP2, RNASEH2A, PPP2RSA, and GOS2) should be investigated further (A Oliviera [Mayo Clinic, Rochester, MN], pers. comm.). Enrichments in immune-related pathways in WD tumors also suggest tumor-infiltrating T cells, macrophages, and others are more prevalent in these tumors, although this effect may be due to overall lower purity levels of WD samples, which would require more in-depth studies. Of note, Matushansky et al. (2008) reported that WD was most similar to Day 21 of mature adipocyte differentiation. We did not observe these adipocytic genes to be differentially expressed compared to normal fat.
In conclusion, circular extrachromosomal DNA that encodes cancer genes is far more common in cancer cells and probably plays a central role in generating the heterogeneity that constitutes advanced cancers because of the stochastic nature of neochromosomal replication with breakage-fusion-bridge cycles (Turner et al. 2017). Much larger functional screens will be required to determine whether these are passenger or driver events. Future studies are needed to investigate the putative mechanisms of HMGA2 that may reveal therapeutic implications in combining inhibitors of CDK4/MDM2 and HMGA2 in DD liposarcoma. Given the few genes that were differentially expressed in the development of DD liposarcoma, there may be additional epigenetic mechanisms that play a role in the progression of these tumors.
METHODS
Samples
Retrospective samples were collected and de-identified under a protocol wherein consent was waived that was approved under the institutional review board at UT MD Anderson Cancer Center. Seventeen patients with primary or recurrent tumors that had both WD and DD components along with their matched normals (nine frozen: five primary and four recurrent; and eight formalin-fixed paraffin-embedded [FFPE]: four primary and four recurrent) were retrieved and re-reviewed for pathology classification by sarcoma pathologist before submission for sequencing (Table 1; Supplemental Table 1). Examples of histology are shown in Figure 1A,B. For all the patients with the exception of DWN1, the tumors examined were from the same date of collection (Table 1). For DWN1, the WD was recurrent after the DD recurrence (Table 1). See Supplemental Methods for purity assessment (Supplemental Fig. 1A).
Whole-Exome Sequencing
For DNA extraction of the frozen samples (<30 mg), the Frozen Tissue protocol from the QIAamp DNA Mini kit was followed (QIAGEN) with an elution volume of 60–100 µL. More details can be found in the Supplemental Methods. The FFPE samples were deparaffinized (see Supplemental Methods) before using the QIAamp DNA FFPE Tissue kit to extract DNA (QIAGEN). Protocol instructions were followed with two exceptions: overnight incubation at 56°C to ensure full lysis of the cells and elution volumes of 25–50 µL. All DNA samples were quantified using PicoGreen or the Tapestation 2200 (Agilent Technologies) and the quality confirmed using gel electrophoresis.
DNA samples were submitted for 76-bp short-read paired-end whole-exome sequencing on Illumina HiSeq 2000 (Illumina) after SureSelect Human All Exon V4 library preparation (Agilent Technologies) (See Supplemental Methods). Median coverage for tumors is 198× and for matched normal is 101× (Supplemental Table 6A). Driver mutations were defined as those somatic variants with minor allele frequencies (MAFs) of <0.01 in both ESP6500 (Exome Variant Server, NHLBI GO Exome Sequencing Project) and 1000Genomes (1000 Genomes Project Consortium et al. 2015) databases, as well as restricting calls to those that were nonsynonymous, stop-gains, stop-losses, and splicing changes. These calls were also compared against a published compilation of known cancer genes (Cheng et al. 2014), composed of CancerGenes (Higgins et al. 2007), Network of Cancer Genes (D'Antonio et al. 2012), and the Sanger Cancer Gene Census in COSMIC (Forbes et al. 2011). Only frozen samples were used in the mutation analysis because the variants in the FFPE samples were largely derived from nonreproducible C > T/G > A sequencing artifacts that were caused by the formalin fixation, and true positives were indistinguishable (Supplemental Fig. 1A; Schweiger et al. 2009). However, we believe that the shared mutations between paired DD and WD FFPE samples are true positives and their numbers are comparable to those of the frozen samples (Supplemental Table 1B).
