

Abstract
The measurement of select circulating metabolites such as creatinine, glucose, and cholesterol are integral to clinical medicine, with implications for diagnosis, prognosis, and treatment. Metabolomics studies in Nephrology research seek to build on this paradigm, with the goal to identify novel markers and causal participants in the pathogenesis of kidney disease and its complications. This article reviews three themes pertinent to this goal. Each is rooted in long-established principles of human physiology, with recent updates enabled by metabolomics and other tools. First, the kidney has a broad and heterogeneous impact on circulating metabolites, with progressive loss of kidney function resulting in a multitude of small molecule alterations. Second, an increasing number of circulating metabolites have been shown to possess functional roles, in some cases acting as ligands for specific G-protein coupled receptors. Third, circulating metabolites traffic through varied, and sometimes complex, inter-organ circuits. Taken together, these themes emphasize the importance of viewing renal metabolomics at the systems-level, recognizing the diverse origins and physiologic effects of blood metabolites. However, how to synthesize these themes and how to establish clinical relevance remain uncertain and will require further investigation.
Keywords: Metabolomics, Metabolism, Systems Biology
The metabolome refers to the global collection of small molecules–e.g. sugars, amino acids, organic acids, nucleotides, acylcarnitines, lipids, bile acids, etc–in a cell or biologic specimen.1 This includes many molecules, such as creatinine, urea, uric acid, lactic acid, glucose, triglycerides and cholesterol that are routinely measured in clinical practice. A consideration of a few of these molecules is instructive. Creatinine is released primarily from muscle and is excreted by the kidney. Blood levels of creatinine are the most commonly utilized biomarker of renal failure, but the molecule itself is not harmful. Glucose has many sources, including diet, as well as gluconeogenesis within the liver and kidney. It is freely filtered at the glomerulus, but then undergoes almost complete tubular reabsorption such that its blood levels are not directly affected by renal failure.2, 3 It is both a fundamental fuel required throughout the body, but also deleterious if chronically elevated. Cholesterol can be absorbed through diet or synthesized and released from the liver. It circulates in the context of lipoprotein particles and thus does not undergo glomerular filtration; as with glucose, prolonged elevations are harmful. Altogether, these examples demonstrate several important features of circulating metabolites, including the heterogeneous impact of renal failure on blood levels, the potential for both salutary and adverse functional effects, and their complex inter-organ traffic. As will be detailed in this article, each of these features or themes has been reinforced and expanded with the application of metabolomics across clinical, basic, and physiologic studies.
The impact of kidney function on the metabolome
The kidneys generate ~140 liters of glomerular filtrate per day, only to reabsorb the vast majority of filtered sodium and water, glucose, amino acids, and other urinary constituents. Although this continuous cycle of filtration and reabsorption seems energetically wasteful, it permits very high rates of clearance of freely soluble waste products such as creatinine and urea. In addition to glomerular filtration, normal renal excretory function includes tubular secretion, a critical mechanism for the excretion of protein-bound or lipophilic waste products that do not readily cross the glomerular filtration barrier. Finally, the kidneys also take up and catabolize some circulating small molecules, such as asymmetric dimethylarginine (ADMA) and S-adenosylhomocysteine (as well as numerous peptides).4–6 Thus, through a combination of filtration, reabsorption, secretion, and metabolism, the kidneys have a broad and heterogeneous impact on circulating metabolite levels, minimizing the loss of desired nutrients while facilitating the excretion of unwanted metabolic waste products.
Metabolites that are normally cleared by the kidneys accumulate as kidney function declines. The relative rise of metabolites, however, varies significantly. For example protein bound metabolites or metabolites that are catabolized within the kidney can increase out of proportion to the increase in creatinine levels.7 This discrepancy is magnified among patients with ESRD on dialysis. For both hemodialysis and peritoneal dialysis, the clearance of freely soluble metabolites such as creatinine is substantially below normal, but the clearance of protein-bound metabolites is even lower, because it is free (un-bound) levels that determine the diffusion gradient across the hemodialysis or peritoneal membrane. For example, one study showed elevations of phenylacetylglutamine (122-fold), hippurate (108-fold), indoxyl sulfate (116-fold), and p-cresol sulfate (41-fold), four metabolites normally cleared by tubular secretion, significantly greater than for urea (5-fold) and creatinine (13-fold) among ESRD patients.8
Building on these long-established principles of renal physiology, metabolomics studies have generated a broader and more detailed view of how kidney function impacts the metabolome. This includes relatively smaller studies of individuals across different stages of CKD,9–11 as well as large studies correlating metabolites with eGFR in population based cohorts.12 In the largest data set published to date, Sekula and colleagues measured ~500 blood metabolites in 1735 participants of the KORA F4 study, followed by replication in 1164 individuals in the TwinsUK registry. Most (≥95%) of the individuals across these studies had an eGFR > 60 mL/min/1.73m2. Despite a relative paucity of CKD in these samples, this “metabolome-wide association study” nevertheless demonstrated the substantial impact of kidney function–in KORA F4, 112 metabolites were significantly associated with eGFR at a conservative Bonferroni adjusted significance threshold, 54 of which replicated in TwinsUK.13 For select metabolites most strongly correlated with eGFR, estimated on the basis of serum creatinine or cystatin C, the authors then examined their association with measured GFR among 200 participants of the AASK study, highlighting c-mannosyltryptophan and pseudouridine (r=0.78 for both) as alternative or complementary markers of kidney function. One potential advantage of these markers is that unlike creatinine, their levels are largely unaffected by sex.
