

Abstract
ERBB2 amplification has been identified in ∼5% of KRAS wild-type colorectal cancers (CRCs). A recent clinical trial showed response to HER2-directed therapy in a subset of ERBB2-amplified metastatic CRCs resistant to chemotherapy and EGFR-directed therapy. With the aim of better understanding mechanisms of resistance to HER2-directed and EGFR-directed therapies, we report the complete molecular characterization of two cases of ERBB2-amplified CRC. PCR-free whole-genome sequencing was used to identify mutations, copy-number alterations, structural variations, and losses of heterozygosity. ERBB2 copy number was also measured by fluorescence in situ hybridization. Single-stranded mRNA sequencing was used for gene expression profiling. Immunohistochemistry and protein mass spectrometry were used to quantify HER2 protein expression. The cases showed ERBB2 copy number of 86 and 92, respectively. Both cases were immunohistochemically positive for HER2 according to CRC-specific scoring criteria. Fluorescence in situ hybridization and protein mass spectrometry corroborated significantly elevated ERBB2 copy number and abundance of HER2 protein. Both cases were microsatellite stable and without mutation of RAS pathway genes. Additional findings included altered expression of PTEN, MET, and MUC1 and mutation of PIK3CA. The potential effects of the molecular alterations on sensitivity to EGFR and HER2-directed therapies were discussed. Identification of ERBB2 amplification in CRC is necessary to select patients who may respond to HER2-directed therapy. An improved understanding of the molecular characteristics of ERBB2-amplified CRCs and their potential mechanisms of resistance will be useful for future research into targeted therapies and may eventually inform therapeutic decision-making.
Keywords: neoplasm of the gastrointestinal tract, neoplasm of the large intestine
INTRODUCTION
Amplification of ERBB2 has been detected in ∼2%–4% of unselected colorectal cancer (CRC) cases and a strong concordance with overexpression of HER2 protein has been demonstrated using immunohistochemistry (IHC) (Ooi et al. 2004; Marx et al. 2010; The Cancer Genome Atlas Network 2012). CRC-specific criteria for assessing HER2 positivity by IHC and fluorescence in situ hybridization (FISH) have been developed (Valtorta et al. 2015). ERBB2 amplification is one of several proposed mechanisms of resistance to epidermal growth factor receptor (EGFR) blockade in KRAS wild-type CRC (Bertotti et al. 2015). The HERACLES trial identified HER2 positivity in 5% of KRAS wild-type metastatic CRC showing resistance to standard therapy, including EGFR-directed therapy (Sartore-Bianchi et al. 2016). Among patients treated with combination trastuzumab and lapatinib, 30% had a partial or complete response and 30% had disease stabilization lasting 16 or more weeks.
Post hoc analysis of the HERACLES trial data indicated that an ERBB2 copy number of >9.45 best identified patients who responded to HER2-directed therapy. However, a subset of patients responded poorly or did not respond to HER2-directed therapy despite high ERBB2 copy number, indicating that additional molecular alterations are likely associated with resistance to HER2-directed therapy.
Alteration of RAS pathway genes is a potential mechanism of resistance to HER2-directed therapy, although the majority of ERBB2-amplified CRCs studied to date appear to have been wild type in RAS pathway genes (Bertotti et al. 2011; Valtorta et al. 2015; Sartore-Bianchi et al. 2016). Patients included in the HERACLES trial were required to have wild-type status of KRAS codons 12 and 13. However, neither KRAS mutations outside of codons 12 and 13 nor mutations in other RAS pathway genes were excluded in patients enrolled in the HERACLES trial. Additional mutational testing following the HERACLES trial revealed a BRAF V600E mutation in one patient whose tumor progressed during the trial and a KRAS A146T mutation in another whose disease was stable. RAS pathway alterations that are not detected in routine molecular testing of CRC have the potential to confer resistance to HER2-directed therapy. Pretreatment of ERBB2-amplified CRC with EGFR-directed therapy may also select for clonal populations of tumor cells harboring RAS pathway alterations that have the potential to confer resistance to HER2-directed therapy. All patients in the HERACLES trial were pretreated with EGFR-directed therapy.
The ongoing Personalized Oncogenomics Group (POG) trial (NCT02155621) has undertaken complete molecular characterization of a total of 60 patients with metastatic CRC, including 29 KRAS wild-type tumors, two of which showed high-level amplification of ERBB2. An improved understanding of the molecular characteristics of ERBB2-amplified CRCs and their potential mechanisms of resistance to HER2-directed therapy may influence the direction of future research on targeted therapies and inform future therapeutic decision-making. With these aims, we report the whole genome and transcriptome molecular profiling of the two ERBB2-amplified CRCs in the POG trial.
RESULTS (CASE 1)
Clinical Presentation
A 45-yr-old man presented with hematochezia and rectal pain. Imaging demonstrated extensive liver and lung lesions, along with soft tissue thickening of the rectum. He had a history of asthma but was otherwise well. His family history was unremarkable apart from an uncle who was diagnosed with CRC.
Colonoscopic biopsy confirmed the diagnosis of primary rectal adenocarcinoma and he received a palliative rectal resection. Histology showed a well-differentiated adenocarcinoma (Fig. 1A) located at the upper rectum with lymphovascular invasion, extension into the mesorectum, extramural tumor deposits, and involvement of 1 out of 28 perirectal lymph nodes. IHC testing of the tumor showed normal mismatch repair (MMR) protein expression. Tumor gene sequencing revealed no mutations in exons 12 or 13 of the KRAS gene.
Figure 1.

Morphologic, immunohistochemical, and fluorescence in situ hybridization (FISH) findings in Case 1. (A) Well-differentiated rectal adenocarcinoma, hematoxylin and eosin staining at 100× magnification. (B) HER2 3+ positivity, immunohistochemical staining at 100× magnification. (C) FISH with HER2 probe and chromosome enumeration probe 17 (CEP 17) showing high-level HER2 amplification.
The patient was commenced on first-line therapy with 5-fluorouracil, irinotecan, and bevacizumab and had an excellent radiologic and biochemical response sustained for just >2 yr before disease progression. Tumor within his liver was then sampled by ultrasound-guided core needle biopsy and submitted to the POG trial for complete molecular characterization.
Genomic Analyses
Whole-genome analysis demonstrated 162 nonsynonymous mutations in protein coding genes, including 157 single-nucleotide variations (SNVs) and five insertion or deletion mutations (indels) representing the 83rd and 68th percentile for mutation burden compared to The Cancer Genome Atlas (TCGA) CRC data set. In keeping with tumor retention of IHC staining for MMR proteins, genomic analysis showed no evidence of MMR deficiency. No increased expression of CD274, the PD-L1 checkpoint inhibitor coding gene, was detected. No specific characteristic mutational signature was identified. Numerous large regional copy-number alterations and losses of heterozygosity were detected throughout the genome. The point mutations, small insertion/deletion mutations, gene copy-number alterations, and expression level alterations that are relevant to the discussion of potential mechanisms of resistance to HER2-directed therapy in Case 1 are summarized in Table 1.
Table 1.
