

Abstract
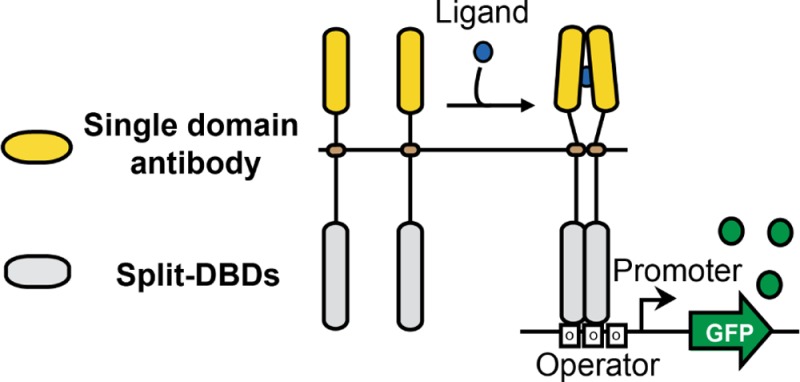
Engineered bacteria promise to revolutionize diagnostics and therapeutics, yet many applications are precluded by the limited number of detectable signals. Here we present a general framework to engineer synthetic receptors enabling bacterial cells to respond to novel ligands. These receptors are activated via ligand-induced dimerization of a single-domain antibody fused to monomeric DNA-binding domains (split-DBDs). Using E. coli as a model system, we engineer both transmembrane and cytosolic receptors using a VHH for ligand detection and demonstrate the scalability of our platform by using the DBDs of two different transcriptional regulators. We provide a method to optimize receptor behavior by finely tuning protein expression levels and optimizing interdomain linker regions. Finally, we show that these receptors can be connected to downstream synthetic gene circuits for further signal processing. The general nature of the split-DBD principle and the versatility of antibody-based detection should support the deployment of these receptors into various hosts to detect ligands for which no receptor is found in nature.
Engineered bacteria offer the potential to dramatically transform and improve approaches to biosensing, diagnostics, and therapeutics.1−5 Bacterial biosensors have been widely used for environmental monitoring and remediation,1,6 and are now being translated into the field of healthcare to detect pathological biomarkers and diagnose diseases.7,8 Bacteria also hold great promise for in vivo diagnostics and therapies. For example, using mouse models, researchers engineered bacteria to detect cancer metastasis,9 monitor gut inflammation,10,11 or specifically target cancer cells.12 In addition, microbiome engineering could help prevent or treat infectious and autoimmune diseases.5
All these applications require sensors enabling bacteria to detect and respond to various signals of interest. Cellular sensors were first engineered by repurposing systems found in nature, like coupling existing transcription factors to a reporter1,6,13 or by rewiring two-component systems.11,14 However, a sensor for every molecule of interest might not exist in nature, limiting the range of detectable molecules and related applications. A pressing challenge is thus to engineer synthetic receptors enabling bacteria to detect novel signals.
Several approaches have been used to engineer synthetic receptors. For example, transcription factor specificity can be switched to other ligands by directed evolution.15−17 While useful, these sensors are limited to recognizing structurally similar molecules and by the difficulty to preserve allosteric control. Synthetic receptors have also been engineered using conditionally stable ligand-binding domains (LBDs) that are degraded in the absence of their ligands.18 Finally, synthetic metabolic pathways can transform nondetectable molecules into ligands recognized by known transcription factors.19 However, all these systems rely on existing LBDs and are limited to the detection of small molecules.
Ideally, synthetic receptors should use versatile LBDs for which recognition specificity can be easily programmed for various applications. In this regard, single-domain antibodies or antibody-like scaffolds are ideal LBDs due to their high stability and solubility, and for which combinatorial libraries can be selected to target many different antigens, from small molecules to proteins.20−23 Consequently, single-domain antibodies like camelid VHHs or scFvs have been used to design mammalian transmembrane receptors such as chimeric antigen receptors (CARs), synthetic notch receptors,24,25 and protease-based transmembrane receptors.26 Despite such progress, to this day all synthetic receptors using antibodies as LBD operate in eukaryotic cells.
We thus aimed at designing a modular bacterial receptor platform using single-domain antibodies as LBDs and following several specifications: (i) receptor activation controls gene expression, so that synthetic gene networks can be connected to ligand detection for further signal processing8 (ii) the receptor mechanism is scalable so that several orthogonal receptors can be designed following the same principle; and (iii) the receptor system can be applied to engineer either cytosolic sensors (for membrane permeable molecules) or transmembrane receptors (for detection of ligands in the extracellular environment).
On the basis of these requirements, we provide a general framework to engineer synthetic prokaryotic receptors by coupling single-domain antibodies undergoing ligand-induced dimerization with DNA-binding domains for which activity is dependent on dimerization.27,28 Using E. coli as a model system, we engineer both transmembrane and cytosolic receptors using a VHH for ligand detection. We demonstrate the scalability of our platform by using the DBDs of two different transcriptional regulators and show that receptor behavior can be optimized by tuning protein expression levels and optimizing interdomain linker regions. Finally, we demonstrate that receptor output can be connected to downstream synthetic gene circuits. These scalable and versatile synthetic bacterial receptors could be deployed in various chassis for applications in diagnostics, therapeutics, and microbiome engineering.
