

Abstract
Hapalindole alkaloids are a structurally diverse class of cyanobacterial natural products defined by their varied polycyclic ring systems and diverse biological activities. These complex metabolites are generated from a common biosynthetic intermediate by the Stig cyclases in three mechanistic steps: a rare Cope rearrangement, 6-exo-trig cyclization, and electrophilic aromatic substitution. Here we report the structure of HpiC1, a Stig cyclase that catalyzes the formation of 12-epi-hapalindole U in vitro. The 1.5-Å structure revealed a dimeric assembly with two calcium ions per monomer and with the active sites located at the distal ends of the protein dimer. Mutational analysis and computational methods uncovered key residues for an acid-catalyzed [3,3]-sigmatropic rearrangement, as well as specific determinants that control the position of terminal electrophilic aromatic substitution, leading to a switch from hapalindole to fischerindole alkaloids.
The hapalindole family of alkaloids are a large and structurally diverse class of natural products from cyanobacteria of the order Stigonematales1. These metabolites are active against a broad range of targets, including antibacterial, antifungal, insecticidal, and antimitotic activities2–7. Each member is classified as a hapalindole, ambiguine, fischerindole, or welwitindolinone on the basis of its core ring system (Supplementary Fig. 1), and these have been the subject of various total syntheses due to their structural complexity and unique biological properties1. Until recently, comparatively little was known regarding the biogenesis of these alkaloids and, in particular, the construction of the tetracyclic core ring system.
Initial reports demonstrated that hapalindoles are derived from cis-indole isonitrile and geranyl pyrophosphate (GPP)8–10, but the biogenesis of the polycyclic ring systems remained elusive. We recently identified an unexpected biosynthetic intermediate, 1, which undergoes a Cope rearrangement that is followed by a cyclization cascade to generate 12-epi-hapalindole U (5) (Fig. 1)11. The Cope rearrangement is a [3,3]-sigmatropic rearrangement that proceeds through a cyclic transition state12. Although this pericyclic reaction is prevalent in organic synthesis13, it has rarely been identified as a biosynthetic transformation14–16. The biosynthesis of 5 was proposed to proceed through a three-part reaction mechanism: (i) Cope rearrangement of 1 to generate intermediate 3, which sets the stereochemistry at positions C11 and C12; (ii) 6-exo-trig cyclization of 3 to intermediate 4, which sets the stereochemistry at positions C10 and C15; and (iii) electrophilic aromatic substitution of 4 to yield 5 upon deprotonation (Fig. 1). This discovery was expanded to include several Stig cyclases and revealed that the variant configurations observed in this class of alkaloids are generated from the central biosynthetic intermediate 1, which is transformed to products in a regio- and stereospecific fashion by various members of the cyclase family (Fig. 1)17–19. Notably, FimC5, which yields the major product 12-epi-fischerindole U (6), generates a product that has a stereochemical configuration identical to that of 5 at C10, C11, C12, and C15, but differs in the site of C-ring attachment to the indole moiety. This indicates that the enzymes direct the site of terminal electrophilic aromatic substitution, thereby providing the regiochemical control that differentiates the hapalindoles and fischerindoles. Both HpiC1 and FimC5 produce low levels of tricyclic 12-epi-hapalindole C (7), a shunt metabolite that is not converted to its tetracyclic congeners by either enzyme. Thus, biogenesis of hapalindole-type metabolites includes a fascinating mechanistic puzzle in regard to how homologous Stig cyclases maintain stereochemical and regiochemical control at each of these biosynthetic steps in the formation of varied hapalindoles and fischerindoles.
Fig. 1. Biogenesis of hapalindole alkaloids.
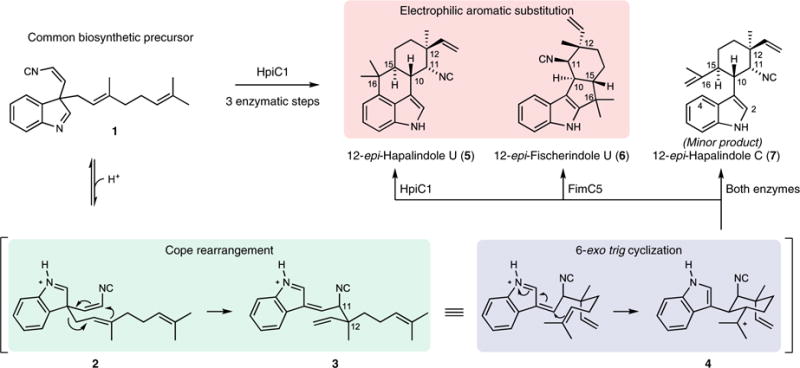
The hapalindole and fischerindole core ring systems arise from the common biosynthetic intermediate 1. Stig cyclases catalyze a Cope rearrangement and subsequent cyclization to generate tetracyclic products and trace levels of tricyclic shunt products. HpiC1 catalyzes formation of 5, while FimC5 catalyzes formation of 6, with identical stereochemistry at C11 and C12 but different C-ring regiochemistry.
In this study, we describe the molecular basis for the Stig cyclase’s ability to control three reactions. As these were originally annotated as unknown proteins8, no information was available regarding the structure of this new type of biosynthetic enzyme. We describe herein the first crystal structure of a Stig cyclase, HpiC1, and show through a mutational analysis, which localizes its active site, the ability to reconfigure its metabolite profile. Density functional theory (DFT) calculations on the mechanism and molecular dynamics simulations provide detailed information on the enzymatic processes that control product formation. These data provide compelling insights into the mechanism of Cope rearrangement, 6-exo-trig cyclization and electrophilic aromatic substitution for this class of natural products.
Results
Structure of HpiC1
Following recent work reporting the function and selectivity of Stig cyclase proteins11,17–19, we sought a structure of HpiC1 to understand the basis for the complex cyclization cascade of 1 and to comprehend how the Stig cyclases catalyze formation of various alkaloid products from this common biosynthetic intermediate. We obtained HpiC1 crystals in four different forms under conditions with varied Ca2+ concentrations. These forms reveal the same overall structure for HpiC1, but differ in their space group, indicating that Ca2+ concentration can influence crystal packing (see below). The 1.7-Å structure in form 1 was solved by selenomethionyl (SeMet) SAD phasing from a HpiC1 W73M/K132M double mutant, as the wild-type protein lacks Met. The SeMet W73M/K132M structure was used to solve structures in the other crystal forms by molecular replacement (Supplementary Table 1). The overall fold of the HpiC1 polypeptide is a flattened β-jelly roll fold (Fig. 2a) composed of two antiparallel β-sheets. The antiparallel pairing of the β6 strands from two monomers creates a continuous β-sheet across an extensive dimer interface, which buries 2,060 Å2 of total surface area (PISA, Fig. 2b)20 and encompasses approximately 20% of the total surface area of each monomer. This arrangement is consistent with that seen using size-exclusion chromatography, wherein HpiC1 and other Stig cyclases migrated as apparent dimers17. HpiC1 shares the highest structural similarity (2.3 Å r.m.s. deviation from DALI21) with the carbohydrate-binding module (CBM) from xylanase in the thermostable bacterium Rhodothermus marinus (PDB ID 2Y64; Fig. 2c)22. These proteins have highly similar tertiary structure and topology, with the most substantial differences occurring at their N termini and in the loop regions between shared β-strands.
Fig. 2. HpiC1 structural overview at 1.5 Å.