Point mutations, small insertion/deletion mutations (indels), gene copy-number alterations, and expression level alterations relevant to potential mechanisms of resistance to HER2-directed therapy in Case 1
| Gene | Chromosome | DNA reference | Protein reference | dbSNP ID | Variant type | Genotype | Predicted effect |
|---|---|---|---|---|---|---|---|
| PIK3CA | 3q26.3 | NM_006218.3:c.112C>T | p.Arg38Cys | 749415085 | SNV_substitution | Heterozygous | Likely pathogenic; strong gain of function |
| APC | 5q22.2 | NM_000038.5:c.847C>T | p.Arg283Ter | 786201856 | SNV_nonsense mutation | Heterozygous | Pathogenic; loss of function |
| APC | 5q22.2 | NM_000038.5(APC):c.3927_3931delAAAGA | p.Ile1309fs | 121913224 | deletion_frameshift | Heterozygous | Pathogenic; loss of function |
| TP53 | 17p13.1 | NM_000546.5:c.919+1G>T | p.Xnspl | 1131691039 | SNV_splice donor | Homozygous | Likely pathogenic; loss of function |
| Gene | Chromosome | DNA reference | Copy change | CNV state | Expression (RPKM) | Expression fold change versus Normal colon | Expression percentile compared to TCGA COAD/READ |
|---|---|---|---|---|---|---|---|
| ERBB2 | 17q12 | NM_004448.3 | +92 | Amplification | 230 | +33 | 98 |
| MET | 7q31.2 | NM_000245.3 | +1 | Gain | 48 | +6.7 | 93 |
| PTEN | 10q23.3 | NM_000314.6 | +1 | Gain | 5.1 | −4.1 | 2 |
RPKM, reads per kilobase of transcript per million mapped reads; TCGA COAD/READ, the mRNA expression level reported by The Cancer Genome Atlas for colon adenocarcinoma and rectum adenocarcinoma.
Receptor Tyrosine Kinase Pathways
HER2 IHC staining performed on the primary rectal resection showed intense (3+), circumferential staining of all tumor cells, interpreted as HER2+ according to CRC-specific scoring criteria (Fig. 1B; Valtorta et al. 2015). All other molecular testing, including FISH and protein mass spectrometry, was performed on tumor sampled from the patient's liver after first-line chemotherapy and bevacizumab. ERBB2 had 92 copy gains and approximately 33-fold increased gene expression, corresponding to 98th percentile expression compared to TCGA CRC data set (Fig. 2). FISH also showed a very high ERBB2 copy number (Fig. 1C). Protein mass spectrometry showed a 5.3-fold increase in HER2 compared to normal colon. Increased mRNA expression of ERBB3 was not detected.
Figure 2.
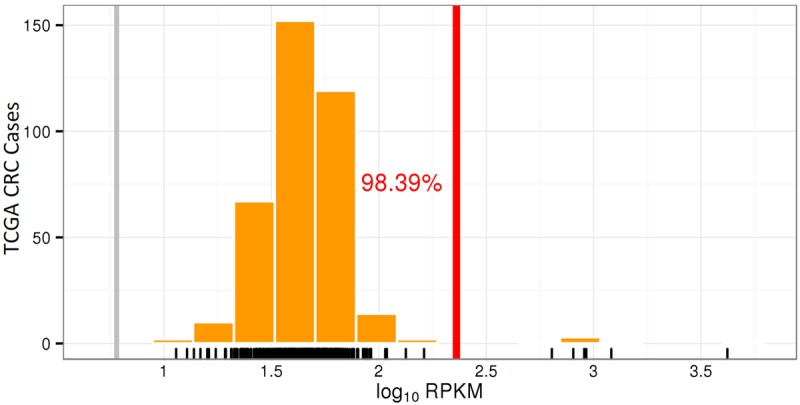
Increased mRNA expression of ERBB2 in Case 1. Histogram showing ERBB2 mRNA expression level in Case 1 (vertical red line) expressed as the common logarithm of reads per kilobase of transcript per million mapped reads (log10 RPKM) compared to cases from the TCGA CRC data set (counted as vertical orange bars).
The receptor tyrosine kinase gene MET showed a single copy gain and 6.7-fold increased gene expression, corresponding to 93rd percentile expression compared to TCGA CRC data set. No mutations in RAS/RAF pathway genes were identified. A gain-of-function mutation in PIK3CA was detected and PTEN showed 4.1-fold reduced gene expression, corresponding to 2nd percentile expression compared to TCGA CRC data set.
Cell Cycle and Apoptosis Pathways
Biallelic deleterious mutations in APC and TP53 are in keeping with tumor evolution through the chromosomal instability pathway of colorectal carcinogenesis. A homozygous loss-of-function mutation of TP53 was identified. Alterations of the Wnt signaling pathway included heterozygous nonsense and frameshift mutations in APC as well as increased expression of downstream targets including MYC, CCND1, and MMP7. Homozygous deletions of cyclin-dependent kinase inhibitors, CDKN2A and CDKN2B, were detected.
Treatment Outcomes
The patient received second-line chemotherapy with 5-fluorouracil and oxaliplatin (FOLFOX). His metastatic disease progressed again after 5 months, and he was switched to a third-line regimen of irinotecan with cetuximab. Upon further progression, he was again switched to regorafenib. Based on the molecular characterization of his tumor there was a consideration of therapy with trastuzumab and lapatinib, but no clinical trial with these agents was available to him at the time of progression.
RESULTS (CASE 2)
Clinical Presentation
Imaging demonstrated bulky liver and pelvic masses and enlarged abdominal lymph nodes in a 49-yr-old woman who had presented with a short history of abdominal pain and distension. She had no concurrent medical problems and no significant family history of malignancy. Colonoscopic biopsy revealed a moderately differentiated colorectal adenocarcinoma in the transverse colon (Fig. 3A). IHC testing of the tumor revealed a normal MMR profile and no BRAF V600E mutation. Tumor gene sequencing revealed no mutations in exons 12 or 13 of the KRAS gene. Tumor tissue was obtained for molecular analysis by a second colonoscopic biopsy prior to the initiation of first-line chemotherapy with 5-fluorouracil, irinotecan (FOLFIRI), and bevacizumab.
Figure 3.

Morphologic and immunohistochemical Findings in Case 2. (A) Moderately differentiated adenocarcinoma of the transverse colon, hematoxylin and eosin staining at 100× magnification. (B) HER2 3+ positivity, immunohistochemical staining at 100× magnification.
Genomic Analyses
Whole-genome analysis revealed 70 nonsynonymous mutations in protein coding genes, including 64 SNVs and 6 indels, representing 23rd and 39th percentile for mutation burden compared to TCGA CRC data set. A mutational signature correlating strongly with patient age at cancer diagnosis was detected (Alexandrov et al. 2013).
Low expression of MMR genes (MLH1, PMS1, PMS2, and MGMT) was identified. However, the tumor showed low mutation burden, no genomic evidence of microsatellite instability, and intact IHC expression of MMR proteins. Increased expression of CD274 was not detected. Numerous large regional copy-number alterations and losses of heterozygosity were detected throughout the genome. The point mutations, small insertion/deletion mutations, gene copy-number alterations, and expression level alterations that are relevant to the discussion of potential mechanisms of resistance to HER2-directed therapy in Case 2 are summarized in Table 2.
Table 2.
