

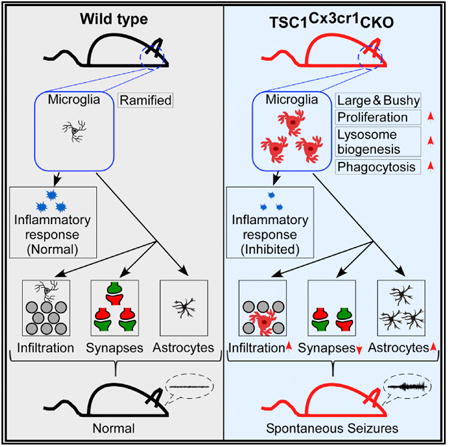
Summary
Microglia are well known to play a critical role in maintaining brain homeostasis. However, their role in epileptogenesis has yet to be determined. Here, we demonstrate that elevated mTOR signaling in mouse microglia leads to phenotypic changes, including an amoeboid-like morphology, increased proliferation, and robust phagocytosis activity, but without a significant induction of pro-inflammatory cytokines. We further provide evidence that these noninflammatory changes in microglia disrupt homeostasis of the CNS, leading to reduced synapse density, marked microglial infiltration into hippo-campal pyramidal layers, moderate neuronal degeneration, and massive proliferation of astrocytes. Moreover, the mice thus affected develop severe early-onset spontaneous recurrent seizures (SRSs). Therefore, we have revealed an epileptogenic mechanism that is independent of the microglial inflammatory response. Our data suggest that microglia could be an opportune target for epilepsy prevention.
Graphical abstract
Zhao et al. reveal that elevated mTOR signaling in microglia propels the cells to acquire a noninflammatory reactive-like phenotype, which leads to disruption of CNS homeostasis. The authors also report that reactive-like microglia promote epileptogenesis independent of the inflammatory response.
Introduction
Epileptogenesis is the process by which a normal brain becomes prone to develop spontaneous recurrent seizures. It typically develops following diverse brain insults or pathological changes such as those caused by brain injury or genetic mutation (e.g., in tuberous sclerosis complex [TSC] genes) (Pitkänen and Lukasiuk, 2011). There is increasing evidence to suggest that microglia may play a role in epileptogenesis (Abraham et al., 2012; Boer et al., 2006; Liu et al., 2014; Sosunov et al., 2012; van Vliet et al., 2012; Zhang et al., 2016). For example, morphologically reactive microglia have been found in the brains of animal models of temporal lobe epilepsy (TLE) (van Vliet et al., 2012) and in surgical resections of epilepsy patients (Liu et al., 2014; Sosunov et al., 2012). Microglia are the main innate immune cells in the CNS and the principal producer of pro-inflammatory cytokines in response to brain injury (Colonna and Butovsky, 2017). Their pro-inflammatory action is postulated to be the etiologic driver of epileptogenesis (Abraham et al., 2012; Aronica et al., 2017; Colonna and Butovsky, 2017).
Apart from being inflammatory mediators, microglia also play a critical role in maintaining CNS homeostasis (Colonna and Butovsky, 2017; Hong et al., 2016; Nimmerjahn et al., 2005). They reside nearly uniformly throughout the entire brain. Their extensive ramifying morphology ensures maximal contacts with neurons for synaptic pruning and clearance of dying cells and debris during brain development and under pathological conditions (Abiega et al., 2016; Nimmerjahn et al., 2005; Schafer et al., 2012; Sierra et al., 2010); thus, microglia are ideally positioned for surveilling and maintaining homeostasis of the CNS. Their homeostatic action has also been implicated in learning and memory in the normal brain (Parkhurst et al., 2013). When a brain is injured, microglia migrate to the site of damage to engulf and eliminate dead cells and debris. They become hyper-proliferated and undergo a morphological change (process shortening), which affects the contacts they make with neurons and non-neuronal cells; consequently, their synaptic surveillance and homeostatic functions are expected to be altered. However, it is not known whether such altered noninflammatory homeostatic changes of microglia contribute to epileptogenesis.
Elevated mTOR signaling is epileptogenic in human TSC and in several other animal models (Crino, 2016). Some focal epilepsies are closely associated with somatic mutations that lead to increased mTOR activity (Jansen et al., 2015). Previous studies have been mainly focused on neurons and astrocytes (Crino, 2016; McMahon et al., 2012; Zhang et al., 2016). It remains unclear if microglial mTOR signaling plays any role in epileptogenesis. mTOR signaling in microglia is elevated in the brains of patients with epilepsy (Liu et al., 2014; Sosunov et al., 2012), pointing to a potential role for mTOR signaling in microglial regulation and epileptogenesis. TSC is a negative regulator of the mTOR pathway (Kwiatkowski et al., 2002); genetic deletion of TSC1 leads to hyperactivation of mTOR. Accordingly, we employed mice with restrictive deletion of TSC1 to model mTOR activation in microglia. Our hypothesis is that perturbation of microglial homeostatic activity drives epileptogenesis.
Results
Elevated mTOR Signaling Changes Microglia Morphology
Immunohistological analysis revealed an increased number of microglia positively stained for phosphorylated s6 in brain tissue obtained from epilepsy surgery (Figure S1A). This finding verifies that mTOR signaling in microglia is upregulated in the epileptic brain (Liu et al., 2014; Sosunov et al., 2012). To elevate mTOR signaling selectively in microglia, we generated TSC1Cx3cr1CKO mice by crossing TSC1flox/flox mice (Kwiatkowski et al., 2002; McMahon et al., 2012) with a Cx3CR1-cre mouse line. The Cx3CR1-cre line drove the expression of Cre recombinase specifically in mouse microglia, as indicated by Tomato fluorescent signal in microglial cells in the brains of Tomatof/f/CX3CR1-cre mice (Figure S1B). In TSC1Cx3cr1CKO mice, TSC1 was deleted in microglia, resulting in increased levels of pS6 (Figures S1C and S1D), which reflects the activation of mTOR (Kwiatkowski et al., 2002; McMahon et al., 2012). The ramified morphology of microglia facilitates their contact with neurons and non-neuronal cells (Nimmerjahn et al., 2005) and is therefore critical for CNS homeostasis. Accordingly, we set out to evaluate whether elevated mTOR signaling alters the morphology of microglia. In contrast to the ramified morphology of microglia seen in Cx3CR1-cre control mouse brain, the microglia in TSC1Cx3cr1CKO mice adopted a bushy or amoeboid-like morphology resembling the microglial reactive-like phenotype (Figures 1A–1C, S1E, and S1F). The microglia became less ramified, displaying an enlarged cell body and short processes with less branching. Fluorescence-activated cell sorting (FACS) analysis confirmed that cells of the microglial population were enlarged in TSC1Cx3cr1CKO mice (Figures 1D, 1E, and S1G). Thus, our data suggest that microglia adopt a reactive-like morphology in the brain when mTOR signaling is activated.
Figure 1. Reactive-like Morphology of Microglia in TSC1Cx3cr1CKO Mice.
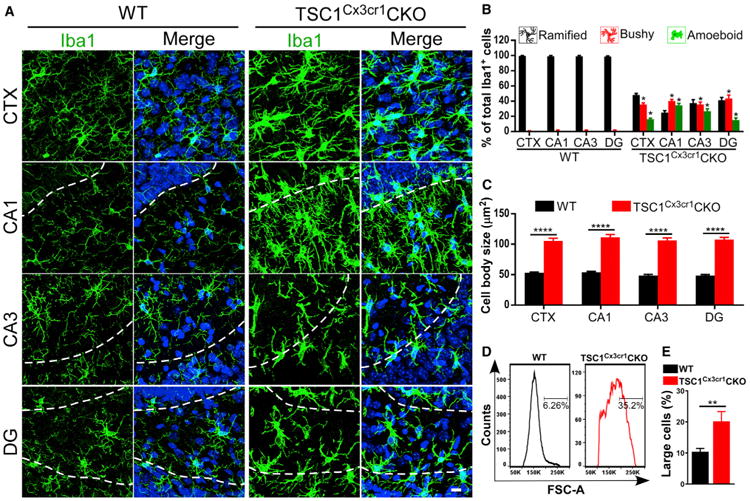
(A) Confocal images acquired from cortex (CTX), hippocampal CA1 and CA3, and the dentate gyrus (DG) of Cx3cr1-cre (hereafter referred to as wild-type [WT]) and TSC1Cx3cr1CKO mouse brains. Dashed lines indicate the borders of the CA1 and CA3 pyramidal layers and the dentate granular layers. Scale bar, 10 μm.
(B) Percentage of microglia displaying a ramified, bushy, or amoeboid morphology in the CTX and hippocampus of WT (n = 6; 3 males and 3 females) and TSC1Cx3cr1CKO (n = 7; 3 males and 4 females) mice.
(C) Quantification of microglial cell-body size in WT (n = 6; 3 males and 3 females) and TSC1Cx3cr1CKO (n = 6; 3 males and 3 females) mice.
(D) FACS analysis of cell sizes of microglia (gated as CD11b+/CD45low & Int) in WT and TSC1Cx3cr1CKO mice.