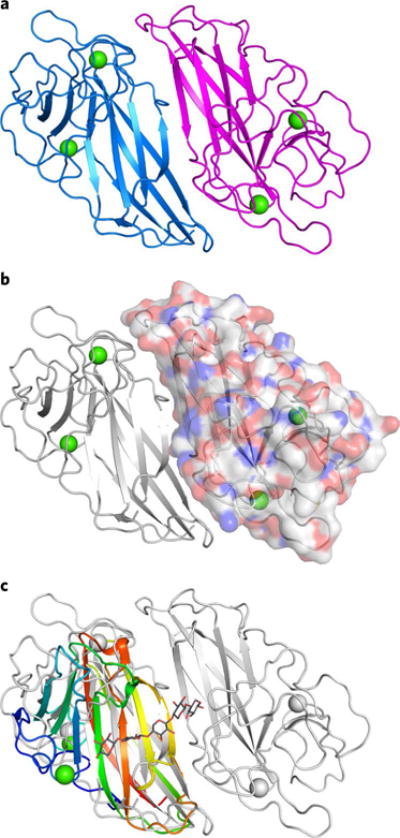
a, Cartoon representation of the HpiC1 homodimer. The subunits are colored blue and magenta; green spheres indicate bound calcium ions. b, Surface representation of a single HpiC1 protomer, colored by atom, indicates 2,060 Å2 of buried surface area between the subunits. c, Superposition of HpiC1 with a xylanase carbohydrate-binding module (CBM) homolog (PDB ID 2Y64, rainbow); CBM is monomeric despite sharing the same fold as HpiC1.
HpiC1 has two integral Ca2+ ions, each with octahedral coordination geometry (Supplementary Fig. 2). These sites were a key starting point for assessing the structural and catalytic role that Ca2+ plays in the Stig cyclase enzymes. In vitro assays conducted in the presence of 5 mM EDTA showed no activity, confirming that Ca2+ is required for catalytic function in HpiC1 (ref. 18). From sequence comparisons, we expect the integral calcium-binding sites and the core dimeric assembly to be maintained in the other Stig cyclase enzymes (Supplementary Fig. 3).
HpiC1 active site identification
The substrate-free HpiC1 structure did not immediately suggest the location(s) of substrate binding and catalysis. Initial localization of the active site was revealed by abnormal electron density in a pocket located at the distal end of each subunit. Polyethylene glycol from the crystallization was modeled into this pocket (Supplementary Fig. 4), composed of numerous aromatic amino acids (Fig. 3). Given the hydrophobicity of substrate 1, we explored whether this hydrophobic pocket could be the active site. This region was probed using AutoDock VINA with 5 (Fig. 1)23, and the major product was chosen based on its defined stereochemistry and rigid scaffold. The top docking solutions had affinities ranging between −9.7 and −9.4 kcal/mol (Supplementary Fig. 5), suggesting that this site possesses an appropriate size and shape to accommodate the hapalindole core.
Fig. 3. Active site of SeMet HpiC1 W73M/K132M.
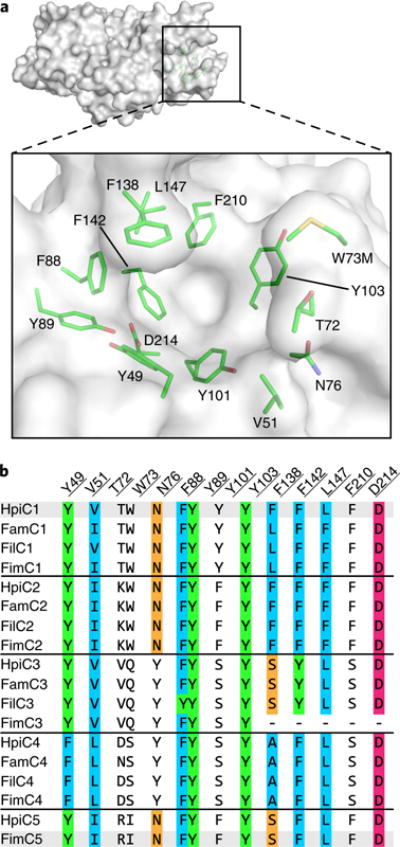
a, Surface representation of the SeMet HpiC1 active site. Key residues are shown as green sticks. Met73 is substituted for the native tryptophan residue. This mutant protein retained wild-type activity. b, Key active site residues shown in an alignment with those from other Stig cyclases. Residues are colored by conservation and side chain composition (ClustalX). The FimC3 gene product is truncated (dashes).
The docking solutions compelled us to further interrogate this region as the putative active site. First, we probed the catalytic role of Asp214 in HpiC1. This residue is 100% conserved in all the currently identified Stig cyclases and is exceptional in that it is the amino acid in the hydrophobic pocket most likely to participate in acid–base chemistry, which can be inferred to promote [3,3]-sigmatropic rearrangments24. The Asp214 carboxylate lacks a counter ion and is hydrogen bonded to the Tyr89 hydroxyl. Substitution of Asp214 to alanine abolished activity (Fig. 4), indicating that it plays a critical role in the catalytic cascade. The corresponding Asn214 and Glu214 variants were also completely inactive (Supplementary Fig. 6). These results provide compelling evidence that this hydrophobic pocket is the enzyme active site.
Fig. 4. In vitro characterization of HpiC1 mutants using 1 as substrate.
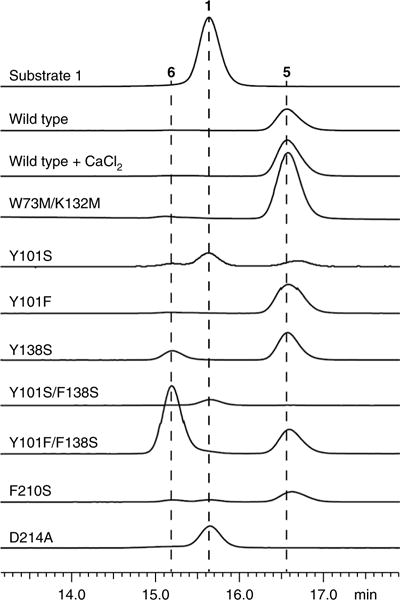
Substitution of the catalytic acid Asp214 with alanine abolished activity. A single mutation, F138S, altered the native product profile of HpiC1 to produce 6, which became predominant in the HpiC1 Y101F/F138S double mutant. Products were monitored by HPLC and confirmed using 1H NMR.
The enzyme conformation around Asp214 varies among the crystal forms (Supplementary Fig. 7). In form 1, Asp214 is shielded by Phe138, whereas in forms 2–4 this residue is shifted to expose Asp214 to the binding pocket. The loop containing Phe138 is also shifted ~3 Å from its position in form 1, although coordination of the calcium ion by the carbonyl oxygen of Phe138 is maintained. The atomic temperature factors in this region also indicate a higher degree of mobility (Supplementary Fig. 8). It is apparent that conformational flexibility is required to perform multiple catalytic steps in a single binding site and to accommodate the broad range of regio- and stereochemical configurations in this class of alkaloids1.
Altered product profile through mutagenesis of HpiC1
Based on the different product profiles of the various Stig cyclases, we examined the conservation of residues in this hydrophobic pocket. A comparison revealed that sequence clustering correlates to similar product profiles. We reasoned that key determinants of cyclase reactivity were contained within these localized sequences (Fig. 3), and these residues were explored by mutagenesis. A comparative analysis between HpiC1 and FimC5 (68% sequence identity with HpiC1; catalyzes production of 6 from 1) was pursued on the basis of their ability to differentially produce hapalindole or fischerindole core ring systems, respectively (Fig. 1)17. Remarkably, substitution of Phe138 in HpiC1 to the corresponding serine from FimC5 led to the generation of a mixture of its major product, hapalindole 5, and the FimC5 major product, fischerindole 6 (Fig. 4). The product ratio in F138S is approximately 1:2 (6:5) and is shifted to 2:1 (6:5) in Y101F/F138S. HpiC1 Y101F had a product profile comparable to that of the wild-type protein, indicating that this effect is driven primarily by Phe138. A corresponding mutation at this position in FimC5, S139F, did not lead to formation of 5; instead, the product profile was shifted toward the production of 7, a minor tricyclic shunt product of the native HpiC1 and FimC5 reactions (Supplementary Fig. 9)17. These data indicate that HpiC1 Phe138 and FimC5 Ser139 both play a key role in directing terminal electrophilic aromatic substitution.
