

Abstract
Marfan syndrome (MFS) is an autosomal dominant disease of the connective tissue due to mutations in the fibrillin-1 gene (FBN1). This study aimed at characterizing microelastic properties of the ascending aorta wall and lung parenchyma tissues from wild type (WT) and age-matched Fbn1 hypomorphic mice (Fbn1mgR/mgR mice) to identify tissue-specific biomechanical effects of aging and disease in MFS. Atomic force microscopy (AFM) was used to indent lung parenchyma and aortic wall tissues, using Hybrid Eshelby Decomposition analysis to extract layer-specific properties of the intima and media. The intima stiffened with age and was not different between WT and Fbn1mgR/mgR tissues, whereas the media layer of mutant aortas showed progressive structural and mechanical degradation with a modulus that was 50% softer than WT by 3.5 months of age. Similarly, mutant mice displayed progressive structural and mechanical deterioration of lung tissue, which was over 85% softer than WT by 3.5 months of age. Chronic treatment with the angiotensin type I receptor antagonist, losartan, attenuated the aorta and lung tissue degradation, resulting in structural and mechanical properties not significantly different from age-matched WT controls. By revealing micromechanical softening of elastin-rich aorta and lung tissues with disease progression in fibrillin-1 deficient mice, our findings support the use of losartan as a prophylactic treatment that may abrogate the life-threatening symptoms of MFS.
Keywords: Atomic Force Microscopy, Vascular Biomechanics, Lung Biomechanics, Losartan, Marfan syndrome
INTRODUCTION
Marfan syndrome (MFS) is a relatively common disease of connective tissue caused by mutations in the extracellular matrix (ECM) protein fibrillin-1.36, 38 Cardiovascular manifestations, especially aortic dilation (aneurysm) with dissection, are the major morbidity and mortality factors in MFS. As the disease progresses, biological dysfunction in all three arterial layers (i.e., intima, media, and adventitia), structural abnormalities in the aortic wall, increasing arterial stiffness, and progressive weakening of the aorta are thought to all contribute to aneurysm and dissection.8, 36, 38 However, incidents of unexpected aneurysm dilation and catastrophic rupture in MFS patients underscore the limits of our understanding. Furthermore, pulmonary complications associated with degradation of the lung parenchyma, such as emphysema and spontaneous pneumothorax, are often misdiagnosed or overlooked.24 Hence, there is an urgent need to investigate MFS disease progression in lung and aorta tissues in order to understand the underlying biomechanics and improve the clinical management of Marfan patients. Unfortunately, there is no proven therapy to regenerate diseased tissues in MFS patients. Drugs that lower high blood pressure are prescribed to slow the rate of aneurysm growth and avoid dissection and rupture of the aortic wall.7, 21 Losartan is an angiotensin II type I receptor (AT1r) blocker that is often prescribed for the treatment of hypertension and which also decreases TGF-β activity.9 Losartan treatment of MFS mice has shown delayed aortic dissection and rupture, improved median survival, and normalized alveolar septation.13, 21 In clinical trials, however, losartan treatment has shown disparate results ranging from protection from aortic dilation in pediatric patients,7 to insignificant effects on aortic root diameter compared to traditionally prescribed β-blocker (anti-hypertension) drugs.25 Direct mechanical testing of MFS tissues with respect to disease progression and treatment could provide needed insight into the controversial efficacy of losartan.
In the absence of an effective therapy, MFS patients are subjected to regular imaging of aortic dimensions; surgical intervention is suggested when aortic diameter reaches 5 cm or if rapid dilation (> 1 cm/year) occurs.31, 44 However, such criteria are not always sufficient to evaluate the risk of dissection. Finite element analysis (FEA) has been implemented to evaluate wall stress and assess aortic aneurysm rupture risk.40, 43 Some FEA studies have incorporated patient-specific material properties of the aorta,40, 43 but these properties can only be measured from the excised end-stage aneurysm tissue. Thus, time-resolved, layer-specific mechanical properties of the aorta could provide needed input variables for computational models and lead to more reliable prediction of rupture risk.
To study the Marfan disease process, genetically modified mouse models have been developed that replicate the clinical spectrum of MFS, from neonatal lethal to mild.23, 38 MFS mice exhibit fragmentation of elastin, destruction of microfibril assembly, over-activation of transforming growth factor beta (TGF-β) signaling, and disorganized collagen networks,27, 33, 38, 42 with systemic vascular abnormalities extending from the aortic tree to peripheral resistance vessels.32, 46 The overexpression of TGF-β, a direct consequence of microfibrillar network breakdown, has also been reported to promote aortic tissue degradation and inhibit post-natal lung development.3 However, intrinsic alteration of the arterial and lung tissue mechanical properties, due to dynamic remodeling in response to the above structural and biochemical changes, has not been fully characterized. Atomic force microscopy (AFM) allows micro-mechanical testing of the intima,37 media,4 and adventitia4, 27 layers of the small, fragile mouse aorta. However, physical dissection of individual layers for testing can cause tissue damage that alters the material properties of interest. Therefore, an AFM micro-indentation method that can extract mechanical properties of the heterogeneously layered arterial wall without physical or chemical isolation would be preferable.
To help fill these gaps in knowledge about the biomechanics of MFS, this study was designed to quantify the micro-mechanical properties of ascending aortic tissue and of lung parenchyma from the upper-right lobes of wild type (WT) and age-matched fibrillin-1-underexpressing mice (Fbn1mgR/mgR mice, herein referred to as MFS mice) in order to identify tissue-specific, biomechanical effects of aging and disease in MFS, and the effects of treatment with the drug losartan. Our findings indicate that microelastic properties of the parenchymal tissue and of the medial layer of the ascending aortic wall, but not the intimal layer, significantly decrease during MFS disease progression, supporting the hypothesis that mechanical degradation occurs primarily in tissues with a normally high elastin content. In addition, the data support the hypothesis that chronic prophylactic treatment with losartan preserves micromechanical properties of MFS aorta and lung tissues. Further investigation of MFS tissue biomechanics could lead to improved management strategies for patients afflicted with Marfan Syndrome.
METHODS
Animal Studies
The Fbn1mgR/mgR mice were used in this study; they express ~20% of the normal amount of fibrillin-1 and develop progressively severe MFS and die from aortic aneurysm and pulmonary insufficiency during early-middle adulthood (median survival: 3 months).34, 38 Using a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the Mount Sinai Medical Center, tissue samples of the ascending aorta and right lung were dissected from MFS mice with pre-symptomatic (0.5 months), early-stage (2 months), and advanced-stage (3.5 months) disease, and of WT littermates. A total of 25 WT and 24 MFS mice were sacrificed for this study, using a mixed population of males and females.
