

Abstract
We use a 0-D photochemical box model and a 3-D global chemistry-climate model, combined with observations from the NOAA Southeast Nexus (SENEX) aircraft campaign, to understand the sources and sinks of glyoxal over the Southeast United States. Box model simulations suggest a large difference in glyoxal production among three isoprene oxidation mechanisms (AM3ST, AM3B, and MCM v3.3.1). These mechanisms are then implemented into a 3-D global chemistry-climate model. Comparison with field observations shows that the average vertical profile of glyoxal is best reproduced by AM3ST with an effective reactive uptake coefficient γglyx of 2 × 10−3, and AM3B without heterogeneous loss of glyoxal. The two mechanisms lead to 0–0.8 μg m−3 secondary organic aerosol (SOA) from glyoxal in the boundary layer of the Southeast U.S. in summer. We consider this to be the lower limit for the contribution of glyoxal to SOA, as other sources of glyoxal other than isoprene are not included in our model. In addition, we find that AM3B shows better agreement on both formaldehyde and the correlation between glyoxal and formaldehyde (RGF = [GLYX]/[HCHO]), resulting from the suppression of δ-isoprene peroxy radicals (δ-ISOPO2). We also find that MCM v3.3.1 may underestimate glyoxal production from isoprene oxidation, in part due to an underestimated yield from the reaction of IEPOX peroxy radicals (IEPOXOO) with HO2. Our work highlights that the gas-phase production of glyoxal represents a large uncertainty in quantifying its contribution to SOA.
1 Introduction
Glyoxal (CHOCHO) is one of the most abundant dicarbonyl compounds in the atmosphere. Its sources include direct emissions from biofuel use and biomass burning, and secondary production from oxidation of various volatile organic compounds (VOCs) [Fu et al., 2008; Hays et al., 2002; Myriokefalitakis et al., 2008]. Glyoxal has a lifetime of about 1–3 hours against photolysis and oxidation by OH at midday [Feierabend et al., 2009; Volkamer et al., 2005; Washenfelder et al., 2011]. It is highly water-soluble, with a Henry’s Law constant of 3.0 – 4.2 × 105 M atm−1 at 298 K [Sander, 2015]. In aerosol water, the solubility increases rapidly at low salt concentrations (up to 5.0 × 108 M atm−1) [Kampf et al., 2013; Waxman et al., 2015], due to the formation of glyoxal hydrate - sulfate complexes (“salting-in”) [Kurtén et al., 2015]. However, this increase is inhibited at high salt concentrations by the kinetic limitation of gas-particle partitioning [Kampf et al., 2013; Knote et al., 2014]. Laboratory and field studies showed that glyoxal readily undergoes heterogeneous uptake to aerosols and cloud droplets to form secondary organic aerosol (SOA) [Carlton et al., 2007; Ervens et al., 2011; Galloway et al., 2009; Galloway et al., 2011b; Liggio et al., 2005a; b; Lim et al., 2005; Volkamer et al., 2007].
Glyoxal can provide important constraints on quantifying VOC emissions and oxidation mechanisms. Observations from aircraft, ground and satellite show that glyoxal is often highly correlated with formaldehyde (HCHO), another product of VOC oxidation [DiGangi et al., 2012; Kaiser et al., 2015; MacDonald et al., 2012; Miller et al., 2014; Stavrakou et al., 2009; Vrekoussis et al., 2010]. The ratio of glyoxal to formaldehyde (surface or tropospheric column concentrations), RGF, varies for different biogenic and anthropogenic VOC precursors, so it can be used to quantify the source strength of local VOC emissions [DiGangi et al., 2012; Kaiser et al., 2015; Miller et al., 2014; Vrekoussis et al., 2010]. The NOx (NOx = NO + NO2) dependence of RGF, although it varies greatly for different VOCs and may offer additional information on VOC oxidation [DiGangi et al., 2012; Kaiser et al., 2015; Vrekoussis et al., 2010]. Thus, it is important to evaluate model performance on RGF and its NOx dependence on regional and global scales, providing critical information for present and future satellite validation [Vrekoussis et al., 2010].
Sources and sinks of glyoxal remain largely uncertain [Ervens et al., 2011; Fu et al., 2008; Galloway et al., 2011a; Myriokefalitakis et al., 2008; Stavrakou et al., 2009; Volkamer et al., 2007; Washenfelder et al., 2011]. Production from isoprene oxidation represents a major source of glyoxal on the global scale [Fu et al., 2008]. Chamber experiments suggest a significant yield (up to 3%) of glyoxal from the first generation of isoprene oxidation under high NOx conditions (500 ppbv NO) [Galloway et al., 2011a; Volkamer et al., 2006]. As we show below, this high yield may not reflect the distribution of β- and δ-isoprene peroxy radicals (ISOPO2) as a function of their lifetimes under ambient conditions [Peeters et al., 2014]. Another large uncertainty lies in the heterogeneous loss of glyoxal to aerosols and cloud droplets, which contributes to SOA mass. Laboratory studies indicate that the uptake of glyoxal depends on aerosol and cloud composition [Corrigan et al., 2008; Jang et al., 2002; Kroll et al., 2005; Liggio et al., 2005a; b; Nozière et al., 2009; Volkamer et al., 2009; Waxman et al., 2015], ambient relative humidity (RH) [Corrigan et al., 2008; Hastings et al., 2005; Kampf et al., 2013; Liggio et al., 2005a] and temperature [Gomez et al., 2015]. By assuming irreversible reactive uptake with an uptake coefficient γglyx = 2.9 × 10−3, Fu et al. [2008] found that this process accounts for 14% of glyoxal loss globally. Volkamer et al. [2007] estimated γglyx of 3.7 × 10−3 in Mexico City, where glyoxal contributes about 15% of ambient SOA. A much lower γglyx (0–8 × 10−4 for the day and (2 ± 1) × 10−4 for the night) was derived in Los Angeles [Washenfelder et al., 2011], where kinetic limitations at the high salt concentrations likely reduce the rate of SOA formation [Kampf et al., 2013; Knote et al., 2014].
The Southeast Nexus (SENEX) aircraft campaign, which took place in June–July of 2013, aimed to improve the understanding of the interactions between biogenic and anthropogenic emissions over the Southeast U.S. It provided a detailed characterization of tropospheric photochemistry (gas and aerosol), including ozone, NOy (NO, NO2 and its atmospheric oxidation products), biogenic VOCs (isoprene, terpene), isoprene oxidation products and organic aerosols [Warneke et al., 2016]. In particular, glyoxal was measured on board the NOAA WP-3D aircraft using a cavity enhanced absorption spectroscopy technique [Min et al., 2015], and formaldehyde was measured using laser induced fluorescence technique [Cazorla et al., 2015]. These measurements provide an unprecedented opportunity to examine the sources and sinks of glyoxal in this region, as well as its contribution to SOA mass.
Here we first examine the glyoxal yield from isoprene oxidation in a box model with three chemical mechanisms (AM3ST, AM3B, and the Master Chemical Mechanism v3.3.1 [Jenkin et al., 2015]). We then use field observations from the SENEX field campaign, interpreted with a chemistry-climate model, to understand the sources and sinks of glyoxal over the Southeast U.S. The comparison of the model to high resolution aircraft data for both glyoxal and formaldehyde provides important new constraints on the potential for glyoxal to form SOA, but also highlights uncertainty in the mechanism for isoprene oxidation, the single largest source of glyoxal in the Southeast U.S.
2 Methods
AM3 is the atmospheric component of the Geophysical Fluid Dynamics Laboratory (GFDL) coupled model CM3. The dynamical core, physical parameterizations, cloud and precipitation processes, and cloud-aerosol interactions in AM3 are described in detail in Donner et al. [2011]. Chemistry in a previous version of AM3 has been described by Naik et al. [2013]. In this work, we nudge the horizontal wind field in the model toward values from the NCEP Global Forecast System (GFS), allowing the model to simulate synoptic conditions corresponding to those sampled during field campaigns [Lin et al., 2012]. We also apply finer vertical grids for convection plumes than the standard AM3 to improve the wet removal of tracers during summer time [Paulot et al., 2015]. The photolysis module has been updated to Fast-JX v7.1 (ftp://128.200.14.8/public/prather/Fast-J), to compute the impacts from aerosols and clouds interactively. Dry deposition velocities prescribed in the model reflect rapid dry deposition of oxidized organic compounds [Nguyen et al., 2015].
