

Abstract
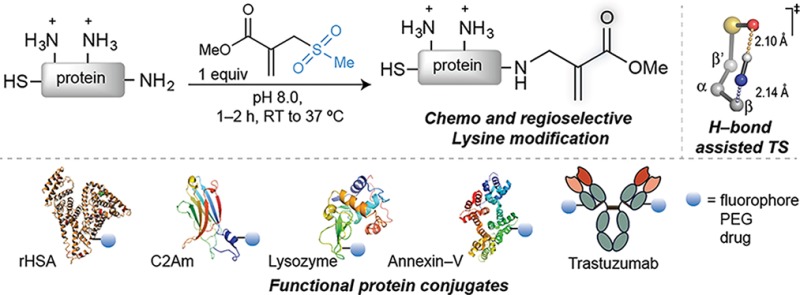
Site-selective chemical conjugation of synthetic molecules to proteins expands their functional and therapeutic capacity. Current protein modification methods, based on synthetic and biochemical technologies, can achieve site selectivity, but these techniques often require extensive sequence engineering or are restricted to the N- or C-terminus. Here we show the computer-assisted design of sulfonyl acrylate reagents for the modification of a single lysine residue on native protein sequences. This feature of the designed sulfonyl acrylates, together with the innate and subtle reactivity differences conferred by the unique local microenvironment surrounding each lysine, contribute to the observed regioselectivity of the reaction. Moreover, this site selectivity was predicted computationally, where the lysine with the lowest pKa was the kinetically favored residue at slightly basic pH. Chemoselectivity was also observed as the reagent reacted preferentially at lysine, even in those cases when other nucleophilic residues such as cysteine were present. The reaction is fast and proceeds using a single molar equivalent of the sulfonyl acrylate reagent under biocompatible conditions (37 °C, pH 8.0). This technology was demonstrated by the quantitative and irreversible modification of five different proteins including the clinically used therapeutic antibody Trastuzumab without prior sequence engineering. Importantly, their native secondary structure and functionality is retained after the modification. This regioselective lysine modification method allows for further bioconjugation through aza-Michael addition to the acrylate electrophile that is generated by spontaneous elimination of methanesulfinic acid upon lysine labeling. We showed that a protein–antibody conjugate bearing a site-specifically installed fluorophore at lysine could be used for selective imaging of apoptotic cells and detection of Her2+ cells, respectively. This simple, robust method does not require genetic engineering and may be generally used for accessing diverse, well-defined protein conjugates for basic biology and therapeutic studies.
Introduction
Site-selective chemical modification of proteins is a versatile strategy for modulating their function and provides diverse structures of biological and therapeutic importance.1−5 In nature, this process occurs under rigorous enzymatic control at a specific residue within the sequence of a protein, mostly guided by complex and exquisitely evolved protein–protein interactions.6 Currently, combined synthetic and biochemical techniques allow the preparation of well-defined protein conjugates bearing the desired modification at a specific site. Site selectivity is typically achieved by targeting highly nucleophilic cysteine residues genetically installed at the protein surface,7−9 uniquely reactive amino acids at the N- or C-terminus,10−12 or noncanonical amino acids bearing side chains with bioorthogonal reactivity.3,13 In one example, it has been shown that the site at which the cysteine is introduced can modulate both the efficacy of the conjugation reaction and therapeutic efficacy of antibody–drug conjugates in tumor-bearing mice.9 However, these methods either rely on extensive sequence engineering or they are limited to specific residues at either terminus of the protein. In addition, most methods require a large excess of often-valuable reagents or require metals or other additives that must later be removed from the reaction mixture.14 Thus, much remains to be learned in order to attain efficiency and selectivity comparable to that of enzymes, particularly when multiple sites are potentially reactive within the same native protein.
When proteins are modified through a reaction with electrophiles, nucleophilic cysteine residues are often targeted. Indeed both native and engineered cysteine residues are known to react rapidly with a variety of electrophilic reagents.4,15 While cysteine contains the most nucleophilic side chains at physiological pH, there are some pitfalls to relying on this residue for selective modification. For example, cysteine is relatively rare and constitutes only 1.9% of residues in human proteins.12,16 Furthermore, cysteine residues are often buried within the folded protein as disulfide bonds critical for structural integrity.15 These residues are not often accessible for modification, and preparing protein mutants with non-native cysteine residues can compromise folding and function. Although there are some isolated examples of chemoselective modification of tyrosine,17 tryptophan,18 and more recently methionine,19 targeting naturally occurring side chains in a chemo- and regioselective fashion remains a great and unexplored challenge.20
Lysine residues, in contrast to cysteine, are much more abundant, making up 5.9% of all sites in human proteins.12,16 Lysine is generally found on the surface of proteins due to the dominant ionic character of the side chain that enhances their hydrosolubility. Thus, native lysine residues are more convenient targets for protein modification than cysteine residues, as chronicled in the long history of bioconjugation at lysine.20,21 However, a general method to label a single lysine residue over all other lysines has been elusive. There are, however, a number of clues in the literature that certain lysine residues can be selectively modified, providing a starting point for developing such a general method. For example, kinetically controlled, regioselective lysine labeling was achieved with some success using substoichiometric amounts of N-hydroxysuccinimide (NHS)-ester activated avidin reagents on two proteins (lysozyme and RNase A) bearing a low copy number of lysine residues (Figure 1a).22 However, the requirement of substoichiometric amounts of reagent means complete conversion is not possible, and challenges in batch-to-batch reproducibility make this approach difficult to generalize to more complex protein systems. In another example, Barbas and co-workers revealed that certain lysine residues in albumin reacted more rapidly with α,β-unsaturated sulfonamides than other lysine residues (Figure 1b).23,24 Recently, Cravatt and co-workers showed that sulfotetrafluorophenyl esters are selective for lysine and, in certain cases, hyper-reactive lysine residues could be specifically modified in a quantitative profiling of lysine reactivity in human cell proteome.25 A number of small molecule inhibitors have been shown to covalently target lysine residues.26 In one example, Kelly and co-workers designed small molecules equipped with stilbenes and showed that these systems can selectively and covalently modify the plasma protein transthyretin with preference for one of the eight lysine ε-amino groups displayed on the protein’s surface.27 Recently, Roush, Rader and co-workers reported the selective modification of a catalytic lysine on an antibody using a β-lactam electrophile.28 Unfortunately, little is known about what governs lysine selectivity and whether any of these methods could be generalized to any given class of proteins.
Figure 1.

Overview of methods for lysine labeling. (a) Kinetically controlled labeling of lysine on proteins using an activated ester electrophile.22 (b) PEG-modified TAK-242, a Toll-like receptor 4 inhibitor reacts with a single lysine on human serum albumin.23 These isolated examples of site-selective lysine modification do not proceed to completion and have been applied to a limited number of proteins. In addition, these approaches were mostly used for the introduction of tags for a further labeling reaction (e.g., copper(I) catalyzed azide–alkyne cycloaddition). (c) This work: Hydrogen bond assisted chemo- and regioselective modification of lysine on native proteins. The addition reaction proceeds through a H-bonded chair-like addition transition state leading to the subsequent spontaneous elimination of methanesulfinic acid; this restores a type 2 alkene that undergoes site-specific modification through aza-Michael addition of suitable synthetic molecules bearing an amine nucleophile.
Therefore, an advance in the chemical modification of lysine is needed, specifically, an understanding of the subtle reactivity differences at each lysine that could guide the design of reagents to target the most reactive lysine residue. This fundamental understanding of lysine reactivity could also aid in the prediction of the site of reaction for a given protein. Such predictive power in kinetically controlled lysine labeling would offer a valuable method for directly modifying a protein and, in principle, be applicable to all classes of proteins in their native form. We began this analysis by first re-examining the basis for reactivity of nucleophilic residues on proteins. To this end, a review of the literature indicated that solvent accessibility is often used to rationalize site-selective amino acid labeling. However, this parameter alone fails to correctly predict reactivity when multiple residues have similarly solvent accessibility.22 On the other hand, it has been known for a long time that the rate of reaction between an amino group and an electrophile in water increases rapidly as the pH of the medium approaches the pKa of the protonated amino group. Along these lines, enzyme active sites are often situated in unique environments that optimize the effective pKa of catalytic groups. In nonpolar, poorly hydrated microenvironments, the neutral form of ionizable groups is favored and the pKa values are shifted with respect to their normal values in water. In these nonpolar microenvironments, cysteine residues have higher pKa’s so that the neutral and less nucleophilic thiol (R–SH) is favored over the charged and highly nucleophilic thiolate (R–S–). Lysine residues, in contrast, have lower pKa’s in these nonpolar microenvironments so that the neutral and nucleophilic amine (R–NH2) is the predominant species, rather than the non-nucleophilic protonated amine (R–NH3+). Of note, the pKa of lysine can vary up to five units within the same protein,29 depending on its local microenvironment and the solvent accessibility, surrounding residues and protein folding.30 Notably, pKa depression of around two units is the basis for substrate lysine activation for E2-mediated nucleophilic conjugation in the small ubiquitin-like modifier (SUMO) pathway. This modulation in the lysine pKa allows catalysis to occur at physiological pH.31 As demonstrated through both computational and experimental mutagenesis, pKa depression was more dependent on desolvation rather than electrostatics, a property governed by the presence of hydrophobic residues nearby the catalytic lysine. Similarly, the reactivity of catalytic lysine residues in aldolases of both natural32,33 and engineered34,35 provenance is enhanced by virtue of a lower pKa in the relatively hydrophobic active site.