MFS mice received either placebo or 0.6 g/L losartan ad libitum in their drinking water starting at 0.5 months of age, and were sacrificed at 3.5 months near the transition between middle- to advanced-stage of disease.13 Sacrificed mice were injected with 100 μL of heparin (Sagent Pharmaceuticals, Schaumburg, IL) to prevent blood clots, then the aortic tree was dissected from the root to the diaphragm and cleaned with forceps to remove the surrounding soft tissues. Live aortic samples were submerged in culture media containing Dulbecco’s modified Eagles medium (DMEM, Mediatech, Inc., Manassas, VA), 1% penicillin/streptomycin solution (Mediatech, Inc., Manassas, VA), and 10% Fetal Bovine Serum (FBS, Atlanta Biological, Norcross, GA)18 and maintained in an incubator at 37°C until testing. Just prior to AFM testing, the ascending thoracic aorta was sliced into a ring section and then cut axially and mounted flat on a 50-mm Petri dish using medical grade double-stick tape (Nearly Me Technologies, Waco, TX) to expose the lumen side of the vessel. The harvested lung sample was de-gassed by rocking in PBS at 4°C for 3~5 hours, and transferred to culture media in an incubator at 37°C for 1 hour before AFM testing. A cranial or medial lobe of the right lung was glued to a petri dish with the pleural surface facing up. Aorta and lung tissue samples were immersed in DMEM-based culture media at 37°C and tested as described below within 3 and 6 hours, respectively, after isolation; no more than one aorta and lung sample per animal was used for micromechanical testing.
Atomic Force Microscopy
Mechanical measurements of live tissue samples were performed using a biological AFM system (MFP-3D-BIO, Asylum Research, Santa Barbara, CA) with environmental control that maintained a testing temperature of 37°C in culture media. The Igor Pro (Wavemetrics, Inc., Portland, OR) software environment allowed hardware control, data acquisition and analysis. Intact aortic tissues were indented on the exposed lumen surface with a trigger force of 0.18 – 0.8 μN at a rate of 2 μm/s to achieve an indentation depth of at least 3 μm using a silicon cantilever (spring constant, k = 8.9 N/m) with a 15-μm borosilicate glass spherical probe tip (Novascan Technologies, Inc., Ames, IA). This indentation depth ensured a broad indentation field, with a sufficient range of apparent inclusion volume fractions to extract individual layer properties using Hybrid Eshelby Decomposition (HED) analysis, described below. Nano-indentation of intact, de-gassed lung parenchyma was performed using a silicon nitride AFM probe (k = 0.1 N/m) with an integrated pyramidal tip (Asylum Research, Santa Barbara, CA), applying a maximum indentation force of 1.5 to 2.5 nN at a 4 μm/s rate. The natural lung surface was relatively smooth, and AFM indentations targeted the highest region of the mounted lung sample to avoid measurement artifacts from contact between the AFM holder and the sample. AFM indentations were performed as force map arrays at 3 to 5 locations per sample; each force map was prescribed as a 6 by 6 array covering a 30 by 30 μm scan area for the aorta, or a 4 by 4 array covering a 20 by 20 μm scan area for the lung. We performed AFM indentation on ascending aorta (sample size, n = 5 ~ 8 animals per group) and upper-right-lobe lung tissues (n = 3 ~ 6 animals) from the same groups, typically yielding hundreds of measurements per group.
Hybrid Eshelby Decomposition (HED)
HED analysis was developed to non-destructively determine component-specific properties of heterogeneous samples from AFM indentation data.2 The HED technique combines pointwise modulus (Epw) analysis,14 modified Eshelby homogenization theory,26 and finite element modeling (FEM), and was used to extract elastic modulus values for the intima (Eint) and media (Emed) layers of intact mouse aorta wall from AFM indentation of the exposed lumen. Tissue-specific models were created using ABAQUS FEM software (v6.10; Simulia, Providence, RI) on a Mac Pro workstation with dual Intel® Xeon® E5520 2.27 GHz and 2.39 GHz processors (Apple, Cupertino, CA) to simulate the AFM indentation response with a 15-μm spherical probe, illustrated as a red intimal layer on top of a grey medial layer (Fig. 1A). The layer thicknesses of the intima and media were determined from Weigert’s elastin staining and histological analysis of aortic sections (Fig. 1B).
Figure 1.
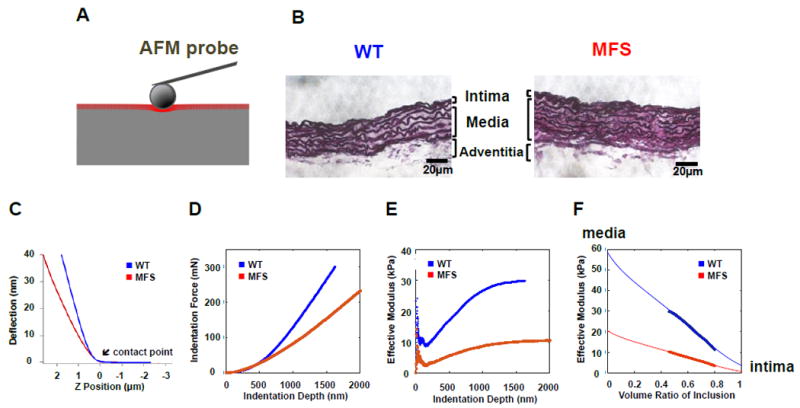
Schematic of atomic force microscope (AFM) indentation and resulting Hybrid Eshelby Decomposition (HED) analysis of WT and MFS aorta at 3.5-months old. (A) Schematic of the double-layered (intima on media) AFM indentation model based on (B) histological analysis of aortic sections with Weigert’s elastin stain, which provided values for the intimal and medial thickness of WT and MFS aortas. Representative experimental AFM indentation responses on WT (blue) and MFS (red) aortas show (C) deflection vs. Z-position, (D) indentation force vs. depth, (E) pointwise elastic modulus vs. depth, and (F) the resulting modulus vs. inclusion volume fraction (feff) curves used to extract the layer-specific aorta elastic properties, Emed (feff = 0) and Eint (feff = 1).
Briefly, AFM indentation uses a reflected laser beam and photodetector system to monitor the deflection of a cantilever probe as its Z-position approaches and contacts the sample (Fig. 1C). Once the contact point is determined, the deflection is multiplied by the cantilever spring constant to obtain the indentation force, and indentation depth is obtained by subtracting the deflection from the Z-position, yielding the indentation force vs. depth relationship (Fig. 1D). The pointwise analysis14 is then used to obtain an apparent elastic modulus from each force-depth pair, revealing depth-dependent changes in elastic properties that reflect heterogeneity of the sample material (Fig. 1E); a pointwise elastic modulus that starts low at shallow depths and increases as the probe indents deeper suggests a composite sample with a soft layer on top of a stiffer substrate. FEM simulation is used to predict the effective inclusion volume fraction (feff) as a function of indentation depth for the given layer geometry (not shown); combined with the pointwise modulus graph, one can plot the modulus vs. feff at matched indentation depths (Fig. 1F). Fitting this data with the modified Eshelby theory26 yields estimates of the substrate modulus (corresponding to feff = 0) and inclusion modulus (at feff = 1), corresponding to Emed and Eint, respectively.