Measurement of peripheral blood levels can provide a catalog of metabolites that increase when kidney function is reduced, but it does not necessarily prove that the elevations are because of reduced renal clearance. Concurrent measurement in urine can be helpful in this regard. For example, Duranton et al. measured amino acids and amino acid derivatives in plasma and urine from 52 individuals across different stages of CKD and plasma only from 25 individuals on dialysis.14 By examining paired plasma and urine, the authors could conclude that uremic elevations in plasma ADMA are due to decreased urinary clearance, whereas elevations in plasma citrulline are due to systemic overproduction. Increased overproduction can result from changes in diet or changes in endogenous metabolism triggered by renal failure.
Direct measurement of metabolite gradients across the renal vasculature is also informative. We applied metabolomics to plasma obtained from the aorta and renal vein of nine individuals who were already scheduled for left and right heart catheterization.15 Indications for cardiac catheterization spanned evaluation of valve disease, dyspnea, and monitoring of orthotopic heart transplants. Mean age was 72.2, mean eGFR was 65.4 mL/min/1.73m2, and the prevalence of comorbidities such as coronary disease (66%), hypertension (100%), and type 2 diabetes (33%) was high. With these caveats, Figure 1 shows the distribution of mean venous to arterial ratio for ~220 metabolites, providing a graphical depiction of how the human kidney impacts these circulating metabolites. Several observations warrant mention. First, many metabolite levels are lower in the renal vein than in the aorta, consistent with some degree of renal clearance (many of the metabolites that do not change or change minimally from aorta to renal vein are lipids). Second, the drop in creatinine levels provides an index for the effect of glomerular filtration, acknowledging that that 16% fall in creatinine in this sample is less than would occur in healthy adults. Third, a substantial number of metabolites fall more than creatinine does from aorta to renal vein, implicating some additional or alternative mechanism for their excretion, such as tubular secretion or intra-organ metabolism. Paired analysis of urine samples was performed to formally demonstrate tubular secretion of kynurenic acid (fractional excretion in urine >100%) and intra-organ metabolism of choline and citrulline (very low fractional excretion).15 Finally, some metabolites are actually higher in the renal vein than in the aorta, a topic that is discussed in more detail in the section on inter-organ metabolite traffic.
Figure 1. Renal Arterio-Venous Metabolite Gradients in Humans.

Mean venous to arterial ratios for ~220 metabolites, ordered left to right on the x-axis from lowest to highest ratio. For any given metabolite, [V]/[A] <1 is consistent with net renal uptake, whether via filtration, secretion, and/or metabolism, whereas [V]/[A] >1 suggests net release by the kidney. Representative metabolites, including creatinine, kynurenic acid, choline, and citrulline are highlighted.
In addition to studies of mild to moderate kidney dysfunction, metabolomics has been utilized to better characterize uremia. Indeed, given the long-standing interest in small molecules as potential uremic toxins, studies of ESRD were among the earliest applications of metabolomics in nephrology research.16–18 These efforts have rapidly added to the compendium of retained metabolites in ESRD, expanding the list to >270 metabolites.19, 20 These results largely confirm the view of uremia as a state of small molecule retention; however, they have also shown that a substantial number of metabolites, particularly lipid metabolites such as low molecular weight triacylglycerols and select phosphatidylcholines, are depleted in uremic blood (Figure 2).16 Metabolomics studies of ESRD have also underscored the fundamental importance of diet and the gut microbiome on modulating the blood metabolome. In an elegant study, Aronov et al. profiled blood obtained from 9 hemodialysis patients with intact colons and 6 hemodialysis patients who had previously undergone colectomy.21 These authors demonstrated normal levels of established uremic metabolites such as indoxyl sulfate and p-cresol sulfate, metabolites that are known to require bacterial metabolism for their synthesis, among individuals without colons. Similarly, many other compounds measured by mass spectrometry, most of uncertain identity, were only elevated among hemodialysis patients with intact colons. Studies of mice with renal failure in germ-free (no gut microbes) versus control conditions have corroborated these findings, implicating gut microbes in the production of many uremic metabolites.22
Figure 2. ESRD is a State of Small Molecule Disarray.

Mean ratio in peripheral plasma of ESRD patients versus control for ~220 metabolites, with polar (left) and lipid (right) metabolites shown separately. Metabolites are ordered from left to right on the x-axis from highest to lowest ratio. Two representative metabolites, kynurenic acid and arginine are highlighted.
Thus, the impact of kidney function on circulating metabolite levels is complex and includes both direct and indirect effects. Loss of kidney function directly results in the accumulation of many metabolites, although the relative degree of accumulation may differ depending on the relative importance of glomerular filtration, tubular secretion, or intra-organ metabolism for each metabolite. In addition, kidney disease can indirectly modify the rate of metabolite production by leading to alterations in diet or the composition of the gut microbiome,23 or alternatively, can alter metabolism in general through incompletely understood effects on insulin resistance and protein energy wasting.24–26 Finally, non-renal mechanisms of clearance increase for some metabolites in the context of renal failure; for example, creatinine excretion through the gastrointestinal tract is known to increase with advancing CKD.27 Regardless of why they occur, the numerous and significant alterations outlined by metabolomics raise the question of whether select metabolites may play a causal role in the morbidity and mortality that accompany CKD.
