

Abstract
Metastatic adenoid cystic carcinomas (ACCs) can cause significant morbidity and mortality. Because of their slow growth and relative rarity, there is limited evidence for systemic therapy regimens. Recently, molecular profiling studies have begun to reveal the genetic landscape of these poorly understood cancers, and new treatment possibilities are beginning to emerge. The objective is to use whole-genome and transcriptome sequencing and analysis to better understand the genetic alterations underlying the pathology of metastatic and rare ACCs and determine potentially actionable therapeutic targets. We report five cases of metastatic ACC, not originating in the salivary glands, in patients enrolled in the Personalized Oncogenomics (POG) Program at the BC Cancer Agency. Genomic workup included whole-genome and transcriptome sequencing, detailed analysis of tumor alterations, and integration with existing knowledge of drug–target combinations to identify potential therapeutic targets. Analysis reveals low mutational burden in these five ACC cases, and mutation signatures that are commonly observed in multiple cancer types. Notably, the only recurrent structural aberration identified was the well-described MYB-NFIB fusion that was present in four of five cases, and one case exhibited a closely related MYBL1-NFIB fusion. Recurrent mutations were also identified in BAP1 and BCOR, with additional mutations in individual samples affecting NOTCH1 and the epigenetic regulators ARID2, SMARCA2, and SMARCB1. Copy changes were rare, and they included amplification of MYC and homozygous loss of CDKN2A in individual samples. Genomic analysis revealed therapeutic targets in all five cases and served to inform a therapeutic choice in three of the cases to date.
Keywords: mouth neoplasm, neoplasm of the eye, tracheal neoplasm
INTRODUCTION
Adenoid cystic carcinomas (ACCs) are relatively uncommon malignant epithelial neoplasms that can arise in a variety of gland-bearing organs most commonly in the head and neck region. They account for ∼1% of all head and neck malignancies and 10% of salivary gland tumors (Coca-Pelaz et al. 2015). ACCs demonstrate three main histologic patterns: solid, cribriform, and tubular (Cheng et al. 1992). Low-grade (grade 1–2) ACCs exhibit a combination of cribriform and tubular patterns and are associated with slower rates of disease progression, including a 5-yr survival rate of ∼90%, with <40% survival at 15 yr. Conversely, high-grade (grade 3) ACC, based on the presence of a significant proportion of tumor showing solid growth pattern, is associated with advanced-stage and more frequent development of late-onset distant metastases (Liu et al. 2012).
Because of the typically slow growth pattern of ACC, historically it has been difficult to assess the efficacy of systemic therapy in metastatic disease, and at present there are no reliable therapeutic options for long-term disease control of ACC (Chae et al. 2015; Coca-Pelaz et al. 2015). Molecular and genome-sequencing studies have begun to elucidate the genetic landscape of ACC in efforts to understand its pathophysiology and potentially alter its clinical course (Ross et al. 2014; Bell et al. 2016; Rettig et al. 2016).
The use of whole-genome sequencing to inform clinical decision-making is an active area of research. Through the Personalized Oncogenomics project (POG) at the British Columbia Cancer Agency (Vancouver, Canada), we have established a pipeline that generates a comprehensive molecular analysis of individual cancer patients with advanced malignancies (Laskin et al. 2015). The aims of these analyses are both to further elucidate and catalog cancer genomes in a metastatic setting and to provide patients with rational treatment options by using genomic data to identify available systemic therapies or active clinical trials. By performing whole-genome and transcriptome sequencing of five patients with ACC, we confirmed a role for the well-characterized MYB- and MYBL1-NFIB gene fusions in ACC pathogenesis and identified actionable targets that helped to inform therapy decisions in three of the five patients.
RESULTS
Clinical and Histological Data
Between 2012 and September 2017, five patients with ACC were enrolled in POG with an average age of 46.2 (SD 9.68) at time of diagnosis. Of the five cases, three patients presented with tumors originating from the head and neck. The other two cases presented with primary tumors at the trachea (POG 1 and POG 2), which are rare but described in the literature (Gondivkar et al. 2011). Interestingly, none of our cases had tumors originating from the major or minor salivary glands, though POG 3 with primary ACC of the hard palate had evidence of invasion into the salivary gland. All patients had evidence of metastatic disease prior to POG analysis. Primary treatments included localized debulking surgery (in four of five cases, including laser bronchoscopy in POG 1) and radiation therapy (in all five cases, including brachytherapy in POG 1). Patient 4 received systemic therapy involving vinorelbine followed by gemcitabine and carboplatin prior to enrollment in the POG program. The average time from diagnosis to enrollment in POG was 8.6 yr ranging from 1 to 24 yr (SD 9.07), thereby highlighting the indolent but persistent nature of ACCs (Table 1).
Table 1.
Patient characteristics
| Patient | Sex | Age at Dx | Year of Dx | Location of primary | Metastases sites | Treatment before POG biopsy | Year of POG consult | Biopsy site |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 45 | 2009 | Distal trachea | Lung | Multiple laser bronchoscopies, XRT (45 Gy), brachytherapy | 2013 | Trachea |
| 2 | F | 44 | 1991 | Trachea | Lung | Partial tracheotomy, XRT (55 Gy), lung met wedge resection | 2015 | Lung nodule |
| 3 | M | 34 | 2007 | Right hard palate | Lung | Resection, XRT | 2016 | Lung nodule |
| 4 | M | 61 | 2015 | Left maxillary sinus/orbit | Lung, liver, T11 vertebra | Left orbital exenteration and reconstruction, XRT (60 Gy) | 2016 | Liver |
| 5 | M | 47 | 2011 | Floor of mouth | Tongue, mylohyoid, myoglossus, lung | XRT (70 Gy) | 2016 | Floor of mouth |
Dx, diagnosis; POG, Personalized Oncogenomics program; XRT, radiotherapy.
Biopsies were obtained from either primary site recurrences or metastases for POG analysis. Notably, histopathologic evaluation of our samples correlated with their clinical course. For example, biopsy of POG 1 revealed a grade 2 ACC characterized by cribriform and tubular patterns, and no evidence of perineural or lymphovascular invasion, which correlates with slower progression over several years (Fig. 1A). Conversely, biopsy of POG 4 revealed a grade 3 ACC characterized by cribriform and solid patterns as well as extensive perineural and lymphovascular invasion, which is reflected in its rapid progression and distant metastases to the bone (Fig. 1B).
Figure 1.

Hematoxylin and eosin staining images of representative POG ACC cases at 100× original magnification: (A) POG 1, (B) POG 2.
