

Abstract
The integrity of the interferon (IFN)-γ circuit is necessary to mount an effective immune response to intra-macrophagic pathogens, especially Mycobacteria. Inherited monogenic defects in this circuit that disrupt the production of, or response to, IFN-γ underlie a primary immunodeficiency known as Mendelian susceptibility to mycobacterial disease (MSMD). Otherwise healthy patients display a selective susceptibility to clinical disease caused by poorly-virulent mycobacteria such as BCG (bacille Calmette-Guérin) vaccines and environmental mycobacteria, and more rarely by other intra-macrophagic pathogens, particularly Salmonella and M. tuberculosis. There is high genetic and allelic heterogeneity, with 19 genetic etiologies due to mutations in 10 genes that account for only about half of the patients reported. An efficient laboratory diagnostic approach to suspected MSMD patients is important, because it enables the establishment of specific therapeutic measures that will improve the patient’s prognosis and quality of life. Moreover, it is essential to offer genetic counseling to affected families. Herein, we review the various genetic and immunological diagnostic approaches that can be used in concert to reach a molecular and cellular diagnosis in patients with MSMD.
Keywords: Mycobacteria, intracellular pathogens, interferon gamma, primary immunodeficiency, diagnosis, MSMD
Introduction
Mendelian susceptibility to mycobacterial disease (MSMD) is a primary immunodeficiency (PID) characterized by a selective predisposition in otherwise healthy individuals to disease when infected by bacille Calmette-Guérin (BCG) vaccines or environmental mycobacteria [1,2]. It is included in the PID classification by the IUIS (International Union of Immunology Societies) in the VIth group of defects in Intrinsic and Innate immunity [3]. Immunity to mycobacteria relies on the IFN-γ (interferon) circuit (Figure 1), as shown by the study of mice both in vitro and in vivo, and by the study of humans with MSMD. Pattern recognition receptors are important sensors of mycobacteria after infection; however, their role in generating a protective response is apparently redundant [4,5]. After bacilli/us phagocytosis, antigen-presenting cells (APC), including macrophages, are activated and produce tumor necrosis factor (TNF)-α, interferon-stimulated gene (ISG) 15, and interleukin (IL)-12p70, which induce T helper (Th) cells to produce IFN-γ and differentiate into Th1 cells. This creates a positive loop between the T cell and the APC, which enhances the former’s microbicidal capacity through production of oxygen reactive species (ROS) [6–10].
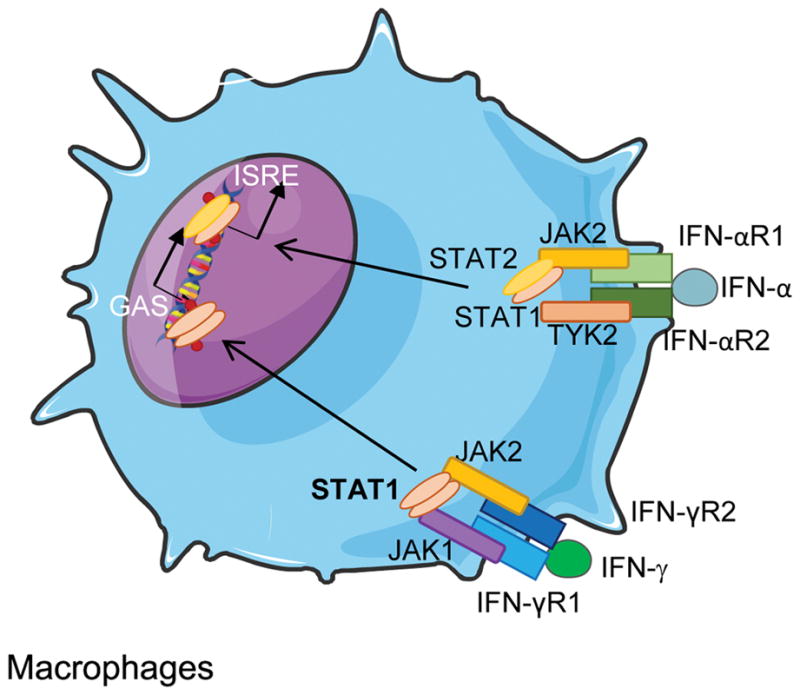
Figure 1. IFN-γ circuit.

Summary of molecules implicated in the IFN-γ circuit. Molecules represented with bold characters are known to cause of MSMD. GAS: γ-interferon activated site; GAF: γ-activated factor.
IL-12 (IL-12p70) is a heterodimer composed of a p40 subunit (in common with IL-23) and a p35 subunit that bind IL-12Rβ1 and IL-12Rβ2, respectively, activating both natural killer (NK) and Th cells [11]. Janus-associated kinase 2 (JAK2) binds to IL-12Rβ2 subunit and tyrosine kinase 2 (TYK2) to IL-12Rβ1 subunit. After IL-12p70 binds to the IL-12 receptor (IL-12Rβ1-IL-12Rβ2 dimer), TYK2 and JAK2 come closer and JAKs are trans-phosphorylated, thereby phosphorylating the receptor chains. Signal transducer and activator of transcription 4 (STAT4) binds to phosphorylated IL-12Rβ2, becomes auto-phosphorylated, and dimerizes. Then, STAT4 homodimers translocate to the nucleus, where they bind to the IFNG promoter, inducing its transcription [12]. In parallel, secreted free ISG15 from APCs also promotes IFN-γ production by T cells and CD3-CD56+ NK cells, which are considered the key ISG15-responder leukocytes [7,13]. Thus, ISG15 and IL-12p70 act synergistically to induce IFN-γ production.
IFN-γ response in APCs, especially in macrophages, is mediated by its binding to IFN-γ receptor (IFN-γR) 1 and IFN-γR2, followed by internalization and signalization via the receptor complex. After IFN-γ binding, the two subunits of the receptor, as well as JAK1 (bonded to IFN-γR1) and JAK2 (bonded to IFN-γR2) come closer. JAK1 and JAK2 then cross-phosphorylate and phosphorylate IFN-γR2, creating a docking site for STAT1. After binding, STAT1 is activated by phosphorylation of tyrosine 701 and dimerizes, forming γ-activated factor (GAF) and translocating to the nucleus where it binds to γ-interferon-activated site (GAS) of ISG, promoting its expression [14,15]. MSMD is caused by monogenic defects in different steps of this circuit (Figure 1), which impair the production of, or the response to, IFN-γ, thereby disrupting protective immunity to mycobacterial infection.