Histological Analysis
Tissue structures were quantified from histological sections of aorta and lung samples prepared for visualizing the architecture. Mounting of the tissue for AFM testing precluded using the identical sample for histological sectioning. Ascending aorta samples were obtained from adjacent tissue rings, or if this was not possible due to the small dimensions of the mouse aorta, then from separate animals under matched conditions. For the lung, if the cranial lobe was used for AFM testing, then the medial lobe was used for histology and vice versa. Ascending aorta tissues were fixed in 3.7% paraformaldehyde, embedded in paraffin, cut into 7-μm sections, and stained with Weigert for elastic fibers in the medial layer of the vessel wall. De-gassed lung tissues were fixed in 3.7% paraformaldehyde, embedded in Tissue-Tek O.C.T. optimal cutting temperature compound (VWR International, Radnor, PA), cryo-sectioned into 10-μm sections, and stained with Hematoxylin and Eosin (H&E). High-resolution images (2560 × 1920 pixels) of the ascending thoracic aorta and of alveolar regions without prominent vessels were acquired using a digital color camera (DS-Fi1, Nikon Instruments, Melville, NY) mounted on a Zeiss inverted microscope (Axiovert 40 CFL, Carl Zeiss Microscopy, Thornwood, NY) with a 40x objective.
Aortic media layer thickness, elastic fiber area fraction, and lung tissue area fraction, were quantified using ImageJ software. The color threshold tool was used to define the area fraction of elastic fibers as the ratio of dark-blue elastic lamellae relative to the total area of the medial layer of aorta. Lung area fraction was obtained based on a thresholded area of the lung alveolar region relative to the total area of the image. Images were obtained at 5 locations per tissue section and 3-7 sections per animal, with measurements averaged for 3-9 animals per condition.
Statistical Analysis
Histograms of mechanical properties represent all the indentations collected from each group; for the associated bar graphs, an average modulus value was obtained for each animal, and the mean ± SD were calculated based on the numbers of animals per group. The aorta and lung data were analyzed by two-way analysis of variance (ANOVA) for effects of genotype (WT vs. MFS) and age (0.5, 2.0, 3.5 months, and 3.5 months plus Losartan treatment) with post-hoc pairwise contrast of means using MATLAB 8.4.0 (R2014b; MathWorks, Natick, MA). A probability value of p<0.05 was considered statistically significant.
RESULTS
Structural alterations of aortic tissue
WT and MFS ascending aortas exhibited similar structural properties in 0.5-month-old mice, but mutant aortas diverged from WT as the disease progressed from early- (2.0-month) to advanced-stage (3.5-month), showing severe fragmentation of the elastic fibers in end-stage disease aortas compared to WT aortas (Fig. 2A). The medial thickness of WT ascending aortas increased modestly with age and plateaued in adulthood, while the area fraction of elastic fibers in the media was maintained at approximately 70 to 80% at all ages. In MFS mice, initial signs of medial thickening and degeneration of elastic fibers appeared at the early disease stage and worsened as the disease progressed. Quantitatively, the 3.5-month mutant media thickenend 41%, on average, and had a 45% loss in elastic fiber area fraction compared to 3.5-month WT mice (Fig. 2B and C).
Figure 2.
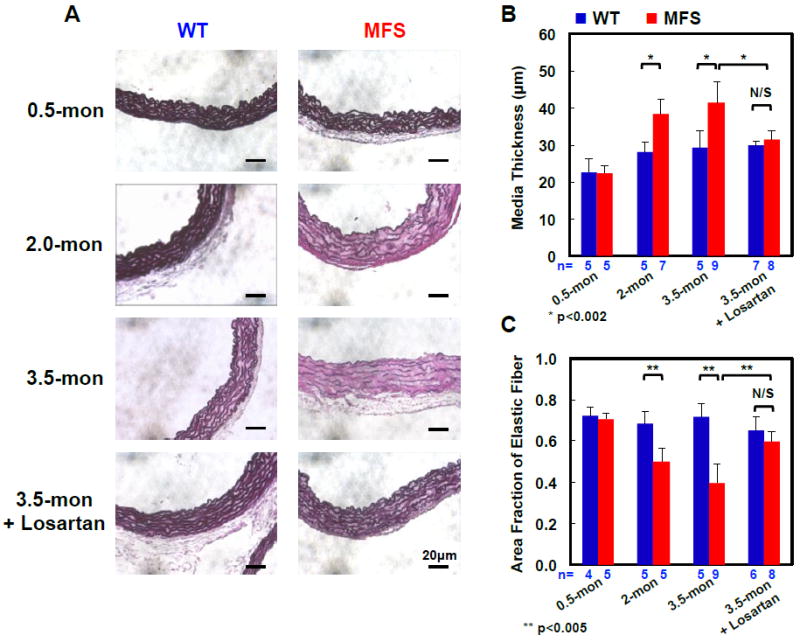
Structural variations of WT and MFS ascending aortas from 0.5, 2.0, and 3.5 month old mice and 3.5-mon Losartan-treated mice. (A) Weigert elastin staining used to quantify medial thickness and elastic area fraction. Compared to WT, the medial thickness of MFS increased (B) and the area fraction of the elastic fibers decreased (C) as the animals aged. Losartan treatment stabilized the aortic structure; no significant differences were observed between the treated MFS and treated WT controls or age-matched untreated WT in terms of media thickness or elastic fiber area fraction. Scale bar = 20 μm for all images. The number of animals (n) per group is noted in (B) and (C). * p < 0.002 and ** p< 0.005
We also examined tissue from mice treated with losartan, which has progressed to clinical trials for treating Marfan Syndrome despite an incomplete understanding of the effects on tissue structure and mechanics. Chronic losartan treatment preserved the elastic fiber structure and content in the mouse aorta, and maintained the medial thickness so that no significant differences were noted in the structural properties between the losartan treated MFS and WT controls at 3.5-months of age (Fig. 2).
Micromechanical alterations of aortic tissue
We next considered the layer-specific properties of the aortic intima (Eint) and media (Emed) resulting from ECM remodeling in response to MFS disease progression. The histograms of Emed were similar for WT and MFS at the age of 0.5 months (n = 5 for both groups), but differed as the animals aged (Fig. 3A). Adult WT aorta stiffened with age, and displayed a broad distribution of Emed, indicating mechanical heterogeneity that possibly reflected distinct contributions from elastic lamellae, smooth muscle cells, and other constituents in the media. By contrast, mutant Emed stiffened somewhat compared to 0.5-months, but remained softer and more narrowly distributed than age-matched WT (Fig. 3A), consistent with the reports of elastic fiber fragmentation in MFS mice8, 11, 29. The interaction of genotype and age was statistically significant for elastic properties of the media (p = 0.0005). Emed increased significantly with growth from 0.5-months to 2.0-months of age (WT: p<0.0001; MFS: p=0.011). However, Emed was 43% softer for MFS (22.1 ± 6.5 kPa, n = 6) than WT (37.2 ± 2.6 kPa, p < 0.001, n = 8) mice at 2 months of age, and was 52% softer (17.5 ± 1.8 kPa) than WT (37.3 ± 2.3 kPa, p < 0.001 , n = 6 for both) by 3.5 months. Importantly, the losartan-treated MFS mice had a similar distribution of Emed as the WT control (Fig. 3A), with no significant difference in average medial mechanical properties at 3.5 months (p = 0.79, n = 5 for both) (Fig. 3C).
Figure 3.