Results
Engineering Synthetic Receptors by Coupling Split-DBDs with a Single Domain Antibody
DNA binding of transcriptional regulators is generally dependent on dimerization of the DBD through the LBD.27 Deletion of the LBD/dimerization domain leads to an inactive, monomeric DBD in which function can be restored via dimerization driven by fusing the proteins of interest.28 We first designed a synthetic receptor activated via ligand-induced dimerization by using the DBD of LexA, a well-characterized transcriptional repressor regulating the transcription of genes involved in E. coli SOS response.29 Upon induction of the SOS response, RecA promotes LexA inactivation through self-cleavage at residue 85, a flexible hinge between DNA-binding and dimerization domains. The repressive activity of the monomeric LexA DBD can be restored through fusion with interacting proteins, a feature used in two-hybrid screens.30,31 In order to prevent interference from endogenous E. coli LexA, we used the mutant LexA-408 and its corresponding promoter that is not recognized by the wild type LexA32 (see Supporting Information). As a ligand binding domain, we chose a single-domain VHH camelid antibody that can be dimerized upon binding to caffeine33 (henceforth termed VHH-Caffeine). As a negative control we used a VHH targeting RNase A34 (henceforth termed VHH-Control). We built two chimeric proteins, expressed in E. coli cytoplasm, composed of an N-terminal LexA DBD and a C-terminal VHH, and placed their expression under the control of the pLacO1, induced by isopropyl β-D-1-thiogalactopyranoside (IPTG),49 while the pLexA promoter drives green fluorescent protein (GFP) expression. In the presence of ligand, LexA DBD should dimerize and repress target gene expression (Figure 1A). We expressed all measurements in Relative Promoter Units (RPUs) by normalizing fluorescence intensity to a strain containing a reference construct35 (see Materials and Methods).
Figure 1.

Engineering synthetic receptors using the split-DBD principle (A) Overview of the LexA-based split-repressor system. LexA DBD was fused to VHH-Caffeine. The monomeric chimeric receptor is expressed in the cytosol upon IPTG induction. In the presence of caffeine, the chimeric receptor dimerizes and binds to the LexA operator, blocking expression of the reporter gene. (B) Response of cells harboring plasmids encoding LexA-VHH-Caffeine and LexA-VHH-Control to increasing concentrations of IPTG and caffeine. (C) Fold repression for the two LexA-VHH fusions in response to increasing concentrations of IPTG and caffeine. For each IPTG concentration, fold changes were calculated from (B) relatively to cells grown without caffeine (lower row).
As positive and negative controls, we expressed full-length LexA and LexA DBD. As expected, we observed that full-length LexA mediated-repression increased with IPTG concentration and was not affected by caffeine (Supplementary Figure S1). We also observed, as already reported,30 that the LexA DBD could repress gene expression at high concentration (up to 35% repression at 100 μM IPTG, Supplementary Figure S1). We then characterized the behavior of cells expressing LexA-VHH fusions in response to increasing concentrations of IPTG and caffeine (Figure 1B). We confirmed by Western blot (WB) that both fusion proteins were expressed at similar levels (Supplementary Figure S2).
We first observed that similarly to LexA DBD, LexA-VHH-Caffeine and LexA-VHH-Control displayed a concentration-dependent repression in the absence of caffeine (up to 45% at 100 μM IPTG induction, Figure 1B, lower rows). Both LexA-VHH fusions had repressive activity comparable to LexA-DBD, suggesting that this repression was primarily due to residual DBD activity and not to VHH oligomerization. We then monitored the response of the LexA-VHH fusions to increasing concentrations of caffeine. While no change was detectable for LexA-VHH-Control (Figure 1B, left panel), LexA-VHH-Caffeine had a dose-dependent response to caffeine starting at 25 μM IPTG concentration and 1 μM caffeine (Figure 1B, right panel). These results show that in the presence of its ligand, VHH-Caffeine dimerizes and restores DNA-binding activity, leading to transcriptional regulation. Response to caffeine was homogeneous among the whole cell population (Figure 2, upper panel). We observed a maximum repression of 4.3-fold at 100 μM IPTG induction and 100 μM caffeine (Figure 1C, Figure 2 lower panel). LexA-VHH-Caffeine had a repression activity comparable to full-length LexA at similar protein expression levels (respectively 100 μM and 12.5 μM IPTG concentration, as full-length LexA had a higher expression level for a comparable IPTG concentration). Taken together, these results demonstrate that synthetic receptors can be engineered by coupling split-DBDs with an antibody-domain undergoing ligand-induced dimerization.
Figure 2.

Response of LexA-VHH-Caffeine to increasing concentrations of caffeine at 25, 50, and 100 μM IPTG induction. Upper panel: flow-cytometry data of LexA-VHH-Caffeine response to caffeine at different expression level. Lower panel: titrations curves of LexA-VHH-Caffeine in response to increasing concentrations of caffeine and at different expression level. Error bars: standard deviation between three independent experiments performed in triplicate. *p < 0.05, and **p < 0.01, compared with signal in the absence of caffeine.
A Prokaryotic Transmembrane Receptor Using a Single-Domain Antibody for Ligand Detection
One current limitation of whole-cell biosensors is the difficulty to detect molecules in their extracellular environment. For example, many biomarkers of disease are proteins that cannot cross the cellular membrane.36,37 Having validated the principle of synthetic receptors using split-DBD, we thus sought to apply this method to engineer transmembrane receptors.
To this aim, we chose CadC, an E. coli transmembrane transcriptional activator from the ToxR family. CadC is composed of a N-terminal cytosolic DBD and a C-terminal periplasmic pH sensor domain.50 CadC activates the pCadBA promoter when environmental pH decreases and in the presence of lysine.51 The Lysine permease LysP inhibit CadC activity through interaction with CadC transmembrane domain when the environmental lysine concentration is low.52 Interestingly, dimerization of artificial transmembrane helices bound to the cytosolic CadC DBD is sufficient to restore CadC transcriptional activity.50
In order to assess the potential of CadC as a scaffold for transmembrane receptor engineering, we wanted to (i) disrupt endogenous regulation by environmental pH (via CadC C-terminal pH sensor domain) and by lysine (via interaction between LysP and CadC transmembrane region); and (ii) demonstrate that CadC could be activated through dimerization of a periplasmic domain.