Point mutations, small insertion/deletion mutations (indels), gene copy-number alterations and expression level alterations relevant to potential mechanisms of resistance to HER2-directed therapy in Case 2
| Gene | Chromosome | DNA reference | Protein reference | dbSNP ID | Variant type | Genotype | Predicted effect |
|---|---|---|---|---|---|---|---|
| APC | 5q22.2 | NM_000038.5:c.1682delA | p.Lys561fs | NAa | SNV_frameshift mutation | Heterozygous | Likely pathogenic; loss of function (inferred) |
| APC | 5q22.2 | NM_000038.5:c.3956delC | p. p.Pro1319fs | 1057517558 | SNV_frameshift mutation | Heterozygous | Pathogenic; loss of function |
| TP53 | 17p13.1 | NM_000546.5:c.743G>A | p.Arg248Gln | 11540652 | Deletion_substitution | Homozygous | Pathogenic; loss of function |
| Gene | Chromosome | DNA reference | Copy change | CNV state | Expression (RPKM) | Expression fold change versus Normal colon | Expression percentile compared to TCGA COAD/READ |
|---|---|---|---|---|---|---|---|
| ERBB2 | 17q12 | NM_004448.3 | +86 | Amplification | 810 | +120 | 100 |
| MET | 7q31.2 | NM_000245.3 | +1 | Gain | 25 | +3.6 | 52 |
| PTEN | 10q23.3 | NM_000314.6 | 0 | Neutral | 6.1 | −3.5 | 5 |
| MUC1 | 1q22 | NM_002456.5 | +1 | Gain | 170 | +26 | 88 |
| MTOR | 1p36.2 | NM_004958.3 | 1 | Gain | 11 | +2.1 | 91 |
RPKM, reads per kilobase of transcript per million mapped reads; TCGA COAD/READ, the mRNA expression level reported by The Cancer Genome Atlas for colon adenocarcinoma and rectum adenocarcinoma.
aThis single-nucleotide deletion has not previously been reported. APC loss of function is inferred because this is a frameshift mutation. The finding has been submitted to dbSNP for assignment of a variant ID.
Receptor Tyrosine Kinase Pathways
Several genes involved in the MAP kinase and PI3K–AKT–mTOR pathways had amplification and increased expression. ERBB2 had 86 copy gains and approximately 120-fold increased gene expression, corresponding to 99th percentile expression compared to TCGA CRC data set (Fig. 4). IHC staining with HER2 antibody also showed intense (3+), circumferential staining of all tumor cells, interpreted as HER2+ according to CRC-specific scoring criteria (Fig. 3B).
Figure 4.
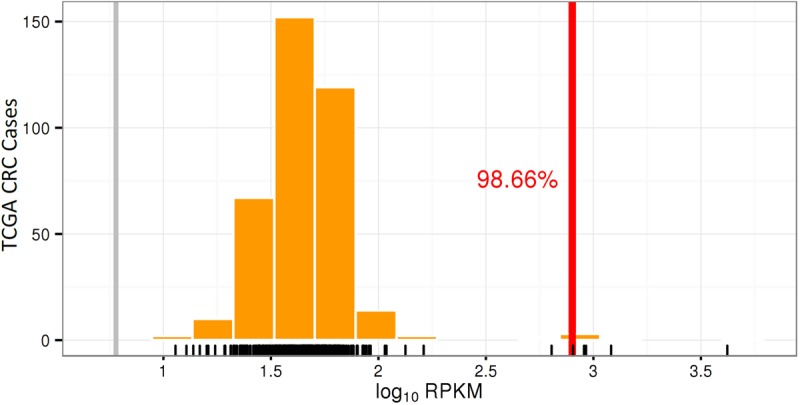
Increased mRNA expression of ERBB2 in Case 2. Histogram showing ERBB2 mRNA expression level in Case 2 (vertical red line) expressed as the common logarithm of reads per kilobase of transcript per million mapped reads (log10 RPKM) compared to cases from the TCGA CRC data set (counted as vertical orange bars).
The receptor tyrosine kinase MET had 3.6-fold increased gene expression, corresponding to 52nd percentile expression compared to TCGA CRC data set. PTEN, a negative regulator of the PI3K–AKT–mTOR pathway, had 3.5-fold decreased gene expression, corresponding to 5th percentile expression compared to TCGA CRC data set. MUC1, a gene that encodes a cell surface mucin protein shown to activate several pathways, including the PI3K–AKT–mTOR, MAP kinase, and Wnt signaling pathways (Kufe 2013), had ∼26-fold increased gene expression, corresponding to 88th percentile expression compared to TCGA CRC data set. MTOR had 2.1-fold increased gene expression, corresponding to 91st percentile expression compared to TCGA CRC data set. The level of ERBB3 expression was very low.
NRAS, BRAF, and MAP3K9 all had increased expression, and NF1, a negative regulator of the MAP kinase pathway, had decreased expression. Downstream transcription factors, MYCN, ETV4, SKP2, and ELK4 all had markedly increased expression. Increased expression of BRD4 and NOTCH1 were inferred drivers of MYCN and SKP2 expression, respectively.
Cell Cycle Pathways
Deregulation of the cell cycle was evident from altered expression of several genes. Increased expression of EZH2 and repression of the cell cycle regulator, CDKN2A, were detected. CDKN1B showed a low level of expression and degradation of its protein product, p27Kip1, is an expected consequence of increased SKP2 expression. Copy gains of CDK6 and CCNE2, as well as increased expression of both genes, were detected. PLK1 and AURKA, facilitators of cell cycle progression, also had copy gains and markedly increased expression.
WNT Pathway and TP53
Biallelic deleterious mutations in APC and TP53 are in keeping with tumor evolution through the chromosomal instability pathway of colorectal carcinogenesis. APC showed biallelic loss-of-function frameshift mutations. CTNNB1 targets, including GAST and BIRC5, had an overall pattern of up-regulation. TP53 harbored a homozygous mutation known to result in mutant p53 protein with both loss of tumor suppressor properties and gain of oncogenic properties (Freed-Pastor and Prives 2012).
Treatment Outcomes
The patient had an excellent clinical, radiological, and biochemical response to her initial chemotherapy. The patient had 38 cycles of leucovorin, 5-fluorouracil, and irinotecan (FOLFIRI) with bevacizumab until radiological progression was noted. She has subsequently initiated therapy with FOLFOX. Upon progression, she may be eligible for a clinical trial of HER2-directed therapy, through the CCTG CAPTUR clinical trial (NCT03297606).
DISCUSSION
The genomic landscape of ERBB2-amplified CRC is not yet described in detail. All tumors in the HERACLES trial tested HER2+ and progressed despite EGFR-directed therapy. A subset of the tumors also did not respond to HER2-directed therapy (Sartore-Bianchi et al. 2016). Most ERBB2-amplified CRCs studied to date have been wild-type in RAS and RAF pathway genes. The reasons that some ERBB2-amplified CRCs do not respond to EGFR, and HER2-directed therapy has not been fully explored. We report the complete molecular characterization of two ERBB2-amplified CRCs identified in the ongoing POG trial with the primary aim of generating hypotheses about the potential mechanisms of resistance to EGFR-directed and HER2-directed therapy. Although neither patient with ERBB2-amplified CRC identified in the POG trial has yet received HER2-directed therapy, the patients may seek government funding or enrollment in a clinical trial to receive trastuzumab and lapatinib.
ERBB2 amplification has been reported to correlate with a lack of response to EGFR-directed therapy (Martin et al. 2013; Jeong et al. 2017) and is a potential explanation for the patient's lack of response to cetuximab in Case 1. Several other molecular alterations have been associated with resistance to EGFR-directed therapy in CRC (Bertotti et al. 2015), although none were detected in Case 1. Although we detected decreased expression of PTEN and an activating mutation in exon 2 of PIK3CA, neither alteration is expected to affect prognosis or to predict response to cetuximab (Karapetis et al. 2014; Llovet et al. 2015) and the College of American Pathologists cites a lack of sufficient evidence to recommend analysis of PIK3CA and PTEN for selection of therapy in CRC, except in the setting of clinical trials (Sepulveda et al. 2017). The patient described in Case 2 has not yet received EGFR-directed therapy but she does not harbor any resistance-associated molecular alterations, other than ERBB2 amplification.