(E) Quantification of the population of microglia with larger cell sizes in WT (n = 4; 2 males and 2 females) and TSC1Cx3cr1CKO (n = 4; 2 males and 2 females) mice. Data are presented as mean ± SEM t test. *p < 0.05, **p < 0.01, and ***p < 0.0001. See also Figure S1.
Elevated mTOR Signaling Increases Microglia Proliferation and Expression of Lysosome Genes
Microglia reside almost uniformly throughout the brain, with some regional differences. We observed that microglia proliferated extensively throughout the entire brain of TSC1CX3CR1CKO mice, with a 3- to 6-fold increase in the cortex and hippocampus (Figures 2A, 2B, and S2A). Moreover, they also lost the uniform distribution pattern, instead forming patches in both the cortex and hippocampus. These data suggest that elevated mTOR signaling promotes the proliferation of microglia. We also performed FACS analysis of monocytes in control and TSC1CX3CR1CKO mouse brains. Monocytes were gated as CD45+/CD11b+/Cx3cr1+/Ly6C+ or CD45+/CD11b+/Cx3cr1+/ CCR2+ (Figures S2B and S2C). We found ∼1.7% of Cx3cr1-positive cells to be CD45+/CD11b+/Cx3cr1+/Ly6C+ and less than 1% to be CD45+/CD11b+/Cx3cr1+/CCR2+ in both control and TSC1CX3CR1CKO mouse brains. Thus, there is no significant difference in either cell population between control and TSC1CX3CR1CKO mice.
Figure 2. Microglia Hyper-proliferation and Induction of Lysosomal Genes in TSC1Cx3cr1CKO Mouse Brains.
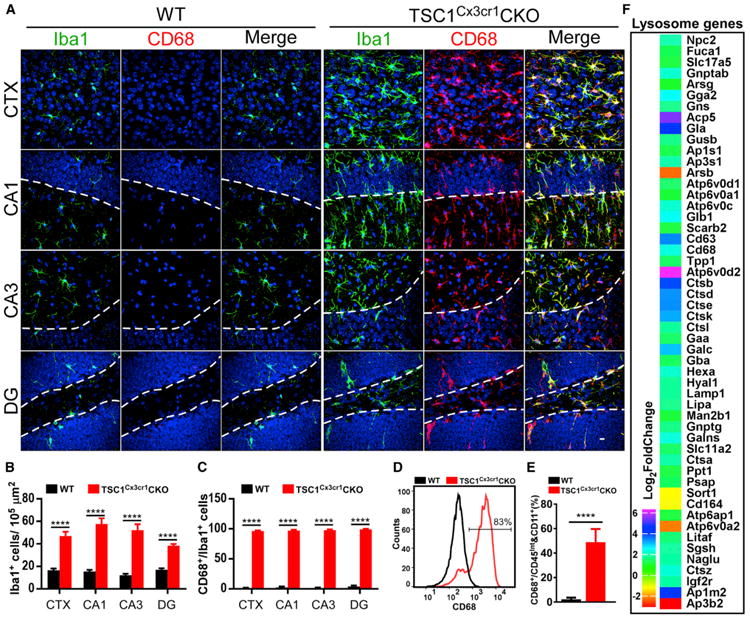
(A) Confocal images acquired from CTX, CA1, CA3, and DG of WT and TSC1Cx3cr1CKO mouse brains. Scale bar, 10 μm.
(B and C) Quantification of Iba1-positive (B) and Iba1/CD68 double-positive (C) microglia in WT (n = 6; 3 males and 3 females) and TSC1Cx3cr1CKO (n = 7; 3 males and 4 females) mice.
(D) FACS analysis of CD68 expression in microglia prepared from WT and TSC1Cx3cr1CKO mouse brains.
(E) Quantification of the CD68-positive cell population in WT (n = 5; 2 males and 3 females) and TSC1Cx3cr1CKO (n = 5; 2 males and 3 females) mice. Data are presented as mean ± SEM (t test). ****p < 0.0001. See also Figure S2.
(F) Analysis of RNA-seq data using the iPathwayguide program revealed altered expression of lysosomal genes in TSC1Cx3cr1CKO microglia (log2 fold change compared to control). Data were from four sets of microglial preparations. p value (padj) < 0.05.
CD68 is a lysosomal protein that is upregulated in microglia in the brains of individuals with epilepsy and in animal models (Boer et al., 2006; Sosunov et al., 2012; van Vliet et al., 2012). It is frequently used as a marker for reactive microglia. We observed marked elevation of CD68 expression in morphologically altered microglia throughout the entire brain (Figures 2A, 2C, and S2A). FACS analysis revealed a CD68-positive population of microglia (CD45low&Int /CD11b+/Cx3cr1+/CD68+) in TSC1CX3CR1CKO mice (Figures 2D and 2E). We used iPathwayGuider (Advaita) to perform RNA-sequencing (RNA-seq) analysis of lysosome genes in microglia purified from control and TSC1Cx3cr1CKO mouse brains (Figures S5B–S5E). We found that the expression of 54 genes was significantly altered in TSC1CX3CR1CKO microglia (Figure 2F). Notably, most genes (49 of 54) were upregulated, whereas only 5 genes were downregulated. This suggests that TSC1CX3CR1CKO microglia undergo a functional adaptation, resembling the increased lysosomal biogenesis observed in activated microglia (Tanaka et al., 2013).
Elevated mTOR Signaling Promotes Phagocytosis
Microglia exhibit phagocytotic activity under both quiescent and reactive states, which is critical in synapse pruning and removal of dead neurons (Nimmerjahn et al., 2005; Schafer et al., 2012; Sierra et al., 2010). Accordingly, we evaluated phagocytotic activity in the brains of TSC1Cx3cr1CKO mice. We employed caspase-3 staining to detect early dying neurons in TSC1CX3CR1CKO brain. We observed increased numbers of caspase-3-positive neurons in the cortex and hippocampus of TSC1CX3CR1CKO mice compared to wild-type mouse brain (Figures 3A, S3A, and S3B). Approximately 50%of the caspase-3 positive neurons in the hippocampus showed direct contact with microglial cells, forming a characteristic phagocytotic cup (Abiega et al., 2016; Sierra et al., 2010), whereas such contacts and cups were much less abundant in wild-type mouse brain (Figures 3B and 3E). These data strongly indicate that microglia with elevated mTOR signaling can effectively phagocytose dying neurons in mouse brain.
Figure 3. TSC1Cx3cr1CKO Microglia Display Increased Phagocytosis Activity.
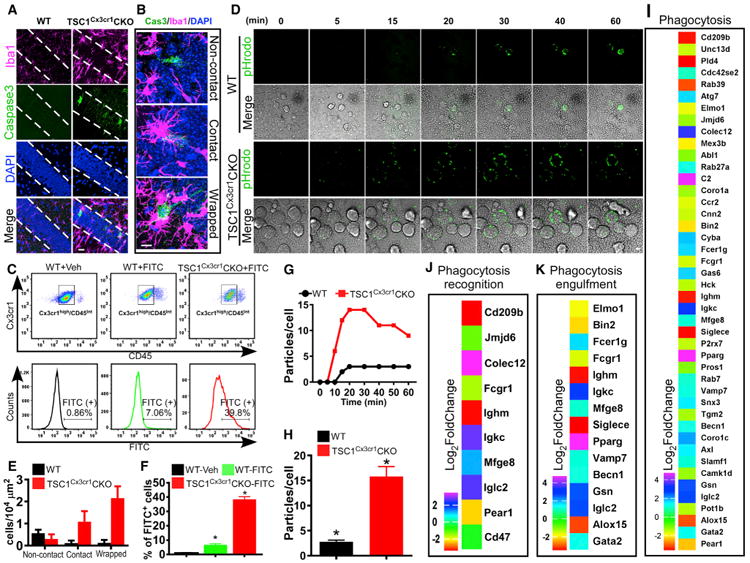
(A) Dying neurons in hippocampal CA1 in TSC1Cx3cr1CKO mice were identified by anti-activated caspase-3 antibody. Scale bar, 20 μm.
(B) Representative high-magnification images showing that dying (activated caspase-3 positive) neurons were isolated, had brief contact with a microglial cell, or were enwrapped by a microglia. Scale bar, 10 μm.
(C) FACS analysis of in vivo phagocytosis of FITC-labeled zymosan particles. The microglial population was identified as CD11b+/Cx3Ce1high/CD45 low/Int cells.
(D) Representative images from in vitro live-imaging of microglia phagocytosis of pHrodo Green zymosan bioparticles. Scale bar, 10 μm.
(E) Quantification of activated caspase-3-positive neurons in CA1 that fall within one of three categories: non-contact, contact, and wrapped. Data were averaged from WT (n = 6; 3 males, 3 females) and TSC1Cx3cr1CKO mice (n = 7; 3 males and 4 females).
(F) Quantification of FITC-positive microglia in the groups of WT- Vehicle (n = 3; 1 male and 2 females), WT- FITC (n = 3; 2 males and 1 female), and TSC1Cx3cr1CKO-FITC (n = 3; 1 male and 2 females) mice.