Mutations guided by sequence alignments were introduced into HpiC1 to identify additional key residues (Fig. 3). The Y101S/F138S variant corresponding to the FamC3, HpiC3, and FilC3 homologs showed no activity with 1 (Fig. 4), which is consistent with the lack of reactivity observed in homodimeric forms of FamC3, HpiC3, and FilC3 (ref. 17). HpiC1 Y101S also showed reduced activity, though to a lesser extent than in combination with F138S. Intriguingly, FamC3 was shown to associate with FamC2 as a heterodimer, and catalyzes the formation of hapalindole H17.
To understand the impact of these mutations, high-resolution structures were determined for HpiC1 variants Y101F, Y101S, F138S, and Y101F/F138S. Whereas our efforts to observe bound ligands by either soaking or co-crystallization only afforded complexes with DMSO and Tris buffer (Supplementary Fig. 4), these structures aided our efforts to interrogate the mechanism of cyclization in HpiC1 using computational methods.
Molecular dynamics of HpiC1
We first applied molecular dynamics (MD) simulations to the substrate-free structures to gain insights into the dynamics of the active site. Starting from the apo-HpiC1 dimeric structure, analysis of the MD trajectories revealed large fluctuations of the loop containing Phe138 (Asn137–Phe150; Supplementary Fig. 10), which is in agreement with the different conformations found for this loop in the crystal structures (Supplementary Fig. 7). MD simulations showed that Asp214, which is essential for enzyme activity, stays preferentially in a conformation in which the Asp214 side chain points toward the inner cavity of the active site, while Phe138 acts as a wall on the side of the active site pocket (Supplementary Fig. 11). This is due, in part, to the Tyr89 hydroxyl hydrogen bond with the Asp214 carboxylic acid group (Supplementary Figs. 11 and 12). Importantly, the predicted pKa for Asp214, estimated from snapshots obtained along 500 ns of MD simulation (Supplementary Fig. 13), is 6.5–7.0, indicating that it can be protonated in an acid–base equilibrium to act as a protonating species during catalysis. An alternative conformation of Phe138 is sampled during the 500-ns trajectory. In this conformation, the Phe138 side chain is displaced and Asp214 becomes inaccessible, generating an inactive conformation (Supplementary Figs. 11 and 12) that is similar to the arrangement observed in the crystal form 1 (Supplementary Fig. 7).
We next considered the lack of activity in the Y101S/F138S mutant. MD simulations for Y101S/F138S showed that Y101S interacts closely with Asp214, as well as with the Tyr89 hydroxyl hydrogen bond (Supplementary Figs. 14 and 15). These two hydrogen bonds favor stabilization of a negatively charged carboxylate group, which would not be protonated in this more polar environment. This is confirmed by the decrease of the predicted pKa value of Asp214 (pKa ≈5.5–6; Supplementary Fig. 13).
As described above, F138S and Y101F/F138S mutants change the product profile in HpiC1, leading to increased formation of fischerindole 6 (Fig. 4). We performed 500-ns MD simulations on both F138S and Y101F/F138S mutants and found critical changes in the shape of the active site. The F138S mutation creates more space around the catalytic Asp214 residue and releases the interaction between the two phenyl rings of Phe138 and Phe210, which results in Phe210 becoming more flexible (Supplementary Fig. 16). This active site reshaping is responsible for the change in the reaction outcome, as discussed below.
Quantum mechanics analysis of the three-part mechanism
We employed DFT calculations to explore the possible reaction mechanism for Stig cyclases, in particular, the Cope rearrangement, which is the first step in the three-part reaction cascade starting from 1 (ref. 11). The instability of 1 has precluded determination of its chiral configuration at the indolenine C3 position. Thus, we computed the Cope rearrangement and cyclization cascade mechanism (Fig. 5) starting from both the (R)-1 and (S)-1 enantiomers (Supplementary Fig. 17).
Fig. 5. Quantum mechanics analysis.
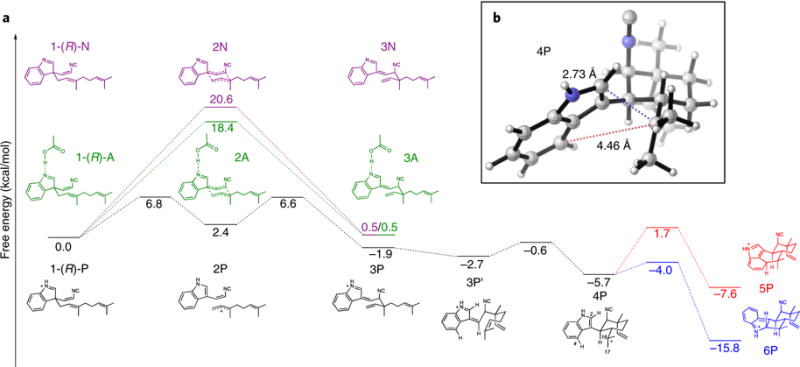
a, Cope rearrangement, 6-exo-trig cyclization, and electrophilic aromatic substitution cascade starting from the R enantiomer of substrate 1 in a near-attack conformation, leading to 5 precursor 5P and 6 precursor 6P. The energetics of the Cope rearrangement are computed with the neutral indolenine (pathway N), the N-protonated indolenine (pathway P), and the indolenine forming a hydrogen bond with acetic acid (pathway A). b, Optimized geometry of key intermediate 4P, which undergoes regioselective electrophilic aromatic substitution to form 5 or 6.
Although a typical Cope rearrangement has four possible transition states, two chairs and two boats (Supplementary Figs. 18 and 19), only one chair-like transition state can account for the known stereochemistry at C11 and C12 in the products, and therefore only this transition state was further considered. Given that the active site of HpiC1 contains the essential Asp214 residue, we explored the impact of this residue on accelerating the Cope rearrangement. The conversion of neutral starting material 1-(R)-N to intermediate 3N is concerted, proceeding through a single chair-like transition state, 2N, which lies 20.6 kcal/mol above the near-attack conformation of the starting material. This chair-like transition structure (Supplementary Fig. 20) has dissociative character, breaking and forming partial single bonds of 2.58 Å and 2.53 Å, respectively. The partial negative charge on the isocyanidovinylindolenine fragment of −0.43 e can be stabilized by a hydrogen bonding donor at the indolenine nitrogen, which has a partial negative charge of −0.49 e. Adding an acetic acid molecule to mimic possible hydrogen bonding between Asp214 and the indolenine nitrogen lowers this barrier by 2.2 kcal/mol (a 40-fold rate enhancement), with the conversion of 1-(R)-A to 3 A proceeding through a chair-like transition state, 2A, with a free-energy barrier of 18.4 kcal/mol. These computations suggest that Asp214 can facilitate the Cope rearrangement by hydrogen bonding to the indolenine nitrogen. Fully protonating the indolenine nitrogen, which represents the maximum limit of potential acid catalysis by Asp214, results in a change of mechanism. The conversion from 1-(R)-P to 3P is stepwise and dissociative, with 2P being an intermediate rather than a transition state, and has a much lower overall free-energy barrier of 6.8 kcal/mol. Intermediate 2P is stabilized by full conjugation between the indole and isonitrile groups, as well as by an allylic cation to produce this much lower overall barrier (Fig. 5).