To do so, we first replaced the C-terminal periplasmic pH sensing domain with the self-dimerizing leucine-zipper GCN4. Second, we replaced the wt CadC transmembrane domain (targeted by LysP) by an artificial transmembrane domain composed of 16 Leucine repeat residues. This Leu(16) transmembrane helix was previously shown to support expression of correctly oriented chimeric CadC proteins into E. coli inner membrane.50
We placed the expression of this fusion protein under the control of the pLacO1 promoter induced by IPTG. As a reporter, we used the GFP driven by the pCadBA promoter. We observed that cells expressing CadC-Leu(16)TM-GCN4 produced a strong GFP signal (Supplementary Figure S3). In comparison, cells expressing the same construction but using the wt transmembrane region had a much lower activity. These results confirm that CadC transcriptional activity can be restored via dimerization of a periplasmic domain and that the Leu(16) transmembrane region is better suited than the wt transmembrane domain for the engineering of synthetic CadC receptors.
We then built two CadC-VHH fusion proteins composed of CadC DBD, CadC juxtamembrane domain (JM), the Leu(16) transmembrane region (TM), CadC wild type external linker region (EL), and the VHH LBDs (Figure 3A). We placed the expression of these chimeric proteins under the control of the pLacO1 promoter (Figure 3B). Both fusion proteins had comparable expression levels across the IPTG concentration range (Supplementary Figure S4). We also confirmed by immunofluorescence that CadC-VHH-Caffeine was targeted to the cellular membrane (Supplementary Figure S5). We then measured GFP expression in response to increasing concentrations of IPTG and caffeine.
Figure 3.

A prokaryotic transmembrane receptor using a single-domain antibody for ligand detection. (A) General architecture of synthetic transmembrane receptor using the split-DBD principle. The DBD and Juxtamembrane of the CadC transcriptional activator were fused to an Leu(16)TM, an external linker, and VHH-Caffeine or VHH-Control as LBD. (B) Principle of transmembrane receptor activation. Genes encoding CadC-VHH fusions are placed under the control of the pLacO1 promoter. The N-terminal CadC DBD is located in the cytosol and the C-terminal VHH in the periplasm. In the presence of caffeine, the chimeric receptor CadC-VHH-Caffeine undergoes ligand-induced dimerization and activates downstream reporter gene expression. (C) Response of CadC-VHH-Caffeine and CadC-VHH-Control to increasing concentrations of caffeine at different expression level. (D) Activation fold of the two CadC-VHH fusions. For each IPTG concentration, fold changes were calculated from (C) as in Figure 1.
We first observed a strong response of CadC-VHH-Caffeine to increasing concentrations of caffeine, starting at 25 μM IPTG concentration (Figure 3C). CadC-VHH-Control did not show any response to caffeine. We calculated the signal swing (i.e., the absolute change in fluorescence intensity between inactive and active states) and fold change in response to caffeine (Figure 3C and D). We found that at 100 μM caffeine, the swing increased with increasing receptor expression (swing = 6, 19, and 21 RPU at 25, 50, and 100 μM IPTG, respectively, Figure 3C, right panel). However, the fold change was maximal at 25 μM IPTG (∼10-fold), and decreased with IPTG concentrations above (fold change = 6- and 4.6-fold at 50 and 100 μM IPTG, respectively; Figure 3D, right panel). This decrease in fold change is explained by a higher background noise due to nonspecific activation of the CadC-VHH-Caffeine receptor at high expression levels (from 0.6 to 5.6 RPUs, ∼9-fold increase, Figure 4). On the other hand, CadC-VHH-Control fusion did not show any background noise (Figure 3C; Supplementary Figure S6). Because both receptors were expressed at the same levels, these results suggest that VHH-Caffeine has a higher propensity to oligomerize than the VHH-Control. Therefore, higher expression levels increase receptor sensitivity to caffeine, but also to nonspecific self-activation and a deteriorated signal-to-noise ratio. We also found that cells induced at 25 μM IPTG induction had a bimodal distribution while at 50 μM and 100 μM the response to caffeine was homogeneous over the whole cell population (Figure 4, upper panel). Therefore, receptor expression levels need to be balanced to satisfy two criteria: support homogeneous response while minimizing self-activation and background noise. These results show that the split-DBD principle can be applied to engineer synthetic prokaryotic transmembrane receptors using a single-domain antibody as a LBD.
Figure 4.

Response of CadC-VHH-Caffeine to increasing concentration of caffeine at 25, 50, and 100 μM IPTG induction. Upper panel: flow cytometry data of CadC-VHH-Caffeine response to caffeine at different expression level. Lower panel: titration curves of CadC-VHH-Caffeine response to increasing concentration of caffeine at different expression level. Error bars: standard deviation between three independent experiments performed in triplicate. *p < 0.05, and **p < 0.01, compared with signal in the absence of caffeine.
Optimizing Transmembrane Receptor Signal-to-Noise Ratio through Linkers Engineering
We then wanted to improve the receptor signal-to-noise ratio by decreasing its background noise. We postulated that nonspecific oligomerization and activation could be reduced by altering interdomain linker sequences. We decided to focus on modifying (i) the external linker region, which was shown to influence receptor basal activation rate and signal-to-noise ratios,26 and (ii) the juxtamembrane region (JM), also described to have a strong effect on CadC response.38
We thus designed a library of 12 receptor variants by combining 3 different JM sequences (full-length [FL], partial deletion [PD] and full deletion [FD]), with 4 types of external linkers (wild-type [DTRLPMS, wt], flexible [GGGSG], rigid [EAAAK], and no linker [NL]) (Figure 5A). We first observed that all receptors incorporating the full deletion of the JM region were nonresponsive to caffeine (Figure 5B; Supplementary Figure S7) due to proteolysis (Supplementary Figure S8A). Variants incorporating the partial deletion of the JM region exhibited a significant increase in both background noise and signal intensity upon ligand addition. This increased noise was sequence dependent as all FL and PD variants were expressed at similar levels (Supplementary Figure S8B). We found that the best results were obtained using the CadC variant incorporating the full length JM and no external linker, which displayed a significant reduction of self-activation (Figure 5B). While the swing of this construct was slightly lower, the fold change was more than quadrupled compared to the wt variant (5- vs 22-fold change, respectively, Figure 5B). These data demonstrate that the response properties of synthetic transmembrane receptors can be optimized by varying the amino acid sequences of receptor linker regions.