Inspired by the ways in which enzymes increase the acidity of specific lysine residues, we wondered whether this phenomenon of local pKa variation could guide selective chemical modification of a single lysine on a native protein. Furthermore, we wondered if an electrophilic reagent could be designed so that it can be directed to this most reactive lysine and react selectively with its side chain under strictly kinetic control. In this way, the first general method for regioselective lysine labeling would be realized.
Here, we report the discovery of that reagent: a sulfonyl acrylate that reacts selectively with the most reactive lysine in a protein (Figure 1c). The reagent is directed to the neutral lysine by transient hydrogen bonding between the sulfonyl group and the ε-amino group of the side chain. By proceeding through a low energy, H–bond assisted chairlike addition transition state, the key N–C bond is formed. Subsequent cleavage of the sulfonyl leaving group, which is also facilitated by hydrogen bonding to the lysine ε-amino group, provides an additional thermodynamic driving force. This reaction is highly regioselective, as only one lysine was modified for all proteins examined when using a single molar equivalent of the reagent. The chemoselectivity was also exquisite, with the sulfonyl acrylate reacting only with the most nucleophilic lysine.
This method was demonstrated on five different proteins including a therapeutic antibody without prior sequencing engineering. The simplicity and exquisite chemo- and regioselectivity is rare in protein modification and provides conjugates that retain their natural folding and function. Furthermore, the modified lysine derivative may be further functionalized through a second aza-Michael addition, since the departure of the sulfone leaving group unmasks another electrophile (i.e., an acrylate) that could be selectively modified with synthetic molecules bearing a reactive amine handle. The full reaction sequence is orthogonal to other bioconjugation strategies such as mixed disulfide formation, enabling the construction of modified proteins that bear two distinct modifications at two different sites. This conjugation method provides far-reaching opportunities to selectively tag naturally abundant lysine, without the need of installing noncanonical residues3,4,13 and motifs8 that can potentially compromise the structural integrity and function of the protein.
Results and Discussion
Computer-Assisted Design of Sulfonyl Acrylate 1c for Lysine Modification
Site-selective reactions at solvent-exposed cysteines are frequently performed at slightly acidic pH (pH ≈ 6), thus preventing reaction with solvent-exposed lysines.7,15 At slightly basic pH, on the other hand, lysines become competitive and heterogeneous conjugation mixtures are obtained. While bearing this very well-known observation in mind, we wondered if there was any possibility to make lysines outcompete cysteines in a kinetically controlled process (i.e., using irreversible reactions unlike thermodynamically driven imine ligation, for instance). While solvent-inaccessible cysteine residues in nonpolar microenvironments have higher pKa values thus existing mostly as poorly nucleophilic thiols (water is not available to stabilize the ionic, nucleophilic thiolate, thus requiring a large excess of labeling reagents to be modified), lysine with similar nonpolar surroundings have lower pKa’s (water is not available to stabilize the cationic, non-nucleophilic ammonium) (Figure 2a). Thus, at near-physiological pH (7.5–8.5), some relatively solvent accessible lysine and cysteine residues are mostly in their neutral form, presenting an opportunity for the regio- and chemoselective modification of these neutral and nucleophilic lysine residues. Furthermore, using kinetic control to modify the most reactive lysine would allow as low as a single molar equivalent of reagent—an important prospect when the cargo to be ligated to the protein is valuable (e.g., cytotoxic drugs or fluorophores). A key step in realizing this goal is to identify a reagent that reinforces the intrinsic nucleophilicity of lysine; this would allow complete aza-Michael ligation using equimolar amounts of the reagent even at nearly neutral pH where the fraction of neutral lysine residues is small. As a result, highly chemo- and regioselective lysine modification could be attained by appropriately matching the reagent-enhanced reactivity and microenvironmentally regulated acidity of each lysine amino group (Figure 2a).
Figure 2.

Computer-assisted design of acrylate electrophile reagents for lysine modification. (a) At near physiological pH (7.5–8.0), lysine residues compete with cysteine as Michael donors. At lower values (typically pH 5.5–6.0) lysine residues are mostly protonated and unreactive, while under more basic conditions (pH > 8.5) the more nucleophilic thiolate of cysteine residues usually dominate. Thus, using stoichiometric amounts of the donor at pH 7.5–8.0, the outcome of the kinetically controlled aza-Michael addition reaction will be determined by the relative reactivity of each lysine (kobs), which is in turn determined by their intrinsic nucleophilicity (kLys–NH2) and acidity (Ka). Highly reactive reagents can be designed to amplify the intrinsic nucleophilicity of lysine side chains for aza-Michael ligation, while their acidity is regulated by the local sequence microenvironment. As a result, enhanced reactivities can be obtained for certain individual lysine residues, leading to high kinetic site selectivity. (b) Acrylate electrophile derivatives 1a–d used in this study and transition states (TS, activation energies ΔG‡ in kcal mol–1) calculated with PCM(H2O)/M06-2X/6-31+g(d,p) for the aza-Michael addition of methylamine (abbreviated lysine model). Interatomic distances (in angstrom) for the forming C–N and hydrogen bonds are shown as blue and orange dashed lines, respectively. The β′-sulfone moiety in 1c stabilizes the aza-Michael transition state through precise hydrogen bonding to the reacting amine, providing a ten-million-fold acceleration with respect to methyl methacrylate 1b. (c) Such hydrogen bonding is much weaker with methanethiol (model for cysteine), and thus competitive sulfa-Michael addition transition structures or stable intermediates cannot be located on the relaxed potential energy surface (PES). The origin for this continuously uphill reaction profile with the cysteine model is the inability of the sulfone group present in 1c to stabilize the positive charge developed at the thiol group upon nucleophilic addition. Conversely, the positive charge developed at the amino group of the lysine model is efficiently dissipated by the sulfone in 1c, as revealed by the exothermic reaction profile; the zwitterionic enolate is more stable than the reactants. This constitutes the basis for lysine chemoselectivity in the presence of predominantly protonated cysteines. (d) Complete minimum-energy reaction pathway for the aza-Michael addition reaction of methylamine and sulfonyl acrylate 1c, followed by spontaneous elimination of methanesulfinic acid. The initial addition is the rate-limiting step. Hydrogen bonding between the nucleophilic amino group and the sulfone moiety promotes both the aza-Michael addition and the elimination of methanesulfinic acid.
Type 2 alkenes (i.e., α,β-conjugated carbonyls) are reactive electrophiles able to undergo 1,4-addition with the nucleophilic side chains of cysteine (thiols)15 and, to a lesser extent, lysine (amines).36,37 Among these, acrylates, acrylamides, and maleimides are commonly used as warheads for protein modification and covalent drug attachment, mainly through ligation at cysteine.7,15,38 Water-soluble acrylamides are relatively unreactive,39 for example in aza-Michael addition reactions at protein interfaces,40 when compared to acrylates. The more reactive vinylsulfonamides41,42 have recently been used to design inhibitors of ubiquitin activating enzymes43 and kinases.44 Also, compounds combining both the acrylic acid and sulfone moieties, such as 2-(sulfonylmethyl)acrylic acid derivatives, have been used for disulfide rebridging.45,46
Considering these observations and inspired by Barbas’ prescient report on selective lysine labeling with a Toll-like receptor 4 (TLR4) inhibitor that features a cyclohexene moiety,23 we first performed quantum mechanical calculations of a model aza-Michael addition reaction using several acrylate reagents (see Supporting Figure S3). In Figure 2b, we show the activation barriers (ΔG‡) calculated for representative examples of acrylate-based electrophiles including cyclohexene-acrylate 1a, acrylate 1b, methyl 2-(sulfonylmethyl)acrylate 1c, and N,N-dimethyl-2-[(methylsulfonyl)methyl]acrylamide 1d as electrophiles. To our delight, the ΔG‡ for 1c was calculated to be only 17.5 kcal mol–1, suggesting that reactions of this type would be quite rapid. The source of this improved reactivity is the ability of the sulfone moiety to precisely hydrogen bond to the nucleophilic amine in a favorable chairlike conformation, dissipating the positive charge developed upon addition to the alkene of the acrylate (Figure 2c). This hydrogen bond interaction between the lysine model and the sulfone lowers the energy barrier by 10–16 kcal mol–1 with respect to acrylate 1b and cyclohexene-acrylate 1a. This corresponds to a 107- and 1011-fold acceleration, respectively, a rate enhancement comparable to that of proficient enzymes.47 The N,N-dimethyl amide group in 1d severely distorts and deactivates this reagent with respect to 1c, despite featuring similar hydrogen bonding to the incoming amine (Supporting Figure S4). Importantly, this type of hydrogen bond activation is not possible with the cysteine side-chain thiol due to the less polar character of the S–H bond, providing a means to selectively modify lysine even in the presence of free cysteine residues at near neutral pH (Figure 2c). The subsequent elimination of methanesulfinic acid from the zwitterionic enolate formed upon aza-Michael addition (reminiscent of the elimination in an E1cB mechanism) was calculated to be spontaneous (ΔG‡ ≈ 4 kcal mol–1) (Figure 2d).