Patients required wild-type status of KRAS codons 12 and 13 for inclusion in the HERACLES trial, but, following the trial, plasma-based mutational analysis revealed a BRAF V600E mutation in one patient and a KRAS A146T mutation in another. KRAS A146T mutation has been shown not to affect sensitivity to EGFR-directed therapy in CRC (De Roock et al. 2010). Data on whether or not BRAF V600E mutation predicts a lack of response to EGFR-directed therapy in CRC are inconclusive, and the use of BRAF V600E as a predictive marker in CRC is not currently recommended (Sepulveda et al. 2017). Additional molecular alterations that were not identified over the course of the HERACLES trial may have contributed to the observed resistance to EGFR and HER2-directed therapies.
Strong concordance between ERBB2 amplification and IHC overexpression of HER2 protein has been demonstrated in CRC (Ooi et al. 2004). Both cases in our cohort are ERBB2-amplified, KRAS wild-type CRCs correctly identified as HER2+ by IHC using newly proposed CRC-specific scoring criteria (Valtorta et al. 2015). In Case 1, FISH analysis and protein mass spectrometry also showed numerous ERBB2 copy gains and increased HER2 protein expression.
Quantification of ERBB2 copy number and HER2 protein expression by analyses such as quantitative IHC (Jensen et al. 2017), PCR, FISH, and protein mass spectrometry has a potential advantage over nonquantitative or semiquantitative detection of HER2 positivity. Data from the HERACLES trial suggested that higher levels of ERBB2 amplification are associated with an increased likelihood of response to HER2-directed therapy in CRC (Sartore-Bianchi et al. 2016). Quantification of HER2 protein expression in breast and gastric cancer has also suggested that higher levels of HER2 protein may predict increased likelihood of response to HER2-directed therapy (Bang et al. 2010; Scaltriti et al. 2015). The use of mass spectrometry to quantify the level of HER2 protein expression in CRC is feasible. However, further study will be required to determine whether this relatively novel method of HER2 protein quantification can reliably be used to predict response to HER2-directed therapy.
The very high-level of ERBB2 amplification is a potential driver of tumorigenesis in both cases in our cohort, although ERBB2 amplification and HER2 protein overexpression do not always indicate that HER2-directed therapy will be effective. A subset of patients in the HERACLES trial designated as HER2+ and shown to have high ERBB2 copy numbers suffered disease progression while on HER2-directed therapy (Sartore-Bianchi et al. 2016). Numerous potential mechanisms of resistance to HER2-directed therapy have been identified, mostly through studies involving breast and gastric cancer (Montemurro and Scaltriti 2014).
Although the effect of PI3K pathway activation on the sensitivity of CRC to HER2-driected therapy has not been directly investigated, activating mutations of PIK3CA and decreased expression of PTEN have been identified as potential mechanisms of resistance to trastuzumab and lapatinib in breast cancer (Berns et al. 2007; Wang et al. 2011). A study using breast cancer cell lines and mouse xenografts demonstrated that activating mutations of PIK3CA and decreased expression of PTEN confer resistance to lapatinib that is reversible by a drug that inhibits both PI3K and mTOR (Eichhorn et al. 2008). However, data from randomized controlled trials have been conflicting (Dave et al. 2011; Loi et al. 2013; Perez et al. 2013), and the evidence is insufficient to confirm that PI3K pathway alterations, as observed in Case 1, would confer resistance to HER2-directed therapy. T-DM1, a conjugation of trastuzumab with emtansine, a chemotherapeutic agent, appears to overcome trastuzumab resistance in breast cancer by bypassing PIK3CA activating mutations downstream from the HER2 receptor (Baselga et al. 2016). A trial of T-DM1 in patients with HER2+ CRC who have shown disease progression in the HERACLES trial is currently ongoing (Siena et al. 2016). Although no PIK3CA activating mutation was identified in Case 2, decreased expression of PTEN and increased expression of MTOR, as were identified in Case 2, could limit the effectiveness of HER2-directed therapy even if combined with drugs that act downstream from PIK3CA.
Although mutations of KRAS, NRAS, and BRAF are not often associated with ERBB2 amplification in CRC (Bertotti et al. 2011; The Cancer Genome Atlas Network 2012; Valtorta et al. 2015), they could account for some cases of resistance to HER2-directed therapy. Discordance of KRAS status between primary and metastatic CRC has been reported as low as 2% (Vakiani et al. 2012) but may be as high as 20% (Lee et al. 2015), suggesting that some cases of CRC may have heterogeneity in the mutational status of RAS pathway genes. Furthermore, the effect of chemotherapy and EGFR-directed therapy on the mutational status of RAS pathway genes in CRC is not well studied. Chemotherapy and EGFR-directed therapy may select for tumor cell populations harboring RAS pathway mutations, even in tumors which are identified as wild-type on pretreatment mutational testing. Therefore, reevaluation of the mutational status of RAS pathway genes prior to the initiation of HER2-directed therapy may be warranted in patients who have received chemotherapy and EGFR-directed therapy.
Clinical testing of both primary colorectal tumors in Cases 1 and 2 occurred prior to the initiation of chemotherapy and targeted therapy; both tumors were wild-type for BRAF and KRAS codons 12 and 13. The primary colorectal tumor in Case 1 was identified as HER2+ by IHC prior to the initiation of chemotherapy and targeted therapy. The molecular features of Case 1 were extensively reanalyzed 2 years later, as part of the POG trial, using tissue obtained from a metastatic liver lesion following progression on first-line therapy consisting of 5-fluorouracil, irinotecan, and bevacizumab. ERBB2-amplification and the mutational status of RAS pathway genes in Case 1 was concordant between the colorectal primary tumor and the liver metastasis.
Marx and colleagues have previously identified heterogeneity of ERBB2 amplification, both within primary tumor and at metastatic sites, in four cases of CRC (Marx et al. 2010). However, none of the ERBB2-amplified CRC cases in the study by Marx and colleagues showed <40% of tumor cells positive for ERBB2 amplification in either the primary tumor or at metastatic sites. Future studies correlating heterogeneity of ERBB2 amplification with response to HER2-directed therapy in CRC are warranted. CRC with heterogeneity of ERBB2 amplification has the potential for at least partial response to HER2-directed therapy if a significant proportion of tumor cells are ERBB2-amplified. However, heterogeneity of amplification remains a potential explanation for the lack of response to HER2-directed therapy observed in some cases of ERBB2-amplified CRC, such as those in the HERACLES trial.
Concordance in ERBB2 amplification between tumor sampled before and after treatment in Case 1 suggests that chemotherapy and bevacizumab do not select against tumor cells harboring ERBB2 amplification. Unfortunately, the data collected on Case 1 did not allow for the comparison of ERBB2 amplification in samples collected before and after administration of cetuximab. Comparisons between primary and metastatic tumor and comparisons pre- and posttreatment could not be performed in Case 2 because only tissue biopsied from the patient's primary transverse colon tumor prior to treatment was made available for testing. Future research should include additional ERBB2-amplified CRCs with primary and metastatic tumor sampled before and after administration of relevant chemotherapeutic agents and targeted therapies.