Although the first clinical description of MSMD was published in 1951 [16], it was not until 1996 that the first genetic etiology of MSMD, autosomal recessive (AR) IFN-γR1 deficiency, was described in an infant with fatal BCG infection [17,18]. Afterwards, defects in other genes encoding proteins involved in IFN-γ immunity have been discovered, affecting both IFN-γ production (IL12RB1 [19–21], IL12B [22,23], ISG15 [7,24], NEMO [25], IRF8 [26], and TYK2 [27]), and cellular responses to IFN-γ (IFNGR1 [17,19,28–31], IFNGR2 [32,33], STAT1 [34–36], IRF8 [26] and CYBB [25,37,38]). There are currently 19 different genetic etiologies of MSMD that involve the impact of the mutation (null or hypomorphic), the mode of transmission in the family (dominant or recessive), the expression of the mutant allele (absent or detectable), or the function affected by the mutation (one domain or another, in the case of a detectable protein); the most common defect is IL-12Rβ1 deficiency, and the second most common, IFN-γR1 deficiency [2,27,39]. The number of genetic etiologies is likely to increase in the coming years. With so many forms, the clinical boundaries of MSMD syndrome and of each genetic etiology are not yet fully defined; the disease spectrum ranges from the complete forms of IFN-γR deficiencies in the most severe cases of MSMD, with an outcome that leads to death if hematopoietic stem cell transplantation (HSCT) is not performed [17,40], to other defects (for example, IL-12Rβ1 or IL-12p40 deficiencies), in which patients can be treated with exogenous human recombinant IFN-γ (hrIFN-γ) in addition to antibiotics [20,21]. For this reason, accurate genetic diagnosis, and the distinction between complete and partial defects, as well as the careful description of the immunological signs, are of the utmost importance to ensure the best possible management of MSMD patients.
Published immunological approaches for the molecular and cellular diagnosis of MSMD are diverse. Some are complex and results, even among healthy controls, can be highly variable. [19,21,23,30,39]. Nevertheless, they are necessary, since they facilitate targeted gene sequencing and the prediction of effectiveness of adjuvant therapies such as exogenous hrIFN-γ. Our main aim is to summarize the current warning signs of MSMD, as well as the functional and genetic approaches available for the study of the IFN-γ circuit, both in clinical practice and in research, and their limitations, in order to guide physicians and immunologists in the diagnosis of MSMD.
Infectious spectrum of MSMD
Patients affected with MSMD are otherwise usually healthy and can present a wide range of severity of the disease, from local and recurrent to disseminated and lethal. The severity of the disease depends on the type of underlying defect (complete or partial). Clinical disease is usually caused by environmental mycobacteria (EM), and BCG after infant vaccination, which is the most common, and sometimes the only, infectious event [2,41]. Some patients are also susceptible to Mycobacterium tuberculosis [42]. Different etiologies of MSMD, especially IL-12Rβ1 [2,42,43], IFN-γR1[2,29], STAT1[2,34], and IL-12p40 [2,23] deficiencies, were found in patients with severe tuberculosis (TB) (disseminated/extrapulmonary or recurrent TB). There are currently 23 reported patients with tuberculosis due to inborn errors of IFN-γ, 13 of whom are IL-12Rβ1-deficient; these include six who did not suffer from any other mycobacterial disease (BCG, EM)[2,42,43]. Interestingly, MSMD underlying Mycobacterium tuberculosis infection restricted to the lung has been described not only in IL-12Rβ1 deficiency, but also in IFN-γR1 deficiency [30,44,45]. Besides mycobacteria, there is a wide range of causative organisms of disease that includes Salmonella, fungi (especially Candida), other intra-macrophagic bacteria, and parasites (Leishmania, Toxoplasma [46]). MSMD usually, but not always, manifests in childhood [2].
Interestingly, specific clinical manifestations have been associated with specific gene defects: the correlation of pathogens and/or clinical forms with all described genetic etiologies of MSMD was nicely reviewed by Bustamante et al. [2]. Briefly, patients with IFN-γ production defects caused by mutations in IL12RB1 and IL12B (encoding IL-12Rβ1 and IL-12p40, respectively) commonly suffer from disease caused by Salmonella (recurrent or not) and, to a lesser extent, by Candida. Patients with IFN-γ production defects do not usually present with viral infections. Regarding IFN-γ response defects, the presence of multifocal osteomyelitis should raise the suspicion of a partial autosomal dominant (AD) IFN-γR1, partial AR, or AD STAT1 loss of function (LOF) [36,47–53]. Patients with complete deficiency in IFN-γR1 and IFN-γR2, abolishing IFN-γ response, are more prone to viral diseases such as cytomegalovirus, respiratory syncytial virus and varicella-Zoster virus, among others [2].
Laboratory testing
Who should be tested?
Children or adults without any other hemato-immunological conditions who develop recurrent or severe/disseminated mycobacterial infectious disease caused by BCG, EM, M. tuberculosis, or Salmonella alone or in combination with other intracellular pathogens or viruses should be tested. Specific warning signs of MSMD are presented in Table 1.
Table 1.
MSMD warning signs.
| Sign | Description |
|---|---|
|
| |
| Age at presentation | Usually in childhood, also in adolescence and adulthood |
|
| |
| General state | Otherwise healthy individuals |
|
| |
| Infectious spectrum | Invasive or recurrent infections by: |
| Mycobacteria: | |
| BCG infection (Mycobacterium bovis vaccine strain) | |
| Environmental mycobacteria (M. chelonae, M. fortuitum, M. mageritense, M. peregrinum, M. smegmatis, M. scrofulaceum...) | |
| Mycobacterium tuberculosis | |
| Intramacrophagic bacteria (alone or in combination with mycobacteria): | |
| Salmonella spp. | |
| Listeria monocytogenes/Nocardia spp./Klebsiella spp. | |
| Fungi (in combination with mycobacteria) | |
| Candida spp. | |
| Histoplasma capsulatum/Paracoccides brasilensis/coccicoides spp. | |
| Parasites (alone or in combination with mycobacteria, rare): | |
| Leishmania spp. | |
| Toxoplasma gondii | |
| Virus (in combination with mycobacteria, rare) | |
| Cytomegalovirus, human herpes virus 8, parainfluenza virus type 3, respiratory syncytial virus and varicella zoster virus. | |
|
| |
| Other | Family history of invasive or recurrent mycobacterial infection |
| Undetectable or very low IFN-γ production in Interferon-Gamma Release Assays (IGRAs) (i. e. QuantiFERON-TB Gold In-Tube) | |
Defects in the IFN-γ circuit are not the only PID predisposing to mycobacterial disease [3,42]. Before performing specific MSMD tests, severe combined immunodeficiency, combined immunodeficiency and chronic granulomatous disease must be ruled out [54], because they are more common than MSMD and they confer susceptibility to various infectious diseases including mycobacteria.