Lysine Regioselective Modification on Albumin Using Computationally Designed Sulfonyl Acrylate 1c
At the outset of our studies we envisioned that the innate and subtle differences in the reactivity of each ε-amino side chain could enable regioselective modification of a single lysine residue on proteins. At the same time, the ability of the ε-amino group of lysine to hydrogen bond with the sulfone of acrylate 1c opened the exciting prospect of using this designed reagent to favor reaction with lysine over cysteine. To verify this hypothesis in a protein context, we evaluated the reaction of the four electrophiles 1a–d with a physiologially and therapeutically relevant protein, human serum albumin (HSA). HSA is the most abundant protein found in plasma, and recombinant HSA (rHSA) derivatives have become available and gained wide acceptance as bespoke proteins for formulation, drug delivery, and imaging applications.48,49 Its native sequence features 59 lysine residues, and 35 cysteines, of which 34 are found in the form of disulfide and one, at position 34, exists in the thiol oxidation state. Thus, a recombinant HSA (rHSA, Recombumin from Albumedix) provides an ideal substrate to test the potential chemo- (lysine versus cysteine) and regioselectivity (single lysine modification versus multiple lysine modifications) of our proposed method.
We started by investigating the reaction of acrylate derivatives 1a–d with rHSA. When a mere 1 mol equiv of methyl 2-(sulfonylmethyl)acrylate derivative 1c is reacted with rHSA at low concentration (10 μM) for 1 h at 37 °C and pH 8.0 (Figure 3a,b), a single modification was produced in >95% as assessed by Liquid Chromatography–Mass Spectrometry (LC–MS) (Figure 3c,d). The difference in mass from rHSA to rHSA–1c corresponded to Michael addition followed by E1Cb-type elimination of HSO2Me from the enolate intermediate (Figure 2d). Importantly, our computational data were corroborated experimentally, as electrophiles 1a, 1b, and 1d did not react with rHSA under the same reaction conditions (Supporting Figures S17, 18, 24). The proposed structure was consistent with reactions on a peptide model using the electrophile 1c (Supporting Figure 8).
Figure 3.

HSA lysine regioselective bioconjugation with sulfonyl acrylate reagent 1c. (a) Scheme for the bioconjugation reaction between rHSA and sulfonyl acrylate 1c. General reaction conditions: rHSA was reacted with 1c (1 mol equiv) in TrisHCl (20 mM, pH 8.0) at 37 °C for 1 h. (b) Optimization of reaction conditions with respect to buffer and pH (see also Supporting Table 3). (c,d) Total ion chromatogram, combined ion series, and deconvoluted mass spectrum reconstructed from the ion series using the MaxEnt algorithm before (c) and after (d) the reaction. The region containing all protein is marked with a left right arrow. After the reaction, protein conjugates were purified using size-exclusion chromatography and the concentration of the starting protein and of the purified protein was measured by Bradford protein assay. Complete conversion to the desired rHSA–1c conjugate was observed in >95% yield. (e) MS/MS spectrum of the m/z 712.93 doubly charged ion of the lysine modified peptide GKKLVAASQAALGL from HSA. Modified residue underlined. (f) CD of rHSA and rHSA–1c. (g) Reaction of rHSA–1c with thiol specific Ellman’s reagent shows full conversion of cysteine 34 to the corresponding disulfide. Deconvoluted mass spectrum of rHSA–1c–Ellman’s.
Next, the product of the reaction was then subjected to enzymatic digestion followed by LC–MS/MS analysis of the resulting peptides. We found that the modification occurred in the peptide GKKLVAASQAALGL (modified residue underscored and in bold–total of 91% sequence coverage) as observed in the MS/MS spectrum, which corresponds to lysine 573 (Figure 3e). Lysine 573 was the only residue modified by reagent 1c, indicating a high degree of regioselectivity for the reaction. Additionally, circular dichroism (CD) analysis of rHSA and rHSA–1c showed no alterations in secondary structural content (Figure 3f), which reflects the mildness and efficiency of the conjugation process. Finally, and to demonstrate the chemoselectivity of the reaction for lysine over cysteine, we then incubated the product of the reaction of rHSA with 1c with thiol specific Ellman’s reagent. We observed rapid and complete conversion to the corresponding disulfide product rHSA–1c–Ellmans’ (Figure 3g), indicating that cysteine was not modified when the protein was treated with acrylate 1c in the first step. The same doubly modified protein could also be obtained when performing the reactions in reverse order (Supporting Figure 27).
Lysine 573 is known to play a key role in albumin’s binding to the FcRn receptor.50 Using Surface Plasma Resonance (SPR), we confirmed that modification of lysine 573 using the sulfonyl acrylate reagent 1c leads to a 2-fold reduction in FcRn affinity as expected (Table 1 and Supporting Figure 33). Next, we applied the same reaction conditions to an rHSA mutant featuring a proline residue at position 573, rHSA-K573P, which is known to increase FcRn binding affinity, in order to evaluate the regioselectivity of our lysine modification protocol (Figure 4a). We were pleased to observe the formation of a chemically defined conjugate after reaction of rHSA-K573P with 1c for 1 h at 37 °C as determined by LC–MS (Figure 4b). Tryptic digestion followed by LC–MS/MS of the resulting peptides indicated modification of lysine at position 4 (Figure 4c). Reaction of 1c at position 4 resulted in a conjugate that retained both secondary structural content (Figure 4d) and binding efficiency to FcRn (Table 1 and Figure 4e). These data demonstrate the regioselectivity of the reaction, since when the modified lysine in the native sequence is genetically altered, the reaction occurs predominantly at the next most-reactive lysine site as exemplified here for rHSA.
Table 1. Kinetic Analysis at pH 5.5 of rHSA Derivatives Binding to Human FcRn Receptor.
| Albumina | kon (10–3/ms) | koff (103/s) | kD (μM) |
|---|---|---|---|
| rHSA | 3.92 | 63.3 | 16.13 |
| rHSA–1c | 2.15 | 62.5 | 29.10 |
| rHSA-K573P | 9.75 | 4.88 | 0.501 |
| rHSA-K573P–1c | 7.92 | 4.65 | 0.587 |
Values are mean of duplicates. There are differences observed in FcRn binding kinetics between the albumins. For rHSA/rHSA–1c: FcRn binding is negatively impacted when 1c was installed at position 573. The kon for rHSA–1c is much slower, driving the kD value up to 29.1 μM, which is 2-fold less than rHSA; for rHSA-K573P/rHSA-K573P–1c: The kon when position 4 is modified is slightly slower (7.925) compared to nonmodified rHSA-K573P (9.755), pushing the affinity kD for the human FcRn up slightly.
Figure 4.

Sequence-lysine conjugation effect of albumins on their binding affinity to human FcRn receptor. (a) Scheme for the bioconjugation reaction between mutant rHSA-K573P and sulfonyl acrylate 1c. General reaction conditions: rHSA-K573P was reacted with 1c (1 mol equiv) in TrisHCl (20 mM, pH 8.0) at 37 °C for 1 h. (b) Mass spectrometry characterization of lysine conjugation with 1c. ESI–MS spectra of rHSA-K573P (i) before (red) and (ii) after (gray) conjugation with 1c. (c) MS/MS spectrum of the m/z 416.54 triply charged ion of the lysine modified N-terminal peptide (1–10) DAHKSEVAHR. Modified residue underlined. (f) CD of rHSA-K573P and rHSA-K573P–1c. (d) CD analysis of the nonmodified rHSA-K573P and the conjugate rHSA-K573P–1c. (e) SPR comparison of the binding to human FcRn of rHSA-K573P and rHSA-K573P–1c.