Figure 5.

Optimizing transmembrane receptor signal-to-noise ratio through linker engineering. (A) Schematic diagram of the 12 receptor variants tested. (B) Quantification of the response of 12 receptor variants to 100 μM caffeine upon 50 μM IPTG induction. Error bars: standard deviation between three independent experiments performed in triplicate. *p < 0.05, and **p < 0.01, compared with signal in the absence of caffeine.
Connecting Synthetic Receptors to Synthetic Gene Circuits
Because split-DBDs control transcription, they could be connected to synthetic gene networks and support many applications requiring downstream signal processing.8 As a practical example, we connected the LexA system to a genetic inverter39 based on the BetI repressor40 (Figure 6A). Starting at 50 μM IPTG induction, we observed a marked increased in GFP intensity (Figure 6B). While the swing was lower than with the repressor only system (2.2 RPUs vs 12.2 RPUs at 100 μM IPTG, respectively), the fold change was still significant, (∼4-fold, Figure 6C), due to the very low background signal in the absence of caffeine. Interestingly, we observed no change in GFP signal across increasing concentrations of IPTG. We hypothesize that the inverter module buffers the nonspecific repressive effect from the DBD at high concentrations, probably because of the delay required to degrade the BetI repressor. These data confirm that synthetic receptors using the split-DBD principle can be connected to synthetic gene networks for downstream signal processing.
Figure 6.

Connecting synthetic receptors to downstream genetic circuits. (A) General architecture of LexA-VHH-Caffeine connected to the BetI inverter. LexA-VHH-Caffeine controls BetI expression, which controls the GFP expression. (B) Response of cells containing the LexA-VHH-Caffeine receptor connected to the BetI inverter to increasing concentrations of IPTG and caffeine. (C) Fold activation of the LexA-VHH-Caffeine/BetI inverter circuit.
Discussion
In this work we developed synthetic bacterial receptors that use a single-domain antibody as sensing domain. Our method consists on fusing the DBD of a transcriptional regulator with a single-domain antibody undergoing ligand-induced dimerization. Upon homodimerization, the DNA binding activity of the DBD is restored, leading to transcriptional regulation. We showed that the principle presented here is scalable and can be applied to both transcriptional repressors and activators.
We explored several parameters that can be tuned to improve receptor behavior. We first showed that high receptor expression levels lead to background noise arising from residual affinity of the monomeric DBD for its operator (Supplementary Figure S1A30) or from LBD multimerization. For example, CadC-VHH-Caffeine exhibits a higher background noise than CadC-VHH-Control (Supplementary Figure S6). Because VHH-Caffeine dimerizes upon ligand binding, we hypothesize that noise arises from an intrinsically higher tendency to oligomerize. VHH-Caffeine also has a higher propensity to oligomerize when expressed at the membrane, probably due to increased local concentrations and constrained mobility that enhances the likelihood of interaction between receptors. Therefore, a critical parameter affecting the performance of synthetic receptors activated by ligand-induced dimerization is the propensity of their sensing domain to spontaneously oligomerize. This tendency to oligomerize can be decreased by reducing receptor expression levels, and further engineering of sensing domain solubility/stability could also be used to reduce self-oligomerization. Nevertheless, here we demonstrate that VHHs can support the engineering of functional receptors and are promising sensing domains. The use of other recognition units will be constrained by their tendency to aggregate.
We demonstrate how receptor signal-to-noise ratio can be improved by varying the different linker regions and anticipate that different LBD-ligand complexes with different sizes and geometries will have varying linker requirements relative to length, charge, or flexibility. Transmembrane receptors coupled to new LBDs could be best engineered via high-throughput screening of combinatorial linker libraries using a Flow-Seq approach.41 Alternatively, directed-evolution coupled with stringent selection methods could help design split-DBDs operating in a plug-and-play fashion.42
The synthetic receptors developed here present several advantageous features that should enable many applications. First, through combinatorial library construction and high-throughput screening, synthetic binders can be generated for a nearly unlimited numbers of ligands.20−23 To detect a monomeric antigen, two different antibodies targeting different epitopes could be combined, an approach already used in sandwich ELISA or in other split-protein biosensors.43 Second, many transcription factors have been characterized from which several orthogonal receptors could be derived.40 Third, because our system controls transcription, ligand detection can be connected to downstream genetic circuits. We provide an example by connecting the LexA-based sensor to a genetic inverter (Figure 6). Higher-order signal processing could also be performed, such as genetically encoded logic or signal amplification.8,44,45
We also provide the first example, to the best of our knowledge, of a bacterial transmembrane receptor using versatile single-domain antibodies for ligand detection. Such transmembrane receptors are highly relevant for future applications in which engineered prokaryotic cells need to detect ligands that cannot penetrate into the cytosol, like proteins which are important pathological biomarkers.36,46 Importantly, VHH and other synthetic binders are efficiently expressed and functional at the bacterial membrane. As a proof-of-concept, we successfully expressed a synthetic alpha-rep binder recognizing GFP and found that it could bind recombinant eGFP in E. coli cells having a deficient outer membrane (Supplementary Figure S9). The transmembrane receptor principle developed here is general enough to support deployment into other bacterial hosts, such as Gram-positives. In this case, the receptor sensing domain would be directly exposed on the cell surface and accessible to extracellular ligands.
In conclusion, prokaryotic receptors using single-domain antibodies as sensing domains offer a promising scalable ligand detection platform to support many translational applications of synthetic biology, including sophisticated low-cost diagnosis, environmental monitoring and remediation, and targeted cellular therapeutics.
Materials and Methods
Plasmids and Strains
Sequences for LexA, CadC, and VHHs are provided in Supporting Information. All constructs were cloned into the low-copy plasmid pSB4K547 using Gibson assembly.48 Plasmid maps are available in SM. All experiments were performed using E. coli strain NEB10β (New England Biolabs). Plasmids and materials will be made available through Addgene. Sequences will be deposited into GENBANK.