Biogenic emission of isoprene is computed using the Model of Emissions of Gases and Aerosols from Nature (MEGAN) inventory [Guenther et al., 2006], with a total 15.9 Tg C in North America in June–August of 2013, which is slightly higher than previous estimates of 12.2–14.6 Tg C (June–August of 2006) in the same region [Millet et al., 2008]. We reduce the isoprene emissions estimated by MEGAN by 20% (to 12.7 Tg C), to be consistent with other estimates of isoprene emission over the Southeast U.S. [Warneke et al., 2010]. We do not consider glyoxal production from oxidation of terpenes and aromatics in this work, as their contribution is relatively small compared to isoprene over the Southeast U.S. [Kaiser et al., 2015]. Inclusion of larger isoprene emissions and other VOC sources would proportionally increase the magnitude of glyoxal sinks derived in the analysis below. Anthropogenic emissions in 2013 follow the Representative Concentration Pathway 8.5 (RCP 8.5) projection [Lamarque et al., 2011]. We applied diurnal variation anthropogenic NOx emissions in North America following Mao et al. [2013b]. Anthropogenic NOx emissions in RCP 8.5 are 0.34 Tg N/month over North America, comparable to the estimate from NEI 2011 of 0.29 Tg N/month [Travis et al., 2015]. We reduce these anthropogenic NOx emissions by 25% to 0.26 Tg N/month, to be consistent with recent estimates in this region [Anderson et al., 2014].
2.1 Isoprene chemistry
The standard isoprene mechanism used in AM3, “AM3ST”, is largely based on Mao et al. [2013b] following chamber observations from Paulot et al. [2009a; 2009b] and Crounse et al. [2011]. The main features of this mechanism include: (1) substantial yield (7%) of first generation organic nitrates from δ-ISOPO2, compared to 4.7% from β-ISOPO2; (2) detailed assignment of the fate of first- and second-generation organic nitrates; and (3) isomerization of ISOPO2 using the laboratory-determined rate constants. We further update this chemistry in several aspects. First, we assume a 25% molar yield of glyoxal from ISOPO2 isomerization as the first-generation product [Marais et al., 2016]. Second, the reaction of ISOPO2 with HO2 is updated following St. Clair et al. [2016a] to reflect a higher yield of unsaturated hydroxy hydroperoxides (ISOPOOH) (94%). Third, we adopt a new methylvinyl ketone (MVK) oxidation chemistry in which the glycolaldehyde molar yield is increased from 0.53 to 0.72 under high NOx conditions [Praske et al., 2015]. Fourth, we include fast photolysis of carbonyl organic nitrates [Müller et al., 2014]. Fifth, we adopt a substantially slower ozonolysis rate of isoprene β-hydroxy nitrate (ISOPNB, 3.7 × 10−19 molec−1cm3s−1 [Lee et al., 2014]) than the previous value from Lockwood et al. [2010] (1.06 × 10−16 molec−1cm3s−1). Lastly, we update the NO3-initiated chemistry of isoprene following MCM v3.2 [Jenkin et al., 1997; Saunders et al., 2003]. As illustrated in Figure S1, glyoxal is produced in the first generation of isoprene oxidation, both from the decomposition of alkoxyl radical (DIBOO shown in Figure S1) through the δ-channel under high NOx conditions [Peeters and Nguyen, 2012], and from isomerization of ISOPO2 under low NOx conditions [Stavrakou et al., 2010]. Glyoxal is further produced in later generations from oxidation of carbonyl compounds including glycolaldehyde and isoprene epoxydiol (IEPOX).
In this work, we introduce an additional mechanism, “AM3_beta” (AM3B), to reflect more recent updates to the understanding of isoprene oxidation. Both theoretical and experimental studies indicate that β- and δ-ISOPO2 undergo fast interconversion [Bates et al., 2014; Crounse et al., 2011; Peeters et al., 2014]. One important implication is that oxidation products observed in experimental chambers at ~500 ppbv NO may not reflect the products in ambient air where NO is several orders of magnitude lower. This is due to a higher fraction of δ-ISOPO2 loss via bimolecular reactions than interconversion back to β-ISOPO2 under high NOx conditions. To reflect this change, we only allow β-ISOPO2 to react with NO because δ-ISOPO2 is believed to only account for < 3% of total ISOPO2 under ambient conditions [Peeters et al., 2014]. The yield of β-hydroxyl isoprene nitrate (ISOPNB) is assumed to be 0.1, an average of the suggested values 0.09 [Xiong et al., 2015] and 0.16 [Teng et al., 2015]. The yields of HCHO, MVK and methacrolein (MACR) from β-ISOPO2 + NO are adjusted accordingly for carbon balance. As we show below, this update changes the production of formaldehyde and glyoxal under high NOx conditions.
The Leeds Master Chemical Mechanism (MCM) is a near-explicit chemical mechanism that describes gas-phase VOC chemistry. The latest revision, v3.3.1, includes recent updates for OH-initiated isoprene chemistry [Jenkin et al., 2015], such as isoprene-peroxy radical interconversion and isomerization pathways [Peeters et al., 2014], and the chemistry of IEPOX [Bates et al., 2014; Paulot et al., 2009b]. Consequently, these updates show significant differences in simulated HOx (HOx = OH + HO2), NOx and major oxidation products compared to earlier versions of MCM. To evaluate the performance of MCM v3.3.1 in our global model, we implement a MCM-like mechanism (‘AM3M’) by adjusting the production of glyoxal from major pathways in the AM3B mechanism, to approximate the glyoxal and HCHO yields of MCM v3.3.1 in a highly condensed chemical mechanism suitable for use in a global model (Figure S2).
2.2 Heterogeneous loss of glyoxal and methylglyoxal
Heterogeneous loss onto aerosols and cloud droplets plays an important role in the fate of dicarbonyls. Here we assume this process is irreversible and can be represented by a first-order reactive uptake rate constant k [Mao et al., 2010]
where a is the effective radius of aerosols or cloud droplets, Dg is the gas phase diffusion constant, γ is the reactive uptake coefficient, A is the surface area of aerosols or cloud droplets, and ν is the mean molecular velocity of the gas molecule.
We allow this process to take place on five types of aerosols including sulfate, black carbon (BC), primary organic carbon (POC), sea salt, mineral dust and SOA, with hygroscopic growth included [Mao et al., 2013a]. Given the large uncertainties associated with the uptake of glyoxal, we use field observations to assess the possible range of γglyx. We do not include glyoxal uptake by cloud droplets in this work, because the uptake coefficient by aerosols is much greater, and because large uncertainties are associated with mixing of cloudy and no-cloud volumes within a grid box [Huijnen et al., 2014; Jacob, 2000]. We include heterogeneous loss of methylglyoxal with an uptake coefficient of 7.6 × 10−3 [Zhao et al., 2006], to produce better closure with total organic aerosol (OA) in the model.
The irreversible surface uptake coefficient, γglyx applied here should be viewed as an “effective reactive uptake coefficient”, to simply represent the net heterogeneous loss of glyoxal that does not revert back to the gas-phase, and provide an estimate for its contribution to SOA mass on the regional scale. Previous studies have indicated that this process is likely reversible to some extent [Galloway et al., 2009; Kroll et al., 2005]. We show below that the values of γglyx are dependent on the choice of gas-phase chemistry.
3 NOx-Dependent Glyoxal and HCHO Yields
We first conduct box model simulations to quantify the yield of glyoxal and HCHO from isoprene oxidation as a function of NOx levels, using the Dynamically Simple Model of Atmospheric Chemical Complexity (DSMACC) [Emmerson and Evans, 2009]. We test three mechanisms (AM3ST, AM3B, and MCM v3.3.1) in the model. The model is initialized at 8:00 local time (LT) for mid-latitude summer conditions, with 1 ppbv of isoprene, 60 ppbv of ozone, 150 ppbv of CO, and a choice of 0.01, 0.1 or 1 ppbv of NOx. While NOx is held constant, isoprene is allowed to decay over time. Figure 1 shows the cumulative yields of glyoxal and HCHO per unit carbon from isoprene oxidation at various NOx levels after three days of model integration with their loss processes turned off [Palmer et al., 2003]. We also use tagged tracers for individual pathways (β/δ-ISOPO2 + NO, ISOPO2 + HO2, isomerization of ISOPO2) to compute their contribution in each mechanism.
Figure 1.
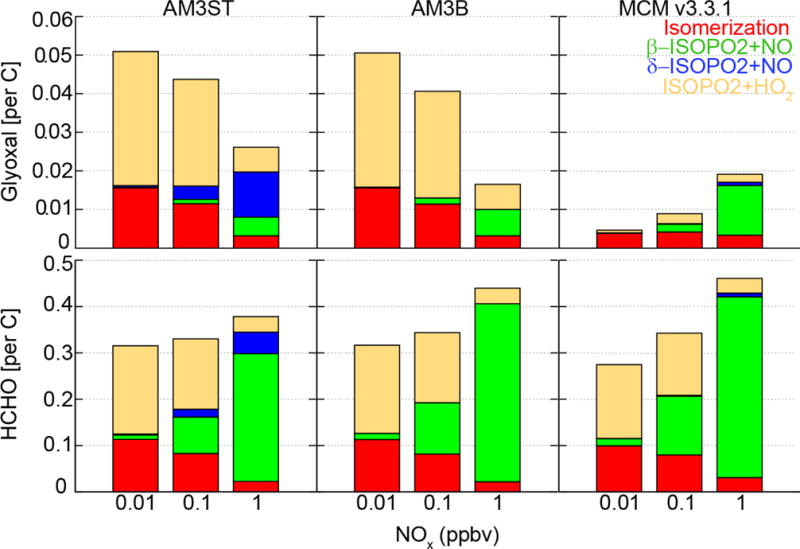
Cumulative yields of glyoxal and HCHO in major pathways from isoprene oxidation at different NOx levels. Glyoxal and HCHO from isoprene oxidized by O3 and by NO3 are not shown due to low production in all the mechanisms.