Our data indicate that when 1c and rHSA react at an equimolar ratio, the reaction proceeds with complete chemo- and regioselectivity after 1 h at 37 °C. Phosphate or TrisHCl buffers at near pH 8.0 were optimal for the lysine modification (Figure 3b, Supporting Table 3 and Supporting Figures 22, 23). As a direct comparison, when an NHS ester derivative is used under the same equimolar reaction conditions for 1 h at 37 °C, incomplete conversion and the formation of conjugates with one and two modified residues were detected by LC–MS (Supporting Figure 35). Complete consumption of the rHSA protein could only be obtained using 10 equiv of the reagent which led to a heterogeneous mixture varying between three to eight modifications (Supporting Figure 36). These experiments show both the superior reactivity and selectivity of the sulfonyl acrylate 1c when compared with conventional NHS ester reagents. We noticed that the reaction also occurs at pH 7.0, though conversion is lower (∼40%) due to an increased protonation of lysine 573. The fact that the modification occurs at all at pH 7.0 reflects the capacity of 1c to augment the intrinsic reactivity of lysine residues toward aza-Michael ligation (large kLys-NH2 in Figure 2a), the local pKa modulation of a specific lysine, and the inability of cysteine residues to react under our optimized conditions. While we note that such chemoselectivity will have to be evaluated for each protein on a case-by-case basis (especially for enzymes containing highly reactive cysteine residues such as cysteine proteases, glutathione transferases, and some oxidoreductases), the results in Figure 3 demonstrate that chemo- and regioselective modification of lysine in the presence of a cysteine is possible at least in certain cases. Additionally, the use of 1 mol equiv is critical. When using excess 1c (10 and 50 mol equiv), additional conjugations are observed (Supporting Figures 20, 21). As expected for a kinetically controlled ligation, it is therefore critical to take care in measuring both protein concentration and the concentration of 1c so that no more than 1 mol equiv is present in the reaction.
This new site-selective reaction is operationally simple and high yielding (the isolated yield of the modified protein was 98% as assessed by Bradford assay). Furthermore, the conjugate rHSA–1c remained stable in both human plasma and in the presence of biological thiols such as glutathione, an important feature for a potentially useful ligation for the preparation of therapeutic conjugates (Supporting Figures 25, 26).
Scope of Modification on Native Proteins
Encouraged by the exquisite selectivity observed on rHSA, we next explored the reaction of 1c with other proteins. Biologically and medically relevant proteins were selected: lysozyme, an endogenously produced glycoside hydrolase that is immunologically important and provides defense against bacteria by damaging their cell walls;51 the C2A domain of Synaptotagmin-I (C2Am)52 and Annexin V,53 both biomarkers used to image apoptotic tissues in patients; and a full IgG antibody, Trastuzumab, which constitutes a frontline treatment to treat breast cancer patients.54 These proteins allowed us to test not only the regioselectivity of the reaction on diverse protein scaffolds but also the chemoselectivity as C2Am and Annexin V have a free cysteine residue that could potentially react with the sulfonyl acrylate 1c.
Markedly, under essentially identical conditions to those used on rHSA (low protein concentration, 1 equiv of sulfonyl acrylate 1c, 1–2 h at 37 °C and pH 8.0), a single modification was observed for all proteins studied (Figure 5a). LC–MS analyses indicated in all cases the addition of 99 Da corresponding to the aza-Michael reaction of a lysine with the alkene of the acrylate followed by spontaneous elimination of the methanesulfonyl group (Figure 5b–e). Moreover, in the case of proteins having free cysteine residues (C2Am and Annexin V) the reactive sulfhydryl side chain remained unreacted and could be subsequently modified through mixed disulfide formation after reaction with the thiol-specific Ellman’s reagent (Supporting Figures 41, 42, 46, 47). For these proteins, cysteine could be also modified first with Ellman’s reagent and then further conjugated through lysine with 1c. These data not only suggest preferential selectivity of lysine for reaction with the sulfonyl acrylate 1c at pH 8.0 but also show that our new method may be used to obtain dually modified proteins at specific cysteine and lysine residues. Again, we note that this chemoselectivity needs to be established in cases where catalytic cysteine residues might compete for modification, but the C2Am and Annexin V experiments are two additional examples where a lysine can be selectively modified in the presence of cysteine.
Figure 5.

Regioselective lysine modification is applicable to a wide-range of native protein scaffolds. (a) Optimized conditions to modify a single lysine on multiple native proteins. (b–d) ESI–MS spectra for three modified proteins showing a homogeneous product for each bioconjugation. Combined ion series and full ESI–MS spectra can be found in the Supporting Information. (e) ESI–MS spectra before (red) and after (blue) conjugation of 1c to the full-length IgG antibody Trastuzumab. The addition of 99 Da occurred exclusively in the light chain of the antibody. (f) Biolayer Interferometry (BLI) curves (in blue) and fitting curves (in red) obtained for Trastuzumab–1c. For the curves of nonmodified, commercial Trastuzumab, see Supporting Figure 60. –– indicates disulfide bonds in Trastuzumab.
Next, we assessed antigen binding of the modified Trastuzumab–1c using biolayer interferometry (BLI) and confirmed that the introduction of a single lysine modification within the light chain of the antibody (Figure 5e) did not affect its ability to recognize the Her2 antigen it was designed to bind to (KD = 16 ± 2.2 nM for Trastuzumab compared KD = 14 ± 1.4 nM of Trastuzumab–1c) (Figure 5f). Finally, we tested the stability of Annexin V–1c both in the presence of 1 mM of reduced glutathione and in human plasma at 37 °C. Importantly, the conjugate remained stable under those conditions for up to 48 h (Supporting Figures 39, 40), which demonstrates the potential utility of our method to create stable conjugates for therapeutic applications. This is a distinct advantage over more traditional conjugation methods such as disulfide formation or maleimide conjugates that are not stable in the presence of thiols.7,55 Moreover, the optimized ligation method is advantageous in at least two additional ways. First, the reaction is highly chemoselective with exclusive reaction at lysine and no side reactions at cysteine. Second, the conjugation method does not require prior sequence engineering and is applicable to proteins in their native constitution. This is critically important for proteins such as antibodies where mutating the sequence to install a uniquely reactive group can compromise their binding affinity. Our modification of the antibody Trastuzumab demonstrates that the protein can be modified directly and retain its full activity.
Theoretical Prediction and Experimental Validation of Modification Site
Based on the hypothesis that the most acidic, reagent-accessible lysine residues would be highly reactive with sulfonyl acrylate 1c, we estimated the lysine pKa of three of the proteins we studied using constant pH molecular dynamics simulations (CpHMD, see Supporting Information). Accurate protein pKa prediction remains a great challenge for computational chemistry,56 and among the many available methods, CpHMD57,58 is perhaps the most successful one since it takes both solvation and protein dynamics into consideration, albeit at a high computational cost.56
Lysozyme has six potentially reactive lysine residues and no cysteine. Based on our calculations, one of these lysines, K33, has a significantly lower pKa (9.5) than the other Lys (pKa ≈ 10) due to its hydrophobic microenvironment imparted by the surrounding residues F34, F38, and W123 (highlighted in orange in Figure 6a). Other methods such as Multi-Conformation Continuum Electrostatics (MCCE)59 predicted an even lower pKa for K33 of 8.1 versus 10–11 for the other lysines. Furthermore, the X-ray structure of hen egg white lysozyme (PDB ID 1G7H) shows a hydrogen bond between the N of K33 and the NH2 group of N37, suggesting that the side chain of K33 is actually deprotonated (i.e., neutral, reactive form, see Supporting Figure 5). Consistent with this computational prediction, enzymatic digestion of the modified lysozyme with 1c followed by LC–MS/MS analysis the resulting peptides (35% sequence coverage that covers all 6 lysine residues) confirmed that lysine 33 is in fact the only modified residue in lysozyme (Figure 6a). Furthermore, and although the simultaneous prediction of the pKa of all 59 lysines in rHSA through CpHMD simulations is computationally very demanding, we were able to calculate the pKa of selected lysine residues such as lysine 64 (targeted by TAK-242;23 pKa 11.07), lysine 573 (targeted by 1c; pKa 10.39), and lysine 574 (adjacent to Lys573; pKa 10.89) (Supporting Table 2). The 0.5–0.7 pKa units difference between these lysine residues agrees well with the observed site selectivity achieved by sulfonyl acrylate 1c toward lysine 573 in rHSA and reinforces the important role of lysine acid–base properties in selective labeling.
Figure 6.

