

Abstract
Nitric oxide (NO) activates soluble guanylyl cyclase in smooth muscle cells to induce vasodilation in the vasculature. However, as hemoglobin (Hb) is an effective scavenger of NO and is present in high concentrations inside the red blood cell (RBC), the bioavailability of NO would be too low to elicit soluble guanylyl cyclase activation in the presence of blood. Therefore, NO bioactivity must be preserved. Here we present evidence suggesting that the RBC participates in the preservation of NO bioactivity by reducing NO influx. The NO uptake by RBCs was increased and decreased by altering the degree of band 3 binding to the cytoskeleton. Methemoglobin and denatured hemoglobin binding to the RBC membrane or cytoskeleton also were shown to contribute to reducing the NO uptake rate of the RBC. These alterations in NO uptake by the RBC, hence the NO bioavailability, were determined to correlate with the vasodilation of isolated blood vessels. Our observations suggest that RBC membrane and cytoskeleton associated NO-inert proteins provide a barrier for NO diffusion and thus account for the reduction in the NO uptake rate of RBCs.
Extensive studies have established the bioactivity of nitric oxide (NO) in vasoregulation through activation of soluble guanylyl cyclase (ref. 1 and references therein). On the other hand, NO is rapidly deactivated by oxygenated hemoglobin (HbO2) and myoglobin (MbO2) to form nitrate. As NO reacts rapidly (k ∼ 107 M−1⋅s−1) with HbO2 and deoxygenated Hb (deoxyHb), infusion of cell-free normoxic Hb (hereafter, Hb denotes both HbO2 and deoxyHb) at μM levels into animal models, human subjects, or isolated blood vessels (2–5) causes significant vessel constriction due to NO scavenging. However, normal blood containing RBC-encapsulated Hb (rbcHb) at an equivalent concentration of about 10 mM shows insignificant NO reactivity under physiological conditions (4, 6, 7). This discrepancy is difficult to explain in view of the high permeability of NO through lipid bilayers. Thus, NO bioactivity in blood must be preserved by a yet unclear mechanism under physiological conditions.
This problem recently was addressed by a NO bioactivity export theory (8). According to this theory, NO enters the RBC and preferentially binds with the free heme on Hb to form heme-nitrosylHb (HbNO) rather than being oxidized by O2-conjugated heme (9). HbNO then transfers the conjugated NO to β-93Cys to form S-nitrosoHb (9). NO bioactivity is then exported out of RBCs through the anion exchange protein, band 3 (or AE1). Although detailed mechanisms of SNO (S-nitrosothiol species) formation and NO bioactivity export from band 3 are still unclear (10, 11), the theory provides an explanation for the preservation of NO bioactivity and highlights the importance of the RBC in preserving NO bioavailability, which has been suggested previously (12).
In parallel to the NO bioactivity export theory, an independent, but not mutually exclusive, explanation that NO bioavailability is preserved by reducing interactions between NO and rbcHb has been proposed. This theory is based on diffusional barriers between the NO-producing endothelium and rbcHb. These barriers include (i) an unstirred boundary layer surrounding each RBC (13), (ii) a RBC-free layer at the vessel wall induced by the flow of blood (4, 14, 15), and (iii) an unknown RBC-intrinsic barrier for NO consumption (12). Independent measurements using NO electrodes (13), microvessel bioassays (4), or competition assays (12) have established that RBCs consume NO at a rate ≈800-fold lower than that of equivalent concentration of free Hb. To account for this observation by the NO export theory alone would require that NO be pumped out of the RBC through a S-nitrosothiol species (SNO)-intermediate at a rate almost as fast as NO influx. Otherwise, the NO influx to RBC needs to be retarded. Given the relatively low rate of transnitrosation reactions (10, 16), the reduction of NO influx is more likely. Therefore, we suspected that the RBC possesses a novel mechanism for preserving NO bioavailability in vivo.
In this article, we provide evidence that suggests that cytoskeletal and associated NO-inert proteins form a significant barrier to retard NO influx into the RBC. This layer of NO-inert protein accounts for a significant part of the difference between the NO reactivity with RBCs and cell-free Hb. Further, by altering the cytoskeleton and associated proteins, the RBC can be used to modulate vessel tone through controlled NO scavenging. This diffusion barrier also may explain S-nitrosothiol species (SNO) formation in RBC and the data reported for oxygen uptake rate of RBC.
Methods
Chemicals.
Spermine NONOate, N-4-[1-(3-aminopropyl)-2-hydroxy-2-nitrosohydrazino]butyl]-1,3-propanediamine was purchased from Alexis (San Diego). Bis(sulfosuccinimidul)-suberate (BS3) was from Pierce. Acetylphenylhydrazine (APHZ), 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS), sodium azide, Sephadex G-25, α cellulose, microcrystalline cellulose, sodium dithionite, EDTA, Hepes, and BSA were purchased from Sigma.
Preparation of Oxyhemoglobin and RBCs.