Other less common PIDs confer susceptibility to various infectious diseases including mycobacteria and should also be ruled out in parallel with MSMD testing: 1) X-linked NF-kB deficiency: anhidrotic ectodermal dysplasia with immunodeficiency (XR-EDA-ID) syndrome. Patients suffering XR-EDA-ID are susceptible to a wide range of pathogens (pyogenic bacteria, viruses) including mycobacteria. Immunologically, these patients present altered NK cell mediated cytotoxicity and TNF-α production after Toll-like receptor (TLR)-4 lipopolysaccharide (LPS) stimulation [2,55–57]; 2) GATA2 deficiency, particularly in otherwise healthy adults with disseminated EM infections [58,59]. Patients with GATA2 deficiency show susceptibility to viral infections and mycobacteria and usually present with severe circulating monocytopenia (78% of patients), and B (86% of patients) and NK (82% of patients) [58,59] lymphopenias. Characteristically, patients with GATA2 deficiency show specific loss of the CD56bright subset [60]. Due to these characteristic myeloid and lymphoid cytopenias, consideration of GATA2 deficiency as a genetic etiology of MSMD is currently open to debate, because MSMD-causing defects occur in otherwise healthy subjects without other significant immune abnormalities except for the defect in the IFN-γ circuit; 3) severe innate PID, predisposing to mycobacteria and viruses (AR STAT1, AR STAT2, AR JAK1, and AR interferon regulatory factor 8 (IRF8) deficiencies [26,61–68]) or mycobacteria and fungi (AR RAR related orphan receptor C (RORC) deficiency [69]).
Beyond PID, other causative conditions such as immunosuppressive drug exposure, including anti-TNFα antibodies, azathioprine, cyclophosphamide, mycophenolate, and cyclosporine, need to be ruled out [70,71]. In addition, long-term potent oral steroids can lead to secondary mycobacterial infection [72]. Also, acquired immunodeficiency by HIV infection [42,71,73] and malignancies such as hairy cell leukemia need to be tested for [74–77]. Finally, patients who have neutralizing anti-IFN-γ autoantibodies can develop MSMD-like clinical manifestations; they are included in group IX of the IUIS classification, which is called PID phenocopies [3]. Patients with neutralizing anti-IFN-γ autoantibodies have impaired IFN-γ production and STAT1 phosphorylation in the presence of autologous serum that is rescued after lavage. This phenomenon has been mostly, but not exclusively, observed in adult Asian populations [78–81].
Baseline IFN-γ in plasma
Detection of baseline IFN-γ in plasma by enzyme-linked immunosorbent assay (ELISA) is a simple technique that can help to rapidly identify patients with complete IFN-γR deficiency [2,82,83]. These patients present with increased levels of IFN-γ in plasma; patients with partial recessive forms of IFN-γR deficiency present with detectable levels of IFN-γ while it is undetectable in other MSMD forms and in healthy controls [30,33,40,82–84]. The threshold to consider a patient with a complete defect as a candidate for HSCT was defined as 2 standard deviations above the mean level in patients with partial AR IFN-γR1 defects (>80 pg/mL), while observed levels in complete IFN-γR deficiency were 150 1700 pg/mL [82,83]. Several years later [82], Sologuren et al. published a case series of partial AR IFN-γR1 defects showing a range of baseline IFN-γ of 51 222 pg/mL, with an outlier of 925 pg/mL [30]. They suggested that the very high concentration of baseline IFN-γ observed in the outlier could reflect an acute mycobacterial disease. Thus, the infectious state of the patient needs to be considered, as baseline IFN-γ plasma levels may vary in acute infection compared with the convalescent phase [30], making it possible that levels may overlap in partial AR IFN-γR1 or IFN-γR2 deficiency in rare cases [30,33,83]. Therefore, if possible, baseline IFN-γ should be measured at least one month after resolution of acute infection. In any case, no IFN-γ is usually detected in the plasma of healthy individuals [82,83]. Plasma samples need to be diluted at least 1:2 to avoid interference from other proteins such as fibrinogen.
To optimize the ELISA technique, IFN-γ measurements on patients’ plasma samples should be batched. Then the cost of an individual determination of IFN-γ can range from 1–12€, depending on the kit used. The selection of the ELISA kit will also determine the hands-on time required (3 h to approximately 6–8 h), depending on whether or not an overnight sensitization step is required. Optimization can lead to an increased response time (turn-around time) when returning the results to the clinician if the number of patients is low.
Cytokine production
The gold standard for the study of IFN-γ circuit integrity, cytokine production, was developed by Feinberg et al. [19]. This assay is based in the measurement of IL-12p40, IL-12p70 and IFN-γ after stimulation of whole blood or peripheral blood mononuclear cells (PBMCs). Stimulation conditions comprise live BCG stimulation at a multiplicity of infection of 20 BCG/leukocyte with or without hrIL-12p70 (20 ng/mL), or hrIFN-γ (5000 IU/mL) co-stimulation for 18 h (for IL-12 measurement) or 48 h (for IFN-γ and IL-12 measurements).
For healthcare practices and laboratories subject to ISO 15189 European regulations, the use of BCG as a stimulus impedes the standardization of the protocol. An alternative that avoids the use of BCG is the use of mitogens as follows: phytohemagglutinin (PHA; 1%) [85] or LPS (from Salmonella minesotta; 100 ng/mL) in combination with hrIL-12p70 or hrIFN-γ (102, 103 and 104 IU/mL) [30]. The output of both BCG and mitogen whole blood or PBMC stimulation is similar (measurement of IL-12p40, IL-12p70 and IFN-γ). Detection of the cytokines produced may be performed with ELISA or multiplex assays by means of flow cytometry (Luminex Technology (Luminex, Austin, TX, USA) or cytometric bead array systems [19,30,40]. As the interval between blood extraction and performance of testing reduces the cytokine production [21], it is important to take this into account when analyzing the results in samples that are assayed 24 h after the blood extraction.
Results obtained from the cytokine production assay will help to distinguish between IFN-γ response defects and IFN-γ production defects. Complete forms of IFN-γR1, IFN-γR2, IL-12Rβ1 or IL-12p40 can be detected with this approach; however, some genetic etiologies of MSMD, such as CYBB or AD IRF8 deficiency, will show normal responses to this stimulation [26,37,86]. IFN-γ production defects are characterized by the absence or low production of IFN-γ after BCG stimulation. If there is no recovery of IFN-γ after hrIL-12p70 co-stimulation, IL-12Rβ1 deficiency should be studied first [21], followed by ISG15 or TYK2 deficiencies [7,27]. Patients with IL-12p40 deficiency produce low or very low levels of IFN-γ in response to BCG stimulation, which can be rescued, at least partially, with exogenous hrIL-12p70. In complete IFN-γ response defects (complete IFN-γR1 and IFN-γR2 deficiencies), there is no response to hrIFN-γ in terms of IL-12 production [19,29,85,87–92]. On the other hand, in partial IFN-γ response defects (partial AR IFN-γR1/IFN-γR2 and partial AD STAT1 LOF deficiency), the response to hrIFN-γ is impaired in a dose-dependent manner, but not abolished [28–30,34,36,45,93–98].