Theoretical calculation of the most reactive lysine residue on three proteins and obtained MS/MS spectra confirming the modified site. The pKa values for lysine and cysteine residues in (a) lysozyme, (b) synaptotagmin C2Am domain, and (c) Annexin V were estimated through constant pH Molecular Dynamics simulations (CpHMD); deeply buried cysteine residues such as C315 in Annexin V could not be fully titrated in the pH 5–14 range; terminal residues cannot be evaluated through the CpHMD method (pKa value not calculated). These values are mapped onto surface representations for each protein (lysine and cysteine highlighted in blue and yellow, respectively). The lowest pKa lysine for each protein is highlighted in red; in agreement with these predictions, the lowest pKa lysine residues are the ones selectively modified as found through enzymatic digestion followed by LC–MS/MS analysis (modified residues underlined). Hydrophobic residues around the reactive lysines (shown in orange) contribute to lower their pKa.
The same phenomenon was observed for C2Am in which K100 shows the lowest calculated pKa (10.1) among the 13 surface-exposed lysines (Figure 6b). As in lysozyme, K100 is located in a small hydrophobic patch (L63-P65-G99-L97-V92). The calculated pKa of the engineered C95, which has been shown to be reactive under physiological conditions,7 is 10.3, suggesting a possible competition between both residues. However, LC–MS/MS analysis after enzymatic digestion (95% sequence coverage) of the protein conjugated with 1 equiv of 1c (Figure 6b) showed selectivity for K100. This result highlights how selective reaction at lysine instead of cysteine can be reinforced by the predicted H-bond interaction in the aza-Michael addition transition state between 1c and primary amines (Figure 2). Additionally, cysteine 95 could be specifically modified after reaction of C2Am with 1c using thiol specific Ellman’s reagent (Supporting Figures 46, 47). In the case of Annexin V, all 22 lysine residues are relatively solvent accessible and thus have calculated pKa’s in the 10–11 range, with K57, K300, and K308 showing slightly depressed pKa’s (Figure 6c). Enzymatic digestion of Annexin V–1c followed by LC–MS/MS analysis gave only 59% sequence coverage and indicated lysine K300 as the one modified (Figure 6c). This result is in line with the theoretical calculations despite the smaller differences in the calculated pKa’s and low sequence coverage. In addition, Annexin V features a cysteine residue at position 315 which is deeply buried in a hydrophobic region of the protein; the pKa of this buried cysteine was calculated to be >14, suggesting very low reactivity at near physiological pH. Indeed, C315 has been shown to react with other alkenes only when a very large excess (1000 to 10000 equiv) is used.60 As in the case of C2Am, the free C315 in Annexin V could also be modified to full conversion upon reaction with thiol specific Ellman’s reagent indicating that this cysteine does not react with 1c under the standard conditions (Supporting Figures 41, 42).
The site-selective, differential labeling of lysine and cysteine in the same protein is uncommon. In fact, we are aware of only one other example where lysine has been modified regioselectively in the presence of cysteine, and Barbas’ report was limited to a single protein.23 The examples of site-selective lysine modification in the presence of reduced cysteine residues illustrated above are novel in the general capability and rational predictability of the site of modification. The unique architecture of the sulfonyl acrylate derivative 1c enables this improvements through its highly selective recognition of the most reactive lysine residues (i.e., those with lower pKa’s) through precise hydrogen bonding in the aza-Michael addition transition state.
Precise Protein Labeling for Bioimaging
The spontaneous elimination of methanesulfinic acid after reaction of 1c with a lysine on a protein restores a type 2 alkene that may, in principle, serve as a handle for further attachment of various cargoes such as polyethylene glycol (PEG) polymers, fluorophores, or drugs. In particular, we considered that synthetic molecules bearing a nucleophilic amine would provide a suitable nucleophile for aza-Michael addition ligation since acrylates are good Michael acceptors for amines. Importantly, such a reaction, and unlike thiol-based Michael addition, would be fully compatible with disulfide bonds and free cysteine residues on proteins thus enabling a noninvasive second-stage chemoselective ligation.
To test this hypothesis, we first examined the aza-Michael addition of benzylamine and a PEG3NH2 polymer, of which the latter is present in many commercially available dyes and drugs. We found that both amines could be successfully conjugated after 1 h of reaction at room temperature with alkene-tagged protein Annexin V–1c, as assessed by LC–MS (Supporting Figures 43, 44). These results demonstrate that the activated alkene generated upon site-selective reaction of the sulfonyl acrylate 1c with a lysine residue may be used to further append synthetic molecules to proteins through a second aza-Michael ligation using low quantities of amine reagents (1 mM of reagent for a 10 μM protein concentration).
Next, we decided to demonstrate the utility of our method to produce fluorescently labeled proteins for bioimaging.5 As a model protein, we chose C2Am as it has been validated in vivo for detecting apoptotic tissue, using a variety of methods including magnetic resonance imaging (MRI)61 and single photon emission computed tomography (SPECT).62 C2Am–1c, which displays the acrylate moiety, reacted rapidly over 1 h with 1 mM of the commercially available PEGylated fluorophore FITC-PEG3NH2 to afford a precisely labeled fluorescent conjugate, C2Am–1c–FITC, as detected by SDS-PAGE (Figure 7a, b). We then tested if this C2Am derivative retains its inherent functionality of binding to the phosphatidylserine (PS) phospholipid on the surface of apoptotic cells.52 The use of C2Am–1c–FITC allowed ready visualization of HEK293 cells rendered apoptotic by treatment with actinomycin D (Figure 7c). Blocking studies where treated cells were first incubated with nonlabeled C2Am and then with the fluorescent surrogate C2Am–1c–FITC showed a significant decrease of the mean fluorescence intensities (MFI). This control experiment verified that the specific affinity of C2Am–1c–FITC for apoptotic cells is retained after functionalization with the fluorophore (Figure 7d). Colocalization of C2Am–1c–FITC with the membrane confirms the high specificity of the probe for the externalized PS phospholipid, as verified by confocal microscopy (Figure 7e). The combined advantages of chemoselectivity and efficiency allow precise labeling of wild-type sequences without perturbing protein secondary structure, as assessed by CD (Supporting Figure 49) and localization (detection of apoptotic cells). In addition, the fluorescence of the synthetic probe is fully retained after conjugation to the protein. This simple method increases the current repertoire for fluorescent protein labeling for bioimaging while avoiding some of the limitations usually associated with these processes such as fluorescence quenching or compromised protein activity and localization.
Figure 7.

Site-selective fluorescence labeling of the lysine–acrylate 1c derivative through aza-Michael ligation: application for the selective imaging and detection of apoptotic positive cells. (a) Scheme for the site-selective labeling of C2Am–1c with commercially available FITC–PEG3NH2. General reaction conditions: C2Am–1c was reacted with FITC–PEG3NH2 (100 mol equiv) in TrisHCl (20 mM, pH 8.0) at room temperature for 1 h. (b) Treatment of C2Am–1c with FITC–PEG3NH2 gave a single new fluorescent band as detected by SDS-PAGE that is consistent with site-selective incorporation of FITC. Lanes 1 and 2, Coomassie staining. Lanes 3 and 4, fluorescence. (c) Epifluorescence images of nonapoptotic (control) and apoptotic HEK293 cells after labeling with C2Am–1c–FITC. Blocking studies were performed by preincubating apoptotic cells with a 10× excess of nonfluorescent C2Am before incubation with C2Am–1c–FITC. (d) The mean fluorescent intensity (M.F.I.) of apoptotic cells incubated with C2Am–1c–FITC (black bar) or blocked with nonfluorescent C2Am before incubation with C2Am–1c–FITC (gray bar). (e) Confocal images of apoptotic cells treated with C2Am–1c–FITC. Zoom in and Z-stacking showing staining of the cellular membrane. Membranes of apoptotic cells are shown in green, while the nuclei in blue. S.D., standard deviation; a.u., arbitrary units; FITC, fluorescein isothiocyanate; PEG, polyethylene glycol.