The second proposed step in the biosynthesis of 5 and 6 is the 6-exo-trig cyclization of intermediate 3 to 4, which sets the stereochemistry at C10 and C15 in the products. In gas-phase DFT optimizations, it was possible to locate a transition state for this cyclization only when the indolenine nitrogen was protonated. Without protonation, the zwitterionic character of the possible transition state leads to bond formation between negatively charged position C3 of the indolenine and positively charged position C16 to generate a cyclobutane ring. By contrast, the protonated species undergoes facile cyclization from 3P to intermediate 4P through a low-lying transition state. This suggests that protonation is crucial and that Asp214 may catalyze cyclization, and possibly the preceding Cope rearrangement, in this way.
The third step is electrophilic aromatic substitution of intermediate 4P, whereby two different transition states lead to the two major products. Electrophilic aromatic substitution at C4 of the indole yields intermediate 5P, which gives 5 upon deprotonation, whereas electrophilic aromatic substitution at C2 of the indole yields intermediate 6P, which gives 6 upon deprotonation. Deprotonation at C16 can lead to formation of tricyclic 7 (Fig. 1), which is generated as a trace product by HpiC1. Quantum mechanics calculations show that formation of 6, which has the lower energy transition state, should be intrinsically favored. Thus, the regioselectivity of electrophilic aromatic substitution to generate 5 appears to be controlled by the HpiC1 active site as opposed to inherent energetics of the system.
Molecular dynamics simulations with modeled ligands
To understand enzymatic control of the Cope rearrangement, we conducted 500-ns MD simulations on wild-type HpiC1 with both 1-(R)-P and 1-(S)-P bound into the active site (Supplementary Fig. 21). Both enantiomers retain the key hydrogen bond interaction between the protonated indolenine NH and the Asp214 residue (Supplementary Fig. 21). However, the C11–C12 distance, which corresponds to the C–C bond formed during the Cope rearrangement, is shorter (~3.5 Å) for the 1-(R)-P substrate than for the 1-(S)-P substrate (>4.0 Å). In addition, only 1-(R)-P is stabilized by the enzyme active site in a near-attack conformation that leads to the correct stereochemistry at positions 11 and 12 (Fig. 6a,b; Supplementary Figs. 21 and 22). Based on these observations, the R enantiomer of 1 is the most plausible natural substrate.
Fig. 6. Molecular dynamics simulations of the active site.
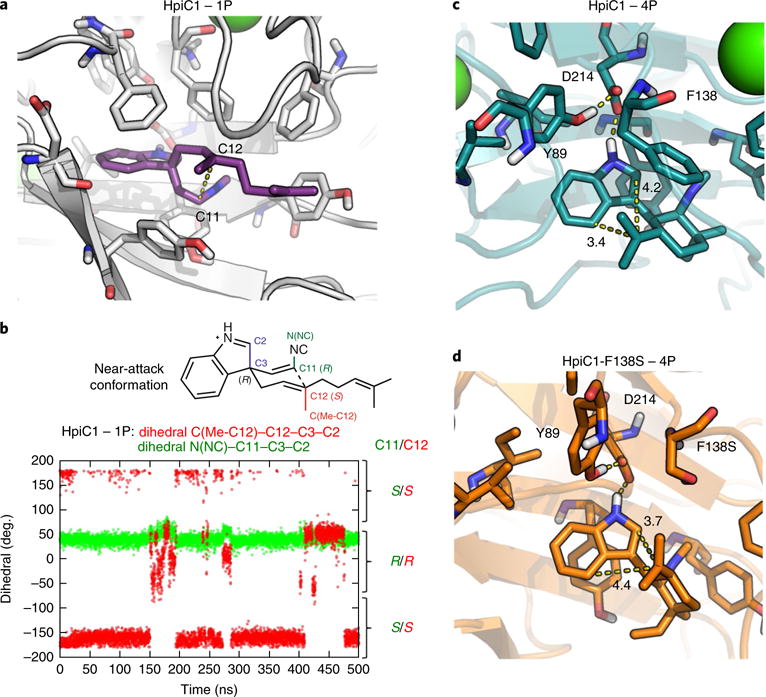
a,b, Representative snapshots of substrate 1-(R)-P (violet) bound into HpiC1 (a) and dihedral angles explored during the 500 ns of MD simulation for substrate 1-(R)-P bound into HpiC1 (b). Dihedral-1 (N(NC)-C11-C3-C2) and dihedral-2 (C(Me-C12)-C12-C3-C2) define the relative orientation of substituents at the C11 and C12 positions, respectively, during the MD simulation. The right axis indicates the final stereochemistry of C11 and C12 expected after the Cope rearrangement coming from the given near-attack conformation of 1-(R)-P, as shown in b and Supplementary Fig. 22. 1-(R)-P mainly explores one conformation during the MD trajectory, leading to R configuration at C11 and S configuration at C12. c,d, Representative snapshots obtained from 500 ns of MD simulations of the active site for intermediate 4P bound to HpiC1 (c; Supplementary Figs. 23 and 24) and F138S (d; Supplementary Fig. 26). Distances (in Ångstroms) show that the conformation adopted by intermediate 4P in the HpiC1 wild-type enzyme moves C16 closer to C4 to form the hapalindole product, while the F138S mutant enables the exploration of a conformation of intermediate 4P in which C16 gets closer to C2 to allow fischerindole product formation.
To understand how HpiC1 could control the differentiation of hapalindole and fischerindole products, we performed MD simulations with intermediates before electrophilic aromatic substitution. We considered the two intermediate precursors 4P and 10P, which derive from the 1-(R)-P and 1-(S)-P starting materials, respectively. As 4P or 10P can each generate both the 5 and 6 products, we analyzed the binding of both intermediates. MD simulations with the two docked intermediates show that 4P effectively maintains the hydrogen bond between the protonated NH-indole and Asp214 more than 10P, which rapidly dissociates during the simulation (Supplementary Fig. 23). These results reinforce the idea that (R)-1 is the most plausible natural substrate. Moreover, MD simulations showed that when 4P is bound into the active site, it adopts a conformation in which the distance between C4 and C16 (~3.5 Å; corresponds to hapalindole formation) is shorter than the distance between C2 and C16 (~4.0 Å; corresponds to fischerindole formation) (Fig. 6c and Supplementary Fig. 23). Finally, at around 200 ns, a conformational change of the Phe138 side chain reverses this trend to disrupt the interaction of 4P and Asp214 (Supplementary Figs. 23 and 24), highlighting the key role of Phe138 in controlling the site selectivity of the reaction.
The critical role of Phe138 was investigated through MD simulations on the F138S and Y101F/F138S mutants using 4P In both trajectories, the distance between C2 and C16 leading to fischerindole formation is shortened compared to that in the wild-type enzyme, becoming closer (~3.5 Å) than the distance between C4 and C16 (~4.0 Å). This leads to hapalindole formation, consistent with the experimentally observed product ratios (Fig. 6d and Supplementary Fig. 26). The absence of the bulky Phe138 residue near C2 allows the intermediate to adopt a slightly different conformation, enabling formation of 6 without completely suppressing generation of 5 (Fig. 6d and Supplementary Fig. 27).
As with the terpene cyclases25–27, aromatic side chains in HpiC1 are appropriately positioned to form cation–π interactions with the carbocation of 4P. Examination of the MD trajectories shows that Phe138 and Phe142 can interact with the C16 carbocation, whereas Phe88 and Phe210 are either too far away or poorly oriented for interaction (Supplementary Fig. 25)28. An aromatic amino acid Phe142 is conserved in all known Stig cyclases, whereas Phe138 varies with different product outcomes, as observed in our mutational analysis and calculations.