Novel functional roles for circulating metabolites
As noted, glucose and cholesterol are paradigmatic examples of how elevations in blood metabolites can exert toxicity. Decades of research have examined the potential deleterious effects of metabolites retained in renal failure, particularly in uremia, where traditional cardiovascular risk factors are less informative as biomarkers and ineffective as treatment targets.28 Although many candidate metabolite toxins have been examined, definitive proof of toxicity has remained elusive. Table 1 summarizes some of the mechanisms by which alterations in the metabolome may exert adverse effects. Here, this article considers two topics in more depth, the identification of trimethylamine-N-oxide (TMAO) as a vascular toxin, and the growing recognition that some metabolites exert hormone-like effects as ligands for specific G-protein coupled receptors (GPCRs).29
Table 1.
Potential Adverse Effects of Metabolite Alterations
| Direct cellular toxicity | A major focus of uremia research, with metabolites such as p-cresol sulfate and indoxyl sulfate shown to impact cell proliferation, cell senescence, fibrosis, and inflammation/oxidative stress (see below).61 |
| Inflammation and oxidative stress | Metabolites such as p-cresol sulfate and indoxyl sulfate can also increase leukocyte activation and adhesion, leukocyte free radical production, and renal cell cytokine expression, and lower endothelial cell glutathione levels.61 |
| Enzyme inhibition | As substrates and products of defined biochemical reactions, some metabolites can modulate specific enzyme actions; e.g. ADMA inhibits nitric oxide synthase, preventing the synthesis of NO from arginine.62 |
| Central nervous system activity | The presence of organic ion transporters at the blood-brain barrier, in some cases the same as expressed in renal tubules,63, 64 suggests that the accumulation of some metabolites that undergo active renal secretion are neurotoxic.65 |
| Protein modification | Metabolites can covalently modify proteins in circulation and in tissue, altering structure and function. Examples include glycation (due to hyperglycemia and oxidative stress) and carbamylation (a urea derived modification).66 |
| Metabolite depletion | Some metabolites are actually depleted in renal failure, for example due to reduced dietary intake, loss of renal biosynthesis, or increased clearance for patients on dialysis. Carnitine depletion has been hypothesized to contribute to skeletal and cardiac myopathy in ESRD.67 |
| GPCR signaling | Several metabolites have emerged as extracellular signaling molecules that are ligands at specific GPCRs, with actions pertinent to blood pressure, metabolism, and inflammation.29 |
TMAO was initially identified as a cardiovascular disease risk marker through a metabolomics analysis of plasma obtained from 75 individuals who subsequently had a myocardial infarction or stroke, or died, and 75 age- and gender-matched controls who did not.30 Several studies since have replicated TMAO’s association with cardiovascular outcomes among individuals with normal kidney function, including a prospective study of 4007 patients undergoing elective coronary angiography.31 TMAO is largely excreted by the kidneys and is significantly elevated in renal failure, but its association with cardiovascular outcomes in the context of CKD has been mixed.32–34 TMAO is derived from the gut microbial conversion of dietary choline and L-carnitine into trimethylamine and hepatic oxidation of trimethylamine to TMAO. A functional role for TMAO in atherogenesis has been hypothesized based on studies showing that TMAO inhibits reverse cholesterol transport, increases platelet hyperresponsiveness, and promotes macrophage foam cell formation in mice–in turn, the use of antibiotics to stop gut microbial production of TMAO is cardioprotective in animals.35, 36 Thus, TMAO is a potential circulating cardiovascular toxin, with blood levels determined by a combination of diet, gut microbial, hepatic, and renal function.
While TMAO was initially highlighted through a metabolomics study of human disease, the potential functional role of other metabolites has emerged from the recognition that they are ligands at specific GPCRs in vitro and in model systems. Many of these metabolites have traditionally been defined in terms of their intracellular bioenergetic functions, but more recent observations have outlined roles as extracellular signaling molecules. For example, He et al. discovered that the citric acid cycle intermediate succinate binds the previously “orphan” GPR91.37 Previously thought to function exclusively as an intermediate in the citric acid cycle, circulating succinate is now recognized to act through this receptor in the kidney to trigger renin release and hypertension in rodents.37, 38 Similarly, Pluznick et al. have shown that circulating short chain fatty acids bind to Olfr78, a receptor expressed in the renal juxtaglomerular apparatus, to mediate renin secretion in mice.39 The short chain fatty acids acetate, propionate, and butyrate are products of the gut microbial fermentation of polysaccharides such as cellulose and starch, providing a potential link between the gut microbiome and blood pressure homeostasis. Bile acids, also modulated by gut bacteria, have emerged as systemic metabolic regulators functioning beyond their traditional role in lipid absorption within the gut lumen. Watanabe et al. have shown that bile acids signal through TGR5 to promote intracellular thyroid hormone activation, whereas others have shown that bile acid signaling through FXR (an intracellular receptor rather than a GPCR) mediates the weight loss effects of sleeve gastrectomy in mice.40, 41 Finally, the tryptophan metabolite kynurenic acid has been identified as a ligand for GPR35, a previously orphan receptor highly expressed on leukocytes,42 and signaling through this receptor induces monocyte firm arrest to endothelium in vitro.43
Of the examples detailed here, only kynurenic acid is an established uremic solute, and its effects through GPR35 raise the possibility it makes a functional contribution to aberrant inflammation in kidney disease. It is likely that future studies will reveal unanticipated biologic effects for other uremic metabolites as well. In addition, metabolite ligands of GPCRs are of interest even if not significantly elevated in renal failure. That these metabolites have cognate cell surface receptors means that even relatively modest concentration changes that might occur with acute or chronic renal dysfunction may be biologically meaningful. They also outline novel treatment approaches. For example, administration of short-chain fatty acids has been shown to protect against ischemic AKI in mice, although it is unclear if this effect is dependent on agonism through Olfr78, other GPCRs, or the fact that they can also inhibit histone deacetylases inside the nucleus.44 Taken together, the recognition that circulating metabolites directly impact a range of biologic processes including blood pressure, metabolism, and inflammation—at least in animal models—enhances interest in circulating metabolites as causal factors in human kidney disease and its complications.