Polymerase Chain Reaction
PCR amplifications were performed in a 40 μL reaction mixture consisting of 0.1–10 ng of template DNA fragment, 1 μL of each forward/reverse primer (20 μM), and 20 μL of Q5 hot start high-fidelity 2× master mix (NEB). After 30 s of initial denaturation at 98 °C, 35 cycles were conducted with the PCR procedures of 10 s at 98 °C, 30 s at corresponding annealing temperature (different with each primer combination, calculated with NEB Tm calculator: http://tmcalculator.neb.com/#!/), and elongation (2 kb/min) at 72 °C, with a final extension at 72 °C for 10 min. The PCR product was verified by gel electrophoresis, then purified by PCR cleanup kit, and the DNA concentration was determined using a Nanodrop spectrophotometer.
Gibson Assembly
The PCR reactions were digested with 1 μL DpnI (20 units/ml, NEB) in 40 μL CutSmart reaction buffer (NEB) at 37 °C for 1 h to remove template DNA. The resulting mixture was then used for Gibson assembly reaction. In each Gibson assembly reaction, 100 ng of vector DNA fragment and 3–5 fold of insert fragments were incubated with 10 μL of 2× Gibson assembly master mix (NEB) in a final volume of 20 μL at 50 °C for 60 min. To prevent the DNA ligase activity in the reaction mix affecting the following electroporation efficiency, the reaction mix was further heat-inactivated at 80 °C for 15 min.
Electrotransformation
One μL of Gibson assembly products was added into 40 μL NEB10β electro-competent cells and then transferred in Biorad 0.1 cm gap Micropulser electroporation cuvettes. Immediately after electroporation with Biorad Micropulser electroporator using program EC1, 1 mL of prewarmed (37 °C) SOC medium was added into the transformants. The cell culture was further incubated at 37 °C with vigorous shaking for 1 h. The transformants were then plated on the selection plate with antibiotics (e.g., kanamycin) and incubated at 37 °C overnight. The constructs were verified by Sanger sequencing.
Functional Characterization of Synthetic Receptors
Plasmids encoding the different receptors were transformed into chemically competent E. coli NEB10β (New England Biolabs), and plated on LB agar supplemented with 25 μg/mL kanamycin. For each construct, three colonies were picked and inoculated into 3 mL of LB/kanamycin and grown for 16 h. The following day, the cultures were diluted 1:100 into 500 μL LB/kanamycin into 96 deep-well plates, grown at 37 °C for about 1.5 h until O.D. 600 reached 0.3. IPTG and caffeine were then added at different concentrations. Plates were then incubated at 37 °C for 16 h with shaking and analyzed by flow cytometry. All experiments were performed at least 3 times in triplicate. Data points and error bars represent the mean and standard deviation, respectively. The signal outputs with significant difference verified by paired sample t test (p-value < 0.05) marked by asterix. We used as an in vivo reference standard a reference promoter (J23101) driving GFP, and expressed all values in Relative Promoter Units (RPUs). We chose to work within a range of caffeine concentration from 0 to 100 μM as we observed a nonspecific receptor activation caused by excess amount of caffeine at a 1 mM concentration (Supplementary Figure S10 and S11)
Flow Cytometry Analysis
Flow cytometry was performed using an Attune NxT cytometer coupled with high-throughput autosampler (Thermo Fisher Scientific). 30 000 cells were collected for each data point. Flow cytometry data were analyzed using FlowJo (Treestar Inc., Ashland, USA). All raw data values are in Supporting Information.
Calculation of Relative Promoter Units (RPUs)
Fluorescence intensity measurements among different experiments were converted into RPUs by normalizing them according to the fluorescence intensity of an E. coli strain containing a reference construct and grown in parallel for each experiment. We used the constitutive promoter J23101 and RBS_B0032 as our in vivo reference standard and placed superfolder GFP as a reporter gene in plasmid pSB4K5. We quantified the median of fluorescence intensity (MFI) of the flow cytometry data and calculated RPUs according to the following equation:
Calculation of Repression or Activation Fold
Repression and activation folds for the different synthetic receptors at different caffeine concentrations were calculated by dividing the signal intensity of cells grown in the presence of caffeine by the signal signal intensity of cells grown at the same IPTG concentration without caffeine, according to the following equation:
Western Blotting
In order to quantify receptor expression levels by Western-blot, all constructs were fused with a C-terminal c-Myc tag. A 1.5 mL of overnight culture was centrifuged at 13 000g for 5 min to collect the cells. The whole cell extract sample was prepared in 50 μL 1× SDS sample buffer and heated at 95 °C for 10 min. After centrifugation for 10 min at 13 000g, 10 μL of the denatured sample was fractionated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membrane using the Trans-Blot Turbo transfer system (Bio-Rad). The membrane was washed twice with PBS and blocked with 5% nonfat dry milk dissolved in PBST (PBS supplemented with 0.5% Tween 20) at 4 °C for 2 h. After one wash with PBST, the membrane was incubated with two primary antibodies: anti-c-Myc, chicken IgY fraction (Life, 1:2500); and anti-GroEL, rabbit polyclonal antibody (Sigma, 1:5000) at 4 °C overnight. Membranes were washed twice with PBST buffer for 10 min and incubated with secondary antibodies at 4 °C for 2 h. After washing twice with PBST, the membranes were imaged on a Typhoon scanner. The band intensities were quantified using ImageJ software. The normalized expression level (NEL) was calculated by following equation:
Preparation of Cell Wall Deficient E. coli
Cell wall (outer membrane) deficient E. coli were grown in osmo-protective medium composed of 2 × MSM medium (40 mM MgCl2, 1 M sucrose and 40 mM maleic acid, pH 7), mixed 1:1 with 2 × Beef Medium (6 g beef extract/L, 20 g bacteriological peptone/L, 10 g yeast extract/L, and 10 g NaCl/L). The overnight cultures of E. coli containing relative construct was diluted 100-fold to Beef/MSM medium with 45 μg/mL cefsulodin to transiently interfere with E. coli outer membrane formation. Cells were grown for 16 h at 30 °C.