Copy-Number Analysis
For all patients, somatic copy-number calls from whole-exome data using matched normals were obtained by first deriving segments using circular binary segmentation (CBS) (Olshen et al. 2004) and then deriving log2 ratio scores with an in-house tool, exomeCN, which is a modified version of HMMcopy (Ha et al. 2012) tuned for our data. For assessing amplifications and deletions, log2 scores > 0.5 were considered gains, whereas log2 scores < −0.5 were considered losses. Focal somatic copy-number alterations were identified using GISTIC2.0 (Mermel et al. 2011) at the 95% confidence level. See Supplemental Methods for comments on somatic copy-number alterations profiles in FFPE samples. Gene-level copy-number assessment was done using customized log2 score cutoffs for each patient to designate the segments as amplifications or deletions. Subsequently, the genes found within the amplified or deleted segments were intersected using Venny (Oliveros 2007–2015).
RNA Sequencing
For all frozen samples except for DWN6-WD, DWN7-WD, and DWN10-WD, total RNA was extracted with the RNeasy Mini Kit. For those three samples, the fat content in the samples interfered with extraction and they required a user-developed protocol from QIAGEN's website, “Purification of total RNA from fatty tissues using the RNeasy Lipid Tissue Mini Kit and MaXtract High Density” (QIAGEN 2007). Complementary DNA (cDNA) was generated from total RNA using the NuGEN Ovation RNA-Seq FFPE System. After quantification and shearing, libraries were then made from cDNA using the NuGEN Ovation Ultralow Library System V2. Libraries from each sample were pooled together in equimolar amounts and sequenced on the Illumina HiSeq 2500 (Illumina, Inc.) (Supplemental Table 6B). See Supplemental Methods for data processing methods.
Differential Copy-Number Alterations between WD and DD Tumors
Fisher's test was used to determine whether the copy number of a gene was differentially altered between WD and DD tumors. To identify whether the copy number of genes within a pathway were differentially altered between WD and DD tumors, an enrichment score of copy-number alteration was derived for a pathway in a sample, which is odds ratio for the ratio of genes with copy-number alterations in a pathway versus the ratio of genes with copy-number changes among all genes. The copy-number alteration enrichment score (CNAPS) for pathway i and sample j is defined as
where NAi,j is the number of copy-number altered genes in pathway i for sample j, NPi is the number of genes in pathway i, TNAi is the number of altered genes in sample j, and N is the number of genes in sample j. A t-test was then used to compare enrichment scores of each pathway between the WD and DD samples to identify pathways that are specifically enriched in WD or DD copy-number alterations. Pathway information in the MSigDB database (Subramanian et al. 2005) (category C2) was used for calculations.
An integration analysis was performed to integrate copy-number and gene expression data in order to identify pathways in which changes in somatic copy number were correlated with differential gene expression changes between WD and DD samples. First, all genes are ordered based on the mean difference in copy-number alteration (log2 ratio) between WD and DD samples. Gene set enrichment analysis (GSEA) (Mootha et al. 2003) was then used to identify pathways in which genes with amplifications or losses between WD and DD samples were enriched (termed GSEA_CNV). The categorized pathways in the MSigDB database (category C2) were used in this analysis. GSEA was also applied to identify differentially expressed pathways between WD and DD samples (termed GSEA_EX) based on the gene list ordered by gene expression fold changes between WD and DD samples. The product-truncated method (Zaykin et al. 2002) was then applied to combine P-values of GSEA_CNV and GSEA_EX for each pathway. Pathways with significant combined P-values (P < 0.05) would have significant difference in both copy-number alterations and gene expression between WD and DD samples. In the truncated product method, the product score W of the two P-values (Pi) that do not exceed a fixed τ value (τ was set to 0.01 both for GSEA association and deregulation analysis) can be calculated as
where I(·) is the indicator function. The probability of W for w < 1 can be evaluated by conditioning on k, the number of pi < τ:
Immune Signature Analysis
Immune signature analysis was performed in gene signature sets of 28 different immune cell types (Bindea et al. 2013) using the R package Gene Set Variation Analysis (GSVA) (Hänzelmann et al. 2013). GSVA calculates the enrichment score of a gene set per sample by comparing the rank distribution of genes in the gene set against the random distribution. Each enrichment score represents the level of the genes in a gene set are harmoniously up- or down-regulated in a sample.