Inter-organ metabolite traffic
Although not as much a focus of metabolomics research as the previous two themes, this topic is discussed here because it is complementary with the overarching focus on circulating metabolites and because it emphasizes the importance of a systems-level view. The kidney’s contribution to inter-organ metabolite circuits is usually considered through its impact on metabolite clearance, whether through filtration, secretion, or catabolism. For example, excess nitrogen from muscle and throughout the body is delivered as amino groups in glutamine, alanine, and other amino acids to the liver, incorporated into urea, and then renally excreted. However, the kidneys play other roles as well, including as the target organ for bioactive metabolites generated elsewhere, such as gut-derived short chain fatty acids or succinate released from other tissues. In addition, the kidneys have anabolic functions, including the synthesis of both proteins (renin and erythropoietin) and metabolites (calcitriol and glucose) with important biologic roles. Under fasting conditions, renal glucose production is responsible for approximately 40% of total body gluconeogenesis.2, 3
Long before the advent of metabolomics, plasma sampling from the renal artery and vein of human volunteers showed that the kidneys are also responsible for the net release of several amino acids, including serine, tyrosine, and arginine.45, 46 These amino acids are not made de novo within the organ, but rather are generated from single enzymatic steps to circulating glycine (hydroxymethyl transfer), phenylalanine (hydroxylation), and citrulline (transamination), respectively.6, 47 The inter-organ exchange required for arginine synthesis is complex; first, ingested glutamine is metabolized within the gut epithelium yielding ammonia and citrulline; second, ammonia is incorporated into urea in the liver whereas citrulline passes through the liver un-metabolized; third, citrulline is taken up by the kidneys where it is transaminated to arginine.48 The kidneys synthesize 2–4 g of arginine per day, which may provide 35%–75% of normal daily arginine intake. Why this should occur in the kidney is unclear. Arginine is an essential component of proteins and plays key roles in other metabolic processes as well. It is tempting to hypothesize that its synthesis in the kidney is related to its role as the precursor for nitric oxide synthase mediated NO production. The kidney also plays a major role in the degradation of the NO synthase inhibitor and arginine derivative ADMA. However, a formal role for renal arginine biosynthesis in the organ’s overall control of systemic blood pressure or vascular reactivity in humans has not been demonstrated.
As previously noted, metabolomic profiling of samples acquired from the aorta and renal vein demonstrates the net release of several metabolites from the kidney. These data recapitulate prior observations with serine, tyrosine, and arginine (Figure 3), and highlight other metabolites that have not been previously described. Although not all of these metabolites are necessarily being synthesized within the organ–for example, the intra-organ breakdown of small peptides or other macromolecules could lead to higher levels of some metabolites in the renal vein relative to the aorta–it is possible that these kinds of studies will reveal new renal anabolic products, perhaps with functional effects throughout the body. Loss of renal anabolic activity, for example in ESRD, could lead to lower levels of these metabolites as has been demonstrated for arginine. Clearly, replication in additional samples followed by in depth mechanistic work in model systems would be required to substantiate these speculations. Regardless, with examples here that span established physiology to hypothesis-generating observations, maintaining a systems-level view will be important for capturing the full discovery potential of metabolomics in kidney disease (Figure 4).
Figure 3. Net Renal Release of Select Metabolites.

Aortic and renal venous levels for creatinine, serine, arginine, and tyrosine, reported as peak areas detected by the mass spectrometer. The uniform decrease in creatinine is a positive control, consistent with renal clearance. Serine and arginine increased from aorta to renal vein in all nine individuals, whereas tyrosine increased in seven individuals (P < 0.001 for all three metabolites).
Figure 4. Selected Examples of Inter-organ Metabolite Traffic.
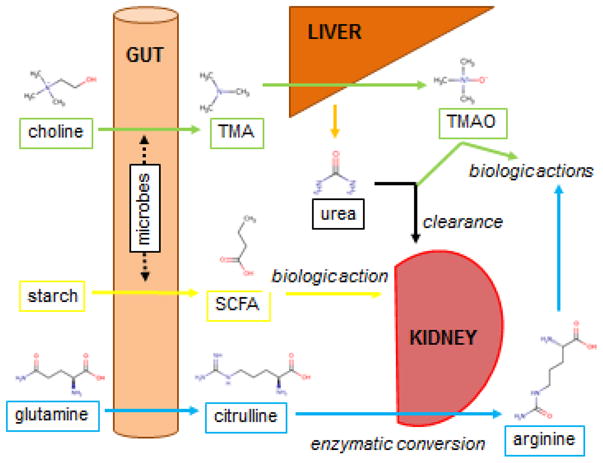
Four distinct circuits are shown. (Green) Dietary choline is converted by gut microbes to trimethylamine (TMA), which undergoes oxidation in the liver to generate trimethylamine-N-oxide (TMAO). TMAO is renally cleared and has toxic cardiovascular effects. (Yellow) Dietary starch is converted by gut microbes to short chain fatty acids (SCFA, butyrate shown), which can then exert biologic effects as ligands for specific GPCRs, including in the kidney. (Blue) Dietary glutamine is converted by enterocytes to citrulline, which can then undergo enzymatic conversion in the kidney to arginine, which then exerts a range of biologic effects. Finally urea, which is synthesized primarily in the liver from excess ammonia derived from throughout the body (not shown) is excreted by the kidney.