A striking difference between the MCM v3.3.1 and AM3 mechanisms is the glyoxal production under low NOx conditions. While the AM3 mechanisms show a large yield of glyoxal from isomerization of ISOPO2 and the oxidation of IEPOX (via the ISOPO2 + HO2 pathway), MCM v3.3.1 shows very little production of glyoxal from these channels. For the isomerization pathway, MCM v3.3.1 assumes 50% of 1,6-H shift isomerization flux produces hydroperoxy-aldehydes (HPALDs), with another 50% produces complex dihydroperoxy formyl peroxy radicals, referred as “di-HPCARPs” by Peeters et al. [2014]. MCM v3.3.1 includes low production of glyoxal from the degradation of HPALDs and di-HPCARPs. On the other hand, the AM3 mechanisms do not include di-HPCARPs, and assume fast photolysis as the only loss process of HPALDs, with prompt yield of glyoxal following Stavrakou et al. [2010].
Another large difference in glyoxal yield comes from the treatment of the ISOPO2 + HO2 pathway, particularly regarding the fate of IEPOX. MCM v3.3.1 assumes a major production of organic peroxides from the reaction of IEPOX peroxy radicals (IEPOXOO) (α-carbonyl peroxy radicals) with HO2, while AM3 mechanisms follow Paulot et al. [2009b] and assume full radical propagation for this reaction, with a 28% yield of glyoxal. As a result, a much higher yield of glyoxal is predicted from the ISOPO2 + HO2 pathway in the AM3 mechanisms compared to MCM v3.3.1. In fact, several studies have confirmed significant OH production from the reaction of HO2 with α-carbonyl peroxy radicals [Dillon and Crowley, 2008; Hasson et al., 2004; Jenkin et al., 2007]. This is also consistent with recent studies of IEPOX kinetics, which show very little formation of peroxides under low NOx conditions [Bates et al., 2014; Jacobs et al., 2013].
Under high NOx conditions, the three mechanisms show better agreement for glyoxal production, though with different pathways contributing. MCM v3.3.1 shows a dominant production from the β-ISOPO2 + NO pathway, mainly due to a higher yield of glyoxal from oxidation of glycolaldehyde (20%), based on Niki et al. [1987]. An even higher yield of glyoxal (29%) was adopted by Galloway et al. [2011a]. In contrast, the AM3 mechanisms apply a lower yield of glyoxal (13%) following Butkovskaya et al. [2006]. Surprisingly, both AM3ST and AM3B show significant production from isomerization of ISOPO2 and the ISOPO2 + HO2 pathway, suggesting the potential importance of these channels for glyoxal production even under high NOx conditions. AM3ST has a major production from the δ-ISOPO2 + NO pathway, leading to 60% higher glyoxal yield than AM3B. MCM v3.3.1 shows a small contribution from the δ-ISOPO2 + NO channel, due to a small fraction of δ-ISOPO2 in the radical pool of ISOPO2 (< 3%). This reflects a different distribution of δ-ISOPO2 and β-ISOPO2 under ambient conditions vs. very high NO chamber conditions [Peeters et al., 2014], consistent with our assumption in AM3B. Overall, the AM3 mechanisms show decreasing glyoxal production with increasing NOx concentration, whereas MCM v3.3.1 shows the opposite.
The HCHO yield and its NOx dependence appear to be more consistent across the different mechanisms. Under low NOx conditions, isomerization of ISOPO2 and the ISOPO2 + HO2 pathway contribute most to HCHO. At NOx = 1 ppbv, the ISOPO2 + NO pathway becomes the primary source of HCHO. The HCHO yield in AM3B is 17% higher than AM3ST and comparable to MCM v3.3.1, mainly due to a higher HCHO yield from the β-ISOPO2 + NO pathway than the δ-ISOPO2 + NO pathway. It should be noted that the cumulative yields shown in Figure 1 represent the potential glyoxal and HCHO produced from sufficient oxidation of 1 ppbv isoprene and the intermediate VOCs. This is different from ambient conditions that isoprene is continuously emitted and the production of glyoxal and HCHO are dependent on OH levels (i.e. the production rate of ISOPO2). Chemical loss of HCHO and glyoxal are similar across the mechanisms (Table S1).
The NOx dependent yields of glyoxal and HCHO from box model results are useful to identify their major production pathways at different NOx levels. This knowledge can be translated to 3D model outputs and help evaluate model performance from these pathways using aircraft observations.
4 Observational Constrains on Glyoxal Production
4.1 Vertical profiles of gaseous and particulate species
We compare the AM3 model predictions for glyoxal, formaldehyde, and isoprene oxidation products to measurements of these species acquired during the SENEX field campaign. All measurements are averaged to 1 minute time resolution. The measurement accuracies for isoprene, formaldehyde, NOx, glyoxal, organic aerosol and aerosol surface area are 25%, 10%, 5%, 5.8%, 50%, and 36% respectively ([Warneke et al., 2016] and the references therein). Although other carbonyl compounds have recently been found to suffer from inlet artifacts arising from catalytic conversion of organic hydroperoxides on metal inlet surfaces [Rivera-Rios et al., 2014], we do not expect such interferences for glyoxal since no metal surfaces are present in the glyoxal sampling line [Min et al., 2015], and since there are currently no known mechanisms for conversion of hydroperoxides to glyoxal. We also expect little interferences for formaldehyde since its residence time during exposure to metal surfaces is very small [Cazorla et al., 2015], This is confirmed by recent laboratory tests, suggesting the interference is < 5% for this specific instrument [St. Clair et al., 2016b]. We exclude biomass burning, urban plumes, stratospheric air (CH3CN ≥ 225 pptv, NOx/NOy > 0.4 or NO2 > 4 ppbv or O3/CO > 1.25 mol/mol, respectively) from our analysis following Hudman et al. [2007], and omit data from the Ozark Mountains, where the model shows a significant positive bias for isoprene (Figure S3). We also exclude nighttime flights from our analysis.
Figure 2 shows the mean vertical profiles of observed and modeled isoprene, glyoxal, HCHO and other related species during SENEX. Model output is sampled along the flight tracks and at the flight time with 1 hour time resolution. We include two model simulations (AM3ST and AM3B). MCM v3.3.1 is not included because of its complexity. Instead, we show AM3M in our global model as a proxy to MCM v3.3.1 in the following sections. Simulated NOx agrees with the observations at all altitudes except for a slight overestimate (~20%) near the surface. Observed isoprene concentration peaks near the surface at 1.2 ppbv and decreases gradually with altitude. Both AM3ST and AM3B show a positive bias of isoprene below 1 km, but no such bias is evident for HCHO. As HCHO in this region is dominantly produced from isoprene and has a relatively longer lifetime than that of isoprene (4 h vs. < 1 h), we attribute this positive bias of modeled isoprene partly to sampling bias and partly to shallow boundary layer and slow vertical mixing in the current model. We further examine modeled OH and J values using ICARTT (2004) [Mao et al., 2013b] and NOMADSS (2013) (https://www.eol.ucar.edu/field_projects/nomadss) field observations over the eastern U.S., and we find that the model agrees with observed OH and J values within 20% (Figure S4 and S5).
Figure 2.
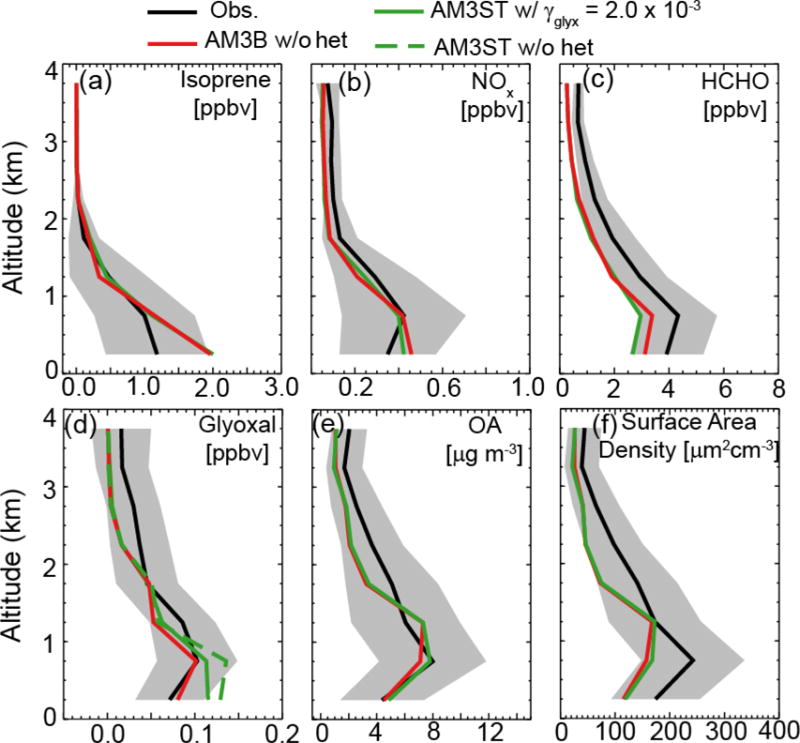
Mean vertical profiles of isoprene, formaldehyde, NOx, glyoxal, organic aerosol (OA) and surface area density of aerosol during SENEX. Grey shades are the standard deviation (σ) of the averaged profiles of the measured tracers. Dashed green line in panel (d) is model estimate without heterogeneous loss of glyoxal by AM3ST. Observed organic aerosol mass and aerosol surface area density are from dry particles.