Discussion
The structure of HpiC1 has provided the first high-resolution insights into a fascinating mechanistic puzzle in which the Stig cyclases are able to generate extensive stereochemical and regiochemical diversity through the common biosynthetic precursor 1. The surprising function of the Stig cyclases11 was not able to be inferred from bioinformatic analysis, and, similarly, structural and mechanistic insights from homology-based tools were unavailable for these remarkable biocatalysts. HpiC1 is not homologous to any characterized terpene cyclase, but instead is most similar to bacterial CBMs, which function primarily to bring various hydrolytic enzymes into contact with their carbohydrate substrates. Target sugar molecules bind the CBMs in an extended cleft at the protein surface, and this is mediated through several amino acids that are not conserved in HpiC1 (Fig. 2c)22. This indicates a divergent functionality in HpiC1 based on a shared protein scaffold. Nevertheless, Stig cyclases and CBMs share several properties including a common fold, thermostability (Supplementary Fig. 28)29, and structural calcium ions, which play a key role in CBM stabilization, substrate recognition, and oligomerization29,30.
The molecular basis for the Ca2+ dependence of HpiC1 was demonstrated through two well-ordered binding sites near the enzyme active site. A paradoxical aspect of this calcium requirement is the observation that low millimolar concentrations of CaCl2 caused HpiC1 to precipitate. This effect was reversible through stoichiometric addition of EDTA, indicative of calcium-dependent higher order oligomerization of HpiC1. However, a requirement for millimolar concentrations of Ca2+ in the reaction to achieve activity has been reported for some cyclases18. There are also fascinating instances of heteromeric association of some Stig cyclases leading to variant stereochemical outcomes of the products compared to the products of their homomeric counterparts17,18. Taken together, these data are indicative of an important, yet complex, structural role for Ca2+ in Stig cyclase catalysis that may involve higher order oligomerization. Because HpiC1 could not be crystallized without supplemental CaCl2, we examined the lattice contacts for evidence of additional calcium binding sites. In crystal forms 1 and 2, which required 200 mM Ca2+ in the well solution, we found two sites in which calcium was able to facilitate formation of higher order complexes. We observed one fully occupied interfacial calcium ion far from the active site in crystal form 1 (Supplementary Fig. 29) and a second Ca2+ at half occupancy in unrelated crystal forms 1 and 2 (Supplementary Fig. 30). This Ca2+ is adjacent to the active sites, indicating that a bridging calcium ion could influence the activity of cyclase oligomers. A functional role of higher order oligomerization in HpiC1 remains unclear, as the addition of supplemental (1–20 mM) Ca2+ enhances the enzymatic activity of HpiC1 but is not required for turnover (Fig. 4). Further structural investigation will be required to understand the assembly of heteromeric Stig cyclase complexes and the relevance of Ca2+ in those cases.
We have established the location of the HpiC1 active site and, using mutational analysis, identified critical residues for catalysis and demonstrated a key relationship between amino acid sequence and product outcome. Most importantly, we identified Asp214 as the source of an active site acid required for catalysis and is consistent with reports of acid-catalyzed Cope rearrangements24. An active site acid is also consistent with the reported three-fold increase in apparent kcat as pH was decreased from 9.0 to 6.0 in the Stig cyclase WelU1 (68% sequence identity with HpiC1)19. Notably, the hydrophobic environment around Asp214 is essential for maintaining a suitable population of the protonated species, as the Y101S/F138S mutation substantially reduces enzyme activity. We also identified a key regiochemical switch at Phe138 that gave rise to production of the fischerindole core in HpiC1. In these studies, the general importance of Phe138 in the HpiC1 homolog FimC5 was also confirmed, whereby a corresponding mutation also affected the product distribution with respect to regiochemistry (Supplementary Fig. 9). Together, these findings will facilitate our efforts to anticipate the product profiles in new cyclases that are identified as additional strains and gene clusters are discovered.
We also explored the HpiC1 active site dynamics using a combination of DFT quantum mechanical calculations of mechanism and modes of catalysis, and MD simulations on the protein. We studied the origins of the three-part catalytic mechanism, and how HpiC1 controls the regiochemistry of product formation by favoring a particular conformation of substrate 1 and the reaction intermediate 4P. We have examined the role of key mutations in HpiC1 that switch the native product outcome from hapalindole 5 to fischerindole 6. Together, these results address several of the catalytic steps in the formation of 5 from (R)-1. Further analysis will be required to establish the mechanistic basis by which the variant Stig cyclases achieve differentiation at the 6-exo-trig cyclization step, where the stereocenters at C10 and C15 are set (for example, Hapalindole U, H, and J series; Supplementary Fig. 1). Structural studies on additional Stig cyclases, mutational analysis across key active site residues, and computational modeling of the reaction intermediates will enable prediction of product profiles, and engineering of new selectivities to diversify further this remarkable family of natural products.
Methods
Methods, including statements of data availability and any associated accession codes and references, are available at https://doi.org/10.1038/s41589-018-0003-x.
Methods
Cloning and mutagenesis of HpiC1 and FimC5
HpiC1 and FimC5 were cloned into pET28 (Novagen) from codon-optimized synthetic genes (IDT gBlocks) with their N-terminal leader peptides truncated17. Site-directed mutagenesis of HpiC1 and FimC5 was performed using a single-primer method based on “Quikchange” mutagenesis (Agilent Genomics). Mutagenic primer sequences are listed in Supplementary Table 2. All mutations were verified by DNA sequencing at the University of Michigan DNA Sequencing Core.
Expression of HpiC1 and FimC5 proteins
HpiC1 and FimC5 and their corresponding active site mutants were overexpressed in Escherichia coli strain BL21(DE3). Cultures from a single colony were used to inoculate 1.5 L terrific broth (TB) supplemented with 50 μg/mL kanamycin. Expression was induced with 0.7 mM isopropyl-β-D-thiogalactopyranoside when cultures reached OD600 ~1.0. After 20 h induction at 18 °C the cells were harvested by centrifugation and stored at −80 °C.
Expression of HpiC1 W73M/K132M selenomethionine derivative
An initial challenge involved the lack of native methionine residues in HpiC1. Therefore, a series of mutants containing methionine substitutions were screened for crystallization. Sites for substitution were selected based on positions containing a native methionine in sequence comparisons with other Stig cyclases. Selenomethionine (SeMet) HpiC1 W73M/K132M was produced by metabolic inhibition31. Briefly, freshly transformed BL21(DE3) cells harboring the hpiC1 gene on pET28 were used to inoculate 3 L M9 minimal medium supplemented with 50 μg/mL kanamycin. An amino acid cocktail containing L-selenomethionine was added when the cells reached OD600 = 1.0. The cells were cooled to 18 °C and shaken for 30 min before induction with 0.7 mM isopropyl-β-D-thiogalactopyranoside. After 20 h induction, the cells were harvested by centrifugation and stored at −80 °C.
Purification of recombinant proteins
All proteins were purified as described previously17. Briefly, 10 g E. coli wet cell mass containing the recombinant cyclase was resuspended in 75 mL lysis buffer (10 mM HEPES pH 7.6, 50 mM NaCl, 10% glycerol). Cells were lysed by the addition of lysozyme (0.5 mg/mL) and sonication, and clarified by centrifugation at 60,000 × g for 25 min. Clarified lysate was loaded by gravity onto 8 mL Ni-NTA Superflow resin (Qiagen). The column was washed with 100 mL lysis buffer containing 20 mM imidazole and 50 mL lysis buffer containing 40 mM imidazole. The proteins were eluted with elution buffer (250 mM imidazole, pH 7.9 and 10% glycerol). Fractions containing the purified cyclase were concentrated using Amicon Ultra 15 centrifugal filters and desalted using PD-10 columns (GE Healthcare) equilibrated with storage buffer (10 mM HEPES pH 7.6, 50 mM NaCl). The purified cyclases were drop-frozen in 30-μL aliquots directly into liquid N2 and stored at −80 °C.