Establishing clinical relevance
A circulating metabolite may be of interest for different reasons, including markedly elevated or depressed levels in CKD, specific hormone-like effects ascribed to the metabolite, or the fact that it is produced by the kidney. Demonstrating the clinical importance of select metabolites, however, is challenging given the multiplicity of metabolite alterations that arise with renal failure, the fact that metabolites may act in concert to co-activate common pathways, and the lack of ideal animal models for kidney disease or its complications.
One approach to prioritizing among numerous metabolites is to identify which have the strongest statistical associations with adverse clinical outcomes. This strategy is feasible because the relatively high throughput of current metabolomics approaches has enabled profiling of large patient cohorts. For example, several groups have measured baseline blood metabolites in >1000 subjects each from the KORA S4/F4Study, the Framingham Heart Study (FHS), and the Atherosclerosis Risk in Communities (ARIC) Study, testing the association of baseline metabolites with the subsequent decline of eGFR to < 60 mL/min/1.73m2.15, 49, 50 Smaller, more selected patient cohorts have been used to examine progression to hard endpoints such as ESRD or death.51 Although each study has nominated select metabolites of interest, methodologic differences across studies have been a barrier to systematic meta-analysis. More specifically, at present all metabolomics platforms provide incomplete coverage of the metabolome, with considerable areas of non-overlap across platforms. As a result, no unequivocal “winner” has emerged from these studies, although partial overlap across data sets has highlighted tryptophan metabolites such as kynurenine and kynurenic acid as potential predictors of GFR loss. In ESRD, elevated levels of p-cresol sulfate, indoxyl sulfate, ADMA, and TMAO have been associated with cardiovascular outcomes or death in some, but not all studies performed to date.52, 53 Metabolite associations with other complications of kidney disease, including malnutrition, inflammation, or uremic symptoms have received less attention to date, and are important areas for future investigation. Finally, there have been relatively fewer studies associating urine metabolites with clinical outcomes. In part, this reflects the lower availability of high quality urine samples (particularly 24 hour collections) in large epidemiologic cohorts, as well as technical challenges introduced by the presence of proteinuria and wide range of urinary dilution/concentration.
Although linking metabolites—in blood or urine—with clinical outcomes is of interest, it is insufficient to prove causality, as confounding variables may underlie the association between select metabolites and outcomes. Mendelian randomization leverages human genetics to test the causality of associations between biomarkers and clinical outcomes. The logic of this approach is illustrated by studies of LDL cholesterol. Because circulating LDL cholesterol is deleterious, genetic variants that increase LDL cholesterol levels are expected to contribute to the risk of cardiovascular disease. Consistent with this, genetic variants that increase LDL cholesterol levels have been shown to be enriched among individuals with cardiovascular disease compared to individuals without cardiovascular disease, and a combined score of these genetic variants is an independent predictor of new onset heart disease.54, 55 Although these studies were not required to prove the atherogenecity of LDL cholesterol, which had already been long-established, they provide proof of principle for the application of this approach to metabolites of uncertain toxicity.
Metabolomic profiling in large cohorts has led to genome wide association studies of the blood metabolome, laying the groundwork for Mendelian randomization analyses.56, 57 As reviewed by Kottgen and colleagues, however, these analyses require not just that the genetic determinants of the metabolite of interest have been identified, but that the genetic determinants have a clinically meaningful impact on metabolite levels and that the genetic determinants only affect disease status via their effect on the metabolite.58 To date, these preconditions have not been met for most of the circulating metabolites measured with current metabolomics approaches.
Conclusion
The term “systems biology” can refer to different analytical and computational techniques, but is often defined in contrast to or as the antithesis of reductionism, which seeks to explain biologic phenomena on the basis of individual parts and their interactions.59 By contrast, systems biology “is about putting together rather than taking apart, integration rather than reduction.”60 From a practical standpoint, this can involve the simultaneous application of multiple omics approaches such as genomics, transcriptomics, proteomics, and metabolomics, along with bioinformatics tools to generate a comprehensive view of biological processes. The focus of this article is a little different, with specific emphasis on the blood metabolome in kidney disease. By referring to a “systems-level” view, however, it purposefully echoes the archetype of systems biology, invoking the need for an integrated perspective that considers the whole organism. The kidneys have complex effects on circulating metabolite levels, which in turn may have diverse functional effects throughout the body. Metabolomics studies have enriched our appreciation for this complexity, and will likely yield further insights on the metabolic inter-relationships across organs, but translating these findings to improved diagnosis, prognosis, and treatment of kidney disease in the clinic will require further study.