Like isoprene, both HCHO and glyoxal decrease with altitude, reflecting their short photochemical lifetimes (4 h and 3.5 h) and their dominant source from isoprene. Our model is able to reproduce their vertical gradients, although the vertical profiles of HCHO and glyoxal appear to be sensitive to the choice of photochemical mechanisms. AM3ST underestimates HCHO by 32% and overestimates glyoxal almost by a factor of two near the surface. With the suppression of δ-ISOPO2 in AM3B, HCHO is increased by 17% and glyoxal is decreased by 38% near the surface, leading to better agreement with observations for both species.
The abundance of glyoxal is also dependent on its heterogeneous loss. By assuming an irreversible reactive uptake on aerosols, an optimal value of γglyx is selected to minimize the difference between modeled and observed glyoxal in the boundary layer. We find that the best agreement is achieved for AM3ST with γglyx of 2 × 10−3 and for AM3B with γglyx of zero (Table S2); we adopt those values in the following analysis.
γglyx derived with AM3B is lower than the value from previous laboratory experiments (2.9 × 10−3 by Liggio et al. [2005b]), likely due to two reasons. First, there could be a missing source of gas-phase glyoxal in the current mechanism, which can be compensated by aerosol uptake and lead to a higher contribution to SOA formation. We find that approximate 30% increase of glyoxal production in AM3B would be required to compensate aerosol uptake of glyoxal with γglyx = 2.9 × 10−3. Second, the effective aerosol uptake coefficient, as we assumed here to represent the net heterogeneous loss of glyoxal, is indeed smaller than the value from the previous study. One possibility is that the aerosol uptake of glyoxal is to some extent reversible [Galloway et al., 2009; Kroll et al., 2005], which would lead to a lower estimate of γglyx. Another possibility is the suppression of glyoxal loss to ambient aerosols that are internally mixed with organic and inorganic components, resulting from its organic coating [Galloway et al., 2011b]. It is also possible that additional production of glyoxal in the particle-phase from other organic compounds reduces the net loss of glyoxal to the aerosol surface. In fact, the negligibly small uptake coefficients of glyoxal (0–8 × 10−4 during the daytime and (2 ± 1) × 10−4 during the nighttime) have also been determined from analysis of glyoxal observations in a previous field study in Los Angeles during the CalNex field campaign [Washenfelder et al., 2011].
Overall, the resulting average contribution of glyoxal to organic aerosols is about 0.8 μg m−3 for AM3ST (Figure S7) and negligible for AM3B in the boundary layer over the Southeast U.S. We emphasize that this is likely the lower limit of glyoxal SOA, as other glyoxal sources such as anthropogenic VOCs and monoterpenes, are not included in our model. Accounting for these additional sources would require a higher sink of glyoxal, and therefore results in higher glyoxal SOA. The estimate from AM3ST is comparable to previous model studies over the same region assuming reversible or irreversible reactive uptake of glyoxal onto aerosols [Knote et al., 2014; Li et al., 2015; Ying et al., 2015], while AM3B is not. We also find that the inclusion of IEPOX aerosol uptake may reduce glyoxal in the boundary layer by less than 15% over the Southeast U.S. (Figure S8). This would lead to an even lower estimate of glyoxal SOA.
4.2 RGF and its dependence on NOx
Comparison of modeled and observed RGF provides additional constraint on the gas-phase chemistry for glyoxal production. Figure 3 shows the co-variation of glyoxal and formaldehyde in the boundary layer during SENEX. The linear regression slope (RGF) derived from observations is 2.4% (R2=0.61) in the boundary layer over the Southeast US, suggesting that the dominant source of both glyoxal and formaldehyde in this region is isoprene [Kaiser et al., 2015]. The AM3ST mechanism overestimates RGF by a factor of two, while better agreement is achieved by the AM3B mechanism, due to the suppressed production of glyoxal and enhanced production of HCHO. Simulations with both mechanisms significantly underestimate the range of HCHO concentrations observed. Without heterogeneous loss of glyoxal, the model with AM3ST significantly overestimates RGF but predicts a similar result as assuming no heterogeneous loss of glyoxal with AM3B.
Figure 3.
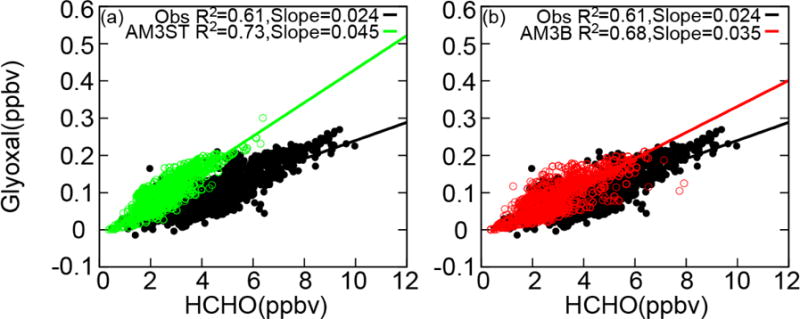
Correlation between glyoxal and formaldehyde below 1.5 km during SENEX. Black dots represent observations, while green and red open circles show AM3ST with γglyx = 2.0 × 10−3 (left) and AM3B without heterogeneous loss of glyoxal (right). Solid lines are linear regression lines, with regression slopes calculated from least-squares fit.
We further examine the NOx dependence of HCHO, glyoxal and RGF during SENEX (Figure 4). A prominent feature of Figure 4 is the high similarity between the observed NOx-dependence of HCHO and glyoxal. Both HCHO and glyoxal are observed to increase with NOx concentrations and to start to level off at 1 ppbv of NOx. Remarkably, RGF shows little variation across NOx concentrations, consistent with Kaiser et al. [2015]. Both AM3ST and AM3B are able to reproduce the NOx dependence of HCHO, although they tend to underestimate HCHO by 1–2 ppbv across NOx regimes, consistent with Wolfe et al. [2016]. AM3B shows better agreement with observations than AM3ST at high NOx level, indicating δ-channel is suppressed under ambient conditions. Using the optimized γglyx values described above (2 × 10−3 for AM3ST and zero for AM3B), our model can roughly capture the NOx dependence of glyoxal. In contrast to Figure 1, neither AM3ST nor AM3B shows high glyoxal concentrations under low NOx conditions, reflecting slower production of glyoxal under such conditions due to low OH radical concentrations and slow ISOPO2 production rate in the ambient atmosphere [Wolfe et al., 2016]. Although the NOx dependent HCHO and glyoxal are individually captured qualitatively, a large positive bias in RGF is apparent for AM3ST across NOx regimes and for AM3B at NOx < 0.5 ppbv. We attribute the bias in AM3ST to the overestimate of glyoxal under low NOx and underestimate of HCHO across NOx regimes. The bias in AM3B is mainly due to the underestimate of HCHO.
Figure 4.
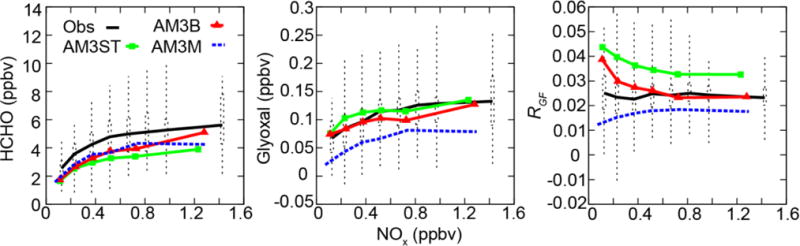
HCHO (ppbv), glyoxal (ppbv), and RGF in each NOx bin below 1.5 km during SENEX. Dots, dashed boxes and whiskers are the mean, interquartile range and lowest and highest of the observations; green squares and red triangles are the mean of AM3ST with γglyx=2.0 × 10−3 and AM3B without heterogeneous loss of glyoxal respectively; blue dashed lines are the mean of AM3M without heterogeneous loss of glyoxal.