Crystallization of SeMet HpiC1 W73M/K132M
Single, diffraction quality crystals of the HpiC1 W73M/K132M selenomethionine derivative were grown in Intelli-Plate 96-2 shallow-well plates (Hampton research) at 20 °C by mixing 1 μL of 11 mg/mL SeMet HpiC1 in storage buffer with 1 μL of a well solution containing 23% PEG 3350, 200 mM CaCl2, 5% trehalose. Sitting droplets were nucleated after 18 h from an earlier spontaneous crystallization event using a cat whisker. Single, rod-shaped crystals grew to approximate dimensions of 50×50×250 μm after 14 d. 8 μL of a cryoprotecting solution containing 10 mM HEPES pH 7.6, 50 mM NaCl, 23% PEG 3350, 200 mM CaCl2, and 9.1% trehalose was added directly to the sitting drops, and the crystals were harvested using nylon loops and vitrified by rapid plunging into liquid nitrogen. SeMet HpiC1 was crystallized in form 1, space group P212121 with unit cell dimensions of a = 44.9 Å, b = 81.1 Å, c = 131.7 Å, and two chains in the asymmetric unit.
Crystallization of Native HpiC1 (P42)
Single, diffraction quality crystals of native HpiC1 were grown in Intelli-Plate 96-2 shallow-well plates (Hampton research) at 20 °C by mixing 1 μL of 20 mg/mL HpiC1 in storage buffer with 1 μL of a well solution containing 22% PEG 4000, 200 mM CaCl2, 100 mM Tris pH 8.5, and 5% ethylene glycol. Sitting droplets were nucleated after 4 h from an earlier spontaneous crystallization event using a cat whisker. Single, rod-shaped crystals grew to approximate dimensions of 50×50×150 μm after 7 d. 8 μL of a cryoprotecting solution containing 10 mM HEPES pH 7.6, 50 mM NaCl, 22% PEG 4000, 200 mM CaCl2, 100 mM Tris pH 8.5, and 15% ethylene glycol was added directly to the sitting drops and the crystals were harvested using nylon loops and vitrified by rapid plunging into liquid nitrogen. In these conditions, HpiC1 native crystallized in Form 2, space group P42 with unit cell dimensions of a = 71.3 Å, b = 71.3 Å, c = 80.6 Å, and two chains in the asymmetric unit.
Crystallization of Native HpiC1 (C2)
Single, diffraction quality crystals of native HpiC1 were grown in Intelli-Plate 96-2 shallow-well plates (Hampton research) at 20 °C by mixing 1 μL of 20 mg/mL HpiC1 in storage buffer and 5% DMSO with 1 μL of a well solution containing 22% PEG 4000, 150 mM CaCl2, 100 mM Tris pH 8.5, and 5% ethylene glycol. Sitting droplets were nucleated after 4 h from an earlier spontaneous crystallization event using a cat whisker. Single, diamond-shaped crystals grew to approximate dimensions of 200×200×100 μm after 7 d. 8 μL of a cryoprotecting solution containing 10 mM HEPES pH 7.6, 50 mM NaCl, 22% PEG 4000, 150 mM CaCl2, 100 mM Tris pH 8.5, 15% ethylene glycol, 5% DMSO was added directly to the sitting drops, and the crystals were harvested using nylon loops and vitrified by rapid plunging into liquid nitrogen. In these conditions, HpiC1 native was crystallized in form 3, space group C2 with unit cell dimensions of a = 113.8 Å, b = 49.5 Å, c = 53.1 Å, α = 90°, β= 110.5°, γ = 90° and one chain in the asymmetric unit.
Crystallization of HpiC1 Y101F
Single, diffraction quality crystals of HpiC1 Y101F were grown in Intelli-Plate 96-2 shallow-well plates (Hampton research) at 20 °C by mixing 1 μL of 15 mg/mL protein in storage buffer with 1 μL of a well solution containing 22% PEG 4000, 150 mM CaCl2, 100 mM Tris pH 8.5, and 5% ethylene glycol. Sitting droplets were nucleated after 4 h from an earlier spontaneous crystallization event using a cat whisker. Single, diamond-shaped crystals grew to approximate dimensions of 250×250×270 μm after 7 d. 8 μL of a cryoprotecting solution containing 10 mM HEPES pH 7.6, 50 mM NaCl, 22% PEG 4000, 150 mM CaCl2, 100 mM Tris pH 8.5, and 15% ethylene glycol was added directly to the sitting drops, and the crystals were harvested using nylon loops and vitrified by rapid plunging into liquid nitrogen. HpiC1 Y101F crystallized in form 3, space group C2 with unit cell dimensions of a = 113.8 Å, b = 49.8 Å, c = 53.4 Å, α = 90°, β= 110.4°, γ = 90° and one chain in the asymmetric unit.
Crystallization of HpiC1 Y101S
Single, diffraction quality crystals of HpiC1 Y101S were grown in Intelli-Plate 96-2 shallow-well plates (Hampton research) at 20 °C by mixing 1 μL of 15 mg/mL protein in storage buffer with 1 μL of a well solution containing 20% MEPEG 5000, 150 mM CaCl2, 100 mM Tris pH 8.5, 5% ethylene glycol. Sitting droplets were nucleated after 4 h from an earlier spontaneous crystallization event using a cat whisker. Single, diamond-shaped crystals grew to approximate dimensions of 250×250×270 μm after 7 d. 8 μL of a cryoprotecting solution containing 10 mM HEPES pH 7.6, 50 mM NaCl, 20% MEPEG 5000, 150 mM CaCl2, 100 mM Tris pH 8.5, 15% ethylene glycol, 5% DMSO was added directly to the sitting drops and the crystals were harvested using nylon loops and vitrified by rapid plunging into liquid nitrogen. HpiC1 Y101S crystallized in form 3, space group C2 with unit cell dimensions of a = 113.9 Å, b = 49.6 Å, c = 53.4 Å, α = 90°, β= 110.3°, γ = 90° and one chain in the asymmetric unit.
Crystallization of HpiC1 F138S and Y101F/F138S
Single, diffraction quality crystals of HpiC1 F138S and Y101F/F138S were grown in Intelli-Plate 96-2 shallow-well plates (Hampton research) at 20 °C by mixing 1 μL of 15 mg/mL protein in storage buffer, 20 mM CaCl2, 5% DMSO with 1 μL of a well solution containing 20% MEPEG 5000, 100 mM Bis-Tris pH 6.5, and 5% ethylene glycol. Sitting droplets were nucleated after 4 h from an earlier spontaneous crystallization event using a cat whisker. Single, plate-shaped crystals grew to approximate dimensions of 50×50×300 μm after 7 d. 8 μL of a cryoprotecting solution containing 10 mM HEPES pH 7.6, 50 mM NaCl, 20% MEPEG 5000, 20 mM CaCl2, 100 mM Bis–Tris pH 6.5, and 15% ethylene glycol, 5% DMSO was added directly to the sitting drops and the crystals were harvested using nylon loops and vitrified by rapid plunging into liquid nitrogen. HpiC1 F138S and HpiC1 Y101F/F138S crystallized in form 4, space group P21 with unit cell dimensions of a = 62.0 Å, b = 47.9 Å, c = 174.2 Å, α = 90°, β = 97.2°, γ = 90° and four chains in the asymmetric unit.