Acknowledgments
This work was supported in part by the National Institutes of Health (U01DK060990)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kalim S, Rhee EP. An overview of renal metabolomics. Kidney Int. 2017;91:61–69. doi: 10.1016/j.kint.2016.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cano N. Inter-relationships between renal metabolism (both in physiology and renal dysfunction) and the liver. Curr Opin Clin Nutr Metab Care. 2001;4:279–285. doi: 10.1097/00075197-200107000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Gerich JE. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: therapeutic implications. Diabet Med. 2010;27:136–142. doi: 10.1111/j.1464-5491.2009.02894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baylis C. Arginine, arginine analogs and nitric oxide production in chronic kidney disease. Nat Clin Pract Nephrol. 2006;2:209–220. doi: 10.1038/ncpneph0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garibotto G, Valli A, Anderstam B, Eriksson M, Suliman ME, Balbi M, et al. The kidney is the major site of S-adenosylhomocysteine disposal in humans. KidneyInt. 2009;76:293–296. doi: 10.1038/ki.2009.117. [DOI] [PubMed] [Google Scholar]
- 6.van de Poll MC, Soeters PB, Deutz NE, Fearon KC, Dejong CH. Renal metabolism of amino acids: its role in interorgan amino acid exchange. Am J Clin Nutr. 2004;79:185–197. doi: 10.1093/ajcn/79.2.185. [DOI] [PubMed] [Google Scholar]
- 7.Suchy-Dicey AM, Laha T, Hoofnagle A, Newitt R, Sirich TL, Meyer TW, et al. Tubular Secretion in CKD. J Am Soc Nephrol. 2016;27:2148–2155. doi: 10.1681/ASN.2014121193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sirich TL, Funk BA, Plummer NS, Hostetter TH, Meyer TW. Prominent accumulation in hemodialysis patients of solutes normally cleared by tubular secretion. J Am Soc Nephrol. 2014;25:615–622. doi: 10.1681/ASN.2013060597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toyohara T, Akiyama Y, Suzuki T, Takeuchi Y, Mishima E, Tanemoto M, et al. Metabolomic profiling of uremic solutes in CKD patients. Hypertens Res. 2010;33:944–952. doi: 10.1038/hr.2010.113. [DOI] [PubMed] [Google Scholar]
- 10.Shah VO, Townsend RR, Feldman HI, Pappan KL, Kensicki E, Vander Jagt DL. Plasma metabolomic profiles in different stages of CKD. Clin J Am Soc Nephrol. 2013;8:363–370. doi: 10.2215/CJN.05540512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mutsaers HA, Engelke UF, Wilmer MJ, Wetzels JF, Wevers RA, van den Heuvel LP, et al. Optimized metabolomic approach to identify uremic solutes in plasma of stage 3–4 chronic kidney disease patients. PloS One. 2013;8:e71199. doi: 10.1371/journal.pone.0071199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goek ON, Doring A, Gieger C, Heier M, Koenig W, Prehn C, et al. Serum metabolite concentrations and decreased GFR in the general population. Am J Kidney Dis. 2012;60:197–206. doi: 10.1053/j.ajkd.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 13.Sekula P, Goek ON, Quaye L, Barrios C, Levey AS, Romisch-Margl W, et al. A Metabolome-Wide Association Study of Kidney Function and Disease in the General Population. J Am Soc Nephrol. 2016;27:1175–1188. doi: 10.1681/ASN.2014111099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duranton F, Lundin U, Gayrard N, Mischak H, Aparicio M, Mourad G, et al. Plasma and urinary amino acid metabolomic profiling in patients with different levels of kidney function. Clin J Am Soc Nephrol. 2014;9:37–45. doi: 10.2215/CJN.06000613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rhee EP, Clish CB, Ghorbani A, Larson MG, Elmariah S, McCabe E, et al. A combined epidemiologic and metabolomic approach improves CKD prediction. J Am Soc Nephrol. 2013;24:1330–1338. doi: 10.1681/ASN.2012101006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rhee EP, Souza A, Farrell L, Pollak MR, Lewis GD, Steele DJ, et al. Metabolite profiling identifies markers of uremia. J Am Soc Nephrol. 2010;21:1041–1051. doi: 10.1681/ASN.2009111132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato E, Kohno M, Yamamoto M, Fujisawa T, Fujiwara K, Tanaka N. Metabolomic analysis of human plasma from haemodialysis patients. Eur J Clin Invest. 2011;41:241–255. doi: 10.1111/j.1365-2362.2010.02398.x. [DOI] [PubMed] [Google Scholar]
- 18.Godfrey AR, Williams CM, Dudley E, Newton RP, Willshaw P, Mikhail A, et al. Investigation of uremic analytes in hemodialysate and their structural elucidation from accurate mass maps generated by a multidimensional liquid chromatography/mass spectrometry approach. Rapid Commun Mass Spectrom. 2009;23:3194–3204. doi: 10.1002/rcm.4235. [DOI] [PubMed] [Google Scholar]