To examine the NOx dependence of HCHO, glyoxal and RGF from MCM v3.3.1, the AM3M mechanism that mimics MCM v3.3.1 is then tested in our AM3 global model. Comparison to the SENEX data (Figure 4) shows that AM3M can also well reproduce the NOx dependence of HCHO, similar to the performance of other AM3 mechanisms. However, AM3M largely underestimates glyoxal and RGF across all NOx levels, suggesting additional sources of glyoxal needed in the current MCM mechanism. Inclusion of heterogeneous loss of glyoxal would further reduce glyoxal in AM3M and worsen the comparison. One possible reason for the underestimate of glyoxal in AM3M is the lack of a radical propagating channel for the IEPOXOO + HO2 reaction in MCM v3.3.1, as described above.
RGF provides a useful tool to estimate global production of glyoxal from isoprene oxidation. In particular, the fact that RGF is insensitive to NOx allows us to directly compute its global production rate. Assuming 0.4 HCHO yield per isoprene C (Figure 1, also consistent with Palmer et al. [2003]) and a global constant RGF of 2.4% (according to the measurements during SENEX), we can derive the global production of 23 Tg glyoxal with 500 Tg isoprene emitted annually [Guenther et al., 2006]. This is similar to a previous estimate of 21 Tg yr−1 glyoxal from isoprene [Fu et al., 2008].
5 Budget of Glyoxal in North America
Table 1 summarizes the monthly averaged glyoxal budget over North America (20°~55° N, 60°~130° W) in the boundary layer (0–1.5 km) during June–July of 2013 for AM3ST and AM3B. The total chemical production of glyoxal varies with the chemical mechanisms, from 0.32 Tg/month with AM3B to 0.44 Tg/month with AM3ST. The major sink of glyoxal with both mechanisms is photolysis, followed by aerosol uptake and OH oxidation for AM3ST and OH oxidation and aerosol uptake for AM3B, although the contribution of each pathway varies with mechanisms and γglyx. For example, in AM3ST, heterogeneous loss is an important sink, accounting for 26% of the total chemical loss of glyoxal; however, the contribution is negligible in the AM3B simulations. Deposition accounts for only 1–2% of total glyoxal loss in the model. We show in this work that the estimates of sources and sinks of glyoxal are dependent on the choice of gas-phase chemistry.
Table 1.
Budget of glyoxal over North America (20°~55° N, 60°~130° W) below 1.5 km in June–July of 2013. Percentage is the contribution of each pathway to the total chemical loss of glyoxal.
| AM3ST
|
AM3B
|
|
|---|---|---|
| γglyx (×10−3) | 2.0 | 0 |
| Production (Tg/month) | 0.44 | 0.32 |
| Chemical Loss (Tg/month) | 0.41 | 0.29 |
| Photolysis | 57% | 77% |
| OH | 17% | 23% |
| Uptake by Aerosols | 26% | – |
| Dry Deposition (Tg/month) | 1.2×10−2 | 1.2×10−2 |
| Wet Deposition (Tg/month) | 1.1×10−2 | 1.6×10−2 |
6 Conclusions and Discussion
A 0-D photochemical box model and a 3-D chemistry-climate model applied to data from the Southeast U.S. during the SENEX field campaign provide the first model evaluation of in situ glyoxal aircraft observations.
We find that the three mechanisms (AM3ST, AM3B, and MCM v3.3.1) show similarity in HCHO but large differences in glyoxal, leading to opposite NOx dependence of RGF. Under low NOx conditions, the AM3 mechanisms predict much higher glyoxal yields than MCM v3.3.1, largely due to the significant contribution from isomerization of ISOPO2 and oxidation of IEPOX via the ISOPO2+HO2 pathway. Although the three mechanisms agree better under high NOx conditions, they show different pathways contributing to glyoxal production.
With the constraints from field measurements during SENEX, AM3ST with an effective reactive uptake coefficient γglyx of 2 × 10−3 and AM3B without heterogeneous loss of glyoxal can best reproduce the observed vertical profile. The latter shows better agreement with observed RGF in the boundary layer. These two choices lead to less than 0.8 μg m−3 or negligible of glyoxal SOA in the boundary over the Southeast U.S., accounting for 0–14% of the total SOA in this region (Figure S7). These are likely the lower limit of glyoxal SOA due to missing sources of glyoxal that are not included in our model. Over North America, glyoxal sinks are dominated by photolysis, followed by aerosol uptake and OH oxidation for AM3ST and OH oxidation and aerosol uptake for AM3B. Dry and wet deposition are negligible for both AM3 versions (Table 1).
In the boundary layer, observations suggest a very similar NOx dependence for HCHO and glyoxal, resulting in a nearly constant RGF across NOx levels [Kaiser et al., 2015]. Both AM3ST and AM3B can roughly capture the NOx dependence of HCHO and glyoxal, although AM3ST tends to overestimate RGF at all NOx regimes, likely due to the overestimate of glyoxal at low NOx and underestimate of HCHO across NOx regimes. AM3B shows a positive bias in RGF at NOx < 0.5 ppbv due to insufficient production of HCHO. The MCM v3.3.1 like mechanism (“AM3M”) shows a large underestimate of glyoxal across all NOx levels. One possible reason is the lack of a radical propagating channel for the IEPOXOO + HO2 reaction in MCM v3.3.1.
While glyoxal has been studied extensively over the past decade to understand the magnitude of its heterogeneous uptake, we show here that its gas-phase production is a large source of uncertainty that requires equal consideration. We find that its production from isoprene oxidation varies greatly among different chemical mechanisms. This in turn greatly impacts global estimates of glyoxal and in particular its contribution to SOA, especially in regions with low to moderate NOx levels (Figure 1). Under high NOx conditions, models differ significantly in the production of glyoxal from oxidation of glycolaldehyde, as well as the fate of δ-ISOPO2. Under low NOx conditions, there is very little laboratory evidence available on the production of glyoxal from IEPOX, HPALDs, di-HPCARPs or other intermediate products. Although heterogeneous loss of IEPOX is not included in current work, sensitivity tests show that inclusion of this uptake reduces glyoxal concentrations by 10–20% for the mean vertical profile over the Southeast U.S. Thus, the fate of IEPOX represents another mechanistic uncertainty for predicting glyoxal. Also, there are missing sources that are not represented in our model [Volkamer et al., 2015] would increase modeled glyoxal and thus also inferred SOA. Future laboratory measurements are urgently needed and may have important implications for understanding the contribution of glyoxal to SOA in past and future atmospheres.
Supplementary Material
Key Points.
Box model simulated glyoxal production from three isoprene oxidation mechanisms differ greatly
Aerosol uptake of glyoxal was constrained using airborne in situ measurements and a global model
Model results show that glyoxal contributes 0–14% of SOA in the Southeast U.S. during summer
Acknowledgments
The authors thank Charles A. Brock (NOAA) for providing aerosol size data, Vaishali Naik (UCAR/NOAA) for providing emission inventories from the SENEX campaign, and William Cooke for the help with convection scheme of the AM3 model. JL, JM and LWH acknowledge supports by the NOAA Climate Program Office grant # NA13OAR4310071. KEM, RAW and SSB acknowledge support from the NOAA Atmospheric Chemistry, Climate and Carbon Cycle (AC4) program. JK, FNK, GMW, and TFH are grateful for support from EPA Science to Achieve Results (STAR) program grant 83540601 and NASA grant NNH10ZDA001N-SEAC4RS. J. Kaiser acknowledges support from NASA Headquarters under the NASA Earth and Space Science Fellowship Program – grant NNX14AK97H. RV is grateful for support from NSF EAGER award AGS-1452317. VFM acknowledges support from NSF (AGS-1546136). We thank the staff at the NOAA Aircraft Operations Center and the WP-3D flight crew for the help in instrumenting the aircraft and for conducting the flights. Special thanks go to Songmiao Fan (NOAA) for helpful discussions. This research has not been subjected to any EPA review and therefore does not necessarily reflect the views of the agency, and no official endorsement should be inferred.