Fusion Transcripts Identified by RNA Sequencing
To reduce false positives (Carrara et al. 2013), we applied an integrative analysis of multiple fusion detection methods. We first applied four tools—TopHat-Fusion (Kim and Salzberg 2011), MapSplice (Wang et al. 2010), FusionMap (Ge et al. 2011), and PRADA (Torres-Garcia et al. 2014)—to detect putative fusions in our nine pairs of frozen WD/DD samples. Then, fusions with at least one junction read and at least two supporting mate pair reads that mapped to candidate gene pairs were kept. Finally, only those fusions that were detected by at least two tools are selected. Fisher's exact test was used to determine whether a fusion is significantly enriched in WD or DD liposarcoma.
ADDITIONAL INFORMATION
Data Deposition and Access
Whole-exome and RNA sequencing BAM files have been deposited at the European Genome-phenome Archive (EGA; https://www.ebi.ac.uk/ega/), which is hosted by EGI and CRG, under accession number EGAS00001002807.
Ethics Statement
All samples were collected and de-identified under protocols approved by the institutional review board (IRB) at UT MD Anderson Cancer Center to analyze retrospective and prospective tissues and examine patient data retrospectively with consent waiver.
Acknowledgments
Technical assistance was provided by Cancer Genomics Laboratory, IACS Bioinformatics group, and Sarcoma Translational Pathology Laboratory. Some of the authors (H.C.B., C.-C.W., D.R.I., B.W.F., C.L.R., R.S.B., A.J.L., P.A.F., and N.S.) were members of the Liposarcoma Working group in the Sarcoma Oncology Group at MD Anderson Cancer Center.
Author Contributions
The design and concept of the study were conceived by A.J.L., P.A.F., and N.S. Sample identification, collection, and pathology reviews were conducted by D.R.I., W.-L.W., and A.J.L. Sequencing experiments and preprocessing were performed by C.G., L.L., X.S., and J.Z. Downstream analyses were done by H.C.B., C.-C.W., and A.A. The data analyses were interpreted by H.C.B., C.-C.W., A.A., B.W.F., C.L.R., R.S.B., P.H., A.J.L., P.A.F., and N.S. The manuscript was drafted by H.C.B., C.-C.W., W.-L.W., A.J.L., P.A.F., and N.S. All authors were involved in critical revision of the manuscript and approve of the final version.
Funding
Funding sources include Amshwand Foundation, QuadW Foundation, and Lemuel Allen Jr. to the Sarcoma Oncology Group as well as a Cancer Prevention Research Institute (R120501 to P.A.F., Summer Internship to A.A.), Welch Foundation's Robert A. Welch Distinguished University Chair Award (G-0040 to P.A.F.), and National Institutes of Health grant K12CA088084 Paul Calabresi Career Development Award for Clinical Oncology (C.L.R.).
Competing Interest Statement
The authors have declared no competing interest.