Mutational and Expression Landscape of ACC
Whole-genome and transcriptome sequencing was performed on the five samples, and we identified somatic mutations in 112 genes among the cases (Supplemental Table 1). An average of 21.40 protein coding single-nucleotide variants (SNVs) were identified between samples (ranging from 12 to 39). The number of protein coding small insertions and deletions (indels) ranged from one to seven across the five samples, with an average of 3.40. Notably, POG 5 had a considerably higher number of SNVs than the other samples (Fig. 2A). The only genes identified with greater than one mutation among these five samples were BAP1, manifesting as SNVs in POG 4 and POG 5, and BCOR, manifesting as a truncating SNV in POG 2 and frameshifting indels in POG 1 and POG 5 (Fig. 2B). Notably, BCOR is mutated in <15% of tumors of any type across TCGA, but contains putative loss-of-function mutations in 60% (3 of 5) of samples in our cohort (Fig. 2C). BAP1 is a tumor suppressor that encodes a deubiquitinating enzyme regulating key cellular pathways (Wang et al. 2016), whereas BCOR is a BCL6 corepressor mutated in various sarcomas and hematological diseases (Tanaka et al. 2017).
Figure 2.

Mutational landscape of ACC. (A) Number of single-nucleotide variations (SNVs) and short insertion and deletions (indels) in five cases of ACC. (B) Oncoprint depicting small mutations and recurrent fusions in known cancer-related genes, POG1 to POG5 from right to left. (C) Frameshifting mutations observed in BCOR occur throughout the protein, at amino acids 133, 1303, and 1562.
Despite the low frequency of mutations, sequencing of the entire genomes produces sufficient variants for analysis of mutational signatures (Alexandrov et al. 2013) in ACC. All five cases showed evidence of C>T base changes consistent with deamination associated with natural aging processes (Table 2; Supplemental Fig. 1A). In addition, all but one case showed evidence of signature 5, which has an unknown etiology but is commonly observed across cancer types. Furthermore, four of five cases showed evidence for signature 16, also with no known association. Examining the relationship between signature probabilities, we observed a significant anticorrelation between signatures 5 and 16 in all four cases with elevated signature 16, suggesting bleed of signal between these two similar mutation signatures (Supplemental Fig. 1B; Spearman ρ < −0.6). This provides evidence that the presence of signature 16 may be driven by signature 5 elevation rather than representing an additional source of mutations.
Table 2.
Variant table
| Sample | Gene | Chromosome | HGVS DNA reference | HGVS protein reference | Variant type | Predicted effect | dbSNP/COSMIC ID | Genotype |
|---|---|---|---|---|---|---|---|---|
| POG 1 | BCOR | X | c.398_399insAC | p.Ala134fs | ins | Frameshift | Homozygous | |
| POG 1 | MECOM | 3 | c.1636C>T | p.Gln546* | snv | Truncating | Heterozygous | |
| POG 2 | BCOR | X | c.3961G>T | p.Glu1321* | snv | Truncating | Heterozygous | |
| POG 2 | NOTCH1 | 9 | c.2825G>A | p.Cys942Tyr | snv | Missense | Heterozygous | |
| POG 2 | NOTCH1 | 9 | c.4045_4052del GCTCGTAC | p.Ala1349fs | del | Frameshift | Heterozygous | |
| POG 4 | BAP1 | 3 | c.188C>G | p.Ser63Cys | snv | Missense | COSM96362 | Homozygous |
| POG 4 | FGFR2 | 10 | c.1144T>C | p.Cys382Arg | snv | Missense | rs121913474; COSM915496 | Heterozygous |
| POG 5 | ARID2 | 12 | c.2989C>T | p.Gln997* | snv | Truncating | Heterozygous | |
| POG 5 | BAP1 | 3 | c.374A>C | p.Glu125Ala | snv | Missense | Heterozygous | |
| POG 5 | BCOR | X | c.4685_4700del GCACTTGGGACTTCTA | p.Gly1562fs | del | Frameshift | Heterozygous | |
| POG 5 | JAK3 | 19 | c.1765G>A | p.Gly589Ser | snv | Missense | Heterozygous | |
| POG 5 | KRAS | 12 | c.34G>C | p.Gly12Arg | snv | Missense | rs121913530 | Heterozygous |
| POG 5 | SMARCA2 | 9 | c.3638G>C | p.Arg1213Pro | snv | Missense | Heterozygous | |
| POG 5 | SMARCB1 | 22 | c.110G>C | p.Arg37Pro | snv | Missense | Heterozygous | |
| POG 5 | STAT3 | 17 | c.1467T>A | p.Asn489Lys | snv | Missense | Heterozygous |
HGVS, Human Genome Variation Society; dbSNP, Single Nucleotide Polymorphism database; COSMIC, Catalogue of Somatic Mutations in Cancer; ins, insertion; snv, single-nucleotide variant; del, deletion.
In addition to a low mutation load, we observed only a low level of copy-number alterations in these tumors, with genomes of three samples predominantly copy neutral with small numbers of deletions, and POG 4 and POG 5 showing in addition some limited regions of amplification (Supplemental Fig. 2). No genes were subject to homozygous deletion or amplification in more than one sample. POG 4 contained a homozygous deletion of CDKN2A/B (p16) and corresponding decrease in expression. POG 5 showed amplification on 8q24 containing MYC and associated increased MYC expression; MYC also showed increased expression in POG 2 and POG 4 but without associated amplification.
We additionally compared gene expression in our five samples to 40 different primary tumor types, including 37 primary (sub)types from the Cancer Genome Atlas (TCGA) (The Cancer Genome Atlas Research Network et al. 2013). The frequency of mutations in our cases was notably low, with percentiles of 36%, 29%, 21%, and 22% for samples 1–4, respectively, relative to TCGA. Sample 5 had a higher mutation rate, presenting in the 65th percentile relative to TCGA tumors. Of note, this comparator set of cancers does not contain any samples of ACC. However, four of the five samples showed similarity to primary breast cancer (BRCA) despite originating from different tissues (Supplemental Fig. 3). POG 3 showed a similar pattern but with increased similarity to thoracic tumors, likely corresponding to the lung nodule biopsy site of this tumor. To examine further the association with breast cancer, we used the AIMS classifier (Paquet and Hallett 2015) to determine if any of the ACCs had further similarities to specific breast cancer subtypes. We found that POG 1 and POG 4 were classified as basal, corresponding to mostly triple negative breast tumors.
Together, this analysis demonstrates that there are no striking mutational processes active in ACC, consistent with the low mutational burden in these tumors. Furthermore, the lack of characterization of ACC in TCGA demonstrates a rationale for in-depth genomic analysis of ACC.