Layer-specific micromechanical properties of WT and MFS ascending aorta. (A) Distributions of elastic properties from WT (blue; representing 232, 223, 260, and 332 measurements) and MFS (red; representing 211, 160, 248, and 202 measurements) at the ages of 0.5, 2.0, and 3.5 months, and 3.5 months with losartan treatment, respectively, for the media and (B) the intima. Mean (±SD) layer-specific elastic moduli based on individual animals versus age and genotype for (C) the elastin-rich media, and (D) the intima. Note, the intima was 5- to 10-fold softer than the media. The numbers of animals (n) represented in each group is noted in (B) and (C). * p < 0.001
For the aortic intimal layer, the values of Eint were typically an order of magnitude softer than Emed. The distribution of Eint for WT and MFS aortic tissue samples coincided for all ages and conditions tested (Fig. 3B). Mean values of Eint tended to increase from about 2 to 5 kPa with age, but showed no significant difference between WT and MFS at matched ages (Fig. 3D), with no significant interaction between genotype and age (p=0.85). Losartan treatment did not affect the intima modulus.
Structural alterations of lung tissue
We also evaluated the structure of MFS lung tissue using histological staining of lung parenchyma. The tissue area fraction of WT lung increased significantly (p<0.0001) from 28.3±3.6% at 0.5 months (n = 4) to 41.1±2.7% at 2.0 months (n = 4), reflecting maturation of alveolar microstructure due to normal lung development (Fig. 4). The adult WT lung was essentially unchanged from 2.0 to 3.5 months (n = 5). By contrast, MFS lung tissue area fraction tended to decrease between 2.0 and 3.5 months (n = 4 and 3, respectively) reflecting extensive deterioration of the alveolar septa (Fig. 4A). This phenomenon resulted from remodeling of elastin-rich connective tissues in response to the genetic defect in fibrillin-1. The MFS lung showed 7%, 28%, and 34% losses in the alveolar tissue area fraction relative to WT lungs in mice aged 0.5, 2.0 and 3.5 months, respectively (Fig. 4B). Losartan treatment prevented degradation of the fibrillin-1 deficient lung (n = 5) and retained normal-looking alveolar structure in treated MFS animals with a similar lung area fraction to WT (p =0.89, n = 5) (Fig. 4).
Figure 4.
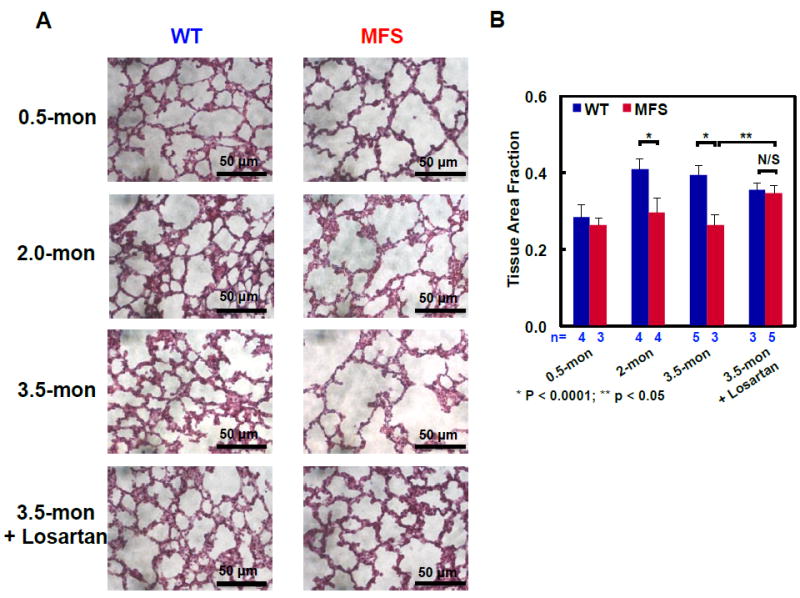
Representative H&E stained sections from WT and MFS mouse lungs at the ages of 0.5, 2.0, and 3.5 months, and in losartan-treated 3.5-month-old mice: (A) Severe airspace enlargement was observed in 3.5-month-old MFS lungs. The drug-treated lungs clearly maintained tissue morphology comparable to the control WT lungs. Scale bar of 50 μm applies to all images. (B) The lung tissue area fraction based on H&E stained sections (sample size, n = 3 to 5 animals as indicated) showed significant tissue deterioration as the diseased progressed in MFS animals. No significant difference in lung alveolar microstructure was observed between the drug-treated MFS and WT control mice. Scale bar = 50 μm for all images. * p< 0.0001 and ** p<0.05
Micromechanical alterations of lung tissue
AFM nanoindentation of the surface of intact lung parenchymal tissue revealed distributions of elastic modulus in WT and MFS lungs that were nearly identical at 0.5-months, but diverged with age and disease progression. The WT lung showed broadening heterogeneity in elastic properties as the animals matured. On the other hand, the MFS lung retained a relatively narrow distribution with a peak value indicating substantially softer elastic properties compared to WT (Fig. 5A), consistent with the microstructural deterioration noted above. To illustrate the local heterogeneity in lung tissue elastic properties, an elastography map was generated from a 6×6 indentation array covering a 20×20-μm region of a 3.5-month-old WT mouse lung, which showed local elastic moduli ranging from 2 to 10 kPa (Fig. 5B). A corresponding elastography map from a 3.5-month-old MFS mouse lung also indicated local tissue heterogeneity, but the elastic moduli were less than 1 kPa (Fig. 5C).
Figure 5.

(A) Lung micromechanics of WT (blue; representing 423, 492, 520, and 319 measurements) and MFS (red; representing 436, 283, 314, and 470 measurements) in mice at ages 0.5, 2.0, and 3.5 months, and in 3.5-month-old mice treated with losartan, respectively. (B) Representative elastography maps show local micromechanical heterogeneity of the intact 3.5-month WT and (C) MFS lungs. Each force map comprises a 6×6 indentation array covering a 20×20-μm scan area. Note different color scales in panels B and C. (D) Mean (±SD) values of Elung from WT and MFS lung tissues (sample size, n, indicates number of animals per group) shows significant mechanical deterioration of MFS lungs at 2.0 and 3.5 months that was essentially prevented by chronic treatment with losartan. * p < 0.001.
Lung mechanical properties based on AFM indentation depended significantly on both the age and genotype (p<0.00005). Mean values of Elung in the WT mice increased with age from 0.72 ± 0.38 kPa at 0.5 months to 4.30 ± 0.91 kPa at 3.5 months (Fig. 5D). Although Elung from MFS mice increased from 0.5 months (0.76 ± 0.25 kPa) to 2 months (1.52 ± 0.28 kPa), the change was much less than in WT lungs, and Elung decreased rather than increased with advancing disease at 3.5 months (0.54 ± 0.40 kPa). The 8-fold softening of the end-stage diseased lung compared to WT (p < 0.0001) is representative of the extensive breakdown of mutant lung microstructures. The walls of the alveolar ducts function as mechanical load-bearing structures that undergo extension during breathing; abnormal weakening of mutant lung tissue could contribute to MFS-related obstructive lung diseases. Chronic losartan treatment preserved lung elastic properties at levels comparable to age-matched WT controls (p = 0.66) (Fig. 5A and D), consistent with the apparently normal tissue architecture observed from histological sections.