- 19.Kikuchi K, Itoh Y, Tateoka R, Ezawa A, Murakami K, Niwa T. Metabolomic analysis of uremic toxins by liquid chromatography/electrospray ionization-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:1662–1668. doi: 10.1016/j.jchromb.2009.11.040. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka H, Sirich TL, Plummer NS, Weaver DS, Meyer TW. An Enlarged Profile of Uremic Solutes. PloS One. 2015;10:e0135657. doi: 10.1371/journal.pone.0135657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aronov PA, Luo FJ, Plummer NS, Quan Z, Holmes S, Hostetter TH, et al. Colonic contribution to uremic solutes. J Am Soc Nephrol. 2011;22:1769–1776. doi: 10.1681/ASN.2010121220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mishima E, Fukuda S, Mukawa C, Yuri A, Kanemitsu Y, Matsumoto Y, et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach. Kidney Int. 2017 doi: 10.1016/j.kint.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 23.Ramezani A, Raj DS. The gut microbiome, kidney disease, and targeted interventions. J Am Soc Nephrol. 2014;25:657–670. doi: 10.1681/ASN.2013080905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu H, Huang X, Arnlov J, Cederholm T, Stenvinkel P, Lindholm B, et al. Clinical Correlates of Insulin Sensitivity and Its Association with Mortality among Men with CKD Stages 3 and 4. Clin J Am Soc Nephrol. 2014;9:690–697. doi: 10.2215/CJN.05230513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeFronzo RA, Alvestrand A, Smith D, Hendler R, Hendler E, Wahren J. Insulin resistance in uremia. J Clin Invest. 1981;67:563–568. doi: 10.1172/JCI110067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carrero JJ, Stenvinkel P, Cuppari L, Ikizler TA, Kalantar-Zadeh K, Kaysen G, et al. Etiology of the protein-energy wasting syndrome in chronic kidney disease: a consensus statement from the International Society of Renal Nutrition and Metabolism (ISRNM) J Ren Nutr. 2013;23:77–90. doi: 10.1053/j.jrn.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 27.Dunn SR, Gabuzda GM, Superdock KR, Kolecki RS, Schaedler RW, Simenhoff ML. Induction of creatininase activity in chronic renal failure: timing of creatinine degradation and effect of antibiotics. Am J Kidney Dis. 1997;29:72–77. doi: 10.1016/s0272-6386(97)90010-x. [DOI] [PubMed] [Google Scholar]
- 28.Mullen W, Saigusa D, Abe T, Adamski J, Mischak H. Proteomics and metabolomics as tools to unravel novel culprits and mechanisms of uremic toxicity: instrument or hype? Semin Nephrol. 2014;34:180–190. doi: 10.1016/j.semnephrol.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 29.Husted AS, Trauelsen M, Rudenko O, Hjorth SA, Schwartz TW. GPCR-Mediated Signaling of Metabolites. Cell Metab. 2017;25:777–796. doi: 10.1016/j.cmet.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 30.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–1584. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaysen GA, Johansen KL, Chertow GM, Dalrymple LS, Kornak J, Grimes B, et al. Associations of Trimethylamine N-Oxide With Nutritional and Inflammatory Biomarkers and Cardiovascular Outcomes in Patients New to Dialysis. J Ren Nutr. 2015;25:351–356. doi: 10.1053/j.jrn.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shafi T, Powe NR, Meyer TW, Hwang S, Hai X, Melamed ML, et al. Trimethylamine N-Oxide and Cardiovascular Events in Hemodialysis Patients. J Am Soc Nephrol. 2017;28:321–331. doi: 10.1681/ASN.2016030374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalim S, Clish CB, Wenger J, Elmariah S, Yeh RW, Deferio JJ, et al. A plasma long-chain acylcarnitine predicts cardiovascular mortality in incident dialysis patients. J Am Heart Assoc. 2013;2:e000542. doi: 10.1161/JAHA.113.000542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell. 2016;165:111–124. doi: 10.1016/j.cell.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, et al. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature. 2004;429:188–193. doi: 10.1038/nature02488. [DOI] [PubMed] [Google Scholar]
- 38.Toma I, Kang JJ, Sipos A, Vargas S, Bansal E, Hanner F, et al. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J Clin Invest. 2008;118:2526–2534. doi: 10.1172/JCI33293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci USA. 2013;110:4410–4415. doi: 10.1073/pnas.1215927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 41.Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P, Myronovych A, Karns R, et al. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature. 2014 doi: 10.1038/nature13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang J, Simonavicius N, Wu X, Swaminath G, Reagan J, Tian H, et al. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J Biol Chem. 2006;281:22021–22028. doi: 10.1074/jbc.M603503200. [DOI] [PubMed] [Google Scholar]
- 43.Barth MC, Ahluwalia N, Anderson TJ, Hardy GJ, Sinha S, Alvarez-Cardona JA, et al. Kynurenic acid triggers firm arrest of leukocytes to vascular endothelium under flow conditions. J Biol Chem. 2009;284:19189–19195. doi: 10.1074/jbc.M109.024042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andrade-Oliveira V, Amano MT, Correa-Costa M, Castoldi A, Felizardo RJ, de Almeida DC, et al. Gut Bacteria Products Prevent AKI Induced by Ischemia-Reperfusion. J Am Soc Nephrol. 2015;26:1877–1888. doi: 10.1681/ASN.2014030288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Owen EE, Robinson RR. Amino acid extraction and ammonia metabolism by the human kidney during the prolonged administration of ammonium chloride. J Clin Invest. 1963;42:263–276. doi: 10.1172/JCI104713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tizianello A, De Ferrari G, Garibotto G, Gurreri G, Robaudo C. Renal metabolism of amino acids and ammonia in subjects with normal renal function and in patients with chronic renal insufficiency. J Clin Invest. 1980;65:1162–1173. doi: 10.1172/JCI109771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brosnan JT. Interorgan amino acid transport and its regulation. J Nutr. 2003;133:2068S–2072S. doi: 10.1093/jn/133.6.2068S. [DOI] [PubMed] [Google Scholar]