Regioselective Antibody Conjugation
The ability to precisely conjugate fluorophores and cytotoxic drugs to antibodies is essential in modern biomedical technology.63 Methods to label antibodies usually rely on the modification of interchain disulfides, genetically encoded free cysteine residues, or noncanonical amino acids.64 Though these methods have been adapted with some success, there are currently several limitations. For instance, modification of the cysteine residues after disulfide reduction is difficult and often leads to heterogeneous mixtures.65 In cases where a free cysteine is genetically installed in antibodies or when a noncanonical is engineered into the protein, there is the risk that altering the native sequence of the antibody can lead to significant loss of binding affinity to the target antigen.8 In contrast, our method can be applied directly to a therapeutic antibody in its native form, without the need for genetic engineering that could compromise its activity. To demonstrate the utility of our regioselective lysine methodology in preparing homogeneous antibody conjugates, we decided to react Trastuzumab–1c with an amine-containing fluorophore (Figure 8a). Much to our delight, the reaction proceeded to completion in the presence of 1 mM of FITC-PEG3NH2 after 1 h at room temperature resulting in a well-defined, fluorescently labeled antibody conjugate (Figure 8b). Importantly, the fluorescent Trastuzumab–1c–FITC conjugate still retained its specificity toward its target antigen (Her2/c-erb-2), as demonstrated by flow cytometry. This experiment allowed the detection of SKBR3 cells (red) that express high levels of Her2-receptors in contrast with cells expressing low levels of this receptor (HepG2 cells, blue) (Figure 8c, d). Our data also indicate that the site-specifically labeled fluorescent antibody we generated preserves its functionality at a concentration range identical to that of its unmodified counterpart.66 The retention of binding activity of the antibody conjugate is a direct result of the mild and direct lysine conjugation method. In contrast to the vast majority of other antibody modification techniques, this method modifies native residues with excellent chemo- and regioselectivity and does not require sequence alteration that can lower binding affinity and specificity toward the target antigen.
Figure 8.

Site-specific aza-Michael installation of FITC–PEG3NH2 and the anticancer drug crizotinib into Trastuzumab–1c preserves Her2 binding affinity and the capacity to selectively detect Her2 positive cells. (a) Scheme for the site-specific labeling of Trastuzumab–1c with commercially available FITC–PEG3NH2. General reaction conditions: Trastuzumab–1c was reacted with FITC–PEG3NH2 (100 mol equiv) in TrisHCl (20 mM, pH 8.0) at room temperature for 1 h. (b) Treatment of Trastuzumab–1c with FITC–PEG3NH2 afforded a new fluorescent band as detected by SDS-PAGE that is consistent with site-selective incorporation of FITC within the light chain of Trastuzumab. Lanes 1–2, Coomassie staining. Lanes 3–4, fluorescence. From the bottom to the top: a band around 20–25 kDa (light chain); a band around 50 kDa (heavy chain). (c, d) Analysis of specificity of Trastuzumab–1c–FITC toward Her2 by flow cytometry. (c) Superposition of contour plots of side-scatter detection versus FITC-equivalent fluorescence intensity, in HepG2 cells (blue, expressing low levels of Her2/c-erb-2), and in SKBR3 cells (red, expressing high levels of Her2/c-erb-2). Controls were treated with nonconjugated Trastuzumab while samples were treated with increasing concentrations of Trastuzumab–1c–FITC (10, 50, and 150 nM). (d) Percentage of FITC-positive single cells, after treatment with fluorescently labeled or nonlabeled Trastuzumab, in both HepG2 cells (blue) and SKBR3 cells (orange). (e) Scheme for the site-specific conjugation of crizotinib to Trastuzumab–1c. General reaction conditions: Trastuzumab–1c (10 μM) was reacted with crizotinib (10 mM, 1000 mol equiv) in TrisHCl (20 mM, pH 8.0) at room temperature for 2 h. (f) Mass spectrometry characterization of site-specific drug conjugation. ESI–MS spectra of the light chain (i) before (red) and (ii) after (gray) conjugation of the anticancer drug crizotinib to Trastuzumab–1c. Calculated mass for the light chain of Trastuzumab–1c–crizotinib, 23986 Da. (g) KD constant derived from BLI experiments for Trastuzumab–1c–crizotinib. For the BLI curves obtained with the Her2 receptor see Supporting Figure 61. (h) CD of Trastuzumab and Trastuzumab–1c–crizotinib. (i, j) HEK293T cells (expressing low levels of Her2/c-erb-2) and SKBR3 cells (expressing high levels of Her2/c-erb-2) were incubated with 10 μM of Trastuzumab–1c–crizotinib and analyzed by flow cytometry.
We have further validated our protocol for the construction of stable and functional antibody conjugates through the conjugation of the anticancer drug crizotinib to the acrylate present in Trastuzumab–1c. Crizotinib is an approved kinase inhibitor for the treatment of ALK-rearranged nonsmall-cell lung carcinoma (NSCLC).67 This drug features a reactive piperidine motif that has been shown to undergo aza-Michael addition to dehydroalanine-tagged proteins.68 When Trastuzumab–1c (10 μM) was treated with an excess (10 mM, 1000 equiv) of crizotinib, complete conversion to an homogeneous conjugate with a single modification within the light chain was observed using LC–MS (Figure 8e,f). No modifications were detected in the heavy chain of the antibody (Supporting Figures 57 and 58). Constant pH molecular simulations (CpHMD) for antibody Trastuzumab (PDB id: 1n8z) showed the lowest pKa value obtained for lysine at position 207 in the light chain, indicating that this residue is likely the one where 1c is preferentially installed (see Supporting Table 2). In addition, and while hydrolysis of the methyl ester was detected under conditions used to modify a cysteine using Ellman’s reagent, in both examples of aza-Michael addition to Trastuzumab–1c the methyl ester remained intact during the two-step antibody conjugation process. Importantly, the conjugate Trastuzumab–1c–crizotinib fully retained antigen binding properties and secondary structural content as determined by BLI and CD, respectively (Figure 8g, h). We also show by flow cytometry analysis and by staining cells with an antihuman IgG Alexa-647 secondary antibody that Trastuzumab–1c–crizotinib retains its specificity toward SKBR3 cells, which express high levels of the Her2 antigen (Figure 8i), as opposed to HEK293 cells, which express low levels of the target antigen (Figure 8j). These data corroborate the mildness and selectivity of the strategy we developed for the construction of stable and functional protein conjugates modified at lysine. As part of our program in targeted cancer therapy we will further evaluate ADCs built using this method for targeted drug delivery in tumor-bearing mice.
Conclusion
We have demonstrated that it is possible to make homogeneous protein conjugates through the chemo- and regioselective modification of lysine. This was achieved by using a sulfonyl acrylate reagent that was designed to selectively target the ε-amino group of the most reactive lysine in the presence of other groups such as the thiols of cysteines. The basis for this chemoselectivity is the transient hydrogen bonding of the lysine amine with the sulfone moiety of the featured reagent occurring in the aza-Michael addition transition state, which significantly decreases its activation barrier and accelerates ligation by several million-fold. This hydrogen bond activation facilitates both the aza-Michael addition and the subsequent expulsion of the methanesulfinate leaving group, which provides an additional thermodynamic driving force. Such an activation mechanism enhances the intrinsic nucleophilicity of neutral lysine residues, enabling aza-Michael ligation even at nearly neutral pH where the fraction of neutral, nucleophilic lysines is small. This reaction proceeds to full conversion with only a molar equivalent of reagent, without perturbing protein secondary structure or impairing protein function. This reaction was deployed successfully on five proteins of diverse structure including a full-length IgG antibody. In all cases a specific lysine was modified and the regiochemical outcome (i.e., which lysine was modified) could be predicted for medium-sized proteins based on computational determination of the pKa of each lysine residue. The modified proteins show stability in human plasma while preserving innate biological activity. Importantly, proteins bearing the acrylate moiety can be further labeled in a site-specific manner in a subsequent aza-Michael ligation with amine-containing synthetic molecules including PEG, fluorophores, and an anticancer drug. We demonstrate that an apoptosis protein biomarker and a therapeutic antibody, when fluorescently labeled through this operationally simple protocol, can be used to detect apoptotic and antigen positive cells, respectively. In addition, our enabling platform for site-selective lysine protein modification is compatible with other conjugation chemistries, as exemplified with the pre- or postmodification of cysteine residues through mixed disulfide formation.
Our discovery of sulfonyl acrylate 1c and its ability to harness the subtle reactivity differences between the many lysine residues on native proteins is a significant advance in protein modification. The majority of other lysine modification methods typically result in a mixture of products, with multiple lysines modified. In contrast, this method can provide a single lysine modification with complete chemo- and regioselectivity. This level of selectivity and synthetic logic in this strategy is akin to the advances in C–H functionalization of complex molecules, where functional groups traditionally viewed as indistinguishable can be modified regioselectively when the innate reactivity of the target group is carefully matched with an appropriate reagent.69−71 Furthermore, this method of protein modification does not require genetic engineering and can be carried out on native protein structures with complete selectivity. This is a critical advance because it means that no sequence perturbations are required that could lead to impaired activity.