Microscopy
Images were acquired using IX71 inverted microscope (Olympus) equipped with RFP (Ex 535/50, Em 610/75) and GFP (Ex 480/40, Em 535/50) filters, and a Phantom Miro LC310 camera. The microscope was operated by the Phantom Camera Control software. An Olympus UIS2 UPLFLN 100× O2PH oil-immersion objective was used. The images were processed using ImageJ (https://imagej.nih.gov/ij/).
Antibody Labeling of CadC-VHH-Caffeine Expressed on Cell Wall Deficient E. coli
Ten μL of E. coli an overnight culture with or without the CadC-VHH-Caffeine plasmid was added diluted into Beef/MSM/kanamycin medium with 45 μg/mL cefsulodin to transiently interfere with E. coli outer membrane formation. IPTG was added to the culture to induce CadC-VHH-Caffeine expression. The cell culture was grown at 30 °C for 16 h. Cells were centrifuged at 850g in 4 °C for 10 min. The pelleted cells were blocked by Beef-MSM medium containing 5% BSA at 4 °C for 1 h. After blocking, the cells were labeled with anti-c-Myc chicken IgY antibody (1:1000) at 4 °C for 1 h. The cells were pelleted and washed twice with Beef-MSM medium containing 5% BSA, then further labeled with goat antichicken antibody conjugated with Alexa 488 at 4 °C for 1 h. After being washed twice with Beef-MSM medium–5% BSA, cells was analyzed by microscopy and flow cytometry.
Acknowledgments
We thank G. Labesse, J. Gracy, L. Ciandrini, G. Cambray, P. Voyvodic, members of the Bonnet lab and of the CBS for fruitful discussions. We thank L. Dumas, L. Lanotte and M. Abkarian for help with microscopy. Support was provided by an ERC Starting Grant “COMPUCELL” to J.B., and ANR “Alphasense” to J.B. and P. Minard. J.B. acknowledges the INSERM Atip-Avenir program and the Bettencourt-Schueller Foundation for continuous support. M.C.G. acknowledges support from the French Infrastructure for Integrated Structural Biology (FRISBI) ANR-10-INSB-05–01.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.7b00266.
Supplemental figures, tables and sequence files for all constructs (PDF)
Author Contributions
H.J.C. and J.B. designed the experiments. H.J.C., P. Mayonove, and A.Z. performed the experiments. P. Minard provided alpha-Reps binding GFP. M.C.G. and A.D.V. purified recombinant GFP. H.J.C. and J.B. interpreted the data. H.J.C., M.C.G., P. Minard and J.B. participated in the elaboration of the manuscript, H.J.C. and J.B. wrote the manuscript. All authors approved the manuscript.
The authors declare the following competing financial interest(s): We have deposited a patent application on the work presented in the manuscript.
Notes
Plasmids and materials will be made available through Addgene. Sequences for CadC-VHH-caffeine and LexA-VHH-caffeine are available on GenBank with accession numbers MG210478 and MG210479, respectively.
Supplementary Material
References
- Yagi K. (2007) Applications of whole-cell bacterial sensors in biotechnology and environmental science. Appl. Microbiol. Biotechnol. 73, 1251–1258. 10.1007/s00253-006-0718-6. [DOI] [PubMed] [Google Scholar]
- Raut N.; O’Connor G.; Pasini P.; Daunert S. (2012) Engineered cells as biosensing systems in biomedical analysis. Anal. Bioanal. Chem. 402, 3147–3159. 10.1007/s00216-012-5756-6. [DOI] [PubMed] [Google Scholar]
- Slomovic S.; Pardee K.; Collins J. J. (2015) Synthetic biology devices for in vitro and in vivo diagnostics. Proc. Natl. Acad. Sci. U. S. A. 112, 14429–14435. 10.1073/pnas.1508521112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñero-Lambea C.; Ruano-Gallego D.; Fernández L. Á. (2015) Engineered bacteria as therapeutic agents. Curr. Opin. Biotechnol. 35, 94–102. 10.1016/j.copbio.2015.05.004. [DOI] [PubMed] [Google Scholar]
- Mimee M.; Citorik R. J.; Lu T. K. (2016) Microbiome therapeutics - Advances and challenges. Adv. Drug Delivery Rev. 105, 44–54. 10.1016/j.addr.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer J. R.; Belkin S. (2010) Where microbiology meets microengineering: design and applications of reporter bacteria. Nat. Rev. Microbiol. 8, 511–522. 10.1038/nrmicro2392. [DOI] [PubMed] [Google Scholar]
- Kumari A.; Pasini P.; Daunert S. (2008) Detection of bacterial quorum sensing N-acyl homoserine lactones in clinical samples. Anal. Bioanal. Chem. 391, 1619–1627. 10.1007/s00216-008-2002-3. [DOI] [PubMed] [Google Scholar]
- Courbet A.; Endy D.; Renard E.; Molina F.; Bonnet J. (2015) Detection of pathological biomarkers in human clinical samples via amplifying genetic switches and logic gates. Sci. Transl. Med. 7, 289ra83. 10.1126/scitranslmed.aaa3601. [DOI] [PubMed] [Google Scholar]