In nuclear factor-kappa B essential modulator (NEMO)-deficient patients, IL-12 production is normal after BCG stimulation but impaired after PHA/CD3 PBMC stimulation [38,99].
It is difficult to establish cut-off values for diagnosis, because published cases cannot always be compared due to differences in the techniques used for cytokine production determination. There have been attempts to study cohorts of IL-12Rβ1 and IL-12p40 patients [19,23,100] functionally: i) in IL-12Rβ1 deficient patients, IL-12p70 production was normal but IFN-γ production was low or null after BCG (4 – 726 pg/mL) and did not increase after IL-12p70 co-stimulation [19,21]; ii) IL-12p40 deficient patients showed no IL-12p70 production and a decreased IFN-γ production that in most cases was undetectable or below 100 pg/mL; only one patient showed IFN-γ production of 1000 pg/mL [19,23]. Patients with complete deficiency of IFN-γR1 or IFN-γR2 showed normal production of IFN-γ but failed to induce IL-12p70 after BCG or BCG + IFN-γ. Expected results of cytokine production in the different genetic forms of the IFN-γ circuit are summarized in Tables 2 and 3.
Table 2.
Diagnostic tests in IFN-γ response defects.
| Gene | Inheritance | Defect complete/ partial (C/P) |
Microbial stimulation (BCG) | Mitogen stimulation | Baseline IFN-γ |
Cytometric determination | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| IFN-γ production |
IL-12 production |
IFN-γ production |
IL-12 production |
IFN-γ R1 | IFN-γ R2 |
IFN-γ binding |
STAT1- phosphorylation in response to IFN-γ |
||||
| IFNGR1 | AR | C | Normal | Does not increase with IFN-γ costimulation | Normal in response to PHA (WB, PBMCs) | Normal or reduced with LPS, no response to IFN-γ (WB, PBMCs) | Very high | Present | NP | NP | Abolished1 |
| AR | C | Normal | Does not increase with IFN-γ costimulation | Normal (WB); low in response to PHA, rescued with IL-12p70 (PBMCs) | No response to IFN-γ | Very high | Absent | NP | NP | Abolished | |
| AD | P | Normal | Increase with IFN-γ costimulation | Normal in response to CD2/CD28 in T cells; low IFN-γ secretion in T cells after IL-12 stimulation | Impaired but not abolished response to LPS + IFN-γ (WB, isolated monocytes) | Normal | Increased | NP | NP | Impaired but not abolished 2 | |
| AR | P | Normal | Does only increase with high IFN-γ costimulation | NP | Low response to low/medium doses of IFN-γ | High | Present | NP | Impaired, dosage dependent | Impaired but not abolished | |
| IFNGR2 | AR | C | Normal | Does not increase with IFN-γ costimulation | NP | NP | High | Present | Present | NP | Abolished |
| AR | C | Normal | Does not increase with IFN-γ costimulation | Low after PHA stimulation; increased to normal levels with IL-12 (PBMCs) | NP | High | Present | Absent | NP | Abolished | |
| AR | P | Normal (WB) | Impaired increase with IFN-γ costimulation | NP | NP | Increased or not | Present (measured in PBMCs) | Low levels | NP | NP | |
| AD | P | NP | NP | NP | NP | NP | NP | NP | NP | Normal in monocytes, T cells and monocyte derived-macrophages | |
| STAT1 | AD | P | Normal increase with IL-12 costimulation | Impaired increase with IFN-γ costimulation | NP | NP | NP | NP | NP | NP | Normal |
| AD | P | Normal increase with IL-12 costimulation | Impaired increase with IFN-γ costimulation | NP | NP | NP | NP | NP | NP | Impaired3 | |
AR: autosomal recessive; AD: autosomal dominant; NP: not published; PBMC: peripheral mononuclear cells; WB: whole blood.
Abolished response: no phosphorylation of STAT1 after IFN-γ stimulation
Impaired response: impaired phosphorylation of STAT1 after IFN-γ stimulation; phosphorylation is present only after high-dose IFN-γ (>105 IU/ml)
Abolished or impaired STAT1 phosphorylation in response to IFN-γ may be due to mutations in the coiled-coil domain and in the DNA-binding domain. Some patients have impaired STAT1-p and impaired binding to DNA.
Table 3.
Diagnostic tests in IFN-γ production defects.
| Gene | Inheritanc e |
Defect complete / partial (C/P) |
Microbial stimulation (BCG) |
Mitogen stimulation | Cytometric determination | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IFN-γ production |
IL-12 production |
IFN-γ production |
IL-12 production |
Baseline IFN-γ |
IL-12R γ 1 presence |
STAT4 phosphorylatio n in response to IL-12p70 |
IFN-γ R1 presenc e |
IFN-γ R2 presenc e |
STAT1 phosphorylatio n in response to IFN-γ |
|||
| IL12B | AR | C | Impaired but not abolished, increases after IL-12p70 costimulation (lower than controls) (WB); impaired and restored after IL-12p70 costimulation (PBMCs) | Abolished IL-12p40, severely impaired IL-12p70 in WB and in PBMCs | Severely impaired/increases after IL-12p70 costimulation (PHA, PBMCs and WB) | Abolished (SAC stimulation, PBMCs), abolished (CD40L, PBMCs) | Not detectabl e | NP | NP | Norma | NP | NP |
| IL12Rβ 1 | AR | C | Low, does not increase after IL-12p70 costimulatio n | Normal | NP | NP | Not detectabl e | Present (normal or impaired) | Abolished | Norma l | Norma l | NP |
| AR | C | Low, does not increase after IL-12p70 costimulatio n | Normal | Abolished or severely impaired (PHA, PHA+IL-12; CD3+CD28; CD2+CD28, PBMCs) | NP | Not detectabl e | Abolishe d | Abolished | Norma l | Norma l | Normal in response to IFN-γ | |
| IRF8 | AD | p | Normal after BCG stimulation (also PPD, in PBMCs, WB) | Normal after BCG (WB) | Normal with PHA (WB) | 1/3 production after R848 stimulation (PBMCs) | NP | NP | NP | NP | NP | NP |
| ISG15 | AR | C | No IFN-γ in response to BCG, partially recovers with IL-12 costimulation (recovers with ISG15 addition, resembles IL-12p40 def) | Normal | NP | NP | NP | Normal | NP | NP | NP | NP |
| CYBB | XR | C | Normal | Normal | NP | NP | NP | NP | NP | NP | NP | NP |
| NEMO | XR | P | NP | Normal | Low levels after PHA/anti-CD3 | NP | NP | NP | NP | NP | NP | NP |
| TYK2 | AR | P | Impaired IFN-γ production, but not abolished, does not recover with IL-12 costimulatio n | Normal | NP | NP | NP | NP1 | NP1 | NP1 | NP1 | NP1 |
AR: autosomal recessive; AD: autosomal dominant; XR: X-linked; NP: not published; PBMC: peripheral mononuclear cells; PHA: phytohemagglutinin, WB: whole blood
Results tested in EBV-B cells are included in Table 4
The main advantage of this technique is that it is the test that most closely assesses the patients’ real immune function. However, it also has limitations: by itself, it only clearly detects complete defects, while partial defect identification can be difficult. To date, the specification of cut-off values to define disease for routine healthcare practice has not been possible. Although it has limitations, cytokine detection after whole blood/PBMC culture is a powerful option with room for improvement.