(G) Representative time course showing the number of phagocytic bioparticles over time.
(H) Average particles per microglia at the 30-min time point (n = 4–6). Data are presented as mean ± SEM (t test). *p < 0.05. See also Figure S3.
(I–K) Analysis of RNA-seq data revealed changes in expression of genes implicated in phagocytosis (I), phagocytosis recognition (J), and phagocytosis engulfment (K) in TSC1Cx3cr1CKO microglia (log2-fold change compared to control). p values (padj) < 0.05.
To better evaluate microglial phagocytosis, we performed an in vivo phagocytosis assay using zymosan bioparticles (Preissler et al., 2015). We injected fluorescein isothiocyanate (FITC)-labeled zymosan particles or vehicle into the hippocampus. Microglia were isolated from the brains of control and TSC1CX3CR1CKO mice 16 hr after injection (Figures 3C, 3F, and S3C–S3F). Microglia were identified as a population of Cx3cr1+/CD11b+/CD45low&Int by FACS analysis (Figures 3C, S3C, and S3D). We found a significant elevation of phagocytosis of FITC-labeled zymosan particles by microglia in brains of TSC1CX3CR1CKO mice as compared to control brains.
We next evaluated phagocytosis in cultured microglia by using pHrodo-labeled zymosan particles, which are conjugated to the pH-sensitive indicator pHrodo green (Figures 3D, 3G, and 3H). This indicator maintains a non-fluorescent state at neutral pH but becomes bright green under acidic conditions (i.e., in phagosomes). Real-time imaging of microglia phagocytosis in vitro revealed that the microglia prepared from TSC1CX3CR1CKO mice began to show green fluorescent punctae within 5 min, whereas it took 10–15 min for control microglia to show zymosan particle uptake (Figures 3D and 3G). Moreover, TSC1CX3CR1CKO microglia displayed a more robust uptake of pHrodo-labeled zymosan particles, 2- to 3-fold, compared to wild-type microglia (Figures 3H and S3G). Together, our data from both in vitro and in vivo studies suggest that elevated mTOR signaling increases phagocytosis by microglia. Analysis of RNA-seq data revealed significant changes in expression of a large set of genes directly or indirectly involved in phagocytosis (Figures 3I–3K). Notably, Colec12, C2, Pparg, Igkc, Iglc2, and Gsn were markedly upregulated, whereas CD209, Ighm, Siglece, Pld4, Rab39, and Alox5 were greatly downregulated. Changes in expression of genes in phagocytosis pathways are consistent with altered phagocytosis activity observed in TSC1CX3CR1CKO microglia.
Elevated mTOR Signaling Suppresses the Microglial Inflammatory Response
Microglia are considered as resident macrophages, mediating the innate immune response and producing pro-inflammatory cytokines/mediators when encountering an injury signal. Accordingly, we evaluated pro-inflammatory cytokine/mediator profiles in TSC1Cx3cr1CKO mouse brains. We observed a moderate induction of tumor necrosis α (TNF-α), interleukin 1β (IL-1β), interferon β (IFN-β), and iNOS in TSC1Cx3cr1CKO mouse brains (Figure 4A). However, this induction predominately occurred in the hippocampus, with little induction in the cortex (Figure 4A). Hippocampal-specific induction of cytokines is intriguing because it suggests that the elevated levels of cytokines/mediators observed in TSC1Cx3cr1CKO mouse brain could be from cells other than microglia, i.e., neurons and astrocytes in the hippocampus. Accordingly, we performed transcriptome analysis of cytokines, chemokines, and other inflammatory mediators (Figures 4D–4H). Microglia in control mouse brains express moderate levels of IL-1a, IL-16, Ccl3, Ccl4, Ccl6, Cxcl16, Tgfa, Tgfb1, Hmgb1, Cox1, and Mmp2, and low but detectable levels of IL-15, IL-18, IL-1b, Ccl2, Ccl9, Ccl12, Tnf, Cox4l1, Cox6a1, Cox6b1, Cox6c, Cox8a, Mmp8, Mmp9, Mmp12, and Mmp14. In TSC1 Cx3cr1CKO microglia, the expression of these genes, in general, was either downregulated or very moderate increased; only Cxcl16 was greatly induced. Cxcl16 is a transmembrane chemokine that regulates interaction among neurons, microglia, and astrocytes (Rosito et al., 2014). There was no significant induction of genes that are known to be highly expressed in activated microglia, including IL-1a, IL-1b, IL-6, Tgfb1, Nos 2 (iNOS), Hmgb1, Cox2, Mmp2, and Mmp8. There was only a moderate increase in TNF-α expression (by 0.7 log2-fold change). However, real-time qPCR analysis of key pro-inflammatory cytokines or mediators using the same RNA preparations confirmed that there was no significant induction of pro-inflammatory cytokines (Figure 4B). The levels of IL-1β, iNOS, and IFN-β were moderately reduced in microglia of TSC1 Cx3cr1CKO mice when compared with microglia from control mice (Figure 4B). These data suggest that microglia are not the main sources of TNF-α, IL-1β, IFN-β, and iNOS in TSC1CX3CR1CKO mouse brains.
Figure 4. Impact of Deletion of TSC1 in Microglia on Production of Cytokines, Chemokines, and Other Inflammatory Mediators.
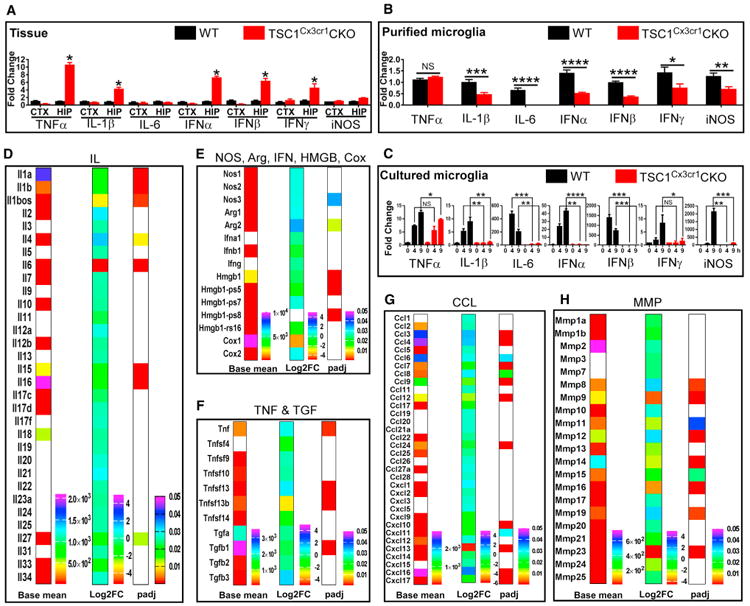
(A) qRT-PCR analysis of cytokine expression in cortical and hippocampal tissues of WT (n = 3; 2 males and 1 female) and TSC1Cx3cr1CKO (n = 3; 2 males and 1 female) mice.
(B) Cytokine expression in purified microglia from control (n = 3; 2 males and 1 female) and TSC1Cx3cr1CKO (n = 3; 2 males and 1 female) mice (n = 3). See also Figures S5B–S5E.
(C) Cytokine expression in cultured microglia prepared from control and TSC1Cx3cr1CKO mice (n = 4-5). Data are presented as mean ± SEM (t test). NS, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
(D–H) Heatmap plot of the expression of IL (D), NOS, Arg, IFN, HMGB, and Cox (E), TNF and TGF (F), CCL (G), and MMP (H). Plot showing basal levels of gene expression in control microglia (Basemean), fold changes (log2FC) in TSC1Cx3cr1CKO microglia, and p values (padj) to evaluate the significance of the changes. Non-color-filled spaces indicate expression was either undetectable or padj value > 0.05. See also Figure S4.
To further clarify the role of mTOR signaling in the microglial inflammatory response, we treated cultured microglia with DNA fragments, which models the innate immune response when microglia phagocytose DNAs from microorganisms or dying cells (Matsuda et al., 2015). DNA treatment stimulated a robust pro-inflammatory response in control microglia, as reflected by robust induction of TNF-α, IL-1β, IFN-β, and iNOS; in contrast, the response of microglia prepared from TSC1Cx3cr1CKO mice was markedly diminished, except in the case of TNF-α induction, for which there was a moderate reduction (Figure 4C). Thus, the in vitro data are generally consistent with the data obtained for microglia purified from TSC1Cx3cr1CKO mouse brain.