Structural Variations
To evaluate structural variations in our five samples, we assessed gene fusion products at the genomic and transcriptomic level (Fig. 3A,B). Previous work has implicated the t(6;9) translocation between the MYB oncogene and the transcription factor NFIB as a prominent driver of ACC pathophysiology found in the majority of tumors (Mitani et al. 2010; Gonda and Ramsay 2016). Our analysis elucidated that four of five samples exhibited the MYB-NFIB translocation at the genome and transcriptome level, with POG 3, POG 4, and POG 5 creating identical fusion products with chromosomal breaks after exon 8 of MYB (Fig. 4A). POG 2 also harbored the MYB-NFIB translocation, but with the chromosomal break after exon 12 (Fig. 4B). Of note, POG 1 exhibited a more recently characterized t(8;9) translocation between MYBL1, a distinct MYB-family transcription factor, and NFIB, demonstrated in both the genome and transcriptome sequencing (Fig. 4C; Brayer et al. 2016; Mitani et al. 2016). Interestingly, the MYB-NFIB fusion product was the only translocation present in the transcriptomes of POG 2 and 3. Furthermore, MYB, MYBL1, and NFIB were not involved in any structural variations with other genes in our cases. Our data therefore confirms the importance of the MYB-NFIB translocation in the pathogenesis of ACC, as a variation of the fusion that was present in the transcriptome of all of our cases.
Figure 3.
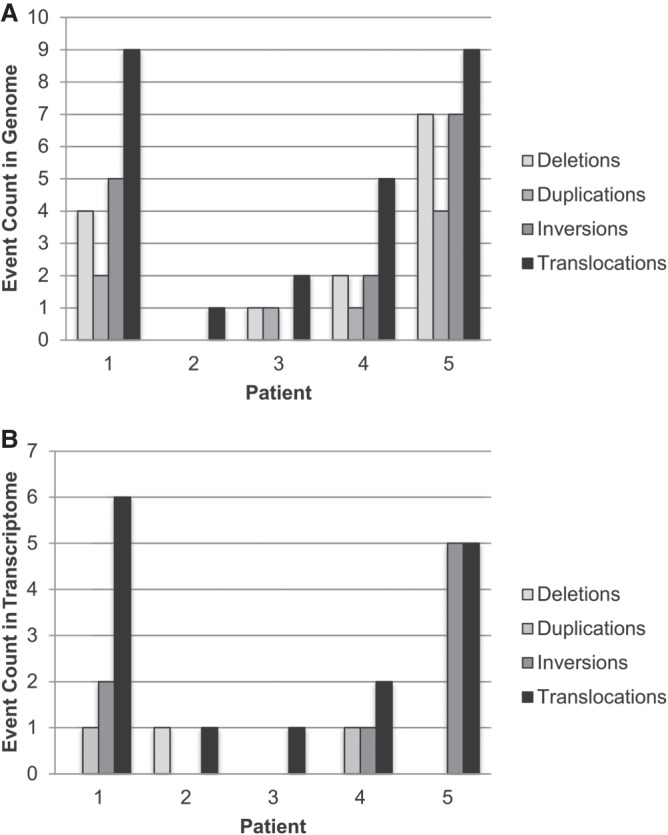
Structural variants in the (A) genome and (B) transcriptome of five cases of ACC.
Figure 4.

MYB-NFIB fusions. (A) Schematic representation of the well-characterized MYB-NFIB fusion (t[6;9]) identified in both the genome and transcriptome of POG 3. POG 4 and 5 created completely identical fusion products with breaks in the same introns. (B) Schematic representation of MYB-NFIB fusion in POG 2, with breakpoint at exon 12. (C) Schematic representation of the lesser characterized MYBL1-NFIB fusion (t[8;9]) identified in POG 1 involving a related, but distinct MYB-family transcription factor. Blue, MYB or MYBL1; green, NFIB; numbered boxes, exons; black boxes, protein coding region; F1, predicted fusion product.
Identification of Potential Therapeutic Targets and Clinical Outcomes
Targeted therapies for advanced ACC are currently under investigation in early phase clinical trials. Because of the rarity of ACC and the relatively low mutational burden, most of drugs in trials have been selected based on observations from preclinical studies. Table 3 highlights some of the targeted agents investigated in phase II trials for advanced ACC. Of note, there were no trials in which complete response was established.
Table 3.
Phase II trials of targeted agents in adenoid cystic carcinoma
| Agent | Molecular target | Authors | N | Partial response | Stable disease |
|---|---|---|---|---|---|
| Imatinib | c-KIT CD117 | Hotte et al. 2005 | 10 | 0 | 2 (20%) |
| Lapatanib | HER-2, EGFR | Agulnik et al. 2007 | 19 | 0 | 15 (79%) |
| Cetuximab | EGFR | Locati et al. 2009 | 23 | 0 | 20 (87%) |
| Bortezomib | Proteosome inhibitor | Argiris et al. 2011 | 25 | 0 | 16 (64%) |
| Sunitinib | VEGFR, c-KIT, PDGFR | Chau et al. 2012 | 13 | 0 | 11 (62%) |
| Gefitinib | EGFR TKI | Jakob et al. 2015 | 19 | 0 | 13 (68%) |
| Axitinib | VEGFR, KIT, PDGFR | Ho et al. 2016 | 33 | 3 (9%) | 25 (76%) |
| Dasatinib | c-KIT, PDGFR | Wong et al. 2016 | 40 (ACC) | 1 (2.5%) | 20 (50%) |
| Nintedanib | VEGFR, FGFR, PDGFR | Kim et al. 2017 | 20 | 0 | 15 (75%) |
| Vorinostat | Histone deacetylase inhibitor | Goncalves et al. 2017 | 30 | 2 (6.7%) | 27 (90%) |
| Dovitinib | VEGFR, PDGFR, FGFR, c-KIT, CSF-1R | Dillon et al. 2017 | 34 | 2 (6%) | 22 (65%) |
HER-2, human epidermal receptor-2; EGFR, epidermal growth factor receptor; VEGFR, vascular endothelial growth factor receptor; PDGFR, platelet-derived growth factor receptor; TKI, tyrosine kinase inhibitor; FGFR, fibroblast growth factor receptor; CSF-1R, colony stimulating factor-1 receptor.