The culture itself takes 48 h, but the hands-on time is limited, and depends on if it is performed in whole blood (30–45 min) or in PBMCs (120–165 min). Depending on the concentration and the source of the stimuli used, the costs may vary. In this technique, an economic limitation may be the acquisition of the stimuli for the first time, because some are expensive, but they can be used for many tests. For the detection of secreted cytokines, the most economic option is to perform ELISA for IFN-γ and for IL-12p70, with an estimated cost from around 6–7€ to 80–100€ per individuala, but it will vary depending on the duplicates run, the assay conditions and the chosen kit. This technique requires the same hands-on time as IFN-γ baseline detection, including the possibility that optimization of the technique by batching of patients can lead to increased response timesb.
Cytometric detection of extracellular receptors
IFN-γR1/IFN-γR2 expression
IFN-γR detection by flow cytometry is a fast technique for the detection of complete forms of AR IFN-γR1 and AR IFN-γR2 deficiency with absent protein expression in the membrane of monocytes; it can be performed in both WB and PBMCs (Figure 2). However, different mutations in IFNGR1 and IFNGR2 can lead to distinct patterns of expression (Table 1). In partial AR IFN-γR1 defects, there is usually a weak expression of the receptor [2,30], and partial AD IFN-γR1 deficiency leads to increased protein expression due to mutations in the recycling motif [2,31]. In case of expression of the receptor, its detection could be affected by the antibody used: for example, IFN-γR1 in cells of patients with the C77Y complete AR IFN-γR1 defect would be detected with the gR99 clone but not with the gR38 clone [28]. Similarly, there are some AR IFN-γR2 defects with protein expression, and partial AD/AR defects show low but detectable IFN-γR2 in the membrane of monocytes [2,33,92]. The currently-available antibodies for the evaluation of IFN-γR2 expression are not optimal.
Figure 2. Diagram of the laboratory analysis of MSMD defects with examples.

IFN-γR and STAT1 phosphorylation detection is performed in whole blood assay. IL-12Rβ2 detection is performed in PBMCs after 72 h stimulation with PHA. STAT4 phosphorylation detection is performed in PBMCs after 72 h stimulation with PHA and at least 48 h of culture in the presence of IL-2 or PHA + IL-2. An example of a healthy control is shown for each technique. Cytokine production is detected after 18 h of culture (for IL-12p70) and 48 h of culture (for IFN-γ and IL-12p70) in the gold standard procedure, BCG with or without IFN-γ or IL-12p70 co-stimulation. Control cohort is shown.
We estimate that cytometric evaluation of IFN-γR1 should allow the identification of approximately 80% of complete AR IFN-γR1 deficiencies; however, normal expression of IFN-γR does not exclude a deficiency. In such cases of expression of normal receptors but suspected MSMD, other techniques that evaluate cellular responses to IFN-γ, such as IL-12p70 production, STAT1 phosphorylation in response to increasing doses of IFN-γ or IFN-γ binding studies, should be used.
Flow cytometry staining for the usual number of samples (1–2 patients and a control) takes about 90 min of hands-on timeb. The cost of antibodies is around 8€ per individuala, but as for all techniques, it can vary depending on the laboratory provider and region.
IL-12Rβ1 expression
IL-12Rβ1 deficiency is the most common genetic form of MSMD [2]. IL-12Rβ1 expression detection with flow cytometry is performed in PBMCs after 72 h of stimulation with PHA [21] (Figure 2). As stated for IFN-γR, not all IL-12Rβ1 described defects have an absence of IL-12Rβ1 in the membrane of activated lymphocytes (Table 3) [21,101]. Only two mutations lead to a detectable but nonfunctional expression of IL-12Rβ1 protein in the membrane; one is a large deletion (700 + 362_1619-944del) in IL12Rβ1 [102], and the other is caused by an N-terminal signal peptide stop-gain homozygous mutation [103]. Cytometric determination of IL-12Rβ1 expression is a powerful and easy-to-perform technique that allows the detection of more than 99% of the described mutations. In the absence of the protein in the membrane of activated lymphocytes, genetic studies of IL12Rβ1 need to be performed, but its presence does not rule out a defect. In such cases, an evaluation of cellular responses to IL-12 is needed.
From receipt of the blood to the acquisition of results, this technique takes 4 days, with a hands-on time of approximately 2 h and 15 min (90 min on day 1 for the PBMCs isolation and stimulation, and approximately 45 min for the staining and acquisition in the cytometer on day 3b. The estimated antibody cost is around 10€ per individuala.
IFN-γ binding studies
Because some defects in IFN-γR do not affect their membrane expression, IFN-γ binding studies can help to evaluate their functionality. These techniques may be performed with radiolabeled 125IFN-γ or by flow cytometry [30–32]. For flow cytometry, PBMCs are first incubated with hrIFN-γ for 30 min, and then washed and incubated for 20 min with an anti-IFN-γ antibody. If the anti-IFN-γ antibody is fluorescence-labeled, cells can be directly acquired with a flow cytometer [32]; otherwise, further steps are needed [30]. With this technique, membrane-expressing IFN-γR1 defects can be easily detected. IFN-γ-binding assays with flow cytometry do not yield consistent results with Epstein-Barr virus-transformed B cells (EBV-B cells), and when using PBMCs, gating on monocytes is required [30]. Some MSMD etiologies (AD IFN-γR1 deficiency) will escape this detection [31].