We also analyzed the receptors involved in inflammatory/innate immune responses (Figures S4A–S4F). Microglia express very high levels of IL-10ra, Ccr5, Tlr7, Tgfbr1, Ifngr1, Trem2, and Cx3cr1 and moderate or detectable levels of IL-10rb, IL-4ra, IL-6ra, IL-13ra1, IL-17ra, IL-21ra, Ccr1, Ccrl2, Tlr2, Tlr4, Tlr13, Tgfbr2, Ifngr1, Tnfrsf1b, Tnfrsf21, and Ifngr2. Several receptors were greatly upregulated in TSC1Cx3cr1CKO microglia, including Il2rg, Il3ra, IL-12rb1, Il12rb2, Cxcr1, Cxcr4, Tlr8, and Tnfrsf10b, while others were markedly downregulated, including Il7r1, Ccr5, Tlr5, Tlr9, TREM2, and Cx3cr1. However, TSC1Cx3cr1CKO microglia remain to express a significant amount of Cx3cr1 proteins because they can be effectively labeled and pulled down by anti-Cx3cr1 antibodies for FACS analysis of microglial population and transcriptional analysis (Figures S2B and S5B–S5E). The exact role of altered expression of these receptors in the innate immune response of TSC1Cx3cr1CKO microglia is unclear. Notably, Tlr9 is known to mediate the DNA-elicited innate immune response in microglia (Matsuda et al., 2015). Downregulation of Tlr9 may explain, in part, the reduced innate immune response in TSC1Cx3cr1CKO microglia. Together, these findings suggest that elevated mTOR signaling in microglia suppresses their pro-inflammatory response.
Elevated mTOR Signaling in Microglia Causes Significant Neuroanatomical Changes
We next set out to examine possible any phenotypic changes other than those involving microglia in the TSC1Cx3cr1CKO mouse brain. We found neither apparent gross abnormalities, including brain size, nor any significant change in overall neuronal density in the cortex and hippocampus (data not shown). In a normal brain, neurons in the hippocampal pyramidal layer and the dentate granules are densely packed, with the soma displaying very minimal direct contact with microglia. However, in TSC1Cx3cr1CKO mice, we observed significant microglial infiltration into hippocampal pyramidal and granule layers (Figures 5A, 5B, and S5A). The mean number of microglia in the hippocampal pyramidal and granule layers was 4–6/μm2 in TSC1Cx3cr1CKO mice versus 2–2.5/μm2 in control mice.
Figure 5. Infiltration of Microglia into Hippocampal Pyramidal and Granular Layers, Reduction of Synapse Density, and Proliferation of Astrocytes in TSC1Cx3cr1CKO Mice.
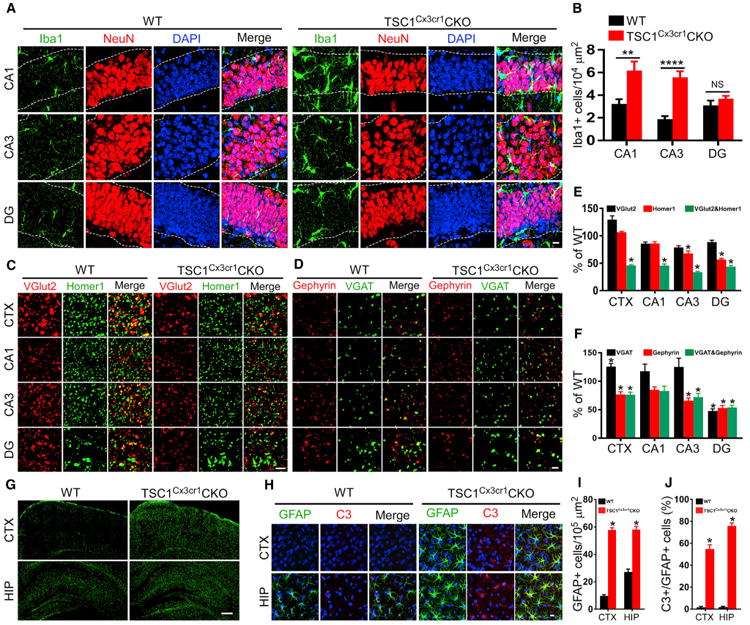
(A) Representative high-magnification images showing infiltration by microglia CA1, CA3, and DG. See also Figure S5A. Scale bar, 10 μm.
(B) Quantification of microglia in CA1, CA3, and DG of control (n = 6; 3 males and 3 females) and TSC1Cx3cr1CKO (n = 7; 3 males and 4 females) mice. Data are presented as mean ± SEM t test. NS, not significant); *p < 0.05, **p < 0.01, and ****p < 0.0001, respectively.
(C and D) Representative super-resolution images of co-immunostaining of vGlut2 (red)/Homer1 (green) (C) and vGAT/Gephyrin (D) in the M1 motor cortex around layer IV (CTX), the hippocampal (HIP) radiatum layer adjacent to pyramidal CA1 (CA1), the stratum lucidum adjacent to CA3 (CA3), and the dentate gyrus (DG) of WT and TSC1Cx3cr1CKO mice. Synapses were identified as yellow dots, reflecting co-localization of the pre-synaptic markers vGlut2 and vGAT with the post-synaptic markers Homer1 and Gephyrin. Scale bar, 2 μm.
(E and F) Quantification of excitatory synapse density in TSC1Cx3cr1CKO mice (n = 7; 3 males and 4 females) presented as percentage of WT (n = 6; 3 males and 3 females) (E) and inhibitory synapse density in TSC1Cx3cr1CKO (n = 5; 3 males and 2 females) and WT (n = 5; 3 males and 2 females) mice (t test) (F). *p < 0.05.
(G) Representative images covering the entire HIP and partial CTX showing a moderate increase of GFAP-positive astrocytes in the HIP and a robust increase in the CTX of TSC1Cx3cr1CKO mice. Scale bar, 200 μm.
(H) Representative high-magnification images of triple staining of Iba1, C3 and DAPI in the HIP and CTX of WT and TSC1Cx3cr1CKO mice. Scale bar, 10 μm.
(I) Quantification of GFAP-positive cells in the HIP and CTX of WT (n = 6; 3 males and 3 females) and TSC1Cx3cr1CKO (n = 7; 3 males and 4 females) mice (t test). ****p < 0.0001.
(J) Quantification of C3 and GFAP double-positive cells in the HIP and CTX of WT (n=6;3 males and 3 females) and TSC1Cx3cr1CKO (n= 7;3 males and 4 females) mice (Mann-Whitney test). *p < 0.05.
Excitatory synapses in the cortex and hippocampus were identified by co-localization of vGlut2 and homer, which are pre- and post-synaptic markers, respectively. We observed a significant reduction in the number of co-localized homer1 and vGlut2 punctae in the cortex and hippocampus of TSC1Cx3cr1CKO mice compared to wild-type mice (Figures 5C and 5E). Inhibitory synapses were also examined by labeling pre- and post-inhibitory synaptic sites with anti-vGAT and antigephyrin, respectively (Figures 5D and 5F). We observed a significant reduction in the density of inhibitory synapses in the hippocampal dentate gyrus and a moderate reduction in the cortex and hippocampal CA3 region in TSC1Cx3cr1CKO mice compared to wild-type mice. Notably, the density of the pre-synaptic marker vGAT was moderately elevated, whereas that of the post-synaptic marker gephyrin was reduced. This may account for the observed reduction of inhibitory synapses.
Astrocytes become reactive and hyperproliferate when they encounter activated microglia (Liddelow et al., 2017). We asked whether astrocytes also exhibit any phenotypic changes. GFAP is a conventional marker for a subset of astrocytes that is strongly expressed in the hippocampus and moderately expressed in the cortex. Notably, GFAP expression is elevated in reactive astrocytes. Accordingly, we performed staining of GFAP to examine this particular subset of astrocytes. In control mouse brain, we observed GFAP-positive astrocytes predominately in the hippocampus, with weakly stained astrocytes in the cortex. In contrast, we saw a marked increase of GFAP-positive astrocytes throughout the entire brain of TSC1Cx3cr1CKO mice (Figures 5G–5I). Moreover, these astrocytes stained positive for complement 3 (C3) (Figures 5H and 5J), which was reported to be present in reactive astrocytes (Liddelow et al., 2017). qPCR revealed that transcripts reported to be preferentially present in reactive astrocytes were increased in TSC1Cx3cr1 CKO mice (Figures S5F–S5I). These data suggest that the astrocytes adopt a reactive-like phenotype.
Compliments has been implicated in synapse pruning (Schafer et al., 2012) and were reported to be upregulated in a status epilepticus model (Schartz et al., 2018). Accordingly, we analyzed RNA-seq data to evaluate the expression profile of compliments and their receptors in microglia. Microglia express high levels of C1q members and a moderate level of C3ar1 (Figure S4F). There was a 1–1.2 log2-fold increase of C1qa and C1qc and 1.8 log2-fold increase of C3ar1 in TSC1Cx3cr1CKO microglia. Cd200R, Megf10, and Mertk have been reported to regulate phagocytosis (Hong et al., 2016). We observed a marked induction of Cd220r2 and Cd220r4 by 5–6 log2-fold in TSC1Cx3cr1 CKO microglia.
Elevated mTOR Signaling in Microglia Is Sufficient to Promote the Development of SRSs
We evaluated whether homeostatic changes have a role in epileptogenesis. We found that TSC1Cx3cr1CKO mice begin to display early-onset limbic and convulsive seizures, and even die, as early as a few days after weaning (∼3.5–4 weeks old). Nearly 100% of the mice developed spontaneous recurrent seizures (SRSs) by 5 weeks of age (Figure 6A). TSC1Cx3cr1CKO mice also exhibited a steep drop in survival over a very narrow period (∼10 days) (Figure 6A). None of the animals survived longer than 5 weeks. Moreover, we noted a clear positive correlation between the onset of seizures and mortality. Video recording confirmed that all deaths were immediately preceded by severe seizures, suggesting that seizures were the primary cause of death.