- Danino T.; Prindle A.; Kwong G. A.; Skalak M.; Li H.; Allen K.; Hasty J.; Bhatia S. N. (2015) Programmable probiotics for detection of cancer in urine. Sci. Transl. Med. 7, 289ra84–289ra84. 10.1126/scitranslmed.aaa3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riglar D. T.; Giessen T. W.; Baym M.; Kerns S. J.; Niederhuber M. J.; Bronson R. T.; Kotula J. W.; Gerber G. K.; Way J. C.; Silver P. A. (2017) Engineered bacteria can function in the mammalian gut long-term as live diagnostics of inflammation. Nat. Biotechnol. 35, 653. 10.1038/nbt.3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daeffler K. N.-M.; Galley J. D.; Sheth R. U.; Ortiz-Velez L. C.; Bibb C. O.; Shroyer N. F.; Britton R. A.; Tabor J. J. (2017) Engineering bacterial thiosulfate and tetrathionate sensors for detecting gut inflammation. Mol. Syst. Biol. 13, 923. 10.15252/msb.20167416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñero-Lambea C.; Bodelón G.; Fernández-Periáñez R.; Cuesta A. M.; Álvarez-Vallina L.; Fernández L. Á. (2015) Programming controlled adhesion of E. coli to target surfaces, cells, and tumors with synthetic adhesins. ACS Synth. Biol. 4, 463–473. 10.1021/sb500252a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su L.; Jia W.; Hou C.; Lei Y. (2011) Microbial biosensors: a review. Biosens. Bioelectron. 26, 1788–1799. 10.1016/j.bios.2010.09.005. [DOI] [PubMed] [Google Scholar]
- Lehning C. E.; Heidelberger J. B.; Reinhard J.; Nørholm M. H. H.; Draheim R. R. (2017) A Modular High-Throughput In Vivo Screening Platform Based on Chimeric Bacterial Receptors. ACS Synth. Biol. 6, 1315. 10.1021/acssynbio.6b00288. [DOI] [PubMed] [Google Scholar]
- Collins C. H.; Leadbetter J. R.; Arnold F. H. (2006) Dual selection enhances the signaling specificity of a variant of the quorum-sensing transcriptional activator LuxR. Nat. Biotechnol. 24, 708–712. 10.1038/nbt1209. [DOI] [PubMed] [Google Scholar]
- Skjoedt M. L.; Snoek T.; Kildegaard K. R.; Arsovska D.; Eichenberger M.; Goedecke T. J.; Rajkumar A. S.; Zhang J.; Kristensen M.; Lehka B. J.; Siedler S.; Borodina I.; Jensen M. K.; Keasling J. D. (2016) Engineering prokaryotic transcriptional activators as metabolite biosensors in yeast. Nat. Chem. Biol. 12, 951–958. 10.1038/nchembio.2177. [DOI] [PubMed] [Google Scholar]
- Taylor N. D.; Garruss A. S.; Moretti R.; Chan S.; Arbing M. A.; Cascio D.; Rogers J. K.; Isaacs F. J.; Kosuri S.; Baker D.; Fields S.; Church G. M.; Raman S. (2016) Engineering an allosteric transcription factor to respond to new ligands. Nat. Methods 13, 177–183. 10.1038/nmeth.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J.; Jester B. W.; Tinberg C. E.; Mandell D. J.; Antunes M. S.; Chari R.; Morey K. J.; Rios X.; Medford J. I.; Church G. M.; Fields S.; Baker D. (2015) A general strategy to construct small molecule biosensors in eukaryotes. eLife 10.7554/eLife.10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libis V.; Delépine B.; Faulon J.-L. (2016) Expanding Biosensing Abilities through Computer-Aided Design of Metabolic Pathways. ACS Synth. Biol. 5, 1076. 10.1021/acssynbio.5b00225. [DOI] [PubMed] [Google Scholar]
- Plückthun A. (2015) Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 55, 489–511. 10.1146/annurev-pharmtox-010611-134654. [DOI] [PubMed] [Google Scholar]
- Guellouz A.; Valerio-Lepiniec M.; Urvoas A.; Chevrel A.; Graille M.; Fourati-Kammoun Z.; Desmadril M.; van Tilbeurgh H.; Minard P. (2013) Selection of specific protein binders for pre-defined targets from an optimized library of artificial helicoidal repeat proteins (alphaRep). PLoS One 8, e71512. 10.1371/journal.pone.0071512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide S.; Koide A.; Lipovšek D. (2012) Target-binding proteins based on the 10th human fibronectin type III domain (10Fn3). Methods Enzymol. 503, 135–156. 10.1016/B978-0-12-396962-0.00006-9. [DOI] [PubMed] [Google Scholar]
- Ducancel F.; Muller B. H. (2012) Molecular engineering of antibodies for therapeutic and diagnostic purposes. MAbs 4, 445–457. 10.4161/mabs.20776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos M.; Levine B. L.; Porter D. L.; Katz S.; Grupp S. A.; Bagg A.; June C. H. (2011) T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Sci. Transl. Med. 3, 95ra73–95ra73. 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsut L.; Roybal K. T.; Xiong X.; Gordley R. M.; Coyle S. M.; Thomson M.; Lim W. A. (2016) Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 164, 780–791. 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz K. A.; Daringer N. M.; Dolberg T. B.; Leonard J. N. (2017) Rewiring human cellular input-output using modular extracellular sensors. Nat. Chem. Biol. 13, 202–209. 10.1038/nchembio.2253. [DOI] [PubMed] [Google Scholar]
- Luscombe N. M.; Austin S. E.; Berman H. M.; Thornton J. M. (2000) An overview of the structures of protein-DNA complexes. Genome Biol. 1, reviews001.1. 10.1186/gb-2000-1-1-reviews001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladant D.; Karimova G. (2000) Genetic systems for analyzing protein–protein interactions in bacteria. Res. Microbiol. 151, 711–720. 10.1016/S0923-2508(00)01136-0. [DOI] [PubMed] [Google Scholar]
- Schlacher K.; Goodman M. F. (2007) Lessons from 50 years of SOS DNA-damage-induced mutagenesis. Nat. Rev. Mol. Cell Biol. 8, 587–594. 10.1038/nrm2198. [DOI] [PubMed] [Google Scholar]
- Schmidt-Dörr T.; Oertel-Buchheit P.; Pernelle C.; Bracco L.; Schnarr M.; Granger-Schnarr M. (1991) Construction, Purification, and Characterization of a Hybrid Protein Comprising the DNA Binding Domain of the LexA Repressor and the Jun Leucine Zipper: A. Biochemistry 30, 9657–9664. 10.1021/bi00104a013. [DOI] [PubMed] [Google Scholar]