This technique, performed in PBMCs, includes four incubation steps. From receipt of the blood sample to acquisition of results in the cytometer, the technique can be performed in approximately 5 h for a patient sample, a health control and a negative control (medium), and the cost of consumables is approximately 34€a. However, this technique was specifically developed to analyze whether a particular mutation in the IFN-γR1 confers a partial recessive or a complete recessive deficiency, and increasing doses of IFN-γ (1–10,000 IU/mL) are required for this analysis; in such a case, the cost and the hours of work may increase to 120€a and 6–6.5 hb, respectively. To our knowledge, only three patients and three healthy controls have been evaluated so far. Therefore, it is particularly difficult to provide sensitivity and reference ranges for this non-radioactive and flow cytometry-based technique. In addition, the antibodies used, and the model of the flow cytometer and its configuration may significantly affect the results. In our hands, mean fluorescence intensity (MFI, binding of the anti-IFN-γ antibody to monocytes) increases 4- to17-fold in cells incubated with as low as 1 IU IFN-γ/mL compared to cells incubated with medium alone. No or a very low MFI is observed in cells from patients with partial AR IFN-γR1 deficiency at the same concentrations of IFN-γ. At high IFN-γ concentrations, binding (MFI) is similar to or only slightly diminished in cells from patients with partial AR IFN-γR1 deficiency compared to cells from healthy controls.
Cytometric detection of phosphorylated STAT molecules
STAT proteins play a crucial role in cytokine signaling. They bind to activated extracellular receptors, and then phosphorylate, dimerize, and translocate to the nucleus to bind to specific DNA regions and activate gene transcription [14,15,104]. The two most relevant STAT molecules implicated in the IFN-γ circuit are STAT1, which is activated after IFN-γ/IFN-α stimulation, and STAT4, which is activated after IL-12p70 stimulation [105]. Flow cytometric determination of STAT1 phosphorylation can be performed in both whole blood and isolated PBMCs, while STAT4 phosphorylation in response to IL-12p70 needs to be performed in activated lymphocytes. First, cells are stimulated with different cytokine concentrations for 15–30 min. Then cells are fixed and permeabilized with special buffers that maintain the phosphorylation state of the cell, are stained with anti-phosphorylated STAT antibodies in conjunction with the extracellular antibodies of choice, and are acquired with a flow cytometer [34,36,102,106] (Figure 2). It should be stressed that when working with anti-STAT antibodies, proper negative controls are mandatory, and, if possible, the results should be corroborated by other techniques such as western blot, to avoid artifacts. Of note, STAT4 phosphorylation evaluation is limited by the lack of a proper antibody to detect total STAT4.
STAT1 phosphorylation
STAT1 phosphorylation is a useful technique to test the response to IFN-γ, as IFNGR mutations may or may not lead to abolished receptor expression on the surface of monocytes [2,28,30,31,33,92]. Complete defects in IFNGR1 and IFNGR2 genes lead to abolished STAT1 phosphorylation in response to hrIFN-γ and normal phosphorylation in response to hrIFN-α, respectively [32,88,92,107], while partial defects lead to impaired, but not abolished, STAT1 phosphorylation in a dose-dependent manner, with normal responses at high doses [30,33,92,94,95,107–109]. When stimulating cells for STAT1 phosphorylation analysis, the IFN-γ dosage is a key factor to consider; a range from 10–105 IU/mL of hrIFN-γ or hrIFN-α is used, with 103 IU/mL and 105 IU/mL being the most common concentrations [30,31,34,35,62,93,96,107].
It is not only mutations affecting STAT1 phosphorylation that cause loss of function. Although Tyr701 phosphorylation is the first step for STAT1 function, mutations on other STAT1 domains implicated in later events can also impair its function. AD STAT1 LOF mutations in the tail segment domain or SH2 domain (with the exception of the M654K mutation [106]) lead to impaired STAT1 phosphorylation in response to hrIFN-γ but not hrIFN-α [35,36,93]. In contrast, mutations in the DNA-binding domain can lead to both normal (E320Q and Q463H mutations [34]) and altered (E157K and G250E mutations [110]) STAT1 phosphorylation. Impaired or abolished phosphorylation to both hrIFN-γ and hrIFN-α suggests a STAT1 deficiency (which is considered a combined immunodeficiency if it is AR or an MSMD if it is AD), while normal phosphorylation does not exclude it. STAT1 phosphorylation after low-dose IFN-γ stimulation will detect almost all IFN-γR defects and approximately 70% of STAT1 defects. Furthermore, it has been recently reported that patients with AD STAT1 gain of function mutations, who usually develop chronic mucocutaneous candidiasis, can also develop mycobacterial infectious disease [110].
STAT1 phosphorylation determination is a very informative technique that can be performed in approximately 4 hb, depending on the number of tubes to be processed, with consumable costs of about 40€ per individuala tested.
STAT4 phosphorylation
STAT4 is an essential part of the downstream signaling cascade that occurs after IL-12 stimulation. After IL-12 binding to the IL-12 receptor, IL-12Rβ1 binds TYK2 and IL-12Rβ2 associates with JAK2, which initiates trans-phosphorylation of the receptors, creating docking sites for STAT4. At these sites, STAT4 is phosphorylated at tyrosine 693, dimerizes, and undergoes nuclear translocation where it binds to its target DNA sequences [12]. The STAT4 phosphorylation cytometric assay needs to be performed in stimulated PBMCs cultured with IL-2 and then stimulated with hrIL-12p70; the whole assay takes approximately 1 week [102,111]. Abolished STAT4 phosphorylation in response to rhIL-12p70 has been observed in both IL-12Rβ1-[101,102] and TYK2-deficient patients [27]. Of interest, STAT4 phosphorylation after IFN-α is normal in IL-12Rβ1-deficient patients [102] and impaired in TYK2 deficient patients [27]. However, STAT4 phosphorylation results must be interpreted with caution due to the lack of a proper STAT4 antibody to assess total STAT4 in the cell with flow cytometry. Bi-allelic mutations in STAT4 have not been described to date. For this reason, STAT4 is meant to help only in the diagnosis of other forms of MSMD.
As explained above, a pre-stimulation step is needed for the detection of phosphorylated STAT4 in response to IL-12p70. For this reason, the technique takes 7 d, with a hands-on time of approximately 90 min on day 1 for PBMC isolation and pre-stimulation, 15 min on day 4 for change of medium, and approximately 4 h for stimulation, staining and acquisition in the flow cytometerb, with a cost of approximately 30€ per individuala.