POG analysis in all five of our ACC cases yielded informative and actionable results, meaning that potential targets or risk factors that could affect treatment plans were identified for all five patients (Table 4). We elucidated potential therapeutic targets by identifying mutated genes or genes that were overexpressed relative to TCGA and through subsequent literature review for drug-targeted combinations. Results were deemed “actionable” by an interdisciplinary tumor board based on options that are clinically available and relevant to the patient as determined by tumor characteristics, patient characteristics (including performance status), and drug availability. A summary was created that allowed clinicians involved to make a treatment decisions (Laskin et al. 2015). Three of the five patients have thus far received POG-informed systemic therapy.
Table 4.
POG analyses in all five adenoid cystic carcinoma cases yielded informative and actionable results
| Patient | Potential therapeutic targets | Actionable targets | Action taken |
|---|---|---|---|
| 1 | KIT, FGFR1/2/L1, CDK1/B1/3/6, GADD45B, CCNB, PLK1, TGFA, EGFR, ERBB2, TGFA, AREG, TUBA1A/A1B/A1C, BIRC5, HMGA1 | FGFR1/2/L1 overexpression | Dovitinib |
| 2 | CDK4/6, CCND1, MYC, ROS1, NTRK3, EIGF1R, GFR, KIT, FGFR1/2, PDGFRA, MET, SMO, GLI3, JAG2, NOTCH1, BCL2, FZD7, BCL2 | NOTCH1 overexpression | BMS-986115 (P1T986115) |
| 3 | MYB, NFIB, EGFR, MET, FGFR1/2, ROS1, CDK6, MAP2K2, MMP7, FZD7 | MET overexpression | MGCD265 (glesatinib) |
| 4 | ARFGEF3, CDKN2A/N2B, IGF1R, MYC, PRAME, SKP2, SCL29A1, FGFR1/2, KIT, ALK, RAS, RAF, NOTCH1, CTNNB1, WNT/FZD | FGFR2 gain of function | None (poor ECOG status) |
| 5 | FOLR1, IGF2, KRAS, MDK, MYC, PRAME, PVRL4, SKP2, PDGFA/B/C, FGFR1/2, KIT, IGF2, NOTCH1, HES1/4, FZD6/7, MMP7 | Aberrations in epigenetic regulators; FGFR1/2 overexpression | Pending; potential for HDAC or FGFR antibody therapy |
Potential targets or risk factors that could affect treatment plans were identified for all five patients. Three of five patients have thus far received POG-informed systemic treatment.
Based on overexpression of FGFR-related genes as determined by POG analysis, and preclinical data suggesting partial response to FGFR inhibition in xenograft models of ACC (Frierson and Moskaluk 2013), Patient 1 was enrolled in a phase II clinical trial with dovitinib (TK1258), a multitargeted tyrosine kinase inhibitor with action against FGFRs 1–3, VEGFR, PDGFR, and c-KIT among other receptor kinases (Chase et al. 2007). This patient withdrew from the trial after 4 months because of intolerable side effects, despite having stable disease (as measured by <20% change in tumor size on CT scan). The patient subsequently underwent further palliative radiotherapy before transitioning to palliative care.
POG analysis of Patient 2 did not suggest any commercially available targeted therapies. However, multiple targets with potentially experimental therapies were elucidated. Analysis revealed a NOTCH1 truncating deletion at amino acid 1349, a single base mutation C942Y of unknown effect, NOTCH1 pathway up-regulation, and up-regulation of downstream targets including BCL2, MYC, and CCND1. NOTCH1 signaling has been shown to occur frequently in ACC and may contribute to cell growth and metastasis in these tumors (Su et al. 2014; Morris et al. 2016). Accordingly, Patient 2 was enrolled in a phase I clinical trial with BMS-986115 (Bristol Myers Squibb), a γ secretase and pan-NOTCH inhibitor. Dose reduction was required early, and the patient withdrew from the trial because of toxicity after 2 months and before a response could be determined.
Patient 3 exhibited aberrations in MET expression and was subsequently enrolled in a phase I clinical trial with MGCD265, a multitarget and ATP-competitive inhibitor of c-MET and VEGFR1/2/3 that has recently been studied in lung cancer models (Engstrom et al. 2017). The patient remained clinically stable for several months while on the study, but was removed after 8 months because of increasing tumor size.
Potential therapeutic targets in phase 1 trials were identified for Patient 4, but the patient was not offered POG-mediated intervention because of declining functional status. POG analysis of Patient 5 revealed aberrations in multiple epigenetic modifiers including ARID2, SMARCA2, and SMARCB1 that would suggest a role for intervention with histone deacetylase inhibitors such as vorinostat, which has recently been studied in ACC in a phase II trial (Goncalves et al. 2017). This patient continues to have stable disease and therefore has not yet undergone any POG-informed therapeutic intervention.
DISCUSSION
Several recent studies have investigated the molecular landscape of ACC with a focus on primary tumors, yet there remains a gap in knowledge regarding characterization of recurrent and metastatic ACC and how to treat these tumors. Through the Personalized Oncogenomics project we have elucidated recurrent mutations among five cases of metastatic ACC and identified therapeutic targets that were implemented into clinical decisions guiding treatment for three of the five cases. It is unfortunate that the drugs in available trials were highly toxic and thus the true effect of inhibiting these active pathways remains unclear.
Approximately 55%–60% of ACCs originate in the head and neck, with the majority arising from the major salivary glands (Khafif et al. 2005; Ross et al. 2014). Accordingly, several molecular sequencing studies of ACC have focused on these subtypes (Stephens et al. 2013; Bell et al. 2016; Morris et al. 2016). Interestingly, none of our patients had cancers derived from the major salivary gland. Two cases originated from the mid respiratory tract, which account for ∼20% of ACCs (Gondivkar et al. 2011). Though our sample size is insufficient to generate broad conclusions, our genomic and transcriptomic analysis help to further characterize less represented subtypes of this relatively rare cancer.
The five cases presented exhibit a low mutational burden in keeping with previous studies (Ho et al. 2013; Morris et al. 2016). Indeed, analysis of mutational signatures in our samples did not elucidate any known mutational process driving tumor growth beyond those commonly observed in many cancer types. Interestingly, however, signature 5 is abundant despite the fact that it has not been found to correlate with age in head and neck cancers (Alexandrov et al. 2015). This, alongside the bleed between signatures 5 and 16, may suggest a yet uncharacterized mutational process or a different role of signature 5 in ACC. Despite infrequency of mutations, whole-genome and transcriptome analyses have revealed a number of alterations that may contribute to ACC tumorigenesis including mutations in NOTCH1 (Rettig et al. 2016) and SMARCA2 (Stephens et al. 2013; Chae et al. 2015). Our study is the first to recognize recurrent mutations in BAP1 and BCOR in ACC and suggests that these alterations may play a role in the pathogenesis of metastatic ACC. BAP1 gene mutations have been implicated in various malignancies including uveal and cutaneous melanoma, renal cell carcinoma, and mesothelioma (Carbone et al. 2013), whereas BCOR-inactivating mutations have been identified in various hematologic malignancies (Tanaka et al. 2017). Thus far neither have been associated with ACC. The low frequency of mutations in ACC limits the identification of potentially useful therapeutic targets, which underscores the importance of identifying novel recurrent mutations that contribute to tumor profiling.