Referees
David M. Thomas
Anonymous
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Bill KL, Casadei L, Prudner BC, Iwenofu H, Strohecker AM, Pollock RE. 2016. Liposarcoma: molecular targets and therapeutic implications. Cell Mol Life Sci 73: 3711–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. 2013. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39: 782–795. [DOI] [PubMed] [Google Scholar]
- Carrara M, Beccuti M, Lazzarato F, Cavallo F, Cordero F, Donatelli S, Calogero RA. 2013. State-of-the-art fusion-finder algorithms sensitivity and specificity. Biomed Res Int 2013: 340620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F, Jia P, Wang Q, Lin CC, Li WH, Zhao Z. 2014. Studying tumorigenesis through network evolution and somatic mutational perturbations in the cancer interactome. Mol Biol Evol 31: 2156–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coindre JM, Pédeutour F, Aurias A. 2010. Well-differentiated and dedifferentiated liposarcomas. Virchows Arch 456: 167–179. [DOI] [PubMed] [Google Scholar]
- Copley MR, Babovic S, Benz C, Knapp DJ, Beer PA, Kent DG, Wohrer S, Treloar DQ, Day C, Rowe K, et al. 2013. The Lin28b-let-7-Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat Cell Biol 15: 916–925. [DOI] [PubMed] [Google Scholar]
- Dalal KM, Kattan MW, Antonescu CR, Brennan MF, Singer S. 2006. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg 244: 381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Antonio M, Pendino V, Sinha S, Ciccarelli FD. 2012. Network of Cancer Genes (NCG 3.0): integration and analysis of genetic and network properties of cancer genes. Nucleic Acids Res 40(Database issue): D978–D983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson MA, Schwartz GK, Keohan ML, D'Angelo SP, Gounder MM, Chi P, Antonescu CR, Landa J, Qin LX, Crago AM, et al. 2016. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: a phase 2 clinical trial. JAMA Oncol 2: 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (ESP) NGESP. Exome Variant Server. Seattle, WA. [Google Scholar]
- Fletcher CDM. 2013. Diagnostic histopathology of tumors, 4th ed. Churchill Livingstone, Edinburgh. [Google Scholar]
- Fletcher CDM, Unni KK, Mertens F. 2002. Pathology and genetics of tumours of soft tissue and bone. World Health Organization Classification of Tumours, Lyon, France. [Google Scholar]
- Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. 2011. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 39(Database issue): D945–D950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garsed DW, Marshall OJ, Corbin VD, Hsu A, Di Stefano L, Schröder J, Li J, Feng ZP, Kim BW, Kowarsky M, et al. 2014. The architecture and evolution of cancer neochromosomes. Cancer Cell 26: 653–667. [DOI] [PubMed] [Google Scholar]
- Ge H, Liu K, Juan T, Fang F, Newman M, Hoeck W. 2011. FusionMap: detecting fusion genes from next-generation sequencing data at base-pair resolution. Bioinformatics 27: 1922–1928. [DOI] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. 2015. A global reference for human genetic variation. Nature 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha G, Roth A, Lai D, Bashashati A, Ding J, Goya R, Giuliany R, Rosner J, Oloumi A, Shumansky K, et al. 2012. Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res 22: 1995–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann S, Castelo R, Guinney J. 2013. GSVA: Gene Set Variation Analysis for microarray and RNA-seq data. BMC Bioinformatics 14: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henricks WH, Chu YC, Goldblum JR, Weiss SW. 1997. Dedifferentiated liposarcoma: a clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol 21: 271–281. [DOI] [PubMed] [Google Scholar]