We are currently exploring extensions of this approach to conjugate two different drugs onto an antibody. This could be done at lysine, for instance, with one drug linked as an ester of the sulfonyl acrylate reagent and another drug attached through aza-Michael addition to the alkene generated after conjugation. Alternatively, the lysine conjugation here could be used in combination with other bioconjugation chemistries directed at different functionalities. The simplicity of the protocol, availability of synthetic reagents, and direct applicability to wild-type protein sequence bode well for routinely accessing site-selectively modified proteins for basic biology and therapeutic applications.
Acknowledgments
We thank Xunta de Galicia (M.J.M.), Spanish Ministry of Education, Culture and Sport under the FPU program (M.M.), the EU (Marie-Sklodowska Curie ITN Protein Conjugates to G.J.L.B.; Marie-Sklodowska Curie IEF to B.L.O.), FCT Portugal (FCT Investigator to G.J.L.B.; SFRH/BPD/103172/2014 Postdoctoral fellowship to P.M.S.D.C.), CNPq Brazil (Fellowship 200456/2015-6 to J.B.B.), D.G.I. MINECO/FEDER (CTQ2015-70524-R and RYC-2013-14706 to G.J.O.; CTQ2015-67727-R and Salvador de Madariaga mobility grant to F.C.), the Australian Research Council (DE150101863 to J.M.C.), and the EPSRC (G.J.L.B.) for funding. We thank Dr. André Neves and Prof. Kevin Brindle for providing the C2Am protein; Drs. Peter Grice and Simon Mccallum for assistance with NMR kinetic and confocal microscopy experiments, respectively; and UR (Beronia cluster) and BiFi (Memento cluster) for computer support. G.J.L.B. is a Royal Society University Research Fellow and the recipient of an European Research Council Starting Grant (TagIt).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b12874.
Methods and any associated references and Supporting Figures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Stephanopoulos N.; Francis M. B. Nat. Chem. Biol. 2011, 7, 876–884. 10.1038/nchembio.720. [DOI] [PubMed] [Google Scholar]
- Spicer C. D.; Davis B. G. Nat. Commun. 2014, 5, 4740. 10.1038/ncomms5740. [DOI] [PubMed] [Google Scholar]
- Lang K.; Chin J. W. Chem. Rev. 2014, 114, 4764–4806. 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- Boutureira O.; Bernardes G. J. L. Chem. Rev. 2015, 115, 2174–2195. 10.1021/cr500399p. [DOI] [PubMed] [Google Scholar]
- Xue L.; Karpenko I. A.; Hiblot J.; Johnsson K. Nat. Chem. Biol. 2015, 11, 917–923. 10.1038/nchembio.1959. [DOI] [PubMed] [Google Scholar]
- Walsh C. T.; Garneau-Tsodikova S.; Gatto G. J. Angew. Chem., Int. Ed. 2005, 44, 7342–7372. 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- Bernardim B.; Cal P. M. S. D.; Matos M. J.; Oliveira B. L.; Martínez-Sáez N.; Albuquerque I. S.; Perkins E.; Corzana F.; Burtoloso A. C. B.; Jiménez-Osés G.; Bernardes G. J. L. Nat. Commun. 2016, 7, 13128. 10.1038/ncomms13128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Welborn M.; Zhu T.; Yang N. J.; Santos M. S.; Van Voorhis T.; Pentelute B. L. Nat. Chem. 2016, 8, 120–128. 10.1038/nchem.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junutula J. R.; Raab H.; Clark S.; Bhakta S.; Leipold D. D.; Weir S.; Chen Y.; Simpson M.; Tsai S. P.; Dennis M. S.; Lu Y.; Meng Y. G.; Ng C.; Yang J.; Lee C. C.; Duenas E.; Gorrell J.; Katta V.; Kim A.; McDorman K.; Flagella K.; Venook R.; Ross S.; Spencer S. D.; Lee Wong W.; Lowman H. B.; Vandlen R.; Sliwkowski M. X.; Scheller R. H.; Polakis P.; Mallet W. Nat. Biotechnol. 2008, 26, 925–932. 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- MacDonald J. I.; Munch H. K.; Moore T.; Francis M. B. Nat. Chem. Biol. 2015, 11, 326–331. 10.1038/nchembio.1792. [DOI] [PubMed] [Google Scholar]
- Casi G.; Huguenin-Dezot N.; Zuberbühler K.; Scheuermann J.; Neri D. J. Am. Chem. Soc. 2012, 134, 5887–5892. 10.1021/ja211589m. [DOI] [PubMed] [Google Scholar]
- Rosen C. B.; Francis M. B. Nat. Chem. Biol. 2017, 13, 697–705. 10.1038/nchembio.2416. [DOI] [PubMed] [Google Scholar]
- Axup J. Y.; Bajjuri K. M.; Ritland M.; Hutchins B. M.; Kim C. H.; Kazane S. A.; Halder R.; Forsyth J. S.; Santidrian A. F.; Stafin K.; Lu Y.; Tran H.; Seller A. J.; Biroc S. L.; Szydlik A.; Pinkstaff J. K.; Tian F.; Sinha S. C.; Felding-Habermann B.; Smider V. V.; Schultz P. G. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 16101–16106. 10.1073/pnas.1211023109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradova E. V.; Zhang C.; Spokoyny A. M.; Pentelute B. L.; Buchwald S. L. Nature 2015, 526, 687–691. 10.1038/nature15739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker J. M.; Bernardes G. J. L.; Lin Y. A.; Davis B. G. Chem. - Asian J. 2009, 4, 630–640. 10.1002/asia.200800427. [DOI] [PubMed] [Google Scholar]
- Kozlowski L. P. Nucleic Acids Res. 2017, 45, D1112–D1116. 10.1093/nar/gkw978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban H.; Gavrilyuk J.; Barbas C. F. J. Am. Chem. Soc. 2010, 132, 1523–1525. 10.1021/ja909062q. [DOI] [PubMed] [Google Scholar]
- Antos J. M.; Francis M. B. J. Am. Chem. Soc. 2004, 126, 10256–10257. 10.1021/ja047272c. [DOI] [PubMed] [Google Scholar]
- Lin S.; Yang X.; Jia S.; Weeks A. M.; Hornsby M.; Lee P. S.; Nichiporuk R. V.; Iavarone A. T.; Wells J. A.; Toste F. D.; Chang C. J. Science 2017, 355, 597. 10.1126/science.aal3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deGruyter J. N.; Malins L. R.; Baran P. S. Biochemistry 2017, 56, 3863–3873. 10.1021/acs.biochem.7b00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means G. E.; Feeney R. E. Bioconjugate Chem. 1990, 1, 2–12. 10.1021/bc00001a001. [DOI] [PubMed] [Google Scholar]
- Chen X.; Muthoosamy K.; Pfisterer A.; Neumann B.; Weil T. Bioconjugate Chem. 2012, 23, 500–508. 10.1021/bc200556n. [DOI] [PubMed] [Google Scholar]
- Asano S.; Patterson J. T.; Gaj T.; Barbas C. F. Angew. Chem., Int. Ed. 2014, 53, 11783–11786. 10.1002/anie.201405924. [DOI] [PubMed] [Google Scholar]
- Patterson J. T.; Wilson H. D.; Asano S.; Nilchan N.; Fuller R. P.; Roush W. R.; Rader C.; Barbas C. F. Bioconjugate Chem. 2016, 27, 2271–2275. 10.1021/acs.bioconjchem.6b00432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker S. M.; Backus K. M.; Lazear M. R.; Forli S.; Correia B. E.; Cravatt B. F. Nat. Chem. 2017, 9, 1181–1190. 10.1038/nchem.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinger J.; Jones K.; Cheeseman M. D. Angew. Chem., Int. Ed. 2017, 56, 15200–15209. 10.1002/anie.201707630. [DOI] [PubMed] [Google Scholar]
- Choi S.; Connelly S.; Reixach N.; Wilson I. A.; Kelly J. W. Nat. Chem. Biol. 2010, 6, 133–139. 10.1038/nchembio.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanna A. R.; Li X.; Walseng E.; Pedzisa L.; Goydel R. S.; Hymel D.; Burke T. R. Jr.; Roush W. R.; Rader C. Nat. Commun. 2017, 8, 1112. 10.1038/s41467-017-01257-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom D. G.; Castañeda C. A.; Cannon B. R.; García-Moreno E. B. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 5260–5265. 10.1073/pnas.1010750108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman M.; Wall J. S. J. Am. Chem. Soc. 1964, 86, 3735–3741. 10.1021/ja01072a025. [DOI] [Google Scholar]