- 48.van de Poll MC, Siroen MP, van Leeuwen PA, Soeters PB, Melis GC, Boelens PG, et al. Interorgan amino acid exchange in humans: consequences for arginine and citrulline metabolism. Am J Clin Nutr. 2007;85:167–172. doi: 10.1093/ajcn/85.1.167. [DOI] [PubMed] [Google Scholar]
- 49.Yu B, Zheng Y, Nettleton JA, Alexander D, Coresh J, Boerwinkle E. Serum metabolomic profiling and incident CKD among African Americans. Clin J Am Soc Nephrol. 2014;9:1410–1417. doi: 10.2215/CJN.11971113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goek ON, Prehn C, Sekula P, Romisch-Margl W, Doring A, Gieger C, et al. Metabolites associate with kidney function decline and incident chronic kidney disease in the general population. Nephrol Dial Transplant. 2013;28:2131–2138. doi: 10.1093/ndt/gft217. [DOI] [PubMed] [Google Scholar]
- 51.Niewczas MA, Sirich TL, Mathew AV, Skupien J, Mohney RP, Warram JH, et al. Uremic solutes and risk of end-stage renal disease in type 2 diabetes: metabolomic study. Kidney Int. 2014;85:1214–1224. doi: 10.1038/ki.2013.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Melamed ML, Plantinga L, Shafi T, Parekh R, Meyer TW, Hostetter TH, et al. Retained organic solutes, patient characteristics and all-cause and cardiovascular mortality in hemodialysis: results from the retained organic solutes and clinical outcomes (ROSCO) investigators. BMC Nephrol. 2013;14:134. doi: 10.1186/1471-2369-14-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shafi T, Meyer TW, Hostetter TH, Melamed ML, Parekh RS, Hwang S, et al. Free Levels of Selected Organic Solutes and Cardiovascular Morbidity and Mortality in Hemodialysis Patients: Results from the Retained Organic Solutes and Clinical Outcomes (ROSCO) Investigators. PloS One. 2015;10:e0126048. doi: 10.1371/journal.pone.0126048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kathiresan S, Melander O, Anevski D, Guiducci C, Burtt NP, Roos C, et al. Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med. 2008;358:1240–1249. doi: 10.1056/NEJMoa0706728. [DOI] [PubMed] [Google Scholar]
- 55.Jansen H, Samani NJ, Schunkert H. Mendelian randomization studies in coronary artery disease. Eur Heart J. 2014;35:1917–1924. doi: 10.1093/eurheartj/ehu208. [DOI] [PubMed] [Google Scholar]
- 56.Atzler D, Schwedhelm E, Zeller T. Integrated genomics and metabolomics in nephrology. Nephrol Dial Transplant. 2014;29:1467–1474. doi: 10.1093/ndt/gft492. [DOI] [PubMed] [Google Scholar]
- 57.Adamski J, Suhre K. Metabolomics platforms for genome wide association studies--linking the genome to the metabolome. Curr Opin Biotech. 2013;24:39–47. doi: 10.1016/j.copbio.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 58.Sekula P, Del Greco MF, Pattaro C, Kottgen A. Mendelian Randomization as an Approach to Assess Causality Using Observational Data. J Am Soc Nephrol. 2016;27:3253–3265. doi: 10.1681/ASN.2016010098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.He JC, Chuang PY, Ma'ayan A, Iyengar R. Systems biology of kidney diseases. Kidney Int. 2012;81:22–39. doi: 10.1038/ki.2011.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Noble D. The music of life: Biology beyond the genome. Oxford University Press; 2006. [Google Scholar]
- 61.Vanholder R, Schepers E, Pletinck A, Nagler EV, Glorieux G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: a systematic review. J Am Soc Nephrol. 2014;25:1897–1907. doi: 10.1681/ASN.2013101062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kielstein JT, Zoccali C. Asymmetric dimethylarginine: a cardiovascular risk factor and a uremic toxin coming of age? Am J Kidney Dis. 2005;46:186–202. doi: 10.1053/j.ajkd.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 63.Deguchi T, Isozaki K, Yousuke K, Terasaki T, Otagiri M. Involvement of organic anion transporters in the efflux of uremic toxins across the blood-brain barrier. J Neurochemistry. 2006;96:1051–1059. doi: 10.1111/j.1471-4159.2005.03550.x. [DOI] [PubMed] [Google Scholar]
- 64.Hosoya K, Tachikawa M. Roles of organic anion/cation transporters at the blood-brain and blood-cerebrospinal fluid barriers involving uremic toxins. Clin Exp Nephrol. 2011;15:478–485. doi: 10.1007/s10157-011-0460-y. [DOI] [PubMed] [Google Scholar]
- 65.Meyer TW, Hostetter TH. Approaches to uremia. J Am Soc Nephrol. 2014;25:2151–2158. doi: 10.1681/ASN.2013121264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kalim S, Karumanchi SA, Thadhani RI, Berg AH. Protein carbamylation in kidney disease: pathogenesis and clinical implications. Am J Kidney Dis. 2014;64:793–803. doi: 10.1053/j.ajkd.2014.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matera M, Bellinghieri G, Costantino G, Santoro D, Calvani M, Savica V. History of L-carnitine: implications for renal disease. J Ren Nutr. 2003;13:2–14. doi: 10.1053/jren.2003.50010. [DOI] [PubMed] [Google Scholar]