DISCUSSION
We observed medial thickening in MFS aorta that correlated with recent in vivo echocardiographic measurements of the proximal ascending aorta diameter, obtained during progression of thoracic aortic aneurysm in the same mouse model.13 We observed a 45% reduction in elastic fiber content in the ascending thoracic aorta at the end-disease state. The observed structural alterations indicated adaptive remodeling of the MFS vessel wall, reflecting elastin fragmentation and compensatory collagen deposition in response to the genetic defect in FBN1. A similar analysis of the descending thoracic aorta in these mice reported a 40% decrease in the percent of medial area occupied by elastic fibers.29 This suggests a fairly uniform structural degradation of the thoracic aorta in MFS, despite anatomical and hemodynamic variations along the aortic tree. However, Bellini et al. recently reported differences in biaxial mechanical properties of the ascending versus descending thoracic aorta that are consistent with a propensity for aneurysms to form in the ascending segment.5 Further studies are required to assess regional variations in vascular mechanics with losartan treatment.
Here we interrogated transmural micromechanical heterogeneity of individual layers of the intact aortic wall for the first time, and showed age-dependent weakening and softening specifically in the media layer in MFS mice. This finding seemingly contrasts with arterial stiffening that was observed in MFS arteries with macroscopic mechanical testing and inferred from ECM alterations.15, 16 Arterial stiffening has been associated with aortic aneurysm in MFS from imaging by transesophageal echocardiography49 and magnetic resonance imaging20, 50 in patients, and using pulse-wave velocity29 and inflation testing28 in animal models. These measurements reflect the composite effects from all layers of the vessel wall, and reflect nonlinear elastic stiffening from the recruitment of collagen fibers during mechanical loading.19 Thus, intrinsic local softening of the media could be overshadowed by the apparent stiffening of over-stretched collagen fibers primarily in the adventitia, both in the context of pressure loading conditions in vivo as well as macroscopic tensile testing in vitro, compared to our AFM measurements of the unloaded vessel wall. A more complete assessment of micromechanics of the ascending aorta wall would include measurements of the adventitia. Indeed, we attempted flipping the sample over to directly indent the adventitia. However, the loose fibrous structure was porous and sticky, interfering with proper contact between the AFM tip and the sample surface and precluding reliable measurements of adventitia microelastic properties.
We implemented AFM indentation to investigate micromechanical properties of the layered aortic wall. AFM indentation involves a finite strain field that is highly localized in an otherwise unloaded sample configuration. We have not considered material anisotropy or nonlinearity in our analysis. Studies of the layer-specific mechanics of the MFS aorta often involve layer separation a priori, either physically45 or chemically16, which can damage key tissue structures and alter intrinsic material properties. Our AFM approach determined the layer-specific mechanics of intact aorta samples, assuming a two-layer configuration of the flattened vessel segments consisting of an exposed intima and an underlying media. Preliminary FEM simulations (not shown) confirmed that the adventitial layer was too distant from the indentation site to significantly impact the force-depth response, and also that the estimates of media elastic properties were robust to experimental uncertainty in the intima layer thickness, which was assumed to have a nominal value of 3 μm for both WT and MFS mice. The micromechanical properties of the spatially heterogeneous aorta derive from its structural composition: the soft tunica intima consists of endothelial cells with a basement membrane, while the tunica media includes vascular smooth muscle cells (SMC) interleaved with elastic lamellae. Our structural analyses revealed variations specifically in the aortic tunica media at different disease states, reflecting MFS-related complications. SMCs in the tunica media confer vascular contractility and mediate mechanosignaling to regulate the bloodstream; consequently, alterations in the local intrinsic mechanics and underlying tissue structure can fundamentally impact vascular biology and physiology.
Newborn MFS mice do not exhibit fragmentation of the medial elastic network and show no indications of disease in early elastogenesis, but pathology progresses as the animals age.29 These findings support our observation of indistinguishable differences in WT and MFS elastin structure and media mechanical properties at 0.5-months of age. The WT aortas stiffened from 0.5 to 2.0 months of age, consistent with documented increases in aortic stiffness with age in non-atherosclerotic animals ranging from mice51 to non-human primates.41 The apparent mechanical stabilization from 2.0 to 3.5 months most likely reflects the relatively short time interval compared to the normal lifespan of the WT mouse. However, these time points were chosen to match different stages of disease progression in the MFS mice, where the 50% survival is less than 3 months in this transgenic model.13 As the MFS animals matured, aortic pathology was seen by mechanical insufficiency within the elastin-rich media, but not in the intimal layer. Elastin fragmentation is reportedly accompanied by compensatory remodeling through deposition of collagen and proteoglycans to restore biomechanical homeostasis.23, 27 Abnormal proteoglycan secretion could alter the absorption and trapping of water molecules in the interstitial region of the medial layer,23 which impacts mechanics and residual stress.1 A more extensive evaluation of the viscoelastic and osmotic properties of the tunica media may elucidate the contribution of proteoglycans in MFS aortic mechanics. Likewise, the mechanics of the tunica intima influence its crucial role in mechanotransduction and signaling. Despite reported endothelial dysfunction during pathological processes such as intimal edema, SMC infiltration and inflammatory responses,10, 38 our data suggest the intrinsic elastic properties of the intima are not adversely affected by the MFS disease process.
Published mechanical testing of dilated human aortas revealed age-dependent differences in mechanical strength: old dilated aortas were stiffer and weaker than young ones,35 but all the samples were obtained at the disease end-stage. It is unethical to harvest human aortic tissue samples at earlier stages of the disease, so the progressive changes in mechanical properties are unknown. Consequently, the assessment for prophylactic surgical intervention with an aortic graft relies on the patient’s aortic diameter,44 which provides a rule of thumb at best. Our time-resolved, layer-specific mechanical properties of the MFS mouse aorta help provide input variables, such as intimal and medial mechanics, necessary for developing computational models of aortic dilation, aneurysm, and rupture. With additional information on adventitia mechanics and physiologic load conditions, such models could eventually provide a tool for predicting the risk and location of lethal aortic rupture based on rational biomechanical analysis.43, 52
In terms of MFS-related lung disease, previous studies have noted changes in alveolar wall thickness and airway dilation resembling the lung pathogenesis of emphysema, which is one of the pulmonary diseases commonly reported in MFS patients.47, 48, 53 In MFS mice, early lung tissue defects have been observed in neonates, and have been linked to the dysregulation of TGF-β activation in the developing lungs, with subsequent inhibition of cellular activities such as proliferation, and stimulation of apoptosis.33 Correspondingly, we measured consistently lower area fraction of lung tissue in untreated MFS mice, with more severe disruption as the disease progressed, compared to age-matched WT mice.