All solutions for preparing proteins were chilled (4°C). The cell lysate was prepared by lysing the purified RBCs with 5 mM sodium phosphate solution, pH 8.0 (5P8) and centrifuging at 22,000 × g, 4°C for 30 min. HbO2 was isolated by passing the cell lysate through Sephadex G-25 fine column (1.5 × 25-cm bed volume). Bovine, human, or porcine blood was collected in heparinized (10 units/ml) tubes. The plasma and buffy coat were removed after centrifugation at 800 × g for 10 min. The cells were resuspended and washed four times in Hepes buffer containing 140 mM NaCl, 20 mM Hepes, 5 mM glucose (pH 7.4), 290 mOsmol/kg. After each wash, the cells were centrifuged at 800 × g for 10 min. The RBCs were purified by filtration through a mixture of α cellulose and microcrystalline cellulose (17).
BS3 Pretreatment.
RBC suspensions [22.5 ml, 15% hematocrit, in Dulbecco's phosphate buffer solution (DPBS) containing 5 mM glucose] in a 30-ml syringe was deoxygenated by bubbling with nitrogen or argon for 2 h. BS3 dissolved in DMSO was then added to the RBC suspension to achieve a final concentration of 1 mM. After 15 min of bubbling nitrogen at 22°C, the syringe was transferred to a nutator (Clay Adams) for 45 min. The treated RBCs were washed twice with DPBS containing 10 mM ethanolamine and reoxygenated by bubbling with air for 15 min. The RBCs were further washed twice with isotonic Hepes buffer containing 5 g/liter BSA and three times with isotonic Hepes buffer.
DIDS Pretreatment.
A RBC suspension (15% hematocrit) was incubated with 100 μM DIDS at 22°C for 1 h. The treated cells were washed twice with isotonic Hepes buffer containing 5 g/liter BSA, and then three times with isotonic Hepes buffer.
Sodium Azide Pretreatment.
RBCs (15% hematocrit) were incubated with 0.1 mM NaN3 for 1 h. Cells were washed five times with Hepes buffer (10-fold dilution). As azide binding of methemoglobin (metHb) alters the spectra, an additional control was used during the competition assay. This control consisted of azide-pretreated RBCs in a 10 μM solution of Hb; the Hb solution was a 50/50 mixture of HbO2 and metHb. A decrease in the amount of metHb in this control is indicative of azide leakage from the treated RBCs caused by inadequate washing of treated cells. Samples exhibiting azide leakage were discarded. Control cells were processed parallel to treated cells.
APHZ Pretreatment.
Fifteen milliliters of washed RBC suspension (15% hematocrit) was exposed to APHZ (15 mM) for 2 h at 37°C. Cells were then washed three times in isotonic Hepes. Detection of Heinz bodies was confirmed by light microscopy after crystal violet staining.
NO Gas Pretreatment.
NO gas (Matheson) was purified by bubbling through deoxygenated NaOH (5 M) as described (18). NO gas was also produced by mixing equal volumes of 1 M HCl and 1 M NaNO2 in a syringe sealed with a septum; the reaction was stopped and higher oxides of nitrogen were removed by adding 1 vol of 5 M NaOH. NO gas (0.1–0.9 ml) was added to 15 ml of RBC suspension (15% hematocrit) contained in a 20-ml syringe. The syringe was placed on a mixer for 1 h. After treatment, the RBCs were washed at least four times in Hepes buffer.
The Competition Assay for Measuring NO Uptake Rate by RBCs.
This procedure has been described (12, 19). A detailed description of the procedure along with a computer program for calculating the kinetic constant is available online (http://www.seas.ucla.edu/∼liaoj). Under this experimental condition, no S-nitrosoHb or heme-nitrosyl Hb was detected by using an independent ozone-based chemiluminescent detection (20). The rate constant (kRBC, or k′RBC) for NO consumption by RBC was defined in the following equation
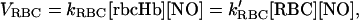 |
where VRBC is the rate of NO consumption by RBCs, [rbcHb], and [NO] are the concentrations of total RBC heme and NO, respectively, based on the total volume as if Hb was not enclosed by the RBC membrane. The first definition allows the comparison between kRBC with kHb (rate constant for the reaction between NO and free HbO2) on the same homogeneous basis. With this definition, kRBC was calculated according to the method reported previously (12, 19). To determine the NO consumption on a per-RBC basis, we used the second definition in the above equation, where [RBC] is the number of RBC per unit volume. Therefore, k′RBC can be calculated from kRBC, k′RBC = kRBC [rbcHb]/[RBC]. The RBC cell density was 3.0 × 106/μl for 15% hematocrit of normal bovine RBCs.
Microvessel Bioassay.