- Dmitrova M.; Younès-Cauet G.; Oertel-Buchheit P.; Porte D.; Schnarr M.; Granger-Schnarr M. (1998) A new LexA-based genetic system for monitoring and analyzing protein heterodimerization in Escherichia coli. Mol. Gen. Genet. 257, 205–212. 10.1007/s004380050640. [DOI] [PubMed] [Google Scholar]
- Thliveris A. T.; Little J. W.; Mount D. W. (1991) Repression of the E coli recA gene requires at least two LexA protein monomers. Biochimie 73, 449–456. 10.1016/0300-9084(91)90112-E. [DOI] [PubMed] [Google Scholar]
- Sonneson G. J.; Horn J. R. (2009) Hapten-induced dimerization of a single-domain VHH camelid antibody. Biochemistry 48, 6693–6695. 10.1021/bi900862r. [DOI] [PubMed] [Google Scholar]
- Koide A.; Tereshko V.; Uysal S.; Margalef K.; Kossiakoff A. A.; Koide S. (2007) Exploring the capacity of minimalist protein interfaces: interface energetics and affinity maturation to picomolar KD of a single-domain antibody with a flat paratope. J. Mol. Biol. 373, 941–953. 10.1016/j.jmb.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz R.; Bujard H. (1997) Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 25, 1203–1210. 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly J. R.; Rubin A. J.; Davis J. H.; Ajo-Franklin C. M.; Cumbers J.; Czar M. J.; de Mora K.; Glieberman A. L.; Monie D. D.; Endy D. (2009) Measuring the activity of BioBrick promoters using an in vivo reference standard. J. Biol. Eng. 3, 4. 10.1186/1754-1611-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrebaeck C. A. K. (2017) Precision diagnostics: moving towards protein biomarker signatures of clinical utility in cancer. Nat. Rev. Cancer 17, 199–204. 10.1038/nrc.2016.153. [DOI] [PubMed] [Google Scholar]
- Tan E. M. (2012) Autoantibodies, autoimmune disease, and the birth of immune diagnostics. J. Clin. Invest. 122, 3835–3836. 10.1172/JCI66510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner E.; White S. H. (2014) Topology, dimerization, and stability of the single-span membrane protein CadC. J. Mol. Biol. 426, 2942–2957. 10.1016/j.jmb.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlundt A.; Buchner S.; Janowski R.; Heydenreich T.; Heermann R.; Lassak J.; Geerlof A.; Stehle R.; Niessing D.; Jung K.; Sattler M. (2017) Structure-function analysis of the DNA-binding domain of a transmembrane transcriptional activator. Sci. Rep. 7, 1051. 10.1038/s41598-017-01031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsch L.; Koller C.; Haneburger I.; Jung K. (2008) The membrane-integrated transcriptional activator CadC of Escherichia coli senses lysine indirectly via the interaction with the lysine permease LysP. Mol. Microbiol. 67, 570–583. 10.1111/j.1365-2958.2007.06070.x. [DOI] [PubMed] [Google Scholar]
- Buchner S.; Schlundt A.; Lassak J.; Sattler M.; Jung K. (2015) Structural and Functional Analysis of the Signal-Transducing Linker in the pH-Responsive One-Component System CadC of Escherichia coli. J. Mol. Biol. 427, 2548–2561. 10.1016/j.jmb.2015.05.001. [DOI] [PubMed] [Google Scholar]
- The MIT Synthetic Biology Working Group, Endy D., Deese I., and Wadey C.. Adventures in Synthetic Biology, November 24, 2005. [Google Scholar]
- Stanton B. C.; Nielsen A. A. K.; Tamsir A.; Clancy K.; Peterson T.; Voigt C. A. (2014) Genomic mining of prokaryotic repressors for orthogonal logic gates. Nat. Chem. Biol. 10, 99–105. 10.1038/nchembio.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon E.; Kalma Y.; Sharp A.; Raveh-Sadka T.; Levo M.; Zeevi D.; Keren L.; Yakhini Z.; Weinberger A.; Segal E. (2012) Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat. Biotechnol. 30, 521–530. 10.1038/nbt.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu J.; Zinkus-Boltz J.; Dickinson B. C. (2017) Evolution of a split RNA polymerase as a versatile biosensor platform. Nat. Chem. Biol. 13, 432–438. 10.1038/nchembio.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stains C. I.; Furman J. L.; Porter J. R.; Rajagopal S.; Li Y.; Wyatt R. T.; Ghosh I. (2010) A general approach for receptor and antibody-targeted detection of native proteins utilizing split-luciferase reassembly. ACS Chem. Biol. 5, 943–952. 10.1021/cb100143m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet J.; Yin P.; Ortiz M. E.; Subsoontorn P.; Endy D. (2013) Amplifying genetic logic gates. Science 340, 599–603. 10.1126/science.1232758. [DOI] [PubMed] [Google Scholar]
- Nielsen A. A. K.; Der B. S.; Shin J.; Vaidyanathan P.; Paralanov V.; Strychalski E. A.; Ross D.; Densmore D.; Voigt C. A. (2016) Genetic circuit design automation. Science 352, aac7341. 10.1126/science.aac7341. [DOI] [PubMed] [Google Scholar]
- Scofield R. H. (2004) Autoantibodies as predictors of disease. Lancet 363, 1544–1546. 10.1016/S0140-6736(04)16154-0. [DOI] [PubMed] [Google Scholar]
- Shetty R. P.; Endy D.; Knight T. F. Jr. (2008) Engineering BioBrick vectors from BioBrick parts. J. Biol. Eng. 2, 5. 10.1186/1754-1611-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D. G.; Young L.; Chuang R.-Y.; Venter J. C.; Hutchison C. A. 3rd; Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.