References
- Anderson DC, et al. Measured and modeled CO and NOy in DISCOVER-AQ: An evaluation of emissions and chemistry over the eastern US. Atmos Environ. 2014;96:78–87. doi: http://dx.doi.org/10.1016/j.atmosenv.2014.07.004. [Google Scholar]
- Bates KH, Crounse JD, St Clair JM, Bennett NB, Nguyen TB, Seinfeld JH, Stoltz BM, Wennberg PO. Gas Phase Production and Loss of Isoprene Epoxydiols. J Phys Chem A. 2014;118(7):1237–1246. doi: 10.1021/jp4107958. [DOI] [PubMed] [Google Scholar]
- Butkovskaya NI, Pouvesle N, Kukui A, Le Bras G. Mechanism of the OH-Initiated Oxidation of Glycolaldehyde over the Temperature Range 233−296 K. J Phys Chem A. 2006;110(50):13492–13499. doi: 10.1021/jp064993k. [DOI] [PubMed] [Google Scholar]
- Carlton AG, Turpin BJ, Altieri KE, Seitzinger S, Reff A, Lim HJ, Ervens B. Atmospheric oxalic acid and SOA production from glyoxal: Results of aqueous photooxidation experiments. Atmos Environ. 2007;41(35):7588–7602. doi: http://dx.doi.org/10.1016/j.atmosenv.2007.05.035. [Google Scholar]
- Cazorla M, Wolfe GM, Bailey SA, Swanson AK, Arkinson HL, Hanisco TF. A new airborne laser-induced fluorescence instrument for in situ detection of formaldehyde throughout the troposphere and lower stratosphere. Atmos Meas Tech. 2015;8(2):541–552. doi: 10.5194/amt-8-541-2015. [DOI] [Google Scholar]
- Corrigan AL, Hanley SW, De Haan DO. Uptake of Glyoxal by Organic and Inorganic Aerosol. Environ Sci Technol. 2008;42(12):4428–4433. doi: 10.1021/es7032394. [DOI] [PubMed] [Google Scholar]
- Crounse JD, Paulot F, Kjaergaard HG, Wennberg PO. Peroxy radical isomerization in the oxidation of isoprene. Phys Chem Chem Phys. 2011;13(30):13607–13613. doi: 10.1039/c1cp21330j. [DOI] [PubMed] [Google Scholar]
- DiGangi JP, et al. Observations of glyoxal and formaldehyde as metrics for the anthropogenic impact on rural photochemistry. Atmos Chem Phys. 2012;12(20):9529–9543. doi: 10.5194/acp-12-9529-2012. [DOI] [Google Scholar]
- Dillon TJ, Crowley JN. Direct detection of OH formation in the reactions of HO2 with CH3C(O)O2 and other substituted peroxy radicals. Atmos Chem Phys. 2008;8(16):4877–4889. doi: 10.5194/acp-8-4877-2008. [DOI] [Google Scholar]
- Donner LJ, et al. The Dynamical Core, Physical Parameterizations, and Basic Simulation Characteristics of the Atmospheric Component AM3 of the GFDL Global Coupled Model CM3. J Clim. 2011;24(13):3484–3519. doi: 10.1175/2011jcli3955.1. [DOI] [Google Scholar]
- Emmerson KM, Evans MJ. Comparison of tropospheric gas-phase chemistry schemes for use within global models. Atmos Chem Phys. 2009;9(5):1831–1845. doi: 10.5194/acp-9-1831-2009. [DOI] [Google Scholar]
- Ervens B, Turpin BJ, Weber RJ. Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies. Atmos Chem Phys. 2011;11(21):11069–11102. doi: 10.5194/acp-11-11069-2011. [DOI] [Google Scholar]
- Feierabend KJ, Flad JE, Brown SS, Burkholder JB. HCO Quantum Yields in the Photolysis of HC(O)C(O)H (Glyoxal) between 290 and 420 nm. J Phys Chem A. 2009;113(27):7784–7794. doi: 10.1021/jp9033003. [DOI] [PubMed] [Google Scholar]
- Fu TM, Jacob DJ, Wittrock F, Burrows JP, Vrekoussis M, Henze DK. Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols. J Geophys Res Atmos. 2008;113(D15):D15303. doi: 10.1029/2007jd009505. [DOI] [Google Scholar]
- Galloway MM, Chhabra PS, Chan AWH, Surratt JD, Flagan RC, Seinfeld JH, Keutsch FN. Glyoxal uptake on ammonium sulphate seed aerosol: reaction products and reversibility of uptake under dark and irradiated conditions. Atmos Chem Phys. 2009;9(10):3331–3345. doi: 10.5194/acp-9-3331-2009. [DOI] [Google Scholar]
- Galloway MM, Huisman AJ, Yee LD, Chan AWH, Loza CL, Seinfeld JH, Keutsch FN. Yields of oxidized volatile organic compounds during the OH radical initiated oxidation of isoprene, methyl vinyl ketone, and methacrolein under high-NOx conditions. Atmos Chem Phys. 2011a;11(21):10779–10790. doi: 10.5194/acp-11-10779-2011. [DOI] [Google Scholar]
- Galloway MM, Loza CL, Chhabra PS, Chan AWH, Yee LD, Seinfeld JH, Keutsch FN. Analysis of photochemical and dark glyoxal uptake: Implications for SOA formation. Geophys Res Lett. 2011b;38:L17811. doi: 10.1029/2011gl048514. [DOI] [Google Scholar]
- Gaston CJ, Riedel TP, Zhang Z, Gold A, Surratt JD, Thornton JA. Reactive Uptake of an Isoprene-Derived Epoxydiol to Submicron Aerosol Particles. Environ Sci Technol. 2014;48(19):11178–11186. doi: 10.1021/es5034266. [DOI] [PubMed] [Google Scholar]
- Gomez ME, Lin Y, Guo S, Zhang R. Heterogeneous Chemistry of Glyoxal on Acidic Solutions. An Oligomerization Pathway for Secondary Organic Aerosol Formation. J Phys Chem A. 2015;119(19):4457–4463. doi: 10.1021/jp509916r. [DOI] [PubMed] [Google Scholar]
- Guenther A, Karl T, Harley P, Wiedinmyer C, Palmer PI, Geron C. Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature) Atmos Chem Phys. 2006;6(11):3181–3210. doi: 10.5194/acp-6-3181-2006. [DOI] [Google Scholar]
- Hasson AS, Tyndall GS, Orlando JJ. A Product Yield Study of the Reaction of HO2 Radicals with Ethyl Peroxy (C2H5O2), Acetyl Peroxy (CH3C(O)O2), and Acetonyl Peroxy (CH3C(O)CH2O2) Radicals. J Phys Chem A. 2004;108(28):5979–5989. doi: 10.1021/jp048873t. [DOI] [Google Scholar]
- Hastings WP, Koehler CA, Bailey EL, De Haan DO. Secondary Organic Aerosol Formation by Glyoxal Hydration and Oligomer Formation: Humidity Effects and Equilibrium Shifts during Analysis. Environ Sci Technol. 2005;39(22):8728–8735. doi: 10.1021/es050446l. [DOI] [PubMed] [Google Scholar]
- Hays MD, Geron CD, Linna KJ, Smith ND, Schauer JJ. Speciation of Gas-Phase and Fine Particle Emissions from Burning of Foliar Fuels. Environ Sci Technol. 2002;36(11):2281–2295. doi: 10.1021/es0111683. [DOI] [PubMed] [Google Scholar]
- Hudman RC, et al. Surface and lightning sources of nitrogen oxides over the United States: Magnitudes, chemical evolution, and outflow. J Geophys Res Atmos. 2007;112(D12) doi: 10.1029/2006JD007912. [DOI] [Google Scholar]
- Huijnen V, Williams JE, Flemming J. Modeling global impacts of heterogeneous loss of HO2 on cloud droplets, ice particles and aerosols. Atmos Chem Phys Discuss. 2014;14(6):8575–8632. doi: 10.5194/acpd-14-8575-2014. [DOI] [Google Scholar]
- Jacob DJ. Heterogeneous chemistry and tropospheric ozone. Atmos Environ. 2000;34(12–14):2131–2159. doi: http://dx.doi.org/10.1016/S1352-2310(99)00462-8. [Google Scholar]
- Jacobs MI, Darer AI, Elrod MJ. Rate Constants and Products of the OH Reaction with Isoprene-Derived Epoxides. Environ Sci Technol. 2013;47(22):12868–12876. doi: 10.1021/es403340g. [DOI] [PubMed] [Google Scholar]
- Jang M, Czoschke NM, Lee S, Kamens RM. Heterogeneous Atmospheric Aerosol Production by Acid-Catalyzed Particle-Phase Reactions. Science. 2002;298(5594):814–817. doi: 10.1126/science.1075798. [DOI] [PubMed] [Google Scholar]
- Jenkin ME, Hurley MD, Wallington TJ. Investigation of the radical product channel of the CH3C(O)O2 + HO2 reaction in the gas phase. Phys Chem Chem Phys. 2007;9(24):3149–3162. doi: 10.1039/B702757E. [DOI] [PubMed] [Google Scholar]
- Jenkin ME, Saunders SM, Pilling MJ. The tropospheric degradation of volatile organic compounds: a protocol for mechanism development. Atmos Environ. 1997;31(1):81–104. doi: http://dx.doi.org/10.1016/S1352-2310(96)00105-7. [Google Scholar]
- Jenkin ME, Young JC, Rickard AR. The MCM v3.3.1 degradation scheme for isoprene. Atmos Chem Phys. 2015;15(20):11433–11459. doi: 10.5194/acp-15-11433-2015. [DOI] [Google Scholar]