The techniques for isolation of porcine coronary microvessels have been described (21). Subepicardial arteriolar branches (50–100 μm in internal diameter in situ) were dissected from the surrounding cardiac tissue, transferred to a vessel chamber, and then cannulated with glass micropipettes on a micromanipulation platform. After cannulation, the vessel and chamber were transferred to the stage of an inverted microscope coupled to a charge-coupled device camera, video micrometer, and video recorder. The intraluminal pressure of the vessels was controlled by connecting the micropipettes to independent reservoir systems. The vessel was incubated at 37°C and pressurized to 60 cm H2O luminal pressure without flow. After developing a basal tone, buffer, RBCs, or treated-RBCs were introduced into the lumen, and the internal diameter of the vessel was continuously recorded. Serotonin (0.1 μM) was added to the vessel bath to induce vessel dilation.
Preparation of Resealed Ghosts Containing Different Hb Concentrations.
Resealed ghosts containing cytosolic Hb less than 200 μM were prepared following the methods of Steck and Kant (22) with some modification. Briefly, 28 ml of 5P8 buffer at 4°C was added to 4 ml of 50% hematocrit RBC suspension in Hepes buffer. The sample was left on ice for 5 min then centrifuged at 22,000 × g (max), 4°C for 40 min. The pellet was washed, resuspended, and centrifuged four times by using 12 ml of 5P8 at 22,000 × g, 4°C for 20 min. The red cell pellet (membrane) was incubated on ice for 30 min in various concentrations of RBC lysate or purified bovine HbO2. A 5 M NaCl solution was used to adjust the osmolarity to 290 mOsm/kg. The ghost membrane was sealed by incubation at 37°C for 40 min. The sealed ghost was centrifuged at 6,700 × g for 20 min and then equilibrated with Hepes buffer at 4°C overnight. For cytosolic Hb higher than 200 μM, RBCs were lysed by gradually adding 8–16 ml of 5P8 to 4 ml of 50% hematocrit RBC suspension. Osmolarity adjustment and the sealing process was the same as that for the low-range Hb preparation.
Viscosity Modification.
The buffer viscosity was increased by the addition of polyethylene glycol (average molecular weight = 8,000; Fisher Scientific) while the osmolarity was held constant at 290 mOsm by adjusting the concentration of NaCl. A falling ball viscometer was used to measure viscosity.
Results
Modification of Band 3 Affects NO Uptake Rate.
Band 3 is responsible for a host of functions ranging from anion transport to supporting the cytoskeleton by binding with ankyrin (23). The NO export theory suggests that band 3 exports NO bioactivity because inhibition of band 3 activity reduced NO bioactivity in nearly anaerobic conditions (1% oxygen saturation) (8). Under normoxic conditions NO bioactivity export by band 3 was not demonstrated (8). To characterize the involvement of band 3 in the RBC intrinsic barrier, band 3 was perturbed with crosslinkers BS3 and DIDS, both of which inhibit anion transport activity and alter band 3 binding with cytoskeletal proteins (24). The NO uptake rate was measured by use of the competition assay (19) under normoxic conditions. The NO uptake rate constant (kRBC) of BS3-treated RBCs decreased ≈50%, whereas kRBC for the DIDS-treated RBCs increased ≈15% (Fig. 1a). Because of the changes in NO uptake that resulted from perturbing band 3, it is highly likely that this membrane-associated protein plays a role in regulating NO uptake, in addition to NO bioactivity export (8). However, opposite effects on NO uptake by BS3 and DIDS (both band 3 inhibitors) suggest that mechanisms other than the anion exchange activity of band 3 may be involved in modulating the NO uptake rate. A difference between the manner in which BS3 and DIDS act on band 3 is that the former shifts the band 3 population toward tetramers (25), which promotes cytoskeleton binding through band 3 binding with ankyrin, whereas the latter shifts the population of band 3 to dimers, which weakens cytoskeleton binding (26). As the state of band 3 directly influences the association of ankyrin, and in turn the cytoskeleton, this submembrane protein layer may retard the diffusion of NO.
Figure 1.

Uptake of NO by RBCs can be altered by perturbing the cytoskeleton and associated proteins. Relative kRBC represents kRBC/kRBC,Control. (a) BS3 treatment, which shifts the band 3 population toward tetramers (increased cytoskeletal binding), decreased the kRBC of porcine RBCs. Treatment of porcine RBCs with DIDS (decreased cytoskeletal binding) increased kRBC by 20%. APHZ (15 mM), which promotes the formation of Heinz bodies that can bind to the cytoskeleton, decreased kRBC of human RBCs by 20%. (b) Treatment of RBCs with 0.1 μM sodium azide increased the NO uptake rate in three different species: porcine, human, and bovine. (c) Treatment of bovine RBCs (15% Hct) with azide (100 μM) increased kRBC 2-fold. Subsequent treatment with NO gas (azide + NO, 300 μM final concentration), which oxidizes membrane-bound and free HbO2, abolished the azide effect. Treatment with NO gas (NO, 300 μM) decreased kRBC by 25%. Subsequent treatment with azide (NO + azide, 100 μM) returned kRBC to the baseline. NO gas treatment oxidized ≈70% of intracellular hemoglobin. (n = 3–7). *, P < 0.05 vs. control; +, P < 0.05. Less than 0.05 μM of NO species (heme-nitrosylHb and nitrosothiol) were formed in the extracellular space.