- Henriksen J, Stabell M, Meza-Zepeda LA, Lauvrak SA, Kassem M, Myklebost O. 2010. Identification of target genes for wild type and truncated HMGA2 in mesenchymal stem-like cells. BMC Cancer 10: 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins ME, Claremont M, Major JE, Sander C, Lash AE. 2007. CancerGenes: a gene selection resource for cancer genome projects. Nucleic Acids Res 35(Database issue): D721–D726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka T, Nakashima Y, Kusuzaki K, Murata H, Nakayama T, Nakamata T, Aoyama T, Okamoto T, Nishijo K, Araki N, et al. 2002. A novel type of EWS-CHOP fusion gene in two cases of myxoid liposarcoma. J Mol Diagn 4: 164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Wang Q, Tang M, Barthel F, Amin S, Yoshihara K, Lang FM, Martinez-Ledesma E, Lee SH, Zheng S, Verhaak RGW. 2018. TumorFusions: an integrative resource for cancer-associated transcript fusions. Nucl Acids Res 46: D1144–D1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Italiano A, Bianchini L, Keslair F, Bonnafous S, Cardot-Leccia N, Coindre JM, Dumollard JM, Hofman P, Leroux A, Mainguené C, et al. 2008. HMGA2 is the partner of MDM2 in well-differentiated and dedifferentiated liposarcomas whereas CDK4 belongs to a distinct inconsistent amplicon. Int J Cancer 122: 2233–2241. [DOI] [PubMed] [Google Scholar]
- Kanojia D, Nagata Y, Garg M, Lee DH, Sato A, Yoshida K, Sato Y, Sanada M, Mayakonda A, Bartenhagen C, et al. 2015. Genomic landscape of liposarcoma. Oncotarget 6: 42429–42444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keung EZ, Rai K. 2016. H3K9me3-mediated repression of KLF6: discovering a novel tumor suppressor in liposarcoma using a systematic epigenomic approach. Mol Cell Oncol 3: e1093691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keung EZ, Akdemir KC, Al Sannaa GA, Garnett J, Lev D, Torres KE, Lazar AJ, Rai K, Chin L. 2015. Increased H3K9me3 drives dedifferentiated phenotype via KLF6 repression in liposarcoma. J Clin Invest 125: 2965–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Salzberg SL. 2011. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol 12: R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight JC, Renwick PJ, Dal Cin P, Van den Berghe H, Fletcher CD. 1995. Translocation t(12;16)(q13;p11) in myxoid liposarcoma and round cell liposarcoma: molecular and cytogenetic analysis. Cancer Res 55: 24–27. [PubMed] [Google Scholar]
- Lahat G, Anaya DA, Wang X, Tuvin D, Lev D, Pollock RE. 2008. Resectable well-differentiated versus dedifferentiated liposarcomas: two different diseases possibly requiring different treatment approaches. Ann Surg Oncol 15: 1585–1593. [DOI] [PubMed] [Google Scholar]
- Laurino L, Furlanetto A, Orvieto E, Dei Tos AP. 2001. Well-differentiated liposarcoma (atypical lipomatous tumors). Semin Diagn Pathol 18: 258–262. [PubMed] [Google Scholar]
- Mariño-Enríquez A, Hornick JL, Dal Cin P, Cibas ES, Qian X. 2014. Dedifferentiated liposarcoma and pleomorphic liposarcoma: a comparative study of cytomorphology and MDM2/CDK4 expression on fine-needle aspiration. Cancer Cytopathol 122: 128–137. [DOI] [PubMed] [Google Scholar]
- Matushansky I, Hernando E, Socci ND, Matos T, Mills J, Edgar MA, Schwartz GK, Singer S, Cordon-Cardo C, Maki RG. 2008. A developmental model of sarcomagenesis defines a differentiation-based classification for liposarcomas. Am J Pathol 172: 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Hemann MT, Bartel DP. 2007. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 315: 1576–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGwire GB, Skidgel RA. 1995. Extracellular conversion of epidermal growth factor (EGF) to des-Arg53-EGF by carboxypeptidase M. J Biol Chem 270: 17154–17158. [DOI] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. 2011. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12: R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meza-Zepeda LA, Berner JM, Henriksen J, South AP, Pedeutour F, Dahlberg AB, Godager LH, Nizetic D, Forus A, Myklebost O. 2001. Ectopic sequences from truncated HMGIC in liposarcomas are derived from various amplified chromosomal regions. Genes Chromosomes Cancer 31: 264–273. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. 2003. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273. [DOI] [PubMed] [Google Scholar]
- Mussi C, Collini P, Miceli R, Barisella M, Mariani L, Fiore M, Casali PG, Gronchi A. 2008. The prognostic impact of dedifferentiation in retroperitoneal liposarcoma: a series of surgically treated patients at a single institution. Cancer 113: 1657–1665. [DOI] [PubMed] [Google Scholar]
- Oliveros JC. 2007–2015. Venny: an interactive tool for comparing lists with Venn's diagrams.