Detection of anti-IFN-γ autoantibodies
Another form of MSMD-like susceptibility to mycobacteria is due to the presence of neutralizing anti-IFN-γ autoantibodies in the blood of affected patients. Although it is not strictly an MSMD-diagnosis technique, we have included it in this review because this condition is a phenocopy of MSMD and should be included in the differential diagnosis of MSMD, especially, but not exclusively, in adults of Asian descent. The most direct approach for detecting IFN-γ autoantibodies is by using an ELISA system and by observing IFN-γ level recovery after the addition of exogenous IFN-γ to patient serum. In both situations, the level of autoantibodies can be titrated by performing an ELISA against anti-IFN-γ-antibodies with different serum dilutions or by increasing the concentration of exogenous IFN-γ in the recovery strategy [78–80,112–114]. In addition, it has been shown recently that undetectable or very low IFN-γ production in the QuantiFERON-TB Gold In-tube assay (Quiagen, Hilden, Germany) is a warning sign for the presence of anti-IFN-γ antibodies[81]. If performed with ELISA, the costa and the timeb needed to perform the test may be similar to those required for the detection of baseline IFN-γ levels in plasma.
Particular considerations in some MSMD
Some genetic defects of MSMD present with characteristic immunological features. For example, in partial AD IRF8 deficiency, there is a loss of CD11c+CD1c+ blood myeloid dendritic cells [26]. MSMD patients with CYBB deficiencies present an abolished respiratory burst in monocyte-derived macrophages in response to purified protein derivative (PPD) or BCG, and in EBV-B cells. However, this oxidative burst defect cannot be detected in a routine dihydrorhodamine test, since monocytes, neutrophils, and monocyte-derived dendritic cells have normal responses [37,115]. Although these are not common tests for MSMD diagnosis, it is important to have these special features in mind in suggestive patients.
Interpretation of results
Cytokine production is the gold standard in the diagnostic pursuit of an inborn error of IFN-γ underlying MSMD. Complete defects often lead to abolished production of, or response to, IFN-γ (Tables 2 and 3). However, although it has not been possible to establish broad cut-off values, it may be possible to have in-house healthy-control range values. In the case of blood samples that have to be shipped, samples from a healthy control are required. As there is great variability in this control group, the lower 10th percentile of the control cohort may define a weak response. Cytokine production data is robust for complete deficiencies but may show limitations in partial defects.
Cytometric determination of receptor presence can be a fast, easy tool to detect complete forms of IFN-γR1, IFN-γR2, and IL-12Rβ1 deficiencies, as their absence confirms the defect. However, the presence of these receptors does not exclude an underlying defect. Functionally, normal phosphorylation of STAT1 in response to IFN-γ rules out complete defects of IFNGR1 and IFNGR2, and virtually all partial defects. Partial IFNGR1 and IFNGR2 may present STAT1-phosphorylation but only at high concentrations of hrIFN-γ. Abolished STAT1 phosphorylation in response to both hrIFN-γ and hrIFN-α is a sign of AR STAT1 defect. If phosphorylation is abolished only after hrIFN-γ stimulation, IFN-γR deficiency must be suspected; in contrast, if it is abolished or impaired after hrIFN-α stimulation, the TYK2 gene may be studied. Abolished STAT4 phosphorylation after IL-12p70 stimulation suggests a defect in IL-12p70 receptor or in TYK2 (Figure 3). The presence or absence of IL-12Rβ1 and STAT1 phosphorylation after hrIFN-α will help to differentiate between these two defects.
Figure 3. IFN-α and IFN-γ signaling.
GAS: γ-interferon activated site; ISRE: interferon-sensitive response element.
Genetic approaches
For a full diagnosis and genetic counseling, genetic studies are needed. Sanger sequencing is a good option if functional tests have identified specific candidate genes. Otherwise, next generation sequencing (NGS) will be less time-consuming and may cost less [116]. For healthcare practice, gene panels with known genes are the option with the best cost-efficiency ratio. However, a great proportion of patients with clinical signs suggestive of MSMD do not show mutations in the known disease-causing genes [2]. In such cases, whole exome sequencing (WES) or whole genome sequencing (WGS) may be required. WGS may reveal mutations in non-coding regulatory regions that would be undetectable by WES, but WGS is more expensive and difficult to interpret than WES. It is important to emphasize that new mutations require further functional confirmation. Different strategies are proposed in order to study the deleterious effects of specific mutations [110]. Recommended guidelines for considering single-patient mutations to be disease-causing have been recently published [117]. An increasingly-used approach for the evaluation of PID (including MSMD) is to start with NGS either with a gene panel or with WES/WGS and then to perform functional tests to confirm the mutations found. Genetic filiation of patients (achievement of a genetic diagnosis) is of utmost importance, as it will condition treatment of the current or future infection and/or prophylaxis. For WES, the cost would be around 500€ but it depends on the coverage. However, analysis of WES studies requires specialized staff. Prices for genetic studies, especially for NGS, are changing rapidly with the development of new technologies and the expansion of their use.
Other useful tests in research
Intracellular detection of IFN-γ producing T-cell blasts can be measured after activation with rhIL-12 in T-cell blasts cultured with PHA; PMA/ionomycin can be used as a positive control for the assay. T-cell blasts are fixed and permeabilized for subsequent intracellular staining with anti-human monoclonal IFN-γ or isotype-matched negative control. Some patients with defects in IFN-γ production (such as IL-12Rβ1 deficiency) may present with normal or only slightly diminished values so that its diagnostic value is limited, particularly in the absence of the analysis of IFN-γ production with PHA or BCG in culture supernatants.
Because samples of primary cells from patients are not infinitely available, some tests have been adapted to the use of patient-derived cell lines. EBV-B cells, herpes virus saimiri-transformed T cells (T-saimiri cells), and immortalized SV-40 fibroblasts are the most common cell lines used [27,30,31,33,107,109,118]. Additional commonly-used cells and techniques are summarized in Table 4. This group of tests requires a laboratory with experience in the field of MSMD and is usually performed in a research laboratory.
Table 4.
Other techniques described to diagnose or confirm different genetic entities.