Heinz Body and metHb Binding to Cytoskeletal Proteins Reduces NO Uptake Rate.
If cytoskeletal proteins are involved in the retardation of NO transport, one should be able to alter the NO uptake rate by perturbing interprotein associations in the cytoskeleton. The binding of several intraerythrocytic proteins, such as Hb, hemichrome, and Heinz bodies, to the membrane has been well documented (27, 28). APHZ is a common reagent for forming Heinz bodies that bind to the cytoskeleton (29). If the cytoskeleton is a “semiporous” barrier, APHZ should decrease the NO uptake rate as the formation of Heinz bodies would fill in some voids. Indeed, treatment of human RBCs with APHZ resulted in a decrease in the NO uptake rate (Fig. 1a). Interestingly, APHZ did not affect the NO uptake rate of bovine RBCs (data not shown) and no Heinz bodies were detected.
MetHb binds more strongly to the membrane and associated proteins than other Hb species (30). Because metHb has a lower reactivity toward NO (than HbO2, deoxygenated Hb, soluble guanylyl cyclase, etc.), its binding to the membrane and cytoskeletal proteins can further retard the diffusion of NO. If metHb contributes to the membrane protein barrier then dissociating metHb from the membrane should increase the NO consumption rate by RBCs. To test this hypothesis, we treated RBCs with sodium azide, which binds to and then dissociates metHb from the membrane (30). After washing several times to remove unbound azide, the treated RBCs were tested with the competition assay. Remarkably, azide pretreatment increased kRBC 2-fold in bovine, 40% in human blood, and about 15% in porcine blood (Fig. 1b). These data support the hypothesis that metHb is involved in the membrane protein barrier. In all of the above competition assays, we simultaneously conducted various control experiments to show that the observed effect is not an artifact caused by increased autooxidation of Hb or metHb reduction. We also have used transmission electron microscopy to show that these chemical modifications did not alter RBC morphology (data not shown). Note that the effect of azide treatment may be underestimated because the RBC was washed several times after azide treatment to avoid azide binding with extracellular metHb, which would skew the absorbance spectrum. After washing, the concentration of azide in the RBC was much lower than the initial concentration.
To demonstrate that membrane-bound metHb could be a component of a dynamic barrier, we treated RBCs with azide to release metHb and then NO gas to increase the amount of membrane-associated metHb and vice versa. We found that NO gas treatment after azide treatment abolished the azide effect and even decreased the NO uptake rate by about 20% (Fig. 1c). Interestingly, we found that treatment of RBCs with NO gas alone elicited about the same decrease ≈20%, and subsequent treatment with azide only returned the NO uptake rate to near baseline. It is known that high concentrations of NO oxidize and denature Hb, forming both metHb and Heinz bodies, both of which are associated with the membrane and cytoskeletal proteins. Because azide can release metHb from the membrane but has little effect on Heinz bodies, it is reasonable to see that it only partially reverses the effect of NO treatment on kRBC.
Functional Role of RBC in Microvascular Regulation.
To test whether the alteration of kRBC demonstrated above has a functional role in vascular regulation, we used isolated porcine coronary microvessels as a bioassay. The isolated artierioles developed basal tone (62 ± 1% of maximum diameter) at 37°C and the diameters were continuously recorded. Serotonin was used to evaluate NO-mediated vascular function. Serotonin is known to exert endothelium-dependent NO-mediated vasodilation in the intact coronary arterioles (31) but produces vasoconstriction in the denuded vessel (32) through direct activation of 5-HT2 serotonergic receptor in vascular smooth muscle cells. When serotonin was added to the vessel without RBC in the lumen, the vessel dilated within a few seconds (Fig. 2a). However, the dilation elicited by serotonin was attenuated when untreated RBCs are present in the lumen. These data are consistent with our previous finding in coronary arterioles (6). When BS3-RBCs (which exhibit a decreased kRBC) were in the lumen, the NO-mediated vessel dilation was enhanced relative to the untreated RBCs. This result shows that the measurement of RBC consumption of NO with the competition assay is physiologically relevant. Consequently, DIDS-treated RBCs (which have an increased NO consumption) caused vessel constriction rather than dilation. A similar effect is also seen when azide-pretreated RBCs (which increased kRBC) were in the lumen (Fig. 2b). Because azide and DIDS elicited an increase (i.e., 15%) in kRBC and correspondingly abolished serotonin-mediated vasodilation, it appears that the vascular function is tightly regulated by NO through a relatively small change in kRBC. As controls, we observed that supernatants from RBCs suspensions (both treated and untreated RBCs) did not change the vessel tone, indicating that the alteration of vessel regulation is not caused by chemical species leaking out of RBCs.