- Kaiser J, et al. Reassessing the ratio of glyoxal to formaldehyde as an indicator of hydrocarbon precursor speciation. Atmos Chem Phys. 2015;15(13):7571–7583. doi: 10.5194/acp-15-7571-2015. [DOI] [Google Scholar]
- Kampf CJ, Waxman EM, Slowik JG, Dommen J, Pfaffenberger L, Praplan AP, Prévôt ASH, Baltensperger U, Hoffmann T, Volkamer R. Effective Henry’s Law Partitioning and the Salting Constant of Glyoxal in Aerosols Containing Sulfate. Environ Sci Technol. 2013;47(9):4236–4244. doi: 10.1021/es400083d. [DOI] [PubMed] [Google Scholar]
- Knote C, et al. Simulation of semi-explicit mechanisms of SOA formation from glyoxal in aerosol in a 3-D model. Atmos Chem Phys. 2014;14(12):6213–6239. doi: 10.5194/acp-14-6213-2014. [DOI] [Google Scholar]
- Kroll JH, Ng NL, Murphy SM, Varutbangkul V, Flagan RC, Seinfeld JH. Chamber studies of secondary organic aerosol growth by reactive uptake of simple carbonyl compounds. J Geophys Res Atmos. 2005;110(D23207) doi: 10.1029/2005JD006004. [DOI] [Google Scholar]
- Kurtén T, Elm J, Prisle NL, Mikkelsen KV, Kampf CJ, Waxman EM, Volkamer R. Computational Study of the Effect of Glyoxal–Sulfate Clustering on the Henry’s Law Coefficient of Glyoxal. J Phys Chem A. 2015;119(19):4509–4514. doi: 10.1021/jp510304c. [DOI] [PubMed] [Google Scholar]
- Lamarque JF, Kyle GP, Meinshausen M, Riahi K, Smith S, van Vuuren D, Conley A, Vitt F. Global and regional evolution of short-lived radiatively-active gases and aerosols in the Representative Concentration Pathways. Clim Change. 2011;109(1–2):191–212. doi: 10.1007/s10584-011-0155-0. [DOI] [Google Scholar]
- Lee L, Teng AP, Wennberg PO, Crounse JD, Cohen RC. On Rates and Mechanisms of OH and O3 Reactions with Isoprene-Derived Hydroxy Nitrates. J Phys Chem A. 2014;118(9):1622–1637. doi: 10.1021/jp4107603. [DOI] [PubMed] [Google Scholar]
- Li J, Cleveland M, Ziemba LD, Griffin RJ, Barsanti KC, Pankow JF, Ying Q. Modeling regional secondary organic aerosol using the Master Chemical Mechanism. Atmos Environ. 2015;102:52–61. doi: http://dx.doi.org/10.1016/j.atmosenv.2014.11.054. [Google Scholar]
- Liggio J, Li SM, McLaren R. Heterogeneous Reactions of Glyoxal on Particulate Matter: Identification of Acetals and Sulfate Esters. Environ Sci Technol. 2005a;39(6):1532–1541. doi: 10.1021/es048375y. [DOI] [PubMed] [Google Scholar]
- Liggio J, Li SM, McLaren R. Reactive uptake of glyoxal by particulate matter. J Geophys Res Atmos. 2005b;110(D10):D10304. doi: 10.1029/2004jd005113. [DOI] [Google Scholar]
- Lim HJ, Carlton AG, Turpin BJ. Isoprene Forms Secondary Organic Aerosol through Cloud Processing: Model Simulations. Environ Sci Technol. 2005;39(12):4441–4446. doi: 10.1021/es048039h. [DOI] [PubMed] [Google Scholar]
- Lin M, et al. Transport of Asian ozone pollution into surface air over the western United States in spring. J Geophys Res Atmos. 2012;117:D00V07. doi: 10.1029/2011jd016961. [DOI] [Google Scholar]
- Lockwood AL, Shepson PB, Fiddler MN, Alaghmand M. Isoprene nitrates: preparation, separation, identification, yields, and atmospheric chemistry. Atmos Chem Phys. 2010;10(13):6169–6178. doi: 10.5194/acp-10-6169-2010. [DOI] [Google Scholar]
- MacDonald SM, Oetjen H, Mahajan AS, Whalley LK, Edwards PM, Heard DE, Jones CE, Plane JMC. DOAS measurements of formaldehyde and glyoxal above a south-east Asian tropical rainforest. Atmos Chem Phys. 2012;12(13):5949–5962. doi: 10.5194/acp-12-5949-2012. [DOI] [Google Scholar]
- Mao J, Horowitz LW, Naik V, Fan S, Liu J, Fiore AM. Sensitivity of tropospheric oxidants to biomass burning emissions: implications for radiative forcing. Geophys Res Lett. 2013a;40(6):1241–1246. doi: 10.1002/grl.50210. [DOI] [Google Scholar]
- Mao J, et al. Chemistry of hydrogen oxide radicals (HOx) in the Arctic troposphere in spring. Atmos Chem Phys. 2010;10(13):5823–5838. doi: 10.5194/acp-10-5823-2010. [DOI] [Google Scholar]
- Mao J, Paulot F, Jacob DJ, Cohen RC, Crounse JD, Wennberg PO, Keller CA, Hudman RC, Barkley MP, Horowitz LW. Ozone and organic nitrates over the eastern United States: Sensitivity to isoprene chemistry. J Geophys Res Atmos. 2013b;118(19):11,256–211,268. doi: 10.1002/jgrd.50817. [DOI] [Google Scholar]
- Marais EA, et al. Aqueous-phase mechanism for secondary organic aerosol formation from isoprene: application to the southeast United States and co-benefit of SO2 emission controls. Atmos Chem Phys. 2016;16(3):1603–1618. doi: 10.5194/acp-16-1603-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CC, Abad GG, Wang H, Liu X, Kurosu T, Jacob DJ, Chance K. Glyoxal retrieval from the Ozone Monitoring Instrument. Atmos Meas Tech Discuss. 2014;7(6):6065–6112. doi: 10.5194/amtd-7-6065-2014. [DOI] [Google Scholar]
- Millet DB, Jacob DJ, Boersma KF, Fu TM, Kurosu TP, Chance K, Heald CL, Guenther A. Spatial distribution of isoprene emissions from North America derived from formaldehyde column measurements by the OMI satellite sensor. J Geophys Res Atmos. 2008;113:D02307. doi: 10.1029/2007jd008950. [DOI] [Google Scholar]
- Min KE, et al. A broadband cavity enhanced absorption spectrometer for aircraft measurements of glyoxal, methylglyoxal, nitrous acid, nitrogen dioxide, and water vapor. Atmos Meas Tech Discuss. 2015;8(10):11209–11254. doi: 10.5194/amtd-8-11209-2015. [DOI] [Google Scholar]
- Müller JF, Peeters J, Stavrakou T. Fast photolysis of carbonyl nitrates from isoprene. Atmos Chem Phys. 2014;14(5):2497–2508. doi: 10.5194/acp-14-2497-2014. [DOI] [Google Scholar]
- Myriokefalitakis S, Vrekoussis M, Tsigaridis K, Wittrock F, Richter A, Brühl C, Volkamer R, Burrows JP, Kanakidou M. The influence of natural and anthropogenic secondary sources on the glyoxal global distribution. Atmos Chem Phys. 2008;8(16):4965–4981. doi: 10.5194/acp-8-4965-2008. [DOI] [Google Scholar]
- Naik V, Horowitz LW, Fiore AM, Ginoux P, Mao J, Aghedo AM, Levy H. Impact of preindustrial to present-day changes in short-lived pollutant emissions on atmospheric composition and climate forcing. J Geophys Res Atmos. 2013;118(14):8086–8110. doi: 10.1002/jgrd.50608. [DOI] [Google Scholar]
- Nguyen TB, Crounse JD, Teng AP, St Clair JM, Paulot F, Wolfe GM, Wennberg PO. Rapid deposition of oxidized biogenic compounds to a temperate forest. Proc Natl Acad Sci. 2015;112(5):E392–E401. doi: 10.1073/pnas.1418702112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niki H, Maker PD, Savage CM, Hurley MD. Fourier transform infrared study of the kinetics and mechanisms for the chlorine-atom- and hydroxyl-radical-initiated oxidation of glycolaldehyde. J Phys Chem. 1987;91(8):2174–2178. doi: 10.1021/j100292a038. [DOI] [Google Scholar]
- Nozière B, Dziedzic P, Córdova A. Products and Kinetics of the Liquid-Phase Reaction of Glyoxal Catalyzed by Ammonium Ions (NH4+) J Phys Chem A. 2009;113(1):231–237. doi: 10.1021/jp8078293. [DOI] [PubMed] [Google Scholar]
- Palmer PI, Jacob DJ, Fiore AM, Martin RV, Chance K, Kurosu TP. Mapping isoprene emissions over North America using formaldehyde column observations from space. J Geophys Res Atmos. 2003;108(D6):4180. doi: 10.1029/2002JD002153. [DOI] [Google Scholar]
- Paulot F, Crounse JD, Kjaergaard HG, Kroll JH, Seinfeld JH, Wennberg PO. Isoprene photooxidation: new insights into the production of acids and organic nitrates. Atmos Chem Phys. 2009a;9(4):1479–1501. doi: 10.5194/acp-9-1479-2009. [DOI] [Google Scholar]
- Paulot F, Crounse JD, Kjaergaard HG, Kürten A, St Clair JM, Seinfeld JH, Wennberg PO. Unexpected Epoxide Formation in the Gas-Phase Photooxidation of Isoprene. Science. 2009b;325(5941):730–733. doi: 10.1126/science.1172910. [DOI] [PubMed] [Google Scholar]