Our AFM micromechanical measurements on intact ex vivo MFS lung tissue showed a 8-fold softening compared to WT at the end-stage disease state. Similar softening effects were recently reported in excised whole lungs from a mouse model of mild MFS, indicating a lower elastance in diseased mice accompanied by emphysema-like structure similar to our findings.48 A mouse model of emphysema showed similar changes in the mechanics of the diseased lung, including a loss of elastin, increased hysteresis and non-linearity, and decreased load-bearing capacity.22 Lung inflation testing has been performed to obtain a pressure-volume relationship that reflects the lung mechanical properties, using sequential images of lung inflation to assess micro- or macro-strain of the parenchyma tissue.6, 39 The interlinked networks of elastin and collagen function as two complementary mechanical systems during lung inflation: at low strain, elastin fibers dominate the stress-bearing role, whereas at high strain, the wavy collagen fibers straighten and sustain the tension imposed on the lung parenchyma.30, 47 In this study, we evaluated lung micromechanics from direct measurements using AFM indentation of lung parenchyma in the deflated state, and assumed that elastic fiber integrity dominates the mechanical response under these conditions. Possible viscoelastic and nonlinear elastic effects were neglected. Interestingly, AFM measurements of ECM scaffolds from decellularized thin sections of MFS lung revealed similar stiffness as WT.48 We suspect the 3D organization of cellular and ECM components both contribute to the disruption of micromechanics in the MFS lung. Our results indicated that the apparent elastic modulus of the lung tissue, Elung, decreased dramatically with disease progression in MFS mice, consistent with the symptoms of obstructive lung disease.
In the in vivo setting, the aorta and lung are constantly in a mechanically loaded state. However, transpulmonary pressure in the lung is 20 to 30-fold lower than blood pressure in the aorta. Thus, the lung is an attractive system in which to evaluate the efficacy of losartan treatment, independent of the confounding influence of its effects on lowering systemic blood pressure. As reported previously, losartan treatment, starting at 0.5 months and continuing throughout the lifetime, attenuated the growth of aortic aneurysms and suppressed medial degradation in mouse models.13, 34 We showed that losartan effectively counteracted the mechanical degradation of the stress-bearing media layer in MFS mice, preserving the mechanical integrity of the tissue. In addition, losartan treatment of MFS mice maintained the tissue structure and mechanical properties of lung parenchyma indistinguishable from age-matched WT controls. These findings strongly support the efficacy of losartan as a prophylactic treatment for MFS, consistent with its reported prevention of lethal aortic dissection and the prolongation of survival rates of diseased animals,13 and reported efficacy in pediatric patients.7
In terms of losartan’s mechanism of action, another study reported that angiotensin II-dependent TGF-β signaling contributes to Loeys-Dietz syndrome (LDS), which has a TGF-β receptor gene mutation and exhibits vascular complications similar to MFS. An overexpression of TGF-β signaling was observed despite malfunction of the diseased TGF-β receptors, implying indirect activation of the signaling pathway.17 Losartan-treated MFS mice exhibited a reduction in latent TGF-β binding molecule (Tsp-1) expression in vivo and a lowered nuclear accumulation of downstream molecule (pSmad2) in diseased skeletal muscle,12 and reduced distal airspace widening in diseased lung parenchyma.21 These protective influences of Angiotensin II receptor blocker (ARB) treatment against the MFS-related skeletal and lung complications support the attenuated activation of the TGF-β pathway as a primary therapeutic mechanism of losartan treatment in MFS mice, beyond a simple improvement in the hemodynamics of the vascular system.
In prior studies, losartan treatment significantly prolonged the lifespan of MFS mice (mgR/mgR) beyond an average of 2.5 months, and delayed, but did not prevent, the eventual fragmentation of elastic fibers and the formation of aortic aneurysms.13 In another study, losartan treatment preserved the structure and breaking strength of diseased thoracic aorta up to 9 months in a mild Marfans mouse model (Fbn1 C1039G/+; average life-span > 1 year); however, endothelial function remained abnormal throughout treatment.55 Therefore, future experiments should examine the long-term effects of losartan on tissue micromechanics in MFS mice beyond 3.5-months old. Furthermore, a combination treatment of losartan with other drugs has been suggested to target organ-specific manifestations in MFS.13, 34 The efficacy of such treatments is often based on molecular expression or histological staining.54 We have shown that direct measurement of intact tissue micromechanics, using AFM indentation, offers a powerful quantitative approach for the evaluation of future combination treatment strategies in small animal models of Marfan Syndrome and related disorders.
CONCLUSION
A FBN1-deficient transgenic mouse model of MFS exhibited irreversible degeneration with compensatory thickening of the tunica media in the ascending aorta, along with parenchymal tissue degradation in the upper lobes of the right lung. AFM indentation revealed age- and disease-dependent micromechanical degradation of the aortic media and lung tissue in MFS, but not of the aortic intima. Chronic, prophylactic treatment with losartan protected the microstructure of the elastin-rich aortic media and lung tissue in the regions tested, and preserved the elastic moduli equivalent to age-matched WT aorta and lung. The findings support early intervention with losartan treatment as a promising strategy for normalizing the properties of aorta and lung tissue in MFS. Losartan could help minimize the associated tissue mechanical degradation and perhaps help delay or prevent some of the most life-threatening disease phenotypes in Marfan syndrome.
Acknowledgments
The authors acknowledge Drs. Evren Azeloglu, Gaurav Kaushik, and Jason Cook for technical support, and Marlon Suarez, Laboratory Manager of the Mount Sinai Pathology Core Facility, for expert assistance with the lung histology. Funding sources include a CUNY PhD Fellowship from the Biomedical Engineering Department at City College of New York (Lee), a Stony Wold-Herbert Fellowship (Lee), and National Institutes of Health grants NIH/NIAMS (P01 AR049698, Ramirez), and NIH/NCRR (S10 RR027609, Costa).