Figure 2.

Chemically treated RBCs modulate serotonin-induced vasodilation. (a) Representative polygraph trace of porcine vessel experiment. When serotonin (0.1 μM) alone is added to the vessel bath, the vessel dilated within a few seconds. The addition of untreated RBCs to the lumen attenuated the serotonin-induced vasodilation. Infusion of DIDS-pretreated RBCs abolished the serotonin-induced vasodilation and elicted 15% vasoconstriction. (b) The vasoactivity of the treated RBCs followed the trends exhibited in the kRBC measurements (Fig. 1). BS3 pretreatment attenuated the inhibition of serotonin-induced vasodilation, indicative of a decreased NO uptake rate. DIDS and azide pretreatments induced vasoconstriction, indicative of an increased NO uptake rate. The hematocrit for all experiments was 50% (n = 3–5); *, P < 0.05 vs. control.
NO Consumption as a Function of Intraerythrocytic Hb.
Although strong evidence linking the cytoskeleton and its associated proteins to the NO consumption rate of the RBC has been presented, intracellular reactions may have a role in regulating NO uptake. To rule out the possibility of intracellular reaction control, we prepared RBC ghosts containing 0–1 mM Hb. Modifed RBCs were achieved either by gradual lyses (0.3–1 mM Hb) or purifying the RBC ghosts and then resealing them in the presence of various concentrations of RBC lysate or purified Hb. The NO consumption rate of the modified RBCs was measured with the competition assay. The modified RBC was sufficiently stable that insignificant lysis occurred. The rate constant for NO consumption by RBCs was calculated based on the number density of RBCs (k′RBC). We found that the NO consumption rate remained constant when the intraerythrocytic Hb concentration decreased from 20 mM to about 0.2 mM. Below 0.2 mM, the NO consumption rate decreased when the intraerythrocytic Hb concentration decreased (Fig. 3). These results demonstrate that the intracellular Hb concentration is not a limiting factor for NO consumption in normal RBCs. The intracellular Hb becomes limiting only when its concentration is lower than 0.2 mM. Thus, intracellular reaction rate is not a limiting factor for NO consumption under physiological conditions.
Figure 3.

NO uptake rate as a function of intraerythrocytic Hb concentration; k′RBC is the NO uptake constant on a per-RBC basis. The NO uptake rate is independent of intracellular Hb for concentrations greater than 0.2 mM (20 mM is physiologic). These data were consistent with the prediction of our mathematical model (12) developed by using independent data and an estimated membrane permeability (415 μm⋅s−1) ≈1,000-fold lower than that of a lipid bilayer (n = 3–7).
Diffusion Resistance in Extracellular Undisturbed Layer Is Not Dominating.
Another possible factor limiting NO consumption by RBCs is the diffusional resistance caused by the undisturbed boundary layer around each RBC (13). If extracellular diffusion is dominating, kRBC measured by the competition assay should be a strong function of viscosity. We therefore measured kRBC in buffers with increased concentrations of polyethylene glycol, which increased the buffer viscosity. The diffusivity of a molecule in liquids is known to be inversely proportional to the viscosity (33). Within small changes (50% increase in viscosity) kRBC was not a function of viscosity (Fig. 4). When the viscosity was increased by 2-fold, kRBC decreased by 20%. These results showed that extracellular diffusion is not the dominating factor in the kRBC measurement.
Figure 4.

Relative kRBC (kRBC/kRBC,Control) as a function of extracellular viscosity. Increasing the extracellular viscosity of RBC solutions by 50% did not significantly change the NO uptake rate (P = 0.24). A 2-fold increase in extracellular viscosity decreased NO uptake by 15 ± 6 (SD) % [*, P < 0.05 versus 1.04 centipoise (cp) sample]. Physiologic blood plasma viscosity is ≈1.23 cp. n = 3–4.
Discussion
The preservation of NO bioactivity in the vasculature is required to reconcile the vasoregulatory role of NO in the presence of mM concentrations of RBC-encapsulated Hb. We and others have shown that diffusion barriers exist between the site of NO production and the RBC-encapsulated Hb (4, 12–14). Here we provide evidence that suggests the possibility of cytoskeleton and associated NO-inert proteins as members of a submembrane resistance to the entry of NO (Fig. 5). As a result of this resistance, kRBC for NO is reduced to 3 × 104 M−1⋅s−1, which is about 3 orders of magnitude lower than kHb (≈107 M−1⋅s−1). The rate constant for NO uptake by RBCs is remarkably close to the value of kRBC for oxygen uptake (6.9 × 104 M−1⋅s−1), which is about 40-fold slower than reaction with free deoxygenated Hb (34). This result is consistent with a previous report that NO and O2 diffuse into the RBC at similar rates (34). It is likely that the same barrier retards both NO and O2 entry into the cell. Limitation of O2 uptake by RBCs was attributed to the extracellular unstirred layer formed in the stopped-flow experiment (34, 35). However, membrane limitation cannot be conclusively ruled out. The existence of membrane limitation is supported by the following results. (i) The kRBC value for NO uptake can be changed by various chemical modifications of RBCs without changing morphology. (ii) Alteration of buffer viscosity (and thus molecular diffusivity) does not change the value of kRBC significantly. (iii) Reduction of intracellular Hb concentration over 2 orders of magnitude does not change kRBC. These results suggest against the possibilities of intracellular Hb limitation and extracellular diffusion limitation. In addition, the competition assay we used has been shown to be insensitive to extracellular diffusion because of the use of homogenous NO donors and the high hematocrit in the assay (12). The existence of this membrane barrier preserves NO bioavailability and might, at least partially, explain the diffusional resistance to oxygen uptake that was previously attributed to an extracellular unstirred layer.