Further highlighting the low mutational burden of ACC, four of our five cases had mutational rates below the 40th percentile relative to TCGA, which is consistent with the well-described indolent course of ACC tumor growth. Of note, mutational burden does not seem to correlate with tumor aggressiveness in our cohort. Although POG 5 had the highest mutation rate, it was not characterized by rapid clinical progression, and thus far the patient has been stably managed with localized treatment. Conversely, POG 4 exhibited the second lowest mutational burden in our samples and was characterized by high-grade solid and cribriform histology and rapid progression with distant metastases to the bone, which is known to be associated with poor outcome (Sung et al. 2003). Interestingly, comparison with tumors in TCGA also revealed similarities in the expression profiles of our cases and breast cancer tissue. This may reflect the histologic and genetic similarity among ACCs irrespective of anatomical origin site that has been previously reported (Marchio et al. 2010; Martelotto et al. 2015). ACC of the breast, which accounts for <1% of breast cancers and displays a triple negative and basal-like phenotype (Wetterskog et al. 2012), demonstrates similarity to salivary gland ACC based on the presence of the MYB-NFIB fusion gene (Persson et al. 2009; Fusco et al. 2016). The recurrent MYB-NFIB fusion demonstrated in our study may therefore rationalize the similarity with TCGA breast cancer samples.
Indeed, our analysis revealed MYB-NFIB fusions in four of five cases, with the fifth case exhibiting the closely related MYBL-NFIB fusion. Prior studies have described MYB-NFIB fusions to be a hallmark genetic event in ACC based on its incidence in 50%–60% of tumors (Mitani et al. 2010; Ho et al. 2013), and its presence in the majority of our cases further highlights its role as a driver mutation. Tumors with MYB and MYBL1 translocation have been shown to have similar gene expression profiles, and the translocation of MYB/MYBL and NFIB activates several target genes associated with apoptosis, cell cycle control, cell adhesion, growth, differentiation, and angiogenesis (Ferrarotto et al. 2016). Among our cases, notable genes downstream from the MYB-NFIB fusion that were altered or overexpressed included KIT, MYC, PRAME, FGFR2, BCL2, and CDK6. The MYB-NFIB fusion has been associated with poorer outcome of salivary ACC (Mitani et al. 2011), and its prevalence among our cohort may suggest that it has a more significant role in the pathogenesis of rare and metastatic ACC, and can provide insight into targeted therapy for these cancers.
Although previous studies have elucidated potential therapeutic targets based on whole-genome and transcriptome sequencing of ACC (Ho et al. 2013; Morris et al. 2016), to our knowledge ours is the first study to implement such findings into direct clinical action. Among the five cases, three were treated with POG-informed therapy; specifically a multitargeted tyrosine kinase inhibitor (dovitinib), a pan-NOTCH inhibitor, and a multitarget and ATP-competitive inhibitor of c-MET and VEGFR1/2/3, all of which were in phase I or II clinical trials at the time. In a recently published phase II clinical trial, Dillon et al. (2017) demonstrated that dovitinib had antitumor activity in patients with recently progressed ACC, but came at the expense of moderate toxicity with one in five patients having to withdraw because of intolerable side effects. Aligning with these observations, Patient 1, who underwent treatment with dovitinib based on overexpression of FGFR- related genes, had to withdraw from the trial. Similarly, Patient 2 chose to cease treatment with a pan-NOTCH inhibitor after requiring several dose reductions. Unfortunately both the pan-NOTCH inhibitor and dovitinib have subsequently halted development primarily because of toxicity and before efficacy could be established. Therefore, other therapeutics will need to be investigated to fulfill the need for targeting these pathways. However, despite these outcomes, our POG analysis identified possible targets within aberrant cancer-related pathways and therapeutic trials that hold potential to be effective for patients based on their genomic landscape.
In summary, our data provide insights into the genetic landscape of rare and metastatic ACC and establish that whole-genome and transcriptome sequencing can effectively yield potential therapeutic options for the treatment of these rare cancers.
METHODS
Oversight, Ethics, and Patient Sample Collection
The study was approved by the University of British Columbia Research Ethics Committee (REB# H14-00681), and written informed consent was obtained from each patient before biopsy and genomic profiling. Consent was also obtained for potential publication of findings. Patient data were de-identified within the research team and identification codes were assigned for communicating clinically relevant information to physicians. Raw sequence data and downstream analytics were maintained within a secure computing environment.
Whole-Genome and Transcriptome Sequencing
Fresh frozen biopsies were obtained and intermittent hematoxylin and eosin (H&E)-stained slides were reviewed for tumor content and cellularity. DNA and RNA extractions were performed on OCT-embedded tumor samples using either the AllPrep DNA/RNA Mini Kit (QIAGEN) or the ALine EvoPure kit (Aline Biosciences) to create genomic and transcriptomic libraries. Additionally, DNA was isolated from peripheral blood for sequencing library construction.
PCR-free libraries were constructed for tumor and normal DNA samples, and strand-specific libraries were constructed for tumor RNA samples, as described previously (Jones et al. 2017). Whole-genome sequencing (WGS) was performed to 80× redundant coverage of the tumor and 40× redundant coverage of the normal, and transcriptome sequencing (RNA-seq) was performed to a depth of 200 million reads, using Illumina HiSeq instruments with 125- or 150-bp paired-end reads (Illumina; http://www.illumina.com) (see Table 5).
Table 5.