Figure 6. Development of Spontaneous Recurrent Seizures in TSC1Cx3cr1CKO and TSC1Cx3cr1-ERT2CKO Mice.
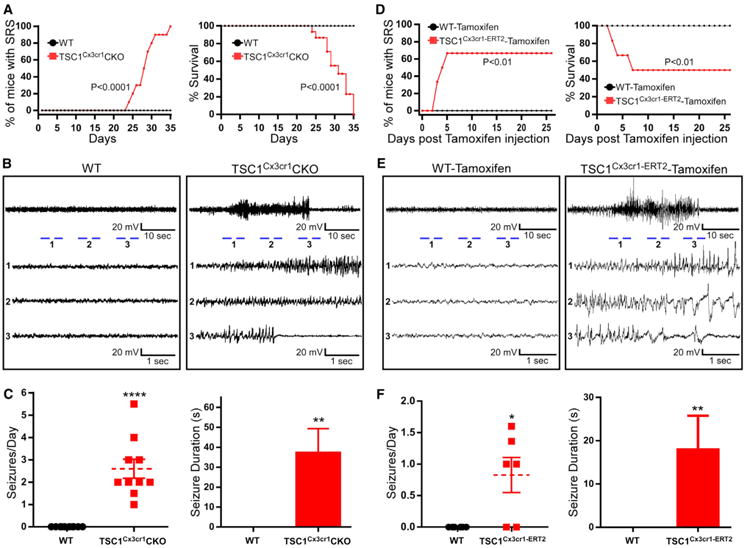
(A) Percentage of mice displaying SRS and mortality in WT (n = 10, 5 males and 5 females) and TSC1Cx3cr1CKO mice (n = 10; 5 males and 5 females). Log-rank (Mantel-Cox) test. p < 0.0001.
(B) Representative electrographic traces of SRS in WT and TSC1Cx3cr1CKO mice.
(C) Average frequency and duration of seizures in WT (n = 10; 5 males, 5 females) and TSC1Cx3cr1CKO (n = 10; 5 males, 5 females) mice. Data are presented as mean ± SEM. Mann-Whitney test. ** and ****p < 0.01 and 0.001, respectively.
(D) Percentage of mice displaying SRS and mortality up to 26 days post tamoxifen treatment in WT (n = 6; 3 males, 3 females) and TSC1Cx3cr1-ERT2 mice (n = 6; 3 males, 3 females). Log-rank (Mantel-Cox) test. p < 0.01.
(E) Representative electrographic traces of SRS recorded from tamoxifen-treated WT and TSC1Cx3cr1-ERT2 mice.
(F) Average seizure frequency and duration of seizures in tamoxifen-treated WT (n=6; 3 males,3 females) and TSC1Cx3cr1-ERT2 (n=6; 3 males and 3 females) mice. Data are presented as mean ± SEM. **p < 0.01 and ****p <0.001 (Mann-Whitney test). See also Figure S6.
To verify that the behavioral seizures observed in TSC1Cx3cr1CKO mice were accompanied by electrographic abnormalities, we performed video/epidural electroencephalogram (EEG) recording on control and TSC1Cx3cr1CKO mice (McMahon et al., 2012). The behavioral seizures were accompanied by stereotypical electrographic seizures, beginning with progressive changes toward higher frequencies and amplitudes, and followed by prolonged postictal suppression of amplitude, a hallmark of electrographic seizures. None of the control animals evidenced any seizures (Figures 6B and 6C). The SRSs in TSC1Cx3cr1CKO mice were quite severe, with frequencies up to 6 episodes per day and in some cases lasting up to 1.5 min (Figure 6C).
Microglia express Cx3cr1 during brain development. Deletion of TSC1 in microglia could impact brain development, resulting in epilepsy. We examined whether elevated mTOR signaling in microglia of the adult brain is still epileptogenic. Inducible TSC1Cx3cr1-CreERT2 mice were generated by crossing Cx3CR1-CreERT2 mice (Parkhurst et al., 2013) with TSC1flox/flox mice (Kwiatkowski et al., 2002). TSC1Cx3cr1-CreERT2 and control mice at 8-10 weeks of age were treated with either vehicle or tamoxifen for 5 days. Mouse brains were harvested 9 days post-tamoxifen treatment. We found a significant elevation of pS6 in microglia of TSC1Cx3cr1-CreERT2 mouse brains, but very few in control animals (Figures S6A and S6C). Moreover, we observed a marked increase in the number of microglia (Figures S6A and S6D), with altered morphology (Figure S6E) and strong induction of CD68 (Figures S6B and S6F) in tamoxifen-treated TSC1Cx3cr1-CreErt mice, which recapitulates what we observed in TSC1Cx3cr1-Cre mice. More importantly, the tamoxifen-treated TSC1Cx3cr1-CreERT2 mice developed SRS as early as three days post cession of tamoxifen treatment. Over the 26 days of continuous video/EEG recording, four of six animals developed SRS (Figures 6D–6F), with an average of 1 to 1.5 seizures per day and an average duration of 20 s (Figure 6F). Three of these mice died from seizures (Figure 6D). None of the tamoxifen-treated control animals developed seizures. Thus, activation of mTOR signaling in microglia of the adult mouse brain is sufficient to drive epileptogenesis.
Discussion
Here, we demonstrated that when mTOR signaling is elevated, microglia adopt a noninflammatory reactive phenotype, including an amoeboid-like morphology, hyperproliferation, robust phagocytosis activity, and expression of lysosome genes, but without significant induction of pro-inflammatory cytokines. Moreover, phenotypic changes of the microglia led to pronounced neuroanatomical alterations, including significant infiltration by microglia into hippocampal pyramidal and dentate granule layers, reduction in synapse density, moderate neurodegeneration, and hyperproliferation of astrocytes. Importantly, the mice developed severe spontaneous seizures. Together, our studies provide compelling evidence supporting a critical role of microglia in CNS homeostasis and revealing an epileptogenic mechanism that is independent of the microglial inflammatory response.
mTOR Signaling Regulates Microglial Morphology
Morphologically, microglia are extensively ramified while in a “resting” state, which facilitates minimal but essential contact simultaneously with multiple neurons and non-neuronal cells for surveillance in the CNS, including synaptic pruning, removal of dead cells, and recruitment of astrocytes. We found that microglia in TSC1Cx3cr1CKO mice displayed striking morphological changes correlating with marked neuroanatomical changes. This supports the notion that maintaining a ramified morphology of microglia could be critical for CNS homeostasis. Mechanistically, it remains to be determined how mTOR signaling regulates microglial morphology. Because mTOR signaling in general promotes cell growth and cell enlargement (Laplante and Sabatini, 2012), this could explain, in part, the increased cell size and thickness of the microglia processes in the brains of TSC1Cx3cr1CKO mice. Some previous studies revealed that GPR-34 deficiency increases the size of the soma and reduces branch length in microglia (Preissler et al., 2015). The small rho GTPase Cdc42 was reported to be involved in LysoPS-induced ramification of microglia (Tokizane et al., 2017). Deletion of TSC2 was found to inhibit Cdc42 activity (Larson et al., 2010). It is conceivable that TSC1 deletion alters microglial morphology via Cdc42.
Elevated mTOR Signaling Promotes the Proliferation of Microglia
Maintaining an appropriate number of microglia is also vital for CNS homeostasis since changing the number alters the interactions of microglia with neurons and astrocytes. We observed that the number of microglia was significantly increased in TSC1Cx3cr1CKO mice at ∼4 weeks of age. Although we can't entirely rule out the possibility that TSC1 deletion may prolong the lifespan of microglia, the observed 2- to 3-fold increase in the number of microglia at 4 weeks is likely due to hyperproliferation because microglia have a long half-life (Askew et al., 2017). It is unclear how elevated mTOR signaling leads to microglial hyperproliferation in an otherwise unperturbed state. One possibility is that microglia become intrinsically hyperproliferative when mTOR signaling is elevated. Alternatively, their hyperproliferation could be extrinsically driven, reflecting a secondary response to some pathological event(s) elicited by the elevated microglial mTOR signaling (e.g., release of inflammation mediators, proliferation of astrocytes, or degeneration of neurons). We found that TSC1Cx3cr1CKO microglia cultured in vitro were hyperproliferative compared to wild-type microglia (data not show), supporting an intrinsic role of mTOR signaling in microglial proliferation.