Figure 5.

Schematic of the hypothetical membrane barrier to NO uptake. The RBC retards small gas molecules such as NO from entering with a submembrane barrier built up around the unique RBC-cytoskeleton. Small molecules are able to cross the barrier through interprotein gaps (pores), but with a much reduced rate. These pores can be loosened or tightened by adjusting the tension in the cytoskeletal structure or plugged with NO-inert proteins. Increasing the tension by strengthening the association of the cytoskeleton to the membrane tightens the cytoskeletal structure and closes the pores, which effectively slows NO entry. Decreasing the tension creates slack in the cytoskeleton, which opens pores and increases the number of entry points for NO. These pores can be plugged with Heinz bodies or metHb (induced by APHZ or NO gas treatment); the metHb plugs can be removed with azide.
The nature of this diffusion resistance is deduced from the above experimental results (Fig. 5). On the cytosolic side of the membrane (submembrane) there is a cytoskeletal network of proteins, including spectrin, metHb, and denatured Hb that are relatively NO-inert (23). This protein network provides surface coverage on the order of magnitude of the RBC surface area (23) and could provide a path sufficiently tortuous to significantly slow the diffusion of small molecules such as NO. RBC cytoskeletal spectrin alone could cover well over half of the cytosolic face of the RBC membrane (23). Normal RBCs contain 1% metHb, which is sufficient to cover the entire membrane (27). When all of the proteins that make up the cytoskeleton (spectrin, band 3, actin, ankyrin, band 4.1, metHb, and denatured Hb) are taken into account, the existence of a dense submembrane layer that completely covers the inner membrane is likely (36). In fact, spectrin at in vivo cytoskeletal concentrations has been found to form a gel in vitro (23).
The submembrane protein network is anchored on the transmembrane protein band 3. Treatment of RBCs with band 3 crosslinkers (BS3 and DIDS) that perturb the cytoskeleton altered the NO uptake rate. BS3 and DIDS are also well-characterized band 3 inhibitors. If NO bioactivity is preserved based on NO export through band 3, inhibition of band 3 activity (by either BS3 or DIDS) should increase the apparent NO consumption rate dramatically. However, DIDS treatment increases kRBC by only 15%, whereas BS3 treatment decreases kRBC by 50%. Because these band 3 inhibitors have opposite effects on NO uptake, inhibition of band 3 activity cannot explain the changes in kRBC observed. Instead, the explanation lies in the effects of BS3 and DIDS on the membrane cytoskeleton. BS3 treatment shifts the band 3 population toward tetramers (25), which enhance binding of band 3 to ankyrin, a protein that anchors the cytoskeleton to the membrane (26). This binding is weakened when the band 3 population is shifted to dimers, as occurs in the case of DIDS treatment (26). These effects on ankyrin binding to band 3 directly impact the NO uptake rate of the RBC and support our theory that the cytoskeleton is a diffusion barrier to physiological concentrations of NO.
Binding of Hb to the cytoskeletal network is well characterized (27, 37, 38). Among the Hb species, the membrane and cytoskeletal network preferentially bind metHb (30), which binds with NO at a much lower rate (k ∼ 104 M−1⋅s−1) (39–41). Because of the relatively low reactivity of metHb with NO, metHb can contribute to a barrier to the diffusion of NO, thus reduce the NO uptake rate. Unbound metHb has little effect on kRBC, as demonstrated previously (13). The involvement of bound metHb and denatured Hb in the diffusion barrier is supported by treatments that increase (APHZ, NO gas) or decrease (azide) the binding of metHb and NO-inert Hb species to the cytoskeleton and associated proteins. Treatment with APHZ or NO gas, which oxidize and denature Hb to form metHb and Heinz bodies, are known to increase the amount of these species bound to the cytoskeletal network (29). As expected, APHZ or NO gas pretreatment decreased kRBC.
Treatment with azide released the metHb from the membrane (30) and thus increased the NO uptake rate constant. Azide is a high affinity ligand for metHb. Once azide binds metHb, the Hb-N3 complex is released from the membrane (30) and should create gaps in the submembrane barrier. When treated with azide followed by washing, the NO uptake rate was found to increase for bovine, porcine, and human RBCs and was further validated with the vessel bioassay. As azide treatment had a common effect over three species, this finding demonstrates the commonality of this mode of NO regulation.