Sequencing coverage of aligned reads
| Sample | Tumor DNA coverage (WGS) | Normal DNA coverage (WGS) | Tumor RNA coverage (RNA-seq) |
|---|---|---|---|
| POG 1 | 73× | 44× | 408 M reads |
| POG 2 | 88× | 45× | 183 M reads |
| POG 3 | 98× | 50× | 70 M reads |
| POG 4 | 88× | 41× | 161 M reads |
| POG 5 | 100× | 50× | 173 M reads |
Somatic Alteration Analysis
An established analytical pipeline was used for identifying somatic nucleotide and copy-number aberrations throughout the genome from tumor DNA in comparison to normal DNA from the same patient, as previously described (Jones et al. 2017). De novo assembly of genomic and transcriptomic data was carried out using ABySS (Robertson et al. 2010) and Trans-ABySS (Birol et al. 2009; Simpson et al. 2009) to detect structural rearrangements and fusions. Copy number and LOH were visualized with Circos (Krzywinski et al. 2009). Gene fusions were evaluated and visualized using MAVIS (http://mavis.bcgsc.ca/). Visualization of gene alterations by sample was through OncoPrinter and MutationMapper, and recurrence of mutations across TCGA was determined through CBioPortal (http://www.cbioportal.org/) (Cerami et al. 2012; Gao et al. 2013). Mutation signatures were computed based on genome-wide single-nucleotide variant calls and trinucleotide contexts as previously described (Alexandrov et al. 2013) and compared to COSMIC reference signatures (http://cancer.sanger.ac.uk/cosmic/signatures). Signature scores represent the fraction of mutations contributed by a particular signature. To examine the relationship between signatures, in particular signatures 5 and 16, we used a hierarchical Bayesian categorical mixture model to explore the posterior distribution of signature exposures (E Zhao, S Jones, in prep.).
Expression Analysis and Comparison with TCGA Data Sets
Expression levels of each gene were derived from aligned RNA-Seq reads as described in Butterfield et al. (2014) (Jones et al. 2017). Gene expression in the ACC samples was compared to available tumor samples from multiple cancer types in the Cancer Genome Atlas (TCGA; https://tcga-data.nci.nih.gov/tcga/) as well as normal samples from Illumina Human Body Map 2.0 to identify genes over- or underexpressed, as described in Jones et al. (2017). Similarity to TCGA cancer types was determined by a machine learning based pan-cancer classifier trained on whole-transcriptome profiles of TCGA primary cancers (J Grewal, S Jones, in prep.)
ADDITIONAL INFORMATION
Data Deposition and Access
The whole-genome sequencing and RNA-seq data for this case are available as .bam files from the European Genome-phenome Archive (EGA; www.ebi.ac.uk/ega/home) as part of the study EGAS00001001159, under accession numbers EGAD00001002032 (Patient 1), EGAD00001002627 (Patient 2), EGAD00001003065 (Patient 3), EGAD00001003640 (Patient 4), and EGAD00001003679 (Patient 5). Somatic variants were deposited in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) under accession numbers SCV000693656 (BCOR, patient 1), SCV000693657 (MECOM, patient 1), SCV000693671 (MYBL1-NFIB fusion, patient 1), SCV000693658 (BCOR, patient 2), SCV000693659 (NOTCH1, patient 2), SCV000693660 (NOTCH1, patient 2), SCV000693672 (MYB-NFIB fusion, patient 2), SCV000693673 (MYB-NFIB fusion, patient 3), SCV000693661 (BAP1, patient 4), SCV000693662 (FGFR2, patient 4), SCV000693674 (MYB-NFIB fusion, patient 4), SCV000693663 (ARID2, patient 5), SCV000693664 (BAP1, patient 5), SCV000693665 (BCOR, patient 5), SCV000693666 (JAK3, patient 5), SCV000693667 (KRAS, patient 5), SCV000693668 (SMARCA2, patient 5), SCV000693669 (SMARCB1, patient 5), SCV000693670 (STAT3, patient 5), and SCV000693675 (MYB-NFIB fusion, patient 5).
Ethics Statement
The study was approved by the University of British Columbia Research Ethics Committee (REB# H14-00681). Written informed consent was obtained from each patient prior to genomic profiling. Patient identity was anonymized within the research team and an identification code was assigned to the case for communicating clinically relevant information to physicians. The patients consented to potential publication of findings. Raw sequence data and downstream analytics were maintained within a secure computing environment.
Acknowledgments
We gratefully acknowledge the participation of our patients and families, the POG team, and the generous support of the BC Cancer Foundation and Genome British Columbia (project B20POG). We also acknowledge previous support from the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI) as part of The Cancer Genome Atlas project (U24 CA143866) and contributions toward equipment and infrastructure from Genome Canada and Genome BC (projects 202SEQ, 212SEQ, 12002), Canada Foundation for Innovation (projects 20070, 30198, 30981, 33408), and the BC Knowledge Development Fund. The results published here are in part based on data obtained from dbGaP (http://www.ncbi.nlm.nih.gov/gap) generated by The Cancer Genome Atlas managed by the NCI and NHGRI (http://cancergenome.nih.gov).
Author Contributions
M.C. and J.L. devised the study. M.C. and E.P. wrote the manuscript with additional text and feedback from all authors. M.C., E.P., J.G., E.Z., M.R.J., Y.S., K.L.M., M.B., and G.A.T. performed the data analysis. Y.M., A.J.M., and R.A.M. supervised the data generation. T.N., E.C., and S.Y. performed the pathology analysis. H.L., D.R., and J.L. were involved in clinical management. S.J.M.J., M.A.M., and J.L. supervised the project.