Data collection and processing
X-ray data were collected at 100 K on beamline 23ID-B at the General Medical Sciences and Cancer Institute Structural Biology Facility at the Advanced Photon Source in Argonne, IL, USA. Diffraction data were integrated and scaled using XDS32. Data collection statistics are given in Supplementary Table 1.
Experimental phasing (SAD) and molecular replacement, model building and refinement
The structure of SeMet HpiC1 W73M/K132M was solved using single-wavelength anomalous diffraction (SAD). Phasing and initial model building were performed using Phenix Autosol33. This resulted in an initial model that could be extended by alternating cycles of manual building in Coot34 and least-squares refinement with Refmac35. The structures for HpiC1 native and Y101F, Y101S, F138S, Y101F/F138S were solved by molecular replacement using Phaser-MR36 with the structure of the HpiC1 SeMet derivative as a search model. Final models were generated by alternating cycles of manual building in Coot34 and refinement in Refmac35 and Phenix33, and were validated using MolProbity37.
Docking 12-epi-hapalindole U with Autodock VINA
12-epi-hapalindole U was docked into the SeMet HpiC1 model using Autodock VINA23. Default parameters for Autodock VINA were used, with the exception of exhaustiveness, which was set to 100.
Molecular graphics
All figures depicting the protein structure were generated with PyMOL (Version 1.8 Schrödinger, LLC).
Chemical synthesis
Indole isonitrile was synthesized as described previously11.
In vitro cyclase assays
In vitro assays were performed with HpiC1 and FimC5 and their corresponding active site mutants as described previously17. Products were analyzed using LC–MS (Shimadzu) using C18 (Agilent) HPLC column and monitored by UV absorbance at 280 nm.
Scaleup, purification, and NMR of F138S product (12-epi-fischerindole U)
The semi-prep scale reaction was performed as described previously17.
Quantum mechanical calculations
Conformational searches of the hapalindole and fischerindole products were performed using the Schrödinger MacroModel software package (release 2017-2, Schrodinger, LLC), and the lowest energy conformation was used for all reported quantum mechanical calculations. All quantum mechanical calculations were performed using the Gaussian 09 (Revision A.02, Gaussian, Inc.) software package. Structures were optimized in the gas phase at the B3LYP38,39/6-31 G(d) level of theory; frequency calculations were used to confirm the presence of local minima (no imaginary frequencies) and transition states (one imaginary frequency) and to calculate free energies at 298 K. To obtain more accurate energetics, single-point energy calculations were performed on the optimized structures at the B3LYP/6-311 + + G(d,p) level of theory using Grimme’s D3(BJ) dispersion correction40,41 and the IEFPCM42–44 solvent model for diethyl ether (ε = 4). The use of the dielectric constant ε = 4 has proven to be a good model to estimate the dielectric permittivity in the enzyme active site, accounting for electronic polarization and small backbone fluctuations45,46.
Molecular dynamics simulations
Molecular dynamics simulations were performed using the GPU code (pmemd)47 of the AMBER 16 package (AMBER 2016, University of California). Parameters for intermediates and substrates were generated within the antechamber module using the general AMBER force field (gaff)48, with partial charges set to fit the electrostatic potential generated at the HF/6-31 G(d) level by the RESP model49. The charges were calculated according to the Merz–Singh–Kollman scheme50,51 using the Gaussian 09 package (Revision A.02, Gaussian, Inc.). Each protein was immersed in a preequilibrated truncated cuboid box with a 10 Å buffer of TIP3P52 water molecules using the leap module, resulting in the addition of around 15,000 solvent molecules. The systems were neutralized by addition of explicit counterions (Na+ and Cl−). All subsequent calculations were done using the widely tested Stony Brook modification of the Amber14 force field (ff14sb)53. A two-stage geometry optimization approach was performed. The first stage minimizes the positions of solvent molecules and ions imposing positional restraints on the solute by a harmonic potential with a force constant of 500 kcal·mol−1·Å−2 and the second stage minimizes all the atoms in the simulation cell. The systems were gently heated using six 50 ps steps, incrementing the temperature by 50 K for each step (0-300 K) under constant-volume and periodic-boundary conditions. Water molecules were treated with the SHAKE algorithm such that the angle between the hydrogen atoms was kept fixed. Long-range electrostatic effects were modeled using the particle-mesh-Ewald method54. An 8 Å cutoff was applied to Lennard–Jones and electrostatic interactions. Harmonic restraints of 10 kcal·mol−1 were applied to the solute and the Langevin equilibration scheme was used to control and equalize the temperature. The time step was kept at 1 fs during the heating stages, allowing potential inhomogeneities to self-adjust. Each system was then equilibrated without restrains for 2 ns with a 2-fs time step at a constant pressure of 1 atm and temperature of 300 K. After the systems were equilibrated in the NPT ensemble, subsequent MD simulations were performed for an additional 500 ns under an NVT ensemble and periodic-boundary conditions.
Life Sciences Reporting Summary
Further information on experimental design is available in the Life Sciences Reporting Summary.
Data availability
Atomic coordinates and structure factors have been deposited to the Protein Data Bank with accession codes 5WPP, 5WPR, 6AL6, 5WPS, 5WPU, 6AL7, and 6AL8.
Supplementary Material
Acknowledgments
The authors thank the National Science Foundation under the CCI Center for Selective C-H Functionalization (CHE-1700982), the National Institutes of Health (CA70375 to R.M.W. and D.H.S.), R35 GM118101, and the Hans W. Vahlteich Professorship (to D.H.S.) for financial support. M.G.-B. thanks the Ramón Areces Foundation for a postdoctoral fellowship. J.N.S. acknowledges the support of the National Institute of General Medical Sciences of the National Institutes of Health under Award Number F32GM122218. Computational resources were provided by the UCLA Institute for Digital Research and Education (IDRE) and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (OCI-1053575). The content does not necessarily represent the official views of the National Institutes of Health. Anton 2 computer time was provided by the Pittsburgh Supercomputing Center (PSC) through Grant R01GM116961 from the National Institutes of Health. The Anton 2 machine at PSC was generously made available by D.E. Shaw Research.
Footnotes
Author contributions
S.A.N. conducted protein preparation and crystallography. S.L. cloned the genes and assayed the enzymes. M.G.-B. conducted molecular dynamics simulations. J.N.S. conducted density functional theory calculations. A.N.L. synthesized substrates. F.Y. performed bioinformatics analyses. S.A.N., S.L., M.G.-B., J.N.S., S.Y., J.L.S., R.M.W., K.N.H. and D.H.S. designed research and conducted data analysis and interpretation. S.A.N., S.L., M.G.-B., J.N.S., J.L.S., K.N.H. and D.H.S. wrote the manuscript.