The implications of a cytoskeletal barrier to the diffusion of NO can be far reaching. We have demonstrated that RBCs can be chemically modified to modulate NO bioactivity, and that these modifications have functional roles in vascular regulation. The isolated vessel experiments not only substantiate the kRBC measurements using competition assays, but also provide evidence that RBCs play a role in vessel regulation by preserving NO bioactivity. Further, this barrier in the submembrane space may contribute to the accumulation of NO in the membrane (42), which may then react with O2 to form nitrosating agents, such as N2O3. These nitrosating agents could then react with the β-93-cys of membrane-bound Hb or band 3 thiol groups to form S-nitrosothiols. Although a dynamic regulation of NO uptake by the RBC is speculative, these results imply that chemically modified RBCs can be used as NO scavengers or NO preservers. Moreover, Hb-based blood substitutes are insufficient and cannot emulate RBCs without a barrier to the diffusion of NO. Thus, the RBC intrinsic barrier to NO may be required to truly emulate RBC functions in the vasculature.
Acknowledgments
This work was supported by National Institutes of Health Grant R01 HL65741.
Abbreviations
- HbO2
oxyhemoglobin
- metHb
methemoglobin
- BS3
bis(sulfosuccinimidul)-suberate
- APHZ
acetylphenylhydrazine
- DIDS
4,4′-diisothiocyanatostilbene-2,2′disulfonic acid
- rbcHb
RBC-encapsulated Hb
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Ignarro L J. Nitric Oxide: Biology and Pathobiology. San Diego: Academic; 2000. [Google Scholar]
- 2.Doherty D H, Doyle M P, Curry S R, Vali R J, Fattor T J, Olson J S, Lemon D D. Nat Biotechnol. 1998;16:672–676. doi: 10.1038/nbt0798-672. [DOI] [PubMed] [Google Scholar]
- 3.Pohl U, Lamontagne D. Cardiovasc Res. 1991;86, Suppl. 2:97–105. doi: 10.1007/978-3-642-72461-9_11. [DOI] [PubMed] [Google Scholar]
- 4.Liao J C, Hein W, Vaughn M W, Huang K T, Kuo L. Proc Natl Acad Sci USA. 1999;96:8757–8761. doi: 10.1073/pnas.96.15.8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel R P. Free Radical Biol Med. 2000;28:1518–1525. doi: 10.1016/s0891-5849(00)00259-8. [DOI] [PubMed] [Google Scholar]
- 6.Lancaster J R., Jr Proc Natl Acad Sci USA. 1994;91:8137–8141. doi: 10.1073/pnas.91.17.8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lancaster J R., Jr Nitric Oxide. 1997;1:18–30. doi: 10.1006/niox.1996.0112. [DOI] [PubMed] [Google Scholar]
- 8.Pawloski J R, Hess D T, Stamler J S. Nature (London) 2001;409:622–626. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- 9.Gow A J, Luchsinger B P, Pawloski J R, Singel D J, Stamler J S. Proc Natl Acad Sci USA. 1999;96:9027–9032. doi: 10.1073/pnas.96.16.9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patel R P, Hogg N, Spencer N Y, Kalyanaraman B, Matalon S, Darley-Usmar V M. J Biol Chem. 1999;274:15487–15492. doi: 10.1074/jbc.274.22.15487. [DOI] [PubMed] [Google Scholar]
- 11.Wolzt M, MacAllister R J, Davis D, Feelisch M, Moncada S, Vallance P, Hobbs A J. J Biol Chem. 1999;274:28983–28990. doi: 10.1074/jbc.274.41.28983. [DOI] [PubMed] [Google Scholar]
- 12.Vaughn M W, Huang K T, Kuo L, Liao J C. J Biol Chem. 2000;275:2342–2348. doi: 10.1074/jbc.275.4.2342. [DOI] [PubMed] [Google Scholar]
- 13.Liu X, Miller M J, Joshi M S, Sadowska-Krowicka H, Clark D A, Lancaster J R., Jr J Biol Chem. 1998;273:18709–18713. doi: 10.1074/jbc.273.30.18709. [DOI] [PubMed] [Google Scholar]
- 14.Vaughn M W, Kuo L, Liao J C. Am J Physiol. 1998;274:H1705–H1714. doi: 10.1152/ajpheart.1998.274.5.H1705. [DOI] [PubMed] [Google Scholar]
- 15.Butler A R, Megson I L, Wright P G. Biochim Biophys Acta. 1998;1425:168–176. doi: 10.1016/s0304-4165(98)00065-8. [DOI] [PubMed] [Google Scholar]