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Agulnik M, Cohen EW, Cohen RB, Chen EX, Vokes EE, Hotte SJ, Winquist E, Laurie S, Hayes DN, Dancey JE, et al. 2007. Phase II study of lapatinib in recurrent or metastatic epidermal growth factor receptor and/or erbB2 expressing adenoid cystic carcinoma and non adenoid cystic carcinoma malignant tumors of the salivary glands. J Clin Oncol 25: 3978–3984. [DOI] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. 2013. Deciphering signatures of mutational processes operative in human cancer. Cell Rep 3: 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, Stratton MR. 2015. Clock-like mutational processes in human somatic cells. Nat Genet 47: 1402–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argiris A, Ghebremichael M, Burtness B, Axelrod RS, Deconti RC, Forastiere AA. 2011. A phase 2 trial of bortezomib followed by the addition of doxorubicin at progression in patients with recurrent or metastatic adenoid cystic carcinoma of the head and neck: a trial of the Eastern Cooperative Oncology Group (E1303). Cancer 117: 3374–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell D, Bell AH, Bondaruk J, Hanna EY, Weber RS. 2016. In-depth characterization of the salivary adenoid cystic carcinoma transcriptome with emphasis on dominant cell type. Cancer 122: 1513–1522. [DOI] [PubMed] [Google Scholar]
- Birol I, Jackman SD, Nielsen CB, Qian JQ, Varhol R, Stazyk G, Morin RD, Zhao Y, Hirst M, Schein JE, et al. 2009. De novo transcriptome assembly with ABySS. Bioinformatics 25: 2872–2877. [DOI] [PubMed] [Google Scholar]
- Brayer KJ, Frerich CA, Kang H, Ness SA. 2016. Recurrent fusions in MYB and MYBL1 define a common, transcription factor-driven oncogenic pathway in salivary gland adenoid cystic carcinoma. Cancer Discov 6: 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield YS, Kreitzman M, Thiessen N, Corbett RD, Li Y, Pang J, Ma YP, Jones SJ, Birol I. 2014. JAGuaR: junction alignments to genome for RNA-seq reads. PLoS One 9: e102398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. 2013. BAP1 and cancer. Nat Rev Cancer 13: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae YK, Chung SY, Davis AA, Carneiro BA, Chandra S, Kaplan J, Kalyan A, Giles FJ. 2015. Adenoid cystic carcinoma: current therapy and potential therapeutic advances based on genomic profiling. Oncotarget 6: 37117–37134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase A, Grand FH, Cross NC. 2007. Activity of TKI258 against primary cells and cell lines with FGFR1 fusion genes associated with the 8p11 myeloproliferative syndrome. Blood 110: 3729–3734. [DOI] [PubMed] [Google Scholar]
- Chau NG, Hotte SJ, Chen EX, Chin SF, Turner S, Wang L, Siu LL. 2012. A phase II study of sunitinib in recurrent and/or metastatic adenoid cystic carcinoma (ACC) of the salivary glands: current progress and challenges in evaluating molecularly targeted agents in ACC. Ann Oncol 23: 1562–1570. [DOI] [PubMed] [Google Scholar]
- Cheng J, Saku T, Okabe H, Furthmayr H. 1992. Basement membranes in adenoid cystic carcinoma. An immunohistochemical study. Cancer 69: 2631–2640. [DOI] [PubMed] [Google Scholar]
- Coca-Pelaz A, Rodrigo JP, Bradley PJ, Vander Poorten V, Triantafyllou A, Hunt JL, Strojan P, Rinaldo A, Haigentz M Jr, Takes RP, et al. 2015. Adenoid cystic carcinoma of the head and neck—an update. Oral Oncol 51: 652–661. [DOI] [PubMed] [Google Scholar]
- Dillon PM, Petroni GR, Horton BJ, Moskaluk CA, Fracasso PM, Douvas MG, Varhegyi N, Zaja-Milatovic S, Thomas CY. 2017. A phase II study of dovitinib in patients with recurrent or metastatic adenoid cystic carcinoma. Clin Cancer Res 23: 4138–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstrom LD, Aranda R, Lee M, Tovar EA, Essenburg CJ, Madaj Z, Chiang H, Briere D, Hallin J, Lopez-Casas PP, et al. 2017. Glesatinib exhibits antitumor activity in lung cancer models and patients harboring MET exon 14 mutations and overcomes mutation-mediated resistance to Type I MET inhibitors in nonclinical models. Clin Cancer Res 23: 6661–6672. [DOI] [PubMed] [Google Scholar]
- Ferrarotto R, Heymach JV, Glisson BS. 2016. MYB-fusions and other potential actionable targets in adenoid cystic carcinoma. Curr Opin Oncol 28: 195–200. [DOI] [PubMed] [Google Scholar]
- Frierson HF Jr, Moskaluk CA. 2013. Mutation signature of adenoid cystic carcinoma: evidence for transcriptional and epigenetic reprogramming. J Clin Invest 123: 2783–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco N, Geyer FC, De Filippo MR, Martelotto LG, Ng CK, Piscuoglio S, Guerini-Rocco E, Schultheis AM, Fuhrmann L, Wang L, et al. 2016. Genetic events in the progression of adenoid cystic carcinoma of the breast to high-grade triple-negative breast cancer. Mod Pathol 29: 1292–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves PH, Heilbrun LK, Barrett MT, Kummar S, Hansen AR, Siu LL, Piekarz RL, Sukari AW, Chao J, Pilat MJ, et al. 2017. A phase 2 study of vorinostat in locally advanced, recurrent, or metastatic adenoid cystic carcinoma. Oncotarget 8: 32918–32929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonda TJ, Ramsay RG. 2016. Adenoid cystic carcinoma can be driven by MYB or MYBL1 rearrangements: new insights into MYB and tumor biology. Cancer Discov 6: 125–127. [DOI] [PubMed] [Google Scholar]
- Gondivkar SM, Gadbail AR, Chole R, Parikh RV. 2011. Adenoid cystic carcinoma: a rare clinical entity and literature review. Oral Oncol 47: 231–236. [DOI] [PubMed] [Google Scholar]
- Ho AS, Kannan K, Roy DM, Morris LG, Ganly I, Katabi N, Ramaswami D, Walsh LA, Eng S, Huse JT, et al. 2013. The mutational landscape of adenoid cystic carcinoma. Nat Genet 45: 791–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AL, Dunn L, Sherman EJ, Fury MG, Baxi SS, Chandramohan R, Dogan S, Morris LG, Cullen GD, Haque S, et al. 2016. A phase II study of axitinib (AG-013736) in patients with incurable adenoid cystic carcinoma. Ann Oncol 27: 1902–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotte SJ, Winquist EW, Lamont E, MacKenzie M, Vokes E, Chen EX, Brown S, Pond GR, Murgo A, Siu LL. 2005. Imatinib mesylate in patients with adenoid cystic cancers of the salivary glands expressing c-kit: a Princess Margaret Hospital phase II consortium study. J Clin Oncol 23: 585–590. [DOI] [PubMed] [Google Scholar]
- Jakob JA, Kies MS, Glisson BS, Kupferman ME, Liu DD, Lee JJ, El-Naggar AK, Gonzalez-Angulo AM, Blumenschein GR Jr. 2015. Phase II study of gefitinib in patients with advanced salivary gland cancers. Head Neck 37: 644–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MR, Lim H, Shen Y, Pleasance E, Ch'ng C, Reisle C, Leelakumari S, Zhao C, Yip S, Ho J, et al. 2017. Successful targeting of the NRG1 pathway indicates novel treatment strategy for metastatic cancer. Ann Oncol 28: 3092–3097. [DOI] [PubMed] [Google Scholar]