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41589-018-0003-x.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bhat V, Dave A, MacKay JA, Rawal VH. The chemistry of hapalindoles, fischerindoles, ambiguines, and welwitindolinones. Alkaloids Chem Biol. 2014;73:65–160. doi: 10.1016/B978-0-12-411565-1.00002-0. [DOI] [PubMed] [Google Scholar]
- 2.Asthana RK, et al. Identification of an antimicrobial entity from the cyanobacterium Fischerella sp isolated from bark of Azadirachta indica (Neem) tree. J Appl Phycol. 2006;18:33–39. [Google Scholar]
- 3.Becher PG, Keller S, Jung G, Süssmuth RD, Jüttner F. Insecticidal activity of 12-epi-hapalindole J isonitrile. Phytochemistry. 2007;68:2493–2497. doi: 10.1016/j.phytochem.2007.06.024. [DOI] [PubMed] [Google Scholar]
- 4.Cagide E, et al. Hapalindoles from the cyanobacterium Fischerella: potential sodium channel modulators. Chem Res Toxicol. 2014;27:1696–1706. doi: 10.1021/tx500188a. [DOI] [PubMed] [Google Scholar]
- 5.Mo S, Krunic A, Chlipala G, Orjala J. Antimicrobial ambiguine isonitriles from the cyanobacterium Fischerella ambigua. J Nat Prod. 2009;72:894–899. doi: 10.1021/np800751j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mo S, Krunic A, Santarsiero BD, Franzblau SG, Orjala J. Hapalindole-related alkaloids from the cultured cyanobacterium Fischerella ambigua. Phytochemistry. 2010;71:2116–2123. doi: 10.1016/j.phytochem.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang X, Smith CD. Microtubule effects of welwistatin, a cyanobacterial indolinone that circumvents multiple drug resistance. Mol Pharmacol. 1996;49:288–294. [PubMed] [Google Scholar]
- 8.Hillwig ML, Zhu Q, Liu X. Biosynthesis of ambiguine indole alkaloids in cyanobacterium Fischerella ambigua. ACS Chem Biol. 2014;9:372–377. doi: 10.1021/cb400681n. [DOI] [PubMed] [Google Scholar]
- 9.Raveh A, Carmeli S. Antimicrobial ambiguines from the cyanobacterium Fischerella sp collected in Israel. J Nat Prod. 2007;70:196–201. doi: 10.1021/np060495r. [DOI] [PubMed] [Google Scholar]
- 10.Stratmann K, et al. Welwitindolinones, unusual alkaloids from the blue-green algae Hapalosiphon welwitschii and Westiella intricata. Relationship to fischerindoles and hapalinodoles. J Am Chem Soc. 1994;116:9935–9942. [Google Scholar]
- 11.Li S, et al. Hapalindole/ambiguine biogenesis is mediated by a Cope rearrangement, C–C bond-forming cascade. J Am Chem Soc. 2015;137:15366–15369. doi: 10.1021/jacs.5b10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cope AC, Hardy EM. The introduction of substituted vinyl groups V A rearrangement involving the migration of an allyl group in a three-carbon system. J Am Chem Soc. 1940;62:441–444. [Google Scholar]
- 13.Ilardi EA, Stivala CE, Zakarian A. [3,3]-Sigmatropic rearrangements: recent applications in the total synthesis of natural products. Chem Soc Rev. 2009;38:3133–3148. doi: 10.1039/b901177n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeClue MS, Baldridge KK, Künzler DE, Kast P, Hilvert D. Isochorismate pyruvate lyase: a pericyclic reaction mechanism? J Am Chem Soc. 2005;127:15002–15003. doi: 10.1021/ja055871t. [DOI] [PubMed] [Google Scholar]
- 15.Luk LY, Qian Q, Tanner ME. A Cope rearrangement in the reaction catalyzed by dimethylallyltryptophan synthase? J Am Chem Soc. 2011;133:12342–12345. doi: 10.1021/ja2034969. [DOI] [PubMed] [Google Scholar]
- 16.Tanner ME. Mechanistic studies on the indole prenyltransferases. Nat Prod Rep. 2015;32:88–101. doi: 10.1039/c4np00099d. [DOI] [PubMed] [Google Scholar]
- 17.Li S, et al. Decoding cyclase-dependent assembly of hapalindole and fischerindole alkaloids. Nat Chem Biol. 2017;13:467–469. doi: 10.1038/nchembio.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu Q, Liu X. Discovery of a calcium-dependent enzymatic cascade for the selective assembly of hapalindole-type alkaloids: On the biosynthetic origin of hapalindole U. Angew Chem Int Edn Engl. 2017;56:9062–9066. doi: 10.1002/anie.201703932. [DOI] [PubMed] [Google Scholar]
- 19.Zhu Q, Liu X. Molecular and genetic basis for early stage structural diversifications in hapalindole-type alkaloid biogenesis. Chem Commun (Camb) 2017;53:2826–2829. doi: 10.1039/c7cc00782e. [DOI] [PubMed] [Google Scholar]
- 20.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 21.Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Schantz L, et al. Structural basis for carbohydrate-binding specificity–a comparative assessment of two engineered carbohydrate-binding modules. Glycobiology. 2012;22:948–961. doi: 10.1093/glycob/cws063. [DOI] [PubMed] [Google Scholar]
- 23.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lutz RP. Catalysis of the Cope and Claisen rearrangements. Chem Rev. 1984;84:205–247. [Google Scholar]
- 25.Wendt KU, Poralla K, Schulz GE. Structure and function of a squalene cyclase. Science. 1997;277:1811–1815. doi: 10.1126/science.277.5333.1811. [DOI] [PubMed] [Google Scholar]
- 26.Starks CM, Back K, Chappell J, Noel JP. Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 27.Lesburg CA, Zhai G, Cane DE, Christianson DW. Crystal structure of pentalenene synthase: mechanistic insights on terpenoid cyclization reactions in biology. Science. 1997;277:1820–1824. doi: 10.1126/science.277.5333.1820. [DOI] [PubMed] [Google Scholar]
- 28.Jenson C, Jorgensen WL. Computational investigations of carbenium ion reactions relevant to sterol biosynthesis. J Am Chem Soc. 1997;119:10846–10854. [Google Scholar]
- 29.Abou-Hachem M, et al. Calcium binding and thermostability of carbohydrate binding module CBM4-2 of Xyn10A from Rhodothermus marinus. Biochemistry. 2002;41:5720–5729. doi: 10.1021/bi012094a. [DOI] [PubMed] [Google Scholar]
- 30.Montanier CY, et al. A novel, noncatalytic carbohydrate-binding module displays specificity for galactose-containing polysaccharides through calcium-mediated oligomerization. J Biol Chem. 2011;286:2499–22509. doi: 10.1074/jbc.M110.217372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 32.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 39.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlationenergy formula into a functional of the electron density. Phys Rev B Condens Matter. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 40.Grimme S, Antony J, Ehrlich S, Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 41.Grimme S, Ehrlich S, Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J Comput Chem. 2011;32:1456–1465. doi: 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- 42.Miertuš S, Scrocco E, Tomasi J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of ab initio molecular potentials for the prevision of solvent effects. Chem Phys. 1981;55:117–129. [Google Scholar]
- 43.Miertus S, Tomasi J. Approximate evaluations of the electrostatic free-energy and internal energy changes in solution processes. Chem Phys. 1982;65:239–245. [Google Scholar]
- 44.Pascual-ahuir JL, Silla E, Tuñon I. GEPOL: An improved description of molecular surfaces. III. A new algorithm for the computation of a solvent-excluding surface. J Comput Chem. 1994;15:1127–1138. [Google Scholar]
- 45.Li L, Li C, Zhang Z, Alexov E. On the dielectric “constant” of proteins: smooth dielectric function for macromolecular modeling and its implementation in DelPhi. J Chem Theory Comput. 2013;9:2126–2136. doi: 10.1021/ct400065j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schutz CN, Warshel A. What are the dielectric “constants” of proteins and how to validate electrostatic models? Proteins. 2001;44:400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- 47.Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2 Explicit solvent particle mesh Ewald. J Chem Theory Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 48.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 49.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges - the RESP model. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 50.Besler BH, Merz KM, Kollman PA. Atomic charges derived from semiempirical methods. J Comput Chem. 1990;11:431–439. [Google Scholar]
- 51.Singh UC, Kollman PA. An approach to computing electrostatic charges for molecules. J Comput Chem. 1984;5:129–145. [Google Scholar]
- 52.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 53.Maier JA, et al. Ff14sb: improving the accuracy of protein side chain and backbone parameters from ff99sb. J Chem Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Darden T, York D, Pedersen L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors have been deposited to the Protein Data Bank with accession codes 5WPP, 5WPR, 6AL6, 5WPS, 5WPU, 6AL7, and 6AL8.