- 16.Hogg N. Anal Biochem. 1999;272:257–262. doi: 10.1006/abio.1999.4199. [DOI] [PubMed] [Google Scholar]
- 17.Beutler E. Red Cell Metabolism: A Manual of Biochemical Methods. New York: Grune & Stratton; 1984. [Google Scholar]
- 18.Feelisch M, Stamler J S. Methods in Nitric Oxide Research. New York: Wiley; 1996. [Google Scholar]
- 19.Vaughn M W, Huang K, Kuo L, Liao J C. Nitric Oxide. 2001;5:18–31. doi: 10.1006/niox.2000.0328. [DOI] [PubMed] [Google Scholar]
- 20.Gladwin M T, Shelhamer J H, Schechter A N, Pease-Fye M E, Waclawiw M A, Panza J A, Ognibene F P, Cannon R O. Proc Natl Acad Sci USA. 2000;97:11482–11487. doi: 10.1073/pnas.97.21.11482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuo L, Chilian W M, Davis M J. Am J Physiol. 1991;261:H1706–H1715. doi: 10.1152/ajpheart.1991.261.6.H1706. [DOI] [PubMed] [Google Scholar]
- 22.Steck T L, Kant J A. Methods Enzymol. 1974;31:172–180. doi: 10.1016/0076-6879(74)31019-1. [DOI] [PubMed] [Google Scholar]
- 23.Bennett V. In: Cell Membranes: Methods and Reviews. Elson E, Frazier W, Glaser L, editors. Vol. 2. New York: Plenum; 1984. pp. 149–195. [Google Scholar]
- 24.Salhany J M. Erythrocyte Band 3 Protein. Boca Raton, FL: CRC; 1990. [Google Scholar]
- 25.Salhany J M, Sloan R L, Cordes K A. J Biol Chem. 1990;265:17688–17693. [PubMed] [Google Scholar]
- 26.Van Dort H M, Moriyama R, Low P S. J Biol Chem. 1998;273:14819–14826. doi: 10.1074/jbc.273.24.14819. [DOI] [PubMed] [Google Scholar]
- 27.Shaklai N, Yguerabide J, Ranney H M. Biochemistry. 1977;16:5593–5597. doi: 10.1021/bi00644a032. [DOI] [PubMed] [Google Scholar]
- 28.Waugh S M, Willardson B M, Kannan R, Labotka R J, Low P S. J Clin Invest. 1986;78:1155–1160. doi: 10.1172/JCI112696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peisach J, Blumberg W E, Rachmilewitz E A. Biochim Biophys Acta. 1975;393:404–418. doi: 10.1016/0005-2795(75)90069-0. [DOI] [PubMed] [Google Scholar]
- 30.Giardina B, Scatena R, Clementi M E, Ramacci M T, Maccari F, Cerroni L, Condo S G. Adv Exp Med Biol. 1991;307:75–84. doi: 10.1007/978-1-4684-5985-2_7. [DOI] [PubMed] [Google Scholar]
- 31.Hein T W, Kuo L. Circ Res. 1998;83:404–414. doi: 10.1161/01.res.83.4.404. [DOI] [PubMed] [Google Scholar]
- 32.Kuo L, Davis M J, Cannon M S, Chilian W M. Circ Res. 1992;70:465–476. doi: 10.1161/01.res.70.3.465. [DOI] [PubMed] [Google Scholar]
- 33.Bird R, Stewart W, Lightfoot E. Transport Phenomena. New York: Wiley; 1960. [Google Scholar]
- 34.Coin J T, Olson J S. J Biol Chem. 1979;254:1178–1190. [PubMed] [Google Scholar]
- 35.Vandegriff K D, Olson J S. J Biol Chem. 1984;259:12619–12627. [PubMed] [Google Scholar]
- 36.Hartwig J H. Protein Profile. 1994;1:706–778. [PubMed] [Google Scholar]
- 37.Fortier N, Snyder L M, Garver F, Kiefer C, McKenney J, Mohandas N. Blood. 1988;71:1427–1431. [PubMed] [Google Scholar]
- 38.Shaklai N, Frayman B, Fortier N, Snyder M. Biochim Biophys Acta. 1987;915:406–414. doi: 10.1016/0167-4838(87)90027-6. [DOI] [PubMed] [Google Scholar]
- 39.Eich R F, Li T, Lemon D D, Doherty D H, Curry S R, Aitken J F, Mathews A J, Johnson K A, Smith R D, Phillips G N, Jr, Olson J S. Biochemistry. 1996;35:6976–6983. doi: 10.1021/bi960442g. [DOI] [PubMed] [Google Scholar]
- 40.Cassoly R. J Mol Biol. 1975;98:581–595. doi: 10.1016/s0022-2836(75)80088-x. [DOI] [PubMed] [Google Scholar]
- 41.Doyle M P, Hoekstra J W. J Inorg Biochem. 1981;14:351–358. doi: 10.1016/s0162-0134(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 42.Liu X, Miller M J S, Joshi M S, Thomas D D, Lancaster J J. Proc Natl Acad Sci USA. 1998;95:2175–2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]