- Khafif A, Anavi Y, Haviv J, Fienmesser R, Calderon S, Marshak G. 2005. Adenoid cystic carcinoma of the salivary glands: a 20-year review with long-term follow-up. Ear Nose Throat J 84: 662, 64–67. [PubMed] [Google Scholar]
- Kim Y, Lee SJ, Lee JY, Lee SH, Sun JM, Park K, An HJ, Cho JY, Kang EJ, Lee HY, et al. 2017. Clinical trial of nintedanib in patients with recurrent or metastatic salivary gland cancer of the head and neck: A multicenter phase 2 study (Korean Cancer Study Group HN14-01). Cancer 123: 1958–1964. [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin J, Jones S, Aparicio S, Chia S, Ch'ng C, Deyell R, Eirew P, Fok A, Gelmon K, Ho C, et al. 2015. Lessons learned from the application of whole-genome analysis to the treatment of patients with advanced cancers. Cold Spring Harb Mol Case Stud 1: a000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Shao C, Tan ML, Mu D, Ferris RL, Ha PK. 2012. Molecular biology of adenoid cystic carcinoma. Head Neck 34: 1665–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locati LD, Bossi P, Perrone F, Potepan P, Crippa F, Mariani L, Casieri P, Orsenigo M, Losa M, Bergamini C, et al. 2009. Cetuximab in recurrent and/or metastatic salivary gland carcinomas: a phase II study. Oral Oncol 45: 574–578. [DOI] [PubMed] [Google Scholar]
- Marchio C, Weigelt B, Reis-Filho JS. 2010. Adenoid cystic carcinomas of the breast and salivary glands (or ‘The strange case of Dr Jekyll and Mr Hyde’ of exocrine gland carcinomas). J Clin Pathol 63: 220–228. [DOI] [PubMed] [Google Scholar]
- Martelotto LG, De Filippo MR, Ng CK, Natrajan R, Fuhrmann L, Cyrta J, Piscuoglio S, Wen HC, Lim RS, Shen R, et al. 2015. Genomic landscape of adenoid cystic carcinoma of the breast. J Pathol 237: 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitani Y, Li J, Rao PH, Zhao YJ, Bell D, Lippman SM, Weber RS, Caulin C, El-Naggar AK. 2010. Comprehensive analysis of the MYB-NFIB gene fusion in salivary adenoid cystic carcinoma: incidence, variability, and clinicopathologic significance. Clin Cancer Res 16: 4722–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitani Y, Rao PH, Futreal PA, Roberts DB, Stephens PJ, Zhao YJ, Zhang L, Mitani M, Weber RS, Lippman SM, et al. 2011. Novel chromosomal rearrangements and break points at the t(6;9) in salivary adenoid cystic carcinoma: association with MYB-NFIB chimeric fusion, MYB expression, and clinical outcome. Clin Cancer Res 17: 7003–7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitani Y, Liu B, Rao PH, Borra VJ, Zafereo M, Weber RS, Kies M, Lozano G, Futreal PA, Caulin C, et al. 2016. Novel MYBL1 gene rearrangements with recurrent MYBL1-NFIB fusions in salivary adenoid cystic carcinomas lacking t(6;9) translocations. Clin Cancer Res 22: 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris LG, Chandramohan R, West L, Zehir A, Chakravarty D, Pfister DG, Wong RJ, Lee NY, Sherman EJ, Baxi SS, et al. 2016. The molecular landscape of recurrent and metastatic head and neck cancers: insights from a precision oncology sequencing platform. JAMA Oncol 10.1001/jamaoncol.2016.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet ER, Hallett MT. 2015. Absolute assignment of breast cancer intrinsic molecular subtype. J Natl Cancer Inst 107: 357. [DOI] [PubMed] [Google Scholar]
- Persson M, Andren Y, Mark J, Horlings HM, Persson F, Stenman G. 2009. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci 106: 18740–18744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig EM, Talbot CC Jr, Sausen M, Jones S, Bishop JA, Wood LD, Tokheim C, Niknafs N, Karchin R, Fertig EJ, et al. 2016. Whole-genome sequencing of salivary gland adenoid cystic carcinoma. Cancer Prev Res (Phila) 9: 265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K, Lee S, Okada HM, Qian JQ, et al. 2010. De novo assembly and analysis of RNA-seq data. Nat Methods 7: 909–912. [DOI] [PubMed] [Google Scholar]
- Ross JS, Wang K, Rand JV, Sheehan CE, Jennings TA, Al-Rohil RN, Otto GA, Curran JC, Palmer G, Downing SR, et al. 2014. Comprehensive genomic profiling of relapsed and metastatic adenoid cystic carcinomas by next-generation sequencing reveals potential new routes to targeted therapies. Am J Surg Pathol 38: 235–238. [DOI] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res 19: 1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Davies HR, Mitani Y, Van Loo P, Shlien A, Tarpey PS, Papaemmanuil E, Cheverton A, Bignell GR, Butler AP, et al. 2013. Whole exome sequencing of adenoid cystic carcinoma. J Clin Invest 123: 2965–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su BH, Qu J, Song M, Huang XY, Hu XM, Xie J, Zhao Y, Ding LC, She L, Chen J, et al. 2014. NOTCH1 signaling contributes to cell growth, anti-apoptosis and metastasis in salivary adenoid cystic carcinoma. Oncotarget 5: 6885–6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung MW, Kim KH, Kim JW, Min YG, Seong WJ, Roh JL, Lee SJ, Kwon TK, Park SW. 2003. Clinicopathologic predictors and impact of distant metastasis from adenoid cystic carcinoma of the head and neck. Arch Otolaryngol Head Neck Surg 129: 1193–1197. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Nakajima-Takagi Y, Aoyama K, Tara S, Oshima M, Saraya A, Koide S, Si S, Manabe I, Sanada M, et al. 2017. Internal deletion of BCOR reveals a tumor suppressor function for BCOR in T lymphocyte malignancies. J Exp Med 214: 2901–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. 2013. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45: 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Papneja A, Hyrcza M, Al-Habeeb A, Ghazarian D. 2016. Gene of the month: BAP1. J Clin Pathol 69: 750–753. [DOI] [PubMed] [Google Scholar]
- Wetterskog D, Lopez-Garcia MA, Lambros MB, A'Hern R, Geyer FC, Milanezi F, Cabral MC, Natrajan R, Gauthier A, Shiu KK, et al. 2012. Adenoid cystic carcinomas constitute a genomically distinct subgroup of triple-negative and basal-like breast cancers. J Pathol 226: 84–96. [DOI] [PubMed] [Google Scholar]
- Wong SJ, Karrison T, Hayes DN, Kies MS, Cullen KJ, Tanvetyanon T, Argiris A, Takebe N, Lim D, Saba NF, et al. 2016. Phase II trial of dasatinib for recurrent or metastatic c-KIT expressing adenoid cystic carcinoma and for nonadenoid cystic malignant salivary tumors. Ann Oncol 27: 318–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.