- Yunus A. A.; Lima C. D. Nat. Struct. Mol. Biol. 2006, 13, 491–499. 10.1038/nsmb1104. [DOI] [PubMed] [Google Scholar]
- Heine A.; DeSantis G.; Luz J. G.; Mitchell M.; Wong C.-H.; Wilson I. A. Science 2001, 294, 369–374. 10.1126/science.1063601. [DOI] [PubMed] [Google Scholar]
- Galkin A.; Kulakova L.; Melamud E.; Li L.; Wu C.; Mariano P.; Dunaway-Mariano D.; Nash T. E.; Herzberg O. J. Biol. Chem. 2007, 282, 4859–4867. 10.1074/jbc.M609534200. [DOI] [PubMed] [Google Scholar]
- Obexer R.; Godina A.; Garrabou X.; Mittl P. R. E.; Baker D.; Griffiths A. D.; Hilvert D. Nat. Chem. 2016, 9, 50–56. 10.1038/nchem.2596. [DOI] [PubMed] [Google Scholar]
- Giger L.; Caner S.; Obexer R.; Kast P.; Baker D.; Ban N.; Hilvert D. Nat. Chem. Biol. 2013, 9, 494–498. 10.1038/nchembio.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulev A. U. Russ. Chem. Rev. 2011, 80, 197. 10.1070/RC2011v080n03ABEH004162. [DOI] [Google Scholar]
- Furman J. L.; Kang M.; Choi S.; Cao Y.; Wold E. D.; Sun S. B.; Smider V. V.; Schultz P. G.; Kim C. H. J. Am. Chem. Soc. 2014, 136, 8411–8417. 10.1021/ja502851h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baslé E.; Joubert N.; Pucheault M. Chem. Biol. 2010, 17, 213–227. 10.1016/j.chembiol.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. Nat. Rev. Drug Discovery 2011, 10, 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Holm L.; Moody P.; Howarth M. J. Biol. Chem. 2009, 284, 32906–32913. 10.1074/jbc.M109.034322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddick J. J.; Cheng J.; Roush W. R. Org. Lett. 2003, 5, 1967–1970. 10.1021/ol034555l. [DOI] [PubMed] [Google Scholar]
- Naven R. T.; Kantesaria S.; Nadanaciva S.; Schroeter T.; Leach K. L. Toxicol. Res. 2013, 2, 235–244. 10.1039/c3tx50027f. [DOI] [Google Scholar]
- Lu X.; Olsen S. K.; Capili A. D.; Cisar J. S.; Lima C. D.; Tan D. S. J. Am. Chem. Soc. 2010, 132, 1748–1749. 10.1021/ja9088549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garske A. L.; Peters U.; Cortesi A. T.; Perez J. L.; Shokat K. M. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 15046–15052. 10.1073/pnas.1111239108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.; Riegger A.; Lamla M.; Wiese S.; Oeckl P.; Otto M.; Wu Y.; Fischer S.; Barth H.; Kuan S. L.; Weil T. Chem. Sci. 2016, 7, 3234–3239. 10.1039/C6SC00005C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaunak S.; Godwin A.; Choi J.-W.; Balan S.; Pedone E.; Vijayarangam D.; Heidelberger S.; Teo I.; Zloh M.; Brocchini S. Nat. Chem. Biol. 2006, 2, 312–313. 10.1038/nchembio786. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Houk K. N. Acc. Chem. Res. 2005, 38, 379–385. 10.1021/ar040257s. [DOI] [PubMed] [Google Scholar]
- Sleep D. Expert Opin. Drug Delivery 2015, 12, 793–812. 10.1517/17425247.2015.993313. [DOI] [PubMed] [Google Scholar]
- Kratz F.; Elsadek B. J. Controlled Release 2012, 161, 429–445. 10.1016/j.jconrel.2011.11.028. [DOI] [PubMed] [Google Scholar]
- Sand K. M. K.; Bern M.; Nilsen J.; Noordzij H. T.; Sandlie I.; Andersen J. T. Front. Immunol. 2015, 5, 682. 10.3389/fimmu.2014.00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld E. F. Annu. Rev. Biochem. 2011, 80, 1–15. 10.1146/annurev.biochem.031209.093756. [DOI] [PubMed] [Google Scholar]
- Alam I. S.; Neves A. A.; Witney T. H.; Boren J.; Brindle K. M. Bioconjugate Chem. 2010, 21, 884–891. 10.1021/bc9004415. [DOI] [PubMed] [Google Scholar]
- Logue S. E.; Elgendy M.; Martin S. J. Nat. Protoc. 2009, 4, 1383–1395. 10.1038/nprot.2009.143. [DOI] [PubMed] [Google Scholar]
- Hudis C. A. N. Engl. J. Med. 2007, 357, 39–51. 10.1056/NEJMra043186. [DOI] [PubMed] [Google Scholar]
- Lyon R. P.; Setter J. R.; Bovee T. D.; Doronina S. O.; Hunter J. H.; Anderson M. E.; Balasubramanian C. L.; Duniho S. M.; Leiske C. I.; Li F.; Senter P. D. Nat. Biotechnol. 2014, 32, 1059–1062. 10.1038/nbt.2968. [DOI] [PubMed] [Google Scholar]
- Alexov E.; Mehler E. L.; Baker N.; Baptista A. M.; Huang Y.; Milletti F.; Erik Nielsen J.; Farrell D.; Carstensen T.; Olsson M. H. M.; Shen J. K.; Warwicker J.; Williams S.; Word J. M. Proteins: Struct., Funct., Genet. 2011, 79, 3260–3275. 10.1002/prot.23189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandogin J.; Brooks C. L. Biophys. J. 2005, 89, 141–157. 10.1529/biophysj.105.061341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandogin J.; Brooks C. L. Biochemistry 2006, 45, 9363–9373. 10.1021/bi060706r. [DOI] [PubMed] [Google Scholar]
- Georgescu R. E.; Alexov E. G.; Gunner M. R. Biophys. J. 2002, 83, 1731–1748. 10.1016/S0006-3495(02)73940-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Link J. M.; Stekhova S.; Yagle K. J.; Smith C.; Krohn K. A.; Tait J. F. Bioconjugate Chem. 2008, 19, 1684–1688. 10.1021/bc800164d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan A. S.; Neves A. A.; de Backer M. M.; Hu D.-E.; Davletov B.; Kettunen M. I.; Brindle K. M. Radiology 2008, 246, 854–862. 10.1148/radiol.2463070471. [DOI] [PubMed] [Google Scholar]
- Wang F.; Fang W.; Zhao M.; Wang Z.; Ji S.; Li Y.; Zheng Y. Nucl. Med. Biol. 2008, 35, 359–364. 10.1016/j.nucmedbio.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Krall N.; da Cruz F. P.; Boutureira O.; Bernardes G. J. L. Nat. Chem. 2016, 8, 103–113. 10.1038/nchem.2393. [DOI] [PubMed] [Google Scholar]
- Chudasama V.; Maruani A.; Caddick S. Nat. Chem. 2016, 8, 114–119. 10.1038/nchem.2415. [DOI] [PubMed] [Google Scholar]
- Lyon R. P.; Bovee T. D.; Doronina S. O.; Burke P. J.; Hunter J. H.; Neff-LaFord H. D.; Jonas M.; Anderson M. E.; Setter J. R.; Senter P. D. Nat. Biotechnol. 2015, 33, 733–735. 10.1038/nbt.3212. [DOI] [PubMed] [Google Scholar]
- Merlin J.-L.; Barberi-Heyob M.; Bachmann N. Ann. Oncol. 2002, 13, 1743–1748. 10.1093/annonc/mdf263. [DOI] [PubMed] [Google Scholar]
- Cui J. J.; Tran-Dubé M.; Shen H.; Nambu M.; Kung P.-P.; Pairish M.; Jia L.; Meng J.; Funk L.; Botrous I.; McTigue M.; Grodsky N.; Ryan K.; Padrique E.; Alton G.; Timofeevski S.; Yamazaki S.; Li Q.; Zou H.; Christensen J.; Mroczkowski B.; Bender S.; Kania R. S.; Edwards M. P. J. Med. Chem. 2011, 54, 6342–6363. 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- Freedy A. M.; Matos M. J.; Boutureira O.; Corzana F.; Guerreiro A.; Akkapeddi P.; Somovilla V. J.; Rodrigues T.; Nicholls K.; Xie B.; Jiménez-Osés G.; Neves A. A.; Bernardes G. J. L. J. Am. Chem. Soc. 2017, 139, 18365–18375. 10.1021/jacs.7b10702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.; Baran P. S. Nature 2009, 459, 824–828. 10.1038/nature08043. [DOI] [PubMed] [Google Scholar]
- Gormisky P. E.; White M. C. J. Am. Chem. Soc. 2013, 135, 14052–14055. 10.1021/ja407388y. [DOI] [PubMed] [Google Scholar]
- Newhouse T.; Baran P. S. Angew. Chem., Int. Ed. 2011, 50, 3362–3374. 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.