References
- 1.Azeloglu EU, Albro MB, Thimmappa VA, Ateshian GA, Costa KD. Heterogeneous transmural proteoglycan distribution provides a mechanism for regulating residual stresses in the aorta. Am J Physiol Heart Circ Physiol. 2008;294:H1197–1205. doi: 10.1152/ajpheart.01027.2007. [DOI] [PubMed] [Google Scholar]
- 2.Azeloglu EU, Kaushik G, Costa KD. Developing a hybrid computational model of AFM indentation for analysis of mechanically heterogeneous samples. Conf Proc IEEE Eng Med Biol Soc. 2009:4273–4276. doi: 10.1109/IEMBS.2009.5334043. [DOI] [PubMed] [Google Scholar]
- 3.Bartram U, Speer CP. The role of transforming growth factor beta in lung development and disease. Chest. 2004;125:754–765. doi: 10.1378/chest.125.2.754. [DOI] [PubMed] [Google Scholar]
- 4.Beenakker JW, Ashcroft BA, Lindeman JH, Oosterkamp TH. Mechanical properties of the extracellular matrix of the aorta studied by enzymatic treatments. Biophys J. 2012;102:1731–1737. doi: 10.1016/j.bpj.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellini C, Korneva A, Zilberberg L, Ramirez F, Rifkin DB, Humphrey JD. Differential ascending and descending aortic mechanics parallel aneurysmal propensity in a mouse model of Marfan syndrome. J Biomech. 2016 doi: 10.1016/j.jbiomech.2015.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brewer KK, Sakai H, Alencar AM, Majumdar A, Arold SP, Lutchen KR, Ingenito EP, Suki B. Lung and alveolar wall elastic and hysteretic behavior in rats: effects of in vivo elastase treatment. J Appl Physiol (1985) 2003;95:1926–1936. doi: 10.1152/japplphysiol.00102.2003. [DOI] [PubMed] [Google Scholar]
- 7.Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, D HC., III Angiotensin II blockade and aortic-root dilation in Marfan’s Syndrome. N Engl J Med. 2008;358:2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bunton T, Biery N, Myers L, Gayraud B, Ramirez F, Dietz H. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res. 2001;19:37–43. doi: 10.1161/01.res.88.1.37. [DOI] [PubMed] [Google Scholar]
- 9.Burnier M, Wuerzner G. Pharmacokinetic evaluation of losartan. Expert Opin Drug Metab Toxicol. 2011;7:643–649. doi: 10.1517/17425255.2011.570333. [DOI] [PubMed] [Google Scholar]
- 10.Chung AW, Yeung KA, Cortes SF, Sandor GG, Judge D, Dietz HC, Breemen Cv. Endothelial dysfunction and compromised eNOS/Akt signaling in the thoracic aorta during the progression of Marfan syndrome. Br J Pharmacol. 2007;150:1075–1083. doi: 10.1038/sj.bjp.0707181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung AW, Yeung KA, Sandor GG, Judge DP, Dietz HC, Breemen Cv. Loss of elastic fiber integrity and reduction of vascular smooth muscle contraction resulting from the upregulated activities of matrix metalloproteinase-2 and -9 in the thoracic aortic aneurysm in Marfan syndrome. Circ Res. 2007;101:512–522. doi: 10.1161/CIRCRESAHA.107.157776. [DOI] [PubMed] [Google Scholar]
- 12.Cohn RD, Erp Cv, Habashi JP, Soleimani AA, Klein EC, Lisi MT, Gamradt M, Rhys CMa, Holm TM, Loeys BL, Ramirez F, Judge DP, Ward CW, Dietz HC. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13:204–210. doi: 10.1038/nm1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cook JR, Clayton NP, Carta L, Galatioto J, E EC, Smaldone S, Nelson CA, Cheng S, Wentworth BM, Ramirez F. Dimorphic Effects of Transforming Growth Factor-β Signaling During Aortic Aneurysm Progression in Mice Suggest a Combinatorial Therapy for Marfan Syndrome. Arterioscler Thromb Vasc Biol. 2015;35:911–917. doi: 10.1161/ATVBAHA.114.305150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costa KD, Yin FC. Analysis of indentation: implications for measuring mechanical properties with atomic force microscopy. J Biomech Eng. 1999;121:462–471. doi: 10.1115/1.2835074. [DOI] [PubMed] [Google Scholar]
- 15.Eberth JF, Taucer AI, Wilson E, Humphrey JD. Mechanics of Carotid Arteries in a Mouse Model of Marfan Syndrome. Ann Biomed Eng. 2009;37:1093–1104. doi: 10.1007/s10439-009-9686-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferruzzi J, Collins M, Yeh A, Humphrey J. Mechanical assessment of elastin integrity in fibrillin-1-deficient carotid arteries: implications for Marfan syndrome. Cardiovasc Res. 2011;92:287–295. doi: 10.1093/cvr195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gallo EM, Loch DC, Habashi JP, Calderon JF, Chen Y, Bedja D, Erp Cv, Gerber EE, Parker SJ, Sauls K, Judge DP, Cooke SK, Lindsay ME, Rouf R, Myers L, Rhys CMa, Kent KC, Norris RA, Huso DL, Dietz HC. Angiotensin II-dependent TGF-β signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J Clin Invest. 2014;124:448–460. doi: 10.1172/JCI69666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gleason RL, Wilsonb E, Humphrey JD. Biaxial biomechanical adaptations of mouse carotid arteries cultured at altered axial extension. J Biomech. 2007;40:766–776. doi: 10.1016/j.jbiomech.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 19.Groenink M, Langerak SE, Vanbavel E, v d Wall EE, Mulder BJ, v d Wal AC, Spaan JA. The influence of aging and aortic stiffness on permanent dilation and breaking stress of the thoracic descending aorta. Cardiovasc Res. 1999;43:471–480. doi: 10.1016/s0008-6363(99)00095-4. [DOI] [PubMed] [Google Scholar]
- 20.Groenink M, Roos Ad, Mulder BJ, Spaan JA, v d Wall EE. Changes in aortic distensibility and pulse wave velocity assessed with magnetic resonance imaging following beta-blocker therapy in the marfan syndrome. Am J Cardiol. 1998;82:203–208. doi: 10.1016/s0002-9149(98)00315-4. [DOI] [PubMed] [Google Scholar]
- 21.Habashi J, Judge D, Holm T, Cohn R, Loeys B, Cooper T, Myers L, Klein E, Liu G, Calvi C, Podowski M, Neptune E, Halushka M, Bedja D, Gabrielson K, Rifkin D, Carta L, Ramirez F, Huso D, Dietz H. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ito S, Ingenito EP, Brewer KK, Black LD, Parameswaran H, Lutchen KR, Suki B. Mechanics, nonlinearity, and failure strength of lung tissue in a mouse model of emphysema: possible role of collagen remodeling. J Appl Physiol 1985. 2005;98:503–511. doi: 10.1152/japplphysiol.00590.2004. [DOI] [PubMed] [Google Scholar]
- 23.Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, Sakai LY, Dietz HC. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. 2004;114:172–181. doi: 10.1172/JCI20641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaartinen V, Warburton D. Fibrillin controls TGF-bold beta activation. Nat Genet. 2003;33:331–332. doi: 10.1038/ng0303-331. [DOI] [PubMed] [Google Scholar]
- 25.Lacro RV, Dietz HC, Mahony L. Atenolol versus Losartan in Marfan’s Syndrome. N Engl J Med. 2015;372:977–981. doi: 10.1056/NEJMc1500128. [DOI] [PubMed] [Google Scholar]
- 26.Li S, Sauer R, Wang G. A circular inclusion in a finite domain I. The Dirichlet-Eshelby problem. Acta Mechanica. 2005;179:67–90. [Google Scholar]