mTOR Signaling Regulates Microglia Phagocytosis
Microglia are the main phagocytes in the CNS. Phagocytosis and degradation of synapses and cells are important for CNS homeostasis. Microglial phagocytosis activity is regulated in response to external triggers, including by glial cell line-derived neurotrophic factor (GDNF), Toll-like receptor (TLR) ligands, uridine diphosphate (UDP), and CD200 receptors, the expression of phagocytosis receptors such as MEGF10 and MERTK, and complement cascades (Hong et al., 2016). We observed moderate induction of C1q members and C3ar1, and a marked induction of Cd200r2 and Cd200r4 in TSC1Cx3cr1CKO microglia. This is consistent with the robust phagocytosis activity observed in TSC1Cx3cr1CKO microglia. In addition, we found that several other genes in the phagocytosis pathway were upregulated, including Colec12 (collectin subfamily member 12; also called scavenger receptor class A, member 4), Igkc (immunoglobulin kappa constant), Iglc2 (immunoglobulin lambda constant 2), C2 (complement c2), Pparg (peroxisome proliferator-activated receptor gamma, also called PPAR-γ), and Gsn (gelsolin). Their biological significance in phagocytosis needs to be determined. Notably, a recent study characterized the microglial phagocytic response in mouse brain following acute or chronic inflammatory challenges and kainic acid (KA) treatment, as well as in human epileptic brains (Abiega et al., 2016). The authors found that acute seizures elicited by KA trigger widespread release of ATP, which in turn impairs microglial phagocytosis. This also occurred in TLE and KA mouse model. This elegant study revealed a mechanism by which an external stimulus regulates microglia-mediated cleanup of apoptotic cells. In contrast, our study focused on microglial mTOR signaling, which is elevated in the epileptic brain. Of note, epileptogenic neurological insults likely trigger many events simultaneously. To better isolate the biological significance of the elevated mTOR signaling in microglia, we utilized a genetic approach to modify the mTOR activity in microglia. The elevated mTOR signaling serves as a primary attack on microglia. Our data demonstrated that elevated mTOR signaling in microglia enhances their capacity for phagocytosis. In our in vitro phagocytosis assay, the particles become visible in mature phagosomes and phagolysosomes, where the pH is low. A previous study revealed that phagosomes in M2 macrophages (less inflammatory form) tend to be acidified rapidly within 10 min, versus those in M1 macrophages (proinflammatory form), which are less amenable to acidification, take approximately 30 min (Canton et al., 2014). We observed that phagosome acidification was accelerated in the microglia prepared from TSC1Cx3cr1CKO mice, suggesting that TSC1Cx3cr1CKO microglia tend to adopt a less inflammatory phenotype (Canton et al., 2014). The accelerated accumulation of particles in phagolysosomes that we observed in TSC1Cx3cr1CKO mice appears to reflect increased phagocytosis rather than a delay in the degradation/clearance of particles. However, this needs to be clarified in future studies. We noticed that the cell sizes of microglia prepared from TSC1Cx3cr1CKO mice were significantly larger than wild-type. Thus, the increased number of phagocytic particles per cell may also reflect the increased capacity of phagocytosis.
Elevated mTOR Signaling Promotes a Reactive-like Morphology of Microglia without Significant Induction of Proinflammatory Cytokines
As innate immune cells in the brain, microglia are potent cytokine/chemokine producers. Microglia-mediated neuroinflammation has been postulated to be one of the plausible causes of epileptogenesis (Aronica et al., 2017). However, many other types of cells, including infiltrated monocytes, also produce pro-inflammatory cytokines/chemokines. It is therefore challenging to pinpoint the exact cellular sources of pro-inflammatory cytokines/chemokines in vivo and thus the exact contribution of microglia. In the TSC1Cx3cr1CKO mouse model, microglia are highly proliferated throughout the entire brain. Purified microglia from TSC1Cx3cr1CKO mice do not display any significant elevation in the expression of pro-inflammatory cytokines. Furthermore, deletion of TSC1 impairs the innate immune response in cultured microglia. Mechanistically, Tlr 9 is downregulated in TSC1Cx3cr1CKO microglia. This may represent an underlying mechanism of impaired innate immune response. TSC1 deletion promotes macrophages to adopt an inflammatory M1 phenotype (Zhu et al., 2014). However, we found that microglial deletion of TSC1 attenuates the pro-inflammatory response. This is a significant departure from what was previously reported in macrophages, which may reflect differences in the signaling cascades involved in microglia and macrophages or the differences in the stimulation (lipopolysaccharide [LPS] versus DNA) employed. Of note, although M1/M2 polarization has been extensively described in macrophages, microglia do not undergo distinct M1/M2 polarization (Ransohoff, 2016). Nevertheless, we found that the elevated mTOR signaling promotes a reactive-like morphology in microglia, without significant induction of proinflammatory cytokines.
Induction of Lysosome Genes in TSC1Cx3cr1CKO Microglia
Quiescent microglia express a nearly undetectable level of lysosomal protein CD68 in an unperturbed brain. The induction of CD68 appears to be persistent for many months after initial neurological insults (van Vliet et al., 2012). We observed strong induction of CD68 in the microglia of TSC1Cx3cr1CKO mice. Thus, the discordance between the induction of CD68, but not of inflammatory cytokines, indicates that microglia with induced CD68 are not necessarily inflammatory. CD68-positive microglia are frequently observed in brains exhibiting chronic spontaneous seizures (Boer et al., 2006; van Vliet et al., 2012). These cells are generally postulated to be pro-inflammatory and the source of epileptogenic pro-inflammatory cytokines and mediators. Future study will determine whether the CD68-positive microglia observed in epileptic brains are true “activated” or “pro-inflammatory” microglia with robust production of inflammatory cytokines. Apart from CD68, many lysosome genes were upregulated, reflecting lysosomal biogenesis. Increased lysosomal biogenesis has been reported in activated microglia (Tanaka et al., 2013). Moreover, the mTOR/autophagy pathway is closely linked to lysosomal biogenesis (Settembre et al., 2011). Given that TSC1 deficit alters mTOR as well as autophagy activity (Laplante and Sabatini, 2012), the induction of lysosomal genes such as CD68 could be a direct consequence of altered mTOR signaling. Notably, a recent study revealed that the Rag-Ragulator complex regulates microglial development and lysosomal activity in zebrafish (Shen et al., 2016). Inactivation of this complex reduces the number of microglia, along with accumulation of lysosomal organelles. This appears to be independent of mTOR activity despite that the Rag-Ragulator complex constitutes an important part of the mTOR pathway. Here, we found that lysosome genes were markedly upregulated in TSC1Cx3cr1CKO microglia, further extending the above findings.
mTOR Signaling in Microglia Regulates the Proliferation of Astrocytes
Apart from microglia, astrocytes too are resident glial cells that play various roles in the brain, including uptake of neuro-transmitters, maintenance of ion homeostasis, and modulation of neuronal functions. Microglia and astrocytes mutually shape each other's behavior, both in the unperturbed healthy brain and under pathological conditions. Pro-inflammatory cytokines and mediators such as TNF-α, IL-1β, and nitric oxide (NO) have been implicated in the phenotypic change of astrocytes (Liddelow et al., 2017). We saw only a moderate induction of TNFα, IL-1β and NO in the hippocampus and no significant induction in the cortex, where GFAP-positive astrocytes were highly proliferated. We saw little induction of cytokines in purified microglia prepared from TSC1Cx3cr1CKO mice. Our data strongly suggest that noninflammatory phenotypic changes of microglia in the TSC1Cx3cr1CKO mouse brain may promote proliferation of astrocytes.
Infiltration of Microglia into the Pyramidal and Granular Layers of the Hippocampus and Neuronal Degeneration
Despite seeing increased caspase-3-positive neurons and engulfed cups in the TSC1Cx3cr1CKO mouse brain, we did not see a significant reduction in the number of neurons in the cortex and the hippocampus, suggesting that the neuronal degeneration is moderate. Reactive microglia appear to possess neuroprotective as well as neurotoxic functions, depending on the nature of the pathological conditions (Colonna and Butovsky, 2017). Thus, reactive-like microglia observed in the TSC1Cx3cr1CKO mouse brain may not be the direct cause of neuronal degeneration. A recent study reported that the A1-type of astrocytes possesses neuron-killing properties (Liddelow et al., 2017). Future studies will determine whether astrocytes adopt a neuron-killing phenotype in TSC1Cx3cr1CKO mouse brain. Typically, neurons are densely packed within the hippocampal pyramidal and granular layers, forming a tightly assembled band of somata. Microglia usually reside away from the pyramidal and granular layers. We observed a marked increase of microglial infiltration into both the pyramidal and granular layers. However, it remains to be determined whether the intrinsic hyperproliferation of microglia accounts for the increased number of microglia in the cell-compacted pyramidal and granular layers. Alternatively, neuronal death could trigger infiltration by microglia into these hippocampal layers.
mTOR Signaling in Microglia in the Regulation of Synaptic Density
Excitatory and, to some extent, inhibitory synapses are reduced in the TSC1Cx3cr1CKO mouse brain. There are several mechanisms that could account for the reduction of synapse density. First, microglia are known to be involved in synapse pruning (Nimmerjahn et al., 2005). Therefore, the increased number of microglia and their phagocytosis activity could account for the reduction in synapse density. In addition, astrocytes also possess potent synapse engulfment properties when they are exposed to certain secreted factors. Astrocytes in the TSC1Cx3cr1CKO brain were hyperproliferative, which may contribute to the reduced synapse density. Microglia prune both excitatory and inhibitory synapses. Synapse pruning has been reported to be dependent on the level of neuronal activity (Schafer et al., 2012). In the present study, we found that there was a significant reduction of excitatory synapse density in the cortex and hippocampus of TSC1Cx3cr1CKO mice. There was also a relatively moderate reduction of inhibitory synapses in the cortex and hippocampal CA3 and no significant reduction in hippocampal CA1. The reduced level of the post-synaptic marker gephyrin appears to account for the reduction of inhibitory synapses.