- Paulot F, Ginoux P, Cooke WF, Donner LJ, Fan S, Lin M, Mao J, Naik V, Horowitz LW. Sensitivity of nitrate aerosols to ammonia emissions and to nitrate chemistry: implications for present and future nitrate optical depth. Atmos Chem Phys Discuss. 2015;15(18):25739–25788. doi: 10.5194/acpd-15-25739-2015. [DOI] [Google Scholar]
- Peeters J, Müller JF, Stavrakou T, Nguyen VS. Hydroxyl Radical Recycling in Isoprene Oxidation Driven by Hydrogen Bonding and Hydrogen Tunneling: The Upgraded LIM1 Mechanism. J Phys Chem A. 2014;118(38):8625–8643. doi: 10.1021/jp5033146. [DOI] [PubMed] [Google Scholar]
- Peeters J, Nguyen TL. Unusually Fast 1,6-H Shifts of Enolic Hydrogens in Peroxy Radicals: Formation of the First-Generation C2 and C3 Carbonyls in the Oxidation of Isoprene. J Phys Chem A. 2012;116(24):6134–6141. doi: 10.1021/jp211447q. [DOI] [PubMed] [Google Scholar]
- Praske E, Crounse JD, Bates KH, Kurtén T, Kjaergaard HG, Wennberg PO. Atmospheric Fate of Methyl Vinyl Ketone: Peroxy Radical Reactions with NO and HO2. J Phys Chem A. 2015;119(19):4562–4572. doi: 10.1021/jp5107058. [DOI] [PubMed] [Google Scholar]
- Pye HOT, et al. Epoxide Pathways Improve Model Predictions of Isoprene Markers and Reveal Key Role of Acidity in Aerosol Formation. Environ Sci Technol. 2013;47(19):11056–11064. doi: 10.1021/es402106h. [DOI] [PubMed] [Google Scholar]
- Rivera-Rios JC, et al. Conversion of hydroperoxides to carbonyls in field and laboratory instrumentation: Observational bias in diagnosing pristine versus anthropogenically controlled atmospheric chemistry. Geophys Res Lett. 2014;41(23):8645–8651. doi: 10.1002/2014GL061919. [DOI] [Google Scholar]
- Sander R. Compilation of Henry’s law constants (version 4.0) for water as solvent. Atmos Chem Phys. 2015;15(8):4399–4981. doi: 10.5194/acp-15-4399-2015. [DOI] [Google Scholar]
- Saunders SM, Jenkin ME, Derwent RG, Pilling MJ. Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds. Atmos Chem Phys. 2003;3(1):161–180. doi: 10.5194/acp-3-161-2003. [DOI] [Google Scholar]
- St Clair JM, Rivera-Rios JC, Crounse JD, Knap HC, Bates KH, Teng AP, Jørgensen S, Kjaergaard HG, Keutsch FN, Wennberg PO. Kinetics and Products of the Reaction of the First-Generation Isoprene Hydroxy Hydroperoxide (ISOPOOH) with OH. J Phys Chem A. 2016a;120(9):1441–1451. doi: 10.1021/acs.jpca.5b06532. [DOI] [PubMed] [Google Scholar]
- St Clair JM, Rivera-Rios JC, Crounse JD, Praske E, Kim MJ, Wolfe GM, Keutsch FN, Wennberg PO, Hanisco TF. Investigation of a potential HCHO measurement artifact from ISOPOOH. Atmos Meas Tech Discuss. 2016b;2016:1–15. doi: 10.5194/amt-2016-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavrakou T, Müller JF, De Smedt I, Van Roozendael M, Kanakidou M, Vrekoussis M, Wittrock F, Richter A, Burrows JP. The continental source of glyoxal estimated by the synergistic use of spaceborne measurements and inverse modelling. Atmos Chem Phys. 2009;9(21):8431–8446. doi: 10.5194/acp-9-8431-2009. [DOI] [Google Scholar]
- Stavrakou T, Peeters J, Müller JF. Improved global modelling of HOx recycling in isoprene oxidation: evaluation against the GABRIEL and INTEX-A aircraft campaign measurements. Atmos Chem Phys. 2010;10(20):9863–9878. doi: 10.5194/acp-10-9863-2010. [DOI] [Google Scholar]
- Teng AP, Crounse JD, Lee L, St Clair JM, Cohen RC, Wennberg PO. Hydroxy nitrate production in the OH-initiated oxidation of alkenes. Atmos Chem Phys. 2015;15(8):4297–4316. doi: 10.5194/acp-15-4297-2015. [DOI] [Google Scholar]
- Travis K, et al. SEAC4RS Science Team Meeting. Pasadena, California: 2015. Declining NOx in the Southeast US and implications for ozone-NOx-VOC chemistry. [Google Scholar]
- Volkamer R, et al. Aircraft measurements of BrO, IO, glyoxal, NO2, H2O, O2–O2 and aerosol extinction profiles in the tropics: comparison with aircraft-/ship-based in situ and lidar measurements. Atmos Meas Tech. 2015;8(5):2121–2148. doi: 10.5194/amt-8-2121-2015. [DOI] [Google Scholar]
- Volkamer R, Barnes I, Platt U, Molina LT, Molina MJ. Remote Sensing of Glyoxal by Differential Optical Absorption Spectroscopy (DOAS): Advancements in Simulation Chamber and Field Experiments, in. In: Barnes I, Rudzinski K, editors. Environmental Simulation Chambers: Application to Atmospheric Chemical Processes. Springer; Netherlands: 2006. pp. 129–141. [DOI] [Google Scholar]
- Volkamer R, Molina LT, Molina MJ, Shirley T, Brune WH. DOAS measurement of glyoxal as an indicator for fast VOC chemistry in urban air. Geophys Res Lett. 2005;32:L08806. doi: 10.1029/2005gl022616. [DOI] [Google Scholar]
- Volkamer R, San Martini F, Molina LT, Salcedo D, Jimenez JL, Molina MJ. A missing sink for gas-phase glyoxal in Mexico City: Formation of secondary organic aerosol. Geophys Res Lett. 2007;34:L19807. doi: 10.1029/2007gl030752. [DOI] [Google Scholar]
- Volkamer R, Ziemann PJ, Molina MJ. Secondary Organic Aerosol Formation from Acetylene (C2H2): seed effect on SOA yields due to organic photochemistry in the aerosol aqueous phase. Atmos Chem Phys. 2009;9(6):1907–1928. doi: 10.5194/acp-9-1907-2009. [DOI] [Google Scholar]
- Vrekoussis M, Wittrock F, Richter A, Burrows JP. GOME-2 observations of oxygenated VOCs: what can we learn from the ratio glyoxal to formaldehyde on a global scale? Atmos Chem Phys. 2010;10(21):10145–10160. doi: 10.5194/acp-10-10145-2010. [DOI] [Google Scholar]
- Warneke C, et al. Biogenic emission measurement and inventories determination of biogenic emissions in the eastern United States and Texas and comparison with biogenic emission inventories. J Geophys Res Atmos. 2010;115(D7):D00F18. doi: 10.1029/2009jd012445. [DOI] [Google Scholar]
- Warneke C, et al. Instrumentation and Measurement Strategy for the NOAA SENEX Aircraft Campaign as Part of the Southeast Atmosphere Study 2013. Atmos Meas Tech Discuss. 2016;2016:1–39. doi: 10.5194/amt-2015-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washenfelder RA, et al. The glyoxal budget and its contribution to organic aerosol for Los Angeles, California, during CalNex 2010. J Geophys Res Atmos. 2011;116(D21):D00V02. doi: 10.1029/2011jd016314. [DOI] [Google Scholar]
- Waxman EM, Elm J, Kurtén T, Mikkelsen KV, Ziemann PJ, Volkamer R. Glyoxal and Methylglyoxal Setschenow Salting Constants in Sulfate, Nitrate, and Chloride Solutions: Measurements and Gibbs Energies. Environ Sci Technol. 2015;49(19):11500–11508. doi: 10.1021/acs.est.5b02782. [DOI] [PubMed] [Google Scholar]
- Wolfe GM, et al. Formaldehyde production from isoprene oxidation across NOx regimes. Atmos Chem Phys. 2016;16(4):2597–2610. doi: 10.5194/acp-16-2597-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong F, et al. Observation of isoprene hydroxynitrates in the Southeastern United States and implications for the fate of NOx. Atmos Chem Phys Discuss. 2015;15(13):17843–17886. doi: 10.5194/acpd-15-17843-2015. [DOI] [Google Scholar]
- Ying Q, Li J, Kota SH. Significant Contributions of Isoprene to Summertime Secondary Organic Aerosol in Eastern United States. Environ Sci Technol. 2015;49(13):7834–7842. doi: 10.1021/acs.est.5b02514. [DOI] [PubMed] [Google Scholar]
- Zhao J, Levitt NP, Zhang R, Chen J. Heterogeneous Reactions of Methylglyoxal in Acidic Media: Implications for Secondary Organic Aerosol Formation. Environ Sci Technol. 2006;40(24):7682–7687. doi: 10.1021/es060610k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.