- 27.Lindeman JH, Ashcroft BA, Beenakker JW, Es Mv, Koekkoek NB, Prins FA, Tielemans JF, Abdul-Hussien H, Bank RA, Oosterkamp TH. Distinct defects in collagen microarchitecture underlie vessel-wall failure in advanced abdominal aneurysms and aneurysms in Marfan syndrome. Proc Natl Acad Sci U S A. 2010;107:862–865. doi: 10.1073/pnas.0910312107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mariko B, Pezet M, Escoubet B, Bouillot S, Andrieu J, Starcher B, Quaglino, Jacob M, Huber P, Ramirez, Faury G. Fibrillin-1 genetic deficiency leads to pathological aging of arteries in mice. J Pathol. 2011;224:33–44. doi: 10.1002/path.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marque V, Kieffer P, Gayraud B, Lartaud-Idjouadiene I, Ramirez F, Atkinson J. Aortic wall mechanics and composition in a transgenic mouse model of Marfan syndrome. Arterioscler Thromb Vasc Biol. 2001;21:1184–1189. doi: 10.1161/hq0701.092136. [DOI] [PubMed] [Google Scholar]
- 30.Mercer RR, Crapo JD. Spatial distribution of collagen and elastin fibers in the lungs. J Appl Physiol (1985) 1990;69:756–765. doi: 10.1152/jappl.1990.69.2.756. [DOI] [PubMed] [Google Scholar]
- 31.Milewicz DM, Dietz HC, Miller DC. Treatment of aortic disease in patients with Marfan Syndrome. Circulation. 2005;111:e150–157. doi: 10.1161/01.CIR.0000155243.70456.F4. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura M, Itoh S, Makita S, Ohira A, Arakawa N, Hiramori K. Peripheral resistance vessel dysfunction in Marfan syndrome. Am Heart J. 2000;139:661–666. doi: 10.1016/s0002-8703(00)90045-0. [DOI] [PubMed] [Google Scholar]
- 33.Neptune E, Frischmeyer P, Arking D, Myers L, Bunton T, Gayraud B, Ramirez F, Sakai L, Dietz H. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 34.Nistala H, Lee-Arteaga S, Carta L, Cook JR, Smaldone S, Siciliano G, Rifkin AN, Dietz HC, Rifkin DB, Ramirez F. Differential effects of alendronate and losartan therapy on osteopenia and aortic aneurysm in mice with severe Marfan syndrom. Hum Mol Genet. 2010;19:4790–4798. doi: 10.1093/hmg/ddq409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okamoto R, Wagenseil J, DeLong W, Peterson S, Kouchoukos N, Sundt Tr. Mechanical properties of dilated human ascending aorta. Ann Biomed Eng. 2002;30:624–635. doi: 10.1114/1.1484220. [DOI] [PubMed] [Google Scholar]
- 36.Pearson G, Devereux R, Loeys B, Maslen C, Milewicz D, Pyeritz R, Ramirez F, Rifkin D, Sakai L, Svensson L, Wessels A, Van Eyk J, Dietz H. Report of the National Heart, Lung, and Blood Institute and National Marfan Foundation Working Group on research in Marfan syndrome and related disorders. Circulation. 2008;118:785–791. doi: 10.1161/CIRCULATIONAHA.108.783753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peloquin J, Huynh J, Williams RM, Reinhart-King CA. Indentation measurements of the subendothelial matrix in bovine carotid arteries. J Biomech. 2011;44:815–821. doi: 10.1016/j.jbiomech.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 38.Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, Biery NJ, Dietz HC, Sakai LY, Ramirez F. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci U S A. 1999;96:3819–3823. doi: 10.1073/pnas.96.7.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perlman CE, Lederer DJ, Bhattacharya J. Micromechanics of alveolar edema. Am J Respir Cell Mol Biol. 2011;44:34–39. doi: 10.1165/rcmb.2009-0005OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polzer S, Gasser TC, Bursa J, Staffa R, Vlachovsky R, Man V, Skacel P. Importance of material model in wall stress prediction in abdominal aortic aneurysms. Med Eng Phys. 2013;35:1282–1289. doi: 10.1016/j.medengphy.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 41.Qiu H, Zhu Y, Sun Z, Trzeciakowski JP, Gansner M, Depre C, Resuello RRG, Natividad FF, Hunter WC, Genin GM, Elson EL, Vatner DE, Meininger GA, Vatner SF. Short communication: vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res. 2010;107:615–619. doi: 10.1161/CIRCRESAHA.110.221846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramirez F, Rifkin D. Extracellular microfibrils: contextual platforms for TGFbeta and BMP signaling. Curr Opin Cell Biol. 2009;21:616–622. doi: 10.1016/j.ceb.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raut SS, Chandra S, Shum J, Finol EA. The role of geometric and biomechanical factors in abdominal aortic aneurysm rupture risk assessment. Ann Biomed Eng. 2013;41:1459–1477. doi: 10.1007/s10439-013-0786-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saliba E, Sia Y. The ascending aortic aneurysm: When to intervene? IJC Heart & Vasculature. 2015;6:91–100. doi: 10.1016/j.ijcha.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sokolis DP, Kritharis EP, Iliopoulos DC. Effect of layer heterogeneity on the biomechanical properties of ascending thoracic aortic aneurysms. Med Biol Eng Comput. 2012;50:1227–1237. doi: 10.1007/s11517-012-0949-x. [DOI] [PubMed] [Google Scholar]
- 46.Syyong H, Chung A, Yang H, van Breemen C. Dysfunction of endothelial and smooth muscle cells in small arteries of a mouse model of Marfan syndrome. Br J Pharmacol. 2009;158:1597–1608. doi: 10.1111/j.1476-5381.2009.00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toshima M, Y YO, Ohtani O. Three-dimensional architecture of elastin and collagen fiber networks in the human and rat lung. Arch Histol Cytol. 2004;67:31–40. doi: 10.1679/aohc.67.31. [DOI] [PubMed] [Google Scholar]
- 48.Uriarte JJ, Meirelles T, d Blanco DG, Nonaka PN, Campillo N, Sarri E, Navajas D, Egea G, Farré R. Early impairment of lung mechanics in a murine model of Marfan Syndrome. PLoS One. 2016;11:e0152124. doi: 10.1371/journal.pone.0152124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vitarelli A, Conde Y, Cimino E, D’Angeli I, D’Orazio S, Stellato S, Padella V, Caranci F. Aortic wall mechanics in the Marfan syndrome assessed by transesophageal tissue Doppler echocardiography. Am J Cardiol. 2006;97:571–577. doi: 10.1016/j.amjcard.2005.09.089. [DOI] [PubMed] [Google Scholar]
- 50.Westenberg JJ, Scholte AJ, Vaskova Z, v d Geest RJ, Groenink M, Labadie G, v d Boogaard PJ, Radonic T, Hilhorst-Hofstee Y, Mulder BJ, Kroft LJ, Reiber JH, Roos Ad. Age-related and regional changes of aortic stiffness in the Marfan syndrome: assessment with velocity-encoded MRI. J Magn Reson Imaging. 2011;34:526–531. doi: 10.1002/jmri.22646. [DOI] [PubMed] [Google Scholar]
- 51.Wheeler JB, Mukherjee R, Stroud RE, Jones JA, Ikonomidis JS. Relation of murine thoracic aortic structural and cellular changes with aging to passive and active mechanical properties. J Am Heart Assoc. 2015;4:e001744. doi: 10.1161/JAHA.114.001744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wisneski AD, Mookhoek A, Chitsaz S, Hope MD, Guccione JM, Ge L, Tseng EE. Patient-specific finite element analysis of ascending thoracic aortic aneurysm. J Heart Valve Dis. 2014;23:765–772. [PMC free article] [PubMed] [Google Scholar]
- 53.Wood J, Bellamy D, Child A, Citron K. Pulmonary disease in patients with Marfan syndrome. Thorax. 1984;39:780–784. doi: 10.1136/thx.39.10.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiong W, Meisinger T, Knispel R, Worth JM, Baxter BT. MMP-2 regulates Erk1/2 phosphorylation and aortic dilatation in Marfan Syndrome. Circ Res. 2012;110:e92–e101. doi: 10.1161/CIRCRESAHA.112.268268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang HC, Kim JM, Chum E, Breemen Cv, Chung AWY. Long-term effects of losartan on structure and function of the thoracic aorta in a mouse model of Marfan syndrome. Br J Pharmacol. 2009;158:1503–1512. doi: 10.1111/j.1476-5381.2009.00443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]