Mechanisms of Epileptogenesis in TSC1Cx3cr1CKO Mice
Although microglia have long been suspected to be involved in epileptogenesis, this is largely based on the assumption that neuronal injury leads to microglial activation and brain inflammation, which in turn causes epilepsy. In other animal models of epileptogenesis, initial neurological insults such as status epilepticus (SE) induced by KA or pilocarpine cause massive neuronal damage. This damage is primarily due to excitotoxicity. However, a direct role of microglia in neurodegeneration and development of SRSs has not been convincingly demonstrated, since microglia activation represents only one part of many possible pathological changes in these models. In the present study, by largely bypassing all other pathological changes elicited by SE, we clearly demonstrated that elevated mTOR signaling in microglia is not only moderately neurodegenerative but also epileptogenic. Nearly all TSC1Cx3cr1CKO mice in our study developed spontaneous seizures by 5 weeks of age. The profound epileptogenic effect of microglial deletion of TSC1 can be independent of brain development, because elevating mTOR signaling in microglia of the adult brain is sufficient to drive reactive-like morphological alterations and trigger spontaneous seizures. Thus, elevated mTOR signaling in microglia could be an epileptogenic mechanism applicable to all stages of life, from the immature to the adult brain.
Our study, however, paints a more complicated picture in terms of how microglial deletion of TSC1 becomes so epileptogenic. First, although pro-inflammatory cytokines are known to be seizure-promoting, in TSC1Cx3cr1CKO mice, we observed only moderate increases in the levels of pro-inflammatory cytokines. These were predominately in the hippocampus and apparently did not originate directly from microglia. Our data do not support a pro-inflammatory role of microglia in epileptogenesis in TSC1Cx3cr1CKO mice. Rather, noninflammatory phenotypic changes in microglia following elevated mTOR signaling are the primary driver that disturbs brain homeostasis and lead to the development of spontaneous seizures. Second, we found that astrocytes hyper-proliferate in TSC1Cx3cr1CKO mice. Given that astrocytes can uptake/release glutamine to modulate neuronal excitability, it is plausible that hyperproliferation of reactive astrocytes, secondary to altered microglia, could contribute to epileptogenesis (Zhang et al., 2016). Moreover, neuronal death occurs in the pyramidal and granular layers of the hippocampus. Microglia and astrocytes infiltrate into these layers to form microinflammatory patches. It is reasonable to postulate that the infiltration by microglia/astrocytes and degeneration of neurons may create comparable pathologic changes echoing those seen in epileptic brains, which could in turn alter neuronal excitability. Finally, we cannot rule out the possibility that reduced synapse density may also play a role in epileptogenesis.
TSC1 deletion either in astrocytes or in neurons, or broadly in early embryonic progenitor cells, leads to severe SRSs with significant mortality (McMahon et al., 2012; Zhang et al., 2016). In the present study, we found that TSC1 deletion in microglia is also epileptogenic. The seizure severity and mortality we observed are at least comparable to, if not greater than, those seen in astrocytic and neuronal TSC1 mouse models (McMahon et al., 2012; Zhang et al., 2016). Thus, our study extends our understanding of the mTOR signaling in epileptogenesis into a completely uncharted territory. Notably, some focal epilepsies have been linked to somatic mutations associated with hyperactivation of the mTOR signaling pathway (Jansen et al., 2015). Future studies will be needed to determine if there is somatic mutation of microglial TSC1 in those disorders.
In summary, we found that elevated mTOR signaling can drive microglia to adopt a noninflammatory reactive-like phenotype. This altered phenotype is associated with the loss of CNS homeostasis; i.e., reduced synapse density, proliferation of astrocytes, moderate neuronal degeneration, and development of SRSs. A microglial cell-mediated inflammatory response has long been the focus of a plausible epileptogenic mechanism. We have identified an epileptogenic role for microglia that is independent of an inflammatory response. Thus, our findings not only establish a clear role for microglia in epileptogenesis, they also point to a noninflammatory epileptogenic mechanism. In light of the observation of elevated mTOR signaling in microglia in epileptic brains, our study further cements the notion that microglia are a potential therapeutic target for epilepsy prevention.
Experimental Procedures
Animals
RosamTomato/meGFP (or RosamT/mG), TSC1flox/flox and Cx3cr1-ERT2 mice were acquired from Jackson Laboratory (Bar Harbor, ME), and Cx3cr1-Cre mice were from the Mutant Mouse Resource and Research Center (MMRRC). All mice were either on a C57/BL6 background or were backcrossed with C57/BL wild-type mice for more than 10 generations (see also Supplemental Experimental Procedures). Both genders were used (see figure legends for more information).
Antibodies, Immunohistochemistry, and Acquisition of Images
All antibodies used for immunohistochemistry, FACS analysis, and cell purification are listed in Table S1, along with vendors, catalog numbers, and dilution factors. Mouse brains were fixed in 4% paraformaldehyde (PFA) in PBS, followed by cryoprotection in 30% sucrose in PBS prior to cryosectioning and histological analyses. Brain sections were permeabilized by 0.3% Triton X-100 in 10% BSA. Sections were incubated with primary antibodies overnight, followed by secondary antibodies. This is the general procedure unless otherwise specifically described. All images were acquired using a Zeiss LSM 880 confocal microscope with Airyscan and processed with Zen black 2.1 or Zen blue lite 2.3 (Carl Zeiss). Detailed methodologies for image acquisition and quantification of cell numbers, synapses, and phagocytosis are described in Supplemental Experimental Procedures.
Microglia Culture, Treatment with Interferon Stimulatory DNA, and In Vitro Phagocytosis Assay
Primary microglia cultures were prepared from postnatal day 1 (P1) or P2 mice. Microglia were either treated with ISD for 4 or 9 hr prior to harvesting total RNA and qPCR analysis or used for microglial uptake assay with pHrodo Green zymosan bioparticles. Images were acquired at 1 frame/min for 61 cycles with the green fluorescence channel and DIC using a 63× oil objective using a Zeiss LSM 880 confocal microscope (Supplemental Experimental Procedures).
Purification of Microglia and Astrocytes, FACs Analysis, and In Vivo Phagocytosis Assay
Mouse brain tissues were digested, followed by neutralization and pipetting, and then passed through a 30-μm cell strainer. Myelin was removed by 70% Percoll centrifugation. Derived single-cell suspensions were either analyzed using LSR II Flow Cytometer (BD Biosciences) or purification of microglia and astrocytes with magnetic beads. For FACS analysis of microglia, cells were first blocked with anti-mouse CD16/CD32 (BioLegend) prior to staining with antibodies against surface or intracellular markers. To purify microglia and astrocytes, cell suspensions were also blocked with anti-mouse CD16/CD32, followed by anti-Cx3cr1-PE (BioLegend) and anti-ACSA-2-PE antibodies (Miltenyi), respectively, along with anti-PE MicroBeads (Miltenyi) to capture bound cells. For the in vivo phagocytosis assay, 0.5 μL of FITC-labeled zymosan particle suspension was infused into the hippocampus via cannula. Microglia were purified by the Percoll centrifugation, stained, and analyzed by FACS analysis (Supplemental Experimental Procedures).
Real-Time PCR and RNA-Seq
Total RNA was extracted using TRIzol Reagent (Life Technologies). RT-PCR was performed in a Step One Plus Real-time PCR System (Applied Biosystems). Four sets of total RNA samples were prepared from control and TSC1Cx3cr1CKO mouse brains and submitted to the Genomics Research Center at University of Rochester for RNA-seq. The primary data analysis for differential expression was completed using DeSeq2-1.10.1 by the genomics center. The heatmap diagrams were produced either using iPathway Guide (Advaita) or GraphPad Prism5 (La Jolla, CA).
Video/EEG Monitoring of Spontaneous Seizures
Video/EEG recording was done according to previously published work (McMahon et al., 2012). 22- to 25-day-old control and TSC1Cx3cr1KO mice were implanted epidurally with three-channel EEG electrodes (Plastics One). Mice were monitored using a video/EEG system 24 hr per day for up to 5 weeks. All electrographic seizures were verified behaviorally by video data to exclude movement artifacts. Six pairs of Cx3cr1-CreERT2+/– and Cx3cr1-CreERT2+/–TSC1f/f mice at 8 to 10 weeks of age were implanted with electrodes 2 or 3 days prior to tamoxifen treatment. Video/EEG recording began immediately after cession of 5-day tamoxifen treatment and continued for up to 26 days.