Trastuzumab is effective against HER2+ breast cancer because it interferes with the dimerization between HER2 and HER3, thereby decreasing the activity of downstream signaling pathways such as the PI3K–AKT–mTOR pathway (Junttila et al. 2009). Lapatinib, an inhibitor of the tyrosine kinase function of HER2, is also used to treat patients with HER2+ metastatic breast cancer whose disease has progressed despite HER2-directed antibody therapy. However, up-regulation of ERBB3, a mechanism of resistance to lapatinib, leads to sustained activation of the PI3K–AKT–mTOR pathway and reduces the effectiveness of PI3K/AKT inhibitors (Chandarlapaty et al. 2011; Garrett et al. 2011; Montemurro and Scaltriti 2014). Both cases in our cohort showed low expression of ERBB3, although neither patient had yet received lapatinib or any other HER2-directed therapy at the time of data collection. Activation of the PI3K pathway may account for the lack of response to trastuzumab and lapatinib in some of the CRC cases in the HERACLES trial. However, the data on the effect of PI3K pathway alterations are presently insufficient to predict response to HER2-directed therapy in HER2+ CRC.
Increased expression of MET, the gene encoding a receptor tyrosine kinase that activates both the PI3K and MAPK pathways, was identified in both cases. High levels of c-MET and HER2 have previously been demonstrated in CRC (Yao et al. 2013). Interaction between HER2 and MET is also a proposed mechanism of resistance to trastuzumab in breast cancer (Shattuck et al. 2008; Paulson et al. 2013). Multiple clinical trials investigating the MET receptor as a therapeutic target in CRC are ongoing and at least one has shown promising initial results (Rimassa et al. 2015). MET-directed therapy as a means of overcoming resistance to HER2-directed therapy warrants further study.
High-level expression of MUC1, the gene encoding a cell surface protein shown to activate the PI3K–AKT–mTOR and MAP kinase pathways and to participate in nuclear signaling alongside Wnt/β-catenin, STAT, and NF-κB (Li et al. 2003; Kufe 2013), was identified in Case 2. Immunohistochemical expression of MUC1 protein correlates with an increased risk of lymph node and distant metastasis in CRC (Zeng et al. 2015) and increased tumor stage in non-MMR deficient CRC (Lugli et al. 2007). High-level MUC1 expression is also a potential mechanism of resistance to HER2-directed therapy. Although interaction between MUC1 and HER2 in CRC has not been extensively studied, a correlation between immunohistochemical expression of MUC1 and HER2 has been identified (Asonuma et al. 2013). GO-203, a peptide inhibitor of MUC1-C, reduces trastuzumab resistance and inhibits survival of breast cancer cells (Raina et al. 2014) and has also been shown to reduce growth of CRC cell cultures and mouse xenografts (Ahmad et al. 2017). A preclinical study suggested that a vaccine strategy targeting MUC1 protein in CRC is feasible and may enhance production of anti-MUC1 antibodies (Zheng et al. 2014). Tumor vaccine strategies targeting MUC1 protein in breast, lung, colorectal, and other cancers have been in clinical trials for the past few years (Wurz et al. 2014); however, many of the trials have now been terminated because of mixed or negative results. If future treatment strategies targeting MUC1 protein are successful, further study of the interaction between MUC1 and HER2 in CRC and other tumors is warranted, as MUC1 expression may affect sensitivity to HER2-directed therapy.
CONCLUSIONS
The POG trial has identified two KRAS wild-type CRCs showing high-level expression of ERBB2 that also tested HER2+ according to newly developed CRC-specific IHC criteria. No data on the effectiveness of HER2-directed therapy are available for either patient in our cohort. However, given the promising results of the HERACLES trial, a trial of HER2-directed therapy would be worthwhile in both patients, particularly once they have exhausted all other lines of proven therapy.
Techniques that quantify the level of HER2 protein expression may be superior to nonquantitative IHC detection of HER2 positivity because CRCs with high-level HER2 protein expression, such as those identified in the POG and HERACLES trials, are thought more likely to respond to HER2-directed therapy. Several molecular alterations that could confer resistance to HER2-directed therapy, such as increased expression of MUC1 and MET, decreased expression of PTEN, and activating mutations in PIK3CA, have been identified in our cohort. Similar molecular alterations may account for the resistance to HER2-directed therapy observed in the HERACLES trial. Despite the potential for resistance, the results of the HERACLES trial indicate that HER2-directed therapy is beneficial for a substantial proportion of patients with HER2+ CRC. Further study into the potential mechanisms of resistance is necessary for the selection of appropriate targeted therapies and for the development of novel treatment approaches in ERBB2-amplified CRC.
METHODS
Genomic and Transcriptomic Analysis
Patient Enrollment
Patients were enrolled in our ongoing study at the BC Cancer Agency in Vancouver, British Columbia, entitled POG, which enrolls patients with metastatic cancers of all subtypes. Individual's cancers undergo whole-genome DNA and RNA sequencing and in-depth bioinformatic analyses to identify somatic variants and gene expression changes that may be targetable cancer “drivers.” Aberrant pathways are matched to drug databases, and this data is reported to the clinician for each individual patient. The study was approved by the University of British Columbia Research Ethics Committee (REB# H14-00681), and written informed consent was obtained from each patient prior to genomic profiling. Patient identity was anonymized within the research team, and an identification code was assigned to the case for communicating clinically relevant information to physicians. The patients consented to potential publication of findings. Raw sequence data and downstream analytics were maintained within a secure computing environment.
Whole-Genome and Transcriptome Sequencing
Fresh tumor biopsy and a blood sample were collected for comprehensive DNA and RNA sequencing as previously described (Laskin et al. 2015). Briefly, the mean redundant depth of coverage for the constitutional and tumor samples was ∼40× and ∼80×, respectively and transcriptomes of ∼200,000,000 reads were generated. Somatic point mutations, small insertions or deletions (indels), and copy-number alterations, detected in the tumor DNA but not in the germline, were identified. De novo assembly of genomic and transcriptomic data was performed to detect rearrangements. Publicly available transcriptome sequencing data from the Illumina BodyMap and TCGA were used to explore the expression profile of human genes and transcripts. A within-sample expression rank was also calculated to further infer significance to outlier gene expression levels. For details regarding the genomic and transcriptomic analyses refer to Supplemental File 1. For information about the number of reads and sequencing coverage for genes listed in Tables 1 and 2, refer to Supplemental File 2.