- Olshen AB, Venkatraman ES, Lucito R, Wigler M. 2004. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5: 557–572. [DOI] [PubMed] [Google Scholar]
- Ozturk N, Singh I, Mehta A, Braun T, Barreto G. 2014. HMGA proteins as modulators of chromatin structure during transcriptional activation. Front Cell Dev Biol 2: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenfuss AT, Thomas DM. 2015. The life history of neochromosomes revealed. Mol Cell Oncol 2: e1000698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedeutour F, Forus A, Coindre JM, Berner JM, Nicolo G, Michiels JF, Terrier P, Ranchere-Vince D, Collin F, Myklebost O, et al. 1999. Structure of the supernumerary ring and giant rod chromosomes in adipose tissue tumors. Genes Chromosomes Cancer 24: 30–41. [PubMed] [Google Scholar]
- QIAGEN. 2007. Purification of total RNA from fatty tissues using the RNeasy® Lipid Tissue Mini Kit and MaXtract High Density. https://www.qiagen.com/at/resources/resourcedetail?id=39519112-355c-4601-bef2-5469f3091fd4&lang=en.
- Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, Heil F, Rueger R, Graves B, Ding M, et al. 2012. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol 13: 1133–1140. [DOI] [PubMed] [Google Scholar]
- Schweiger MR, Kerick M, Timmermann B, Albrecht MW, Borodina T, Parkhomchuk D, Zatloukal K, Lehrach H. 2009. Genome-wide massively parallel sequencing of formaldehyde fixed-paraffin embedded (FFPE) tumor tissues for copy-number- and mutation-analysis. PLoS One 4: e5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Barretina J, Maki RG, Antonescu CR, Singer S, Ladanyi M. 2011a. Advances in sarcoma genomics and new therapeutic targets. Nat Rev Cancer 11: 541–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, DeCarolis PL, Angeles CV, Brenet F, Schultz N, Antonescu CR, Scandura JM, Sander C, Viale AJ, Socci ND, et al. 2011b. Frequent alterations and epigenetic silencing of differentiation pathway genes in structurally rearranged liposarcomas. Cancer Discov 1: 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2017. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell 171: 950–965 e928. Electronic address elizabeth.demicco@sinaihealthsystem.ca; Cancer Genome Atlas Research Network. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Garcia W, Zheng S, Sivachenko A, Vegesna R, Wang Q, Yao R, Berger MF, Weinstein JN, Getz G, Verhaak RG. 2014. PRADA: pipeline for RNA sequencing data analysis. Bioinformatics 30: 2224–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DA, et al. 2017. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543: 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AJ, Banerji U, Mahipal A, Somaiah N, Hirsch H, Fancourt C, Johnson-Levonas AO, Lam R, Meister AK, Russo G, et al. 2017. Phase I trial of the human double minute 2 inhibitor MK-8242 in patients with advanced solid tumors. J Clin Oncol 35: 1304–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al. 2010. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res 38: e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu KR, Shin JH, Kim JJ, Koog MG, Lee JY, Choi SW, Kim HS, Seo Y, Lee S, Shin TH, et al. 2015. Rapid and efficient direct conversion of human adult somatic cells into neural stem cells by HMGA2/let-7b. Cell Rep 10: 441–452. [DOI] [PubMed] [Google Scholar]
- Zaykin DV, Westfall PH, Young SS, Karnoub MA, Wagner MJ, Ehm MG. 2002. Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum Hered 53: 79–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.