Statistical Analysis
Data were analyzed using GraphPad Prism 7 software with appropriate tests for comparisons between wild-type and TSC1Cx3cr1CKO mice. Student's t test was used to test the differences between two groups. A two-way ANOVA followed by Tukey's multiple comparisons was used to examine the differences among multiple groups. A log-rank (Mantel-Cox) test was used to analyze the rates of spontaneous seizures and mortality. The Mann-Whitney test was employed to test the difference in seizure frequency and duration. A p value of < 0.05 was considered significant. Data are shown as the mean ± SEM.
Further details and an outline of resources used in this work can be found in Supplemental Experimental Procedures.
Data and Software Availability
The accession number for the RNA-seq data reported in this paper is GEO: GSE108625.
Supplementary Material
Highlights.
Microglia adopt a noninflammatory reactive-like phenotype upon mTOR activation
Elevation of microglial mTOR activity triggers marked proliferation of astrocytes
Reactive-like microglia drive epileptogenesis independent of inflammatory responses
Acknowledgments
The authors thank Dr. Danielle Califano for assistance with FACS analysis and Julia Nalwalk for proofreading the manuscript. This work was supported by the NIH (grant NS093045 to Y.H. and grants DK088950 and DK099566 to X. Zhao), the Ayco Foundation (Y.H.), and the Crohn's & Colitis Foundation of America (research fellowship 481637 to R.M.).
Footnotes
Supplemental Information: Supplemental Information includes Supplemental Experimental Procedures, six figures, and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.02.004.
Author Contributions: X. Zhao and Y.H. conceived and designed the study. X. Zhao performed immunohistochemistry (IHC), FACS, qPCR, video/EEG recording, image acquisition, live imaging, western blot, primary culture, and data quantification. Y.L. provided assistance on genotyping, qPCR, and quantification of IHC data. S.M. and R.M. provided assistance on animal handling, IHC, and qPCR. J.M. provided assistance on imaging. X. Zhao and Y.H. analyzed data and prepared the manuscript. P.F. provided assistance on statistical analysis. M.A.A., J.Q., A.L.R., M.G., J.C., G.W.M., and X. Zhu provided helpful advice and edited the manuscript.
Declaration of Interests: The authors declare no competing financial interests.
References
- Abiega O, Beccari S, Diaz-Aparicio I, Nadjar A, Layé S, Leyrolle Q, Gómez-Nicola D, Domercq M, Pérez-Samartín A, Sánchez-Zafra V, et al. Neuronal hyperactivity disturbs ATP microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling. PLoS Biol. 2016;14:e1002466. doi: 10.1371/journal.pbio.1002466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham J, Fox PD, Condello C, Bartolini A, Koh S. Minocycline attenuates microglia activation and blocks the long-term epileptogenic effects of early-life seizures. Neurobiol Dis. 2012;46:425–430. doi: 10.1016/j.nbd.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica E, Bauer S, Bozzi Y, Caleo M, Dingledine R, Gorter JA, Henshall DC, Kaufer D, Koh S, Löscher W, et al. Neuroinflammatory targets and treatments for epilepsy validated in experimental models Epilepsia. 2017;58(Suppl 3):27–38. doi: 10.1111/epi.13783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askew K, Li K, Olmos-Alonso A, Garcia-Moreno F, Liang Y, Richardson P, Tipton T, Chapman MA, Riecken K, Beccari S, et al. Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep. 2017;18:391–405. doi: 10.1016/j.celrep.2016.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boer K, Spliet WG, van Rijen PC, Redeker S, Troost D, Aronica E. Evidence of activated microglia in focal cortical dysplasia. J Neuroimmunol. 2006;173:188–195. doi: 10.1016/j.jneuroim.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Canton J, Khezri R, Glogauer M, Grinstein S. Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol Biol Cell. 2014;25:3330–3341. doi: 10.1091/mbc.E14-05-0967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441–468. doi: 10.1146/annurev-immunol-051116-052358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB. The mTOR signalling cascade: paving new roads to cure neurological disease. Nat Rev Neurol. 2016;12:379–392. doi: 10.1038/nrneurol.2016.81. [DOI] [PubMed] [Google Scholar]
- Hong S, Dissing-Olesen L, Stevens B. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol. 2016;36:128–134. doi: 10.1016/j.conb.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen LA, Mirzaa GM, Ishak GE, O'Roak BJ, Hiatt JB, Roden WH, Gunter SA, Christian SL, Collins S, Adams C, et al. PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain. 2015;138:1613–1628. doi: 10.1093/brain/awv045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, Onda H. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet. 2002;11:525–534. doi: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson Y, Liu J, Stevens PD, Li X, Li J, Evers BM, Gao T. Tuberous sclerosis complex 2 (TSC2) regulates cell migration and polarity through activation of CDC42 and RAC1. J Biol Chem. 2010;285:24987–24998. doi: 10.1074/jbc.M109.096917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Reeves C, Michalak Z, Coppola A, Diehl B, Sisodiya SM, Thom M. Evidence for mTOR pathway activation in a spectrum of epilepsy-associated pathologies. Acta Neuropathol Commun. 2014;2:71. doi: 10.1186/2051-5960-2-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Murao N, Katano Y, Juliandi B, Kohyama J, Akira S, Kawai T, Nakashima K. TLR9 signalling in microglia attenuates seizure-induced aberrant neurogenesis in the adult hippocampus. Nat Commun. 2015;6:6514. doi: 10.1038/ncomms7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon J, Huang X, Yang J, Komatsu M, Yue Z, Qian J, Zhu X, Huang Y. Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J Neurosci. 2012;32:15704–15714. doi: 10.1523/JNEUROSCI.2392-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan WB. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155:1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkänen A, Lukasiuk K. Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 2011;10:173–186. doi: 10.1016/S1474-4422(10)70310-0. [DOI] [PubMed] [Google Scholar]
- Preissler J, Grosche A, Lede V, Le Duc D, Krügel K, Matyash V, Szulzewsky F, Kallendrusch S, Immig K, Kettenmann H, et al. Altered microglial phagocytosis in GPR34-deficient mice. Glia. 2015;63:206–215. doi: 10.1002/glia.22744. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19:987–991. doi: 10.1038/nn.4338. [DOI] [PubMed] [Google Scholar]
- Rosito M, Lauro C, Chece G, Porzia A, Monaco L, Mainiero F, Catalano M, Limatola C, Trettel F. Trasmembrane chemokines CX3CL1 and CXCL16 drive interplay between neurons, microglia and astrocytes to counteract pMCAO and excitotoxic neuronal death. Front Cell Neurosci. 2014;8:193. doi: 10.3389/fncel.2014.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schartz ND, Wyatt-Johnson SK, Price LR, Colin SA, Brewster AL. Status epilepticus triggers long-lasting activation of complement C1q-C3 signaling in the hippocampus that correlates with seizure frequency in experimental epilepsy. Neurobiol Dis. 2018;109(Pt A):163–173. doi: 10.1016/j.nbd.2017.10.012. [DOI] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Sidik H, Talbot WS. The Rag-Ragulator complex regulates lysosome function and phagocytic flux in microglia. Cell Rep. 2016;14:547–559. doi: 10.1016/j.celrep.2015.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Over-street-Wadiche LS, Tsirka SE, Maletic-Savatic M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7:483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosunov AA, Wu X, McGovern RA, Coughlin DG, Mikell CB, Goodman RR, McKhann GM., 2nd The mTOR pathway is activated in glial cells in mesial temporal sclerosis Epilepsia. 2012;53(Suppl 1):78–86. doi: 10.1111/j.1528-1167.2012.03478.x. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Matsuwaki T, Yamanouchi K, Nishihara M. Increased lysosomal biogenesis in activated microglia and exacerbated neuronal damage after traumatic brain injury in progranulin-deficient mice. Neuroscience. 2013;250:8–19. doi: 10.1016/j.neuroscience.2013.06.049. [DOI] [PubMed] [Google Scholar]
- Tokizane K, Konishi H, Makide K, Kawana H, Nakamuta S, Kaibuchi K, Ohwada T, Aoki J, Kiyama H. Phospholipid localization implies microglial morphology and function via Cdc42 in vitro. Glia. 2017;65:740–755. doi: 10.1002/glia.23123. [DOI] [PubMed] [Google Scholar]
- van Vliet EA, Forte G, Holtman L, den Burger JC, Sinjewel A, de Vries HE, Aronica E, Gorter JA. Inhibition of mammalian target of rapamycin reduces epileptogenesis and blood-brain barrier leakage but not microglia activation. Epilepsia. 2012;53:1254–1263. doi: 10.1111/j.1528-1167.2012.03513.x. [DOI] [PubMed] [Google Scholar]
- Zhang B, Zou J, Han L, Rensing N, Wong M. Microglial activation during epileptogenesis in a mouse model of tuberous sclerosis complex. Epilepsia. 2016;57:1317–1325. doi: 10.1111/epi.13429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Yang T, Li L, Sun L, Hou Y, Hu X, Zhang L, Tian H, Zhao Q, Peng J, et al. TSC1 controls macrophage polarization to prevent inflammatory disease. Nat Commun. 2014;5:4696. doi: 10.1038/ncomms5696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.