Proteomic Analysis
Tissue Lysis and Enzymatic Digestion
Tissue processing was carried out as described previously (Hughes et al. 2016). FFPE tissue sections (2 × 10-µm scrolls per technical replicate) were provided on glass slides for processing. Tissue was scraped off of each slide and lysed with buffer consisting of 100 mM HEPES pH 8 (CAT#H3375, Sigma-Aldrich), 4% SDS (CAT#L6026, Sigma-Aldrich), 10mM TCEP (CAT#C4706, Sigma-Aldrich), 40mM CAA (CAT#C0267, Sigma-Aldrich), and 1× complete protease inhibitor—EDTA free (CAT#4693159001, Sigma-Aldrich). Mixtures were heated for 90 min at 90°C, and chilled to room temperature for a further 15 min. Prior to digestion, samples were cleaned using a variation on the SP3 protocol (Hughes et al. 2014). Briefly, to each protein mixture to be treated, 200 µg of SP3 beads was added and mixed by pipetting to generate a homogeneous solution. To induce protein binding to the beads, a volume equivalent to that of the sample of acetonitrile was added to achieve a final proportion of 50% by volume. Bead-protein solutions were mixed to ensure a homogeneous distribution of the beads and incubated for a total of 10 min at room temperature. After incubation, tubes were placed on a magnetic rack for 2 min and the supernatant discarded. After incubation, and while on the magnet, the supernatant was removed and discarded. The beads were rinsed twice while on the magnetic rack through addition of 180 µl of freshly prepared 70% absolute ethanol, and the supernatant was discarded each time. Beads were then rinsed one further time while on the magnetic rack with 180 µl of 100% ethanol, and the supernatant discarded. Rinsed beads were reconstituted in aqueous buffer (∼50 µl, 0.2 M HEPES, pH 8) containing a 1:50 (µg:µg) enzyme to protein amount of trypsin/rLysC mix (Promega, CAT#V5071), and briefly sonicated in a water bath (30 sec) to disaggregate the beads. Mixtures were incubated for 14 h at 37°C in a PCR thermocycler. After incubation, the tubes were sonicated briefly (10 sec) in a water bath to resuspend the beads. The supernatants were recovered using a magnetic rack and transferred to fresh 1.5 ml polypropylene microtubes.
TMT Labeling
TMT 6-plex labeling kits were obtained from Pierce. Each TMT label (5 mg per vial) was reconstituted in 500 µl of acetonitrile and frozen at −80°C. Prior to labeling, TMT labels were removed from the freezer and allowed to equilibrate at room temperature. Labeling reactions were carried out through addition of TMT label in two volumetrically equal steps to achieve a 2:1 (µg:µg) TMT label to peptide final concentration, 30 min apart. All incubations were carried out at room temperature. Reactions were quenched by addition of 10 µl of 1 M glycine solution. Labeled peptides were concentrated in a SpeedVac centrifuge to reduce acetonitrile, combined, and run through a SepPak cartridge (Waters, Inc.) for clean-up prior to HPLC fractionation.
HPLC Fractionation
High-pH reversed phase analysis was performed on an Agilent 1100 HPLC system equipped with a diode array detector (254, 260, and 280 nm). Fractionation was performed on a Kinetix EVO C18 column (2.1 × 150-mm, 1.7-µm core shell, 100 Å, Phenomenex). Elution was performed at a flow rate of 0.2 ml per minute using a gradient of mobile phase A (10 mM ammonium bicarbonate, pH 8) and B (acetonitrile), from 3% to 35% over 60 min. Fractions were collected every minute across the elution window for a total of 48 fractions, which were concatenated to a final set of 12 (e.g., 1 + 13 + 25 + 37 = fraction 1). Fractions were dried in a SpeedVac centrifuge and reconstituted in 1% formic acid with 1% DMSO in water prior to MS analysis.
Mass Spectrometry Analysis
Analysis of TMT-labeled peptide fractions was carried out on an Orbitrap Fusion Tribrid MS platform (Thermo Scientific). Samples were introduced using an Easy-nLC 1000 system (Thermo Scientific). Columns used for trapping and separations were packed in-house. Trapping columns were packed in 100 µm internal diameter capillaries to a length of 25 mm with C18 beads (Reprosil-Pur; Dr. Maisch; 3-µm particle size). Trapping was carried out for a total volume of 10 µl at a pressure of 400 bar. After trapping, gradient elution of peptides was performed on a C18 (Reprosil-Pur; Dr. Maisch; 1.9-µm particle size) column packed in-house to a length of 15 cm in 100-µm internal diameter capillaries with a laser-pulled electrospray tip and heated to 45°C using AgileSLEEVE column ovens (Analytical Sales and Service). Elution was performed with a gradient of mobile phase A (water and 0.1% formic acid) and B (acetonitrile and 0.1% formic acid) over 120 min at a flow rate of 300 nl/min.
Data acquisition on the Orbitrap Fusion (control software version 2.1.1565.20) was carried out using a data-dependent method with multinotch synchronous precursor selection MS3 scanning for TMT tags. Survey scans covering the mass range of 350–1500 were acquired at a resolution of 120,000 (at m/z 200), with quadrupole isolation enabled, an S-Lens RF Level of 60%, a maximum fill time of 50 msec, and an automatic gain control (AGC) target value of 5 × 105. For MS2 scan triggering, monoisotopic precursor selection was enabled, charge state filtering was limited to 2–4, an intensity threshold of 5 × 103 was used, and dynamic exclusion of previously selected masses was enabled for 60 sec with a tolerance of 20 ppm. MS2 scans were acquired in the ion trap in Rapid mode after CID fragmentation with a maximum fill time of 150 msec, quadrupole isolation, an isolation window of 1 m/z, collision energy of 30%, activation Q of 0.25, injection for all available parallelizable time turned OFF, and an AGC target value of 4 × 103. Fragment ions were selected for MS3 scans based on a precursor selection range of 400–1200m/z, ion exclusion of 20 m/z low and 5 m/z high, and isobaric tag loss exclusion for TMT. The top 10 precursors were selected for MS3 scans that were acquired in the Orbitrap after HCD fragmentation (NCE 60%) with a maximum fill time of 150 msec, 50,000 resolution, 110–750 m/z scan range, ion injection for all parallelizable time turned OFF, and an AGC target value of 1 × 105. The total allowable cycle time was set to 4 sec. MS1 and MS3 scans were acquired in profile mode, and MS2 in centroid format.
Mass Spectrometry Data Analysis
Data from the Orbitrap Fusion were processed using Proteome Discoverer Software (ver. 2.1.1.21). MS2 spectra were searched using Sequest HT against a combined UniProt Human proteome database appended to a list of common contaminants (24,624 total sequences). Sequest HT parameters were specified as: trypsin enzyme, 2 missed cleavages allowed, minimum peptide length of 6, precursor mass tolerance of 20 ppm, and a fragment mass tolerance of 0.6. Oxidation of methionine and TMT at lysine and peptide amino-termini were set as variable modifications. Carbamidomethylation of cysteine was set as a fixed modification. Peptide spectral match error rates were determined using the target-decoy strategy coupled to Percolator modeling of positive and false distributions (Käll et al. 2007; Spivak et al. 2009). Data were filtered at the peptide spectral match level to control for false discoveries using a q-value cutoff of 0.01 as determined by Percolator. Data sets generated in Proteome Discoverer were exported and analyzed with a combination of scripts built in R designed in-house. Contaminant and decoy proteins were removed from all data sets prior to downstream analysis.
Additional Information
Data Deposition and Access
The whole-genome sequencing and RNA-seq data for these cases are available as .bam files from the European Genome-phenome Archive (EGA; www.ebi.ac.uk/ega/home) as part of the study EGAS00001001159, accession IDs: EGAD00001003660 (Patient 1); EGAD00001003620 (Patient 2). The interpreted sequence variants listed in Tables 1 and 2 have been deposited in the ClinVar public archive (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under the accession numbers SCV000693729 (NM_006218.3:c.112C>T), SCV000693730 (NM_000038.5:c.847C>T), SCV000693731 (NM_000038.5(APC):c.3927_3931delAAAGA), SCV000693732 (NM_000546.5:c.919+1G>T), SCV000693733 (NM_000038.5:c.1682delA and NM_000038.5:c.3956delC), and SCV000693734 (NM_000546.5:c.743G>A).
Ethics Statement
This work, including data deposition, was approved by the Research Ethics Board at the British Columbia Cancer Agency, protocol H14-00681. Written consent was obtained from the patients after discussion with their oncologists.
Acknowledgments
We gratefully acknowledge the participation of our patients and families, the POG team, and the generous support of the BC Cancer Foundation and Genome British Columbia (project B20POG). The results published here are in part based upon data generated by the following projects and obtained from dbGaP (http://www.ncbi.nlm.nih.gov/gap): The Cancer Genome Atlas managed by the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI) (http://cancergenome.nih.gov) and Genotype-Tissue Expression (GTEx) Project, supported by the Common Fund of the Office of the Director of the National Institutes of Health (https://commonfund.nih.gov/GTEx).
Author Contributions
D.R.O. is the primary author of the manuscript. H.W., D.J.R., and H.L. authored the clinical presentations and made substantial edits to the manuscript. M.B. and M.J. performed data collection and analysis and prepared the figures and table. C.S.H. and G.B.M. acquired and analyzed the proteomics data and authored the methodology for the proteomics. S.J.M.J., J.L. and M.M. contributed to the conception and design of the study; J.L. and M.M. are principal investigators of the POG Clinical Trial. S.Y. and D.F.S. performed pathologic analysis, contributed to the conception and design of the study, and made edits to the manuscript. All authors approved the final manuscript.
Funding
This work was supported by the BC Cancer Foundation and Genome British Columbia (project B20POG). We also acknowledge previous support from the NCI and the NHGRI as part of The Cancer Genome Atlas Project (U24 CA143866) and contributions toward equipment and infrastructure from Genome Canada and Genome BC (projects 202SEQ, 212SEQ, 12002), Canada Foundation for Innovation (projects 20070, 30198, 30981, 33408), and the BC Knowledge Development Fund. M.M. wishes to acknowledge support from the Canadian Institutes of Health Research (CIHR) (FDN-143288).
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Ahmad R, Alam M, Hasegawa M, Uchida Y, Al-Obaid O, Kharbanda S, Kufe D. 2017. Targeting MUC1-C inhibits the AKT-S6K1-elF4A pathway regulating TIGAR translation in colorectal cancer. Mol Cancer 16: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asonuma K, Date Y, Tsukuda G, Hirabayashi K, Wada Y, Takahashi H, Kunimura T, Hamatani S, Morohoshi T. 2013. Expression of HER2 and MUC1 in advanced colorectal cancer: frequency and clinicopathological characteristics. Showa Univ J Med Sci 25: 93–100. [Google Scholar]
- Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, et al. 2010. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 376: 687–697. [DOI] [PubMed] [Google Scholar]
- Baselga J, Lewis Phillips GD, Verma S, Ro J, Huober J, Guardino AE, Samant MK, Olsen S, de Haas SL, Pegram MD. 2016. Relationship between tumor biomarkers and efficacy in EMILIA, a phase III study of trastuzumab emtansine in HER2-positive metastatic breast cancer. Clin Cancer Res 22: 3755–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, et al. 2007. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 12: 395–402. [DOI] [PubMed] [Google Scholar]
- Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, Corà D, Di Nicolantonio F, Buscarino M, Petti C, et al. 2011. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov 1: 508–523. [DOI] [PubMed] [Google Scholar]
- Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, Sausen M, Phallen J, Hruban CA, Tokheim C, et al. 2015. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 526: 263–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. 2011. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 19: 58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave B, Migliaccio I, Gutierrez MC, Wu MF, Chamness GC, Wong H, Narasanna A, Chakrabarty A, Hilsenbeck SG, Huang J, et al. 2011. Loss of phosphatase and tensin homolog or phosphoinositol-3 kinase activation and response to trastuzumab or lapatinib in human epidermal growth factor receptor 2-overexpressing locally advanced breast cancers. J Clin Oncol 29: 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P. 2010. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 11: 753–762. [DOI] [PubMed] [Google Scholar]
- Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, Beijersbergen RL, Valero V, Seoane J, Bernards R, et al. 2008. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res 68: 9221–9230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed-Pastor WA, Prives C. 2012. Mutant p53: one name, many proteins. Genes Dev 26: 1268–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sánchez V, Chakrabarty A, Dave B, Cook RS, Pao W, McKinely E, et al. 2011. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci 108: 5021–5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CS, Foehr S, Garfield DA, Furlong EE, Steinmetz LM, Krijgsveld J. 2014. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol Syst Biol 10: 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CS, McConechy MK, Cochrane DR, Nazeran T, Karnezis AN, Huntsman DG, Morin GB. 2016. Quantitative profiling of single formalin fixed tumour sections: proteomics for translational research. Sci Rep 6: 34949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K, Krusenstjerna-Hafstrøm R, Lohse J, Petersen KH, Derand H. 2017. A novel quantitative immunohistochemistry method for precise protein measurements directly in formalin-fixed, paraffin-embedded specimens: analytical performance measuring HER2. Mod Pathol 30: 180–193. [DOI] [PubMed] [Google Scholar]
- Jeong JH, Kim J, Hong YS, Kim D, Kim JE, Kim SY, Kim KP, Yoon YK, Kim D, Chun SM, et al. 2017. HER2 amplification and cetuximab efficacy in patients with metastatic colorectal cancer harboring wild-type RAS and BRAF. Clin Colorectal Cancer 16: e147–e152. [DOI] [PubMed] [Google Scholar]
- Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, Sampath D, Sliwkowski MX. 2009. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 15: 429–440. [DOI] [PubMed] [Google Scholar]
- Käll L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. 2007. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods 4: 923–925. [DOI] [PubMed] [Google Scholar]
- Karapetis CS, Jonker D, Daneshmand M, Hanson JE, O'Callaghan CJ, Marginean C, Zalcberg JR, Simes J, Moore MJ, Tebbutt NC, et al. 2014. PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer—results from NCIC CTG/AGITG CO.17. Clin Cancer Res 20: 744–753. [DOI] [PubMed] [Google Scholar]
- Kufe DW. 2013. MUC1-C oncoprotein as a target in breast cancer: activation of signaling pathways and therapeutic approaches. Oncogene 32: 1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin J, Jones S, Aparicio S, Chia S, Ch'ng C, Deyell R, Eirew P, Fok A, Gelmon K, Ho C, et al. 2015. Lessons learned from the application of whole-genome analysis to the treatment of patients with advanced cancers. Cold Spring Harb Mol Case Stud 1: a000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KH, Kim JS, Lee CS, Kim JY. 2015. KRAS discordance between primary and recurrent tumors after radical resection of colorectal cancers. J Surg Oncol 111: 1059–1064. [DOI] [PubMed] [Google Scholar]
- Li Y, Yu WH, Ren J, Chen W, Huang L, Kharbanda S, Loda M, Kufe D. 2003. Heregulin targets gamma-catenin to the nucleolus by a mechanism dependent on the DF3/MUC1 oncoprotein. Mol Cancer Res 1: 765–775. [PubMed] [Google Scholar]
- Llovet P, Sastre J, Ortega JS, Bando I, Ferrer M, García-Alfonso P, Donnay O, Carrato A, Jiménez A, Aranda E, et al. 2015. Prognostic value of BRAF, PI3K, PTEN, EGFR copy number, amphiregulin and epiregulin status in patients with KRAS codon 12 wild-type metastatic colorectal cancer receiving first-line chemotherapy with anti-EGFR therapy. Mol Diagn Ther 19: 397–408. [DOI] [PubMed] [Google Scholar]
- Loi S, Michiels S, Lambrechts D, Fumagalli D, Claes B, Kellokumpu-Lehtinen PL, Bono P, Kataja V, Piccart MJ, Joensuu H, et al. 2013. Somatic mutation profiling and associations with prognosis and trastuzumab benefit in early breast cancer. J Natl Cancer Inst 105: 960–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli A, Zlobec I, Baker K, Minoo P, Tornillo L, Terracciano L, Jass JR. 2007. Prognostic significance of mucins in colorectal cancer with different DNA mismatch-repair status. J Clin Pathol 60: 534–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin V, Landi L, Molinari F, Fountzilas G, Geva R, Riva A, Saletti P, De Dosso S, Spitale A, Tejpar S, et al. 2013. HER2 gene copy number status may influence clinical efficacy to anti-EGFR monoclonal antibodies in metastatic colorectal cancer patients. Br J Cancer 108: 668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx AH, Burandt EC, Choschzick M, Simon R, Yekebas E, Kaifi JT, Mirlacher M, Atanackovic D, Bokemeyer C, Fiedler W, et al. 2010. Heterogenous high-level HER-2 amplification in a small subset of colorectal cancers. Hum Pathol 41: 1577–1585. [DOI] [PubMed] [Google Scholar]
- Montemurro F, Scaltriti M. 2014. Biomarkers of drugs targeting HER-family signalling in cancer. J Pathol 232: 219–229. [DOI] [PubMed] [Google Scholar]
- Ooi A, Takehaa T, Li X, Suzuki S, Kunitomo K, Iino H, Fujii H, Takeda Y, Dobashi Y. 2004. Protein overexpression and gene amplification of HER-2 and EGFR in colorectal cancers: an immunohistochemical and fluorescent in situ hybridization study. Mod Pathol 17: 895–904. [DOI] [PubMed] [Google Scholar]
- Paulson AK, Linklater ES, Berghuis BD, App CA, Oostendorp LD, Paulson JE, Pettinga JE, Melnik MK, Vande Woude GF, Graveel CR. 2013. MET and ERBB2 are coexpressed in ERBB2+ breast cancer and contribute to innate resistance. Mol Cancer Res 11: 1112–1121. [DOI] [PubMed] [Google Scholar]
- Perez EA, Dueck AC, McCullough AE, Chen B, Geiger XJ, Jenkins RB, Lingle WL, Davidson NE, Martino S, Kaufman PA, et al. 2013. Impact of PTEN protein expression on benefit from adjuvant trastuzumab in early-stage human epidermal growth factor receptor 2–positive breast cancer in the North Central Cancer Treatment Group N9831 trial. J Clin Oncol 31: 2115–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina D, Uchida Y, Kharbanda A, Rajabi H, Panchamoorthy G, Jin C, Kharbanda S, Scaltriti M, Baselga J, Kufe D. 2014. Targeting the MUC1-C oncoprotein downregulates HER2 activation and abrogates trastuzumab resistance in breast cancer cells. Oncogene 33: 3422–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimassa L, Bozzarelli S, Cordio S, Toppo L, Lonardi S, Zaniboni A, Bordonaro R, Liguigli W, Zagonel V, Tronconi MC, et al. 2015. Phase II study of tivantinib (ARQ 197) in combination with cetuximab in EGFR inhibitor-resistant, MET-high, KRAS wild-type (KRASwt) metastatic colorectal cancer (mCRC). Ann Oncol 26(Supplement 4): v108–iv116.26314772 [Google Scholar]
- Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, Bergamo F, Zagonel V, Leone F, Depetris I, Martinelli E, et al. 2016. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol 17: 738–746. [DOI] [PubMed] [Google Scholar]
- Scaltriti M, Nuciforo P, Bradbury I, Sperinde J, Agbor-Tarh D, Campbell C, Chenna A, Winslow J, Serra V, Parra JL, et al. 2015. High HER2 expression correlates with response to the combination of lapatinib and trastuzumab. Clin Cancer Res 21: 569–576. [DOI] [PubMed] [Google Scholar]
- Sepulveda AR, Hamilton SR, Allegra CJ, Grody W, Cushman-Vokoun AM, Funkhouser WK, Kopetz SE, Lieu C, Lindor NM, Minsky BD, et al. 2017. Molecular biomarkers for the evaluation of colorectal cancer: guideline from the American society for clinical pathology, college of American pathologists, association for molecular pathology, and American society of clinical oncology. Arch Pathol Lab Med 141: 625–657. [DOI] [PubMed] [Google Scholar]
- Shattuck DL, Miller JK, Carraway KL III, Sweeney C. 2008. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res 68: 1471–1477. [DOI] [PubMed] [Google Scholar]
- Siena S, Bardelli A, Sartore-Bianchi A, Martino C, Siravegna G, Magri A, Leone F, Zagonel V, Lonardi S, Amatu A, et al. 2016. HER2 amplification as a ‘molecular bait’ for trastuzumab-emtansine (T-DM1) precision chemotherapy to overcome anti-HER2 resistance in HER2 positive metastatic colorectal cancer: the HERACLES-RESCUE trial. J Clin Oncol 34(4_suppl): pTPS774–TPS774. [Google Scholar]
- Spivak M, Weston J, Bottou L, Käll L, Noble WS. 2009. Improvements to the percolator algorithm for Peptide identification from shotgun proteomics data sets. J Proteome Res 8: 3737–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network. 2012. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakiani E, Janakiraman M, Shen R, Sinha R, Zeng Z, Shia J, Cercek A, Kemeny N, D'Angelica M, Viale A, et al. 2012. Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J Clin Oncol 30: 2956–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtorta E, Martino C, Sartore-Bianchi A, Penaullt-Llorca F, Viale G, Risio M, Rugge M, Grigioni W, Bencardino K, Lonardi S, et al. 2015. Assessment of a HER2 scoring system for colorectal cancer: results from a validation study. Mod Pathol 28: 1481–1491. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhang Q, Zhang J, Sun S, Guo H, Jia Z, Wang B, Shao Z, Wang Z, Hu X. 2011. PI3K pathway activation results in low efficacy of both trastuzumab and lapatinib. BMC Cancer 11: 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurz GT, Kao CJ, Wolf M, DeGregorio MW. 2014. Tecemotide: an antigen-specific cancer immunotherapy. Hum Vaccin Immunother 10: 3383–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao YL, Shao J, Zhang C, Wu JH, Zhang QH, Wang JJ, Zhu W. 2013. Proliferation of colorectal cancer is promoted by two signaling transduction expression patterns: ErbB2/ErbB3/AKT and MET/ErbB3/MAPK. PLoS One 8: e78086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Zhang Q, Zhang Y, Lu M, Liu Y, Zheng T, Feng S, Hao M, Shi H. 2015. MUC1 predicts colorectal cancer metastasis: a systematic review and meta-analysis of case controlled studies. PLoS One 10: e0138049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Edil BH, Soares KC, El-Shami K, Uram JN, Judkins C, Zhang Z, Onners B, Laheru D, Pardoll D, et al. 2014. A safety and feasibility study of an allogeneic colon cancer cell vaccine administered with a granulocyte-macrophage colony stimulating factor–producing bystander cell line in patients with metastatic colorectal cancer. Ann Surg Oncol 21: 3931–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.