| Gene | Inheritance | EBV-B cells | Fibroblast (SV40) | Other tests |
|---|---|---|---|---|
| IFNGR1 | AR complete | Abolished GAF DNA binding in response to IFN-γ (EMSA); Abolished IFN-γ binding to IFN-γ receptors (with radiolabeled IFN-γ) | Abolished HLA-II expression response to IFN-γ | No increase of TNF-α secretion after LPS + IFN-γ stimulation in comparison with LPS alone (PBMCs) |
| AR complete | Abolished GAF DNA binding in response to IFN-γ; Abolished IFN-γ binding to IFN-γ receptors | Abolished HLA-II expression response to IFN-γ | Lack of CD64 upregulating capacity after IFN-γ stimulation (monocytes). Abolished STAT1 DNA binding in primary cells | |
| AD | Impaired but not abolished GAF DNA binding in response to IFN-γ, normal IFN-γ binding; Normal levels of IFNGR1 mRNA, increased IFN-γR1 expression | Impaired HLA-II expression in response to IFN-γ at low doses, almost normal at high doses | Impaired but not abolished CD64 upregulation after low/medium IFN-γ stimulation (monocytes); Low STAT1 binding to DNA (PMN); STAT1p after IFN-γ stimulation can be detected. Low LPS+IFN-γ/LPS ratio of TNF-α secretion (PBMCs). Impaired but not abolished STAT1 migration to the nucleus in primary cells. | |
| AR partial | Impaired but not abolished GAF DNA binding in response to IFN-γ, impaired IFN-γ binding; STAT1 translocation after IFN-γ stimulation is impaired but not abolished | NP | CD64 upregulation in response to IFN-γ detectable but low; TNF-α secretion after LPS+IFN-γ impaired but not abolished. Impaired but not abolished STAT1 migration to the nucleus in primary cells. | |
| IFNGR2 | AR complete | Abolished GAF DNA binding in response to IFN-γ | NP | Abolished upregulation of HLA-DR after IFN-γ treatment |
| AR complete | Abolished GAF DNA binding in response to IFN-γ, abolished STAT1p in response to IFN-γ | Abolished GAF DNA binding and no STAT1p in response to IFN-γ | Normal production of TNF-α after PHA/LPS stimulation, no increase with IFN-γ (PBMCs); Abolished upregulation of HLA-DR after IFN-γ stimulation | |
| AR | IFN-γ R2 detection, impaired but not abolished GAF DNA binding and GAF dependent genes (CXCL9, CXCL10, IRF8) induction in response to IFN-γ; impaired nuclear translocation. Normal IFN-γ R1 expression, normal affinity and number of binding sites for IFN-γ | No induction of HLA-II expression in response to IFN-γ; impaired but not abolished GAF DNA binding; IFN-γ R2 retained in the ER; normal response to IFN-γ after kifunensine treatment | Decreased STAT1p and CD4, HLA, CXCL10 induction after IFN-γ stimulation in diverse cell types with overexpression of the mutants. | |
| AD partial | Impaired STAT1-p; impaired but not abolished GAF DNA binding and GAF dependent genes induction in response to IFN-γ | NP | NP | |
| STAT1 | AD partial | Normal/Impaired STAT1p, impaired GAS (but normal ISRE) and DNA binding in response to IFN-γ | Normal/impaired nuclear translocation | Impaired STAT1 DNA binding in primary cells |
| AD partial | Impaired GAS in response to IFN-γ and IFN-α (normal ISRE in response to IFN-α) and impaired DNA binding. | Impaired nuclear translocation | Severely impaired TNF-α production after IFN-γ stimulation in isolated CD14+ monocytes or PBMCs. Impaired STAT1p in U3A cells in heterozygosis, abolished in homozygosis. Impaired STAT1 DNA binding in primary cells. | |
| IL-12B | AR | No IL-12p40/p70 production after PDBu stimulation, normal TNF-α levels | NP | Lower than normal frequency of CD3+IL-17A+ cells ex vivo, but T cells responded to IL-23 by producing IL-17 |
| IL12Rβ1 | AR complete | Present IL-12Rβ1 expression | NP | NP |
| AR complete | Abolished IL-12Rβ1 expression | NP | IL-17-producing T cells ex vivo, T-cell blasts do not express IL-17 in response to IL-23. Normal IL-12Rβ2 expression. Reduced circulating memory Tfh and memory B cells, lower avidity of tetanus toxoid-specific serum antibodies | |
| IRF8 | AD partial | Normal IRF8 expression with low DNA binding in the IL-12B promoter; | NP | Loss of CD11c+CD1c+ blood myeloid DCs |
| ISG15 | AR complete | Absence of ISG15, normal response to IFN-α | Absence of ISG15 | Impaired but not abolished IFN-γ production by T and NK cells; Normal TNF-α production in response to BCG |
| CYBB | XR partial | Normal O-, H2O2 production after PMA stimulation on neutrophils, monocytes | NP | Abolished respiratory burst in monocyte derived-macrophages in response to PPD, BCG, IFN-γ, normal in monocytes, neutrophils and monocyte derived-dendritic cells |
| NEMO | XR partial | Normal NEMO expression | Normal NEMO expression | Normal NEMO expression in monocytes and T cells. PHA stimulation in T cell/Monocytes co-culture system: low production IFN-γ, IL-12p40, IL-12p70, Lower IL10 production after TNF-α stimulation (WB); |
| TYK2 | AR complete | Normal STAT1p and GAS in response to IFN-γ; diminished cell surface expression of IL-10R2, and IL-12Rβ1, normal IFN-γR1 & IFN-γR2. Abolished IL-23 response (STAT3-p, IFN-γ production), abolished pSTAT3, impaired production of ISG15 in response to IFN-γ; impaired pSTAT3 and EMSA after IL-10 stimulation, normal pSTAT3 and EMSA after IL-6/IL-21/IL-27 stimulation. | Impaired pSTAT1 and EMSA (GAS and ISRE) in response to IFN-γ; impaired STAT3p after IL-10 stimulation, normal STAT3p after IL-6 stimulation | Impaired response to IL-10 after LPS and TNF-α stimulation of PBMCs; normal IL17+ cells in PBMCs after PMA/Iono stimulation; impaired IL17A and F production in naive T cells. In HVS transformed T cells: Abolished STAT4 phosphorylation, GAS and IFN-γ production in response to IL-12p70, abolished pSTAT1 in response to IFN-α |
AR: autosomal recessive; AD: autosomal dominant; XR: X-linked; NP: not published; GAF: γ activated factor, GAS: γ interferon-activated site; ISRE: interferon-sensitive response element; PDBu: phorbol 12,13-dibutyrate; STAT1p: STAT1 phosphorylation.
Defects in IFN-γ response are caused by diverse genetic etiologies. Assessment of the effect of the different mutations in the response to IFN-γ is crucial to determine treatment and patient management; for this reason, techniques other than STAT1 phosphorylation may be needed. For example, expression of activation markers such as HLA-DR and CD64 after stimulation with different hrIFN-γ concentrations may be used to determine response to IFN-γ, both in primary cells and in transformed SV40-fibroblasts and EBV-B cells [45,97,107], as not all defects in STAT1 lead to altered phosphorylation in response to hrIFN-γ. To prove that a mutation in STAT1 with normal phosphorylation is pathogenic, other tests showing defective response to IFN-γ are needed. The electrophoretic mobility shift assay (EMSA) is useful to detect forms of IFN-γR and AD STAT1 LOF deficiencies with the presence of phosphorylation, as it reveals STAT1 translocation and DNA binding. In addition, it can help to identify AD STAT1 gain of function deficiencies [35,93]. In the same line, it is possible to study induction of GAS in response to IFN-γ. Specifically, expression of CXCL9 and CXCL10, among others, or the activation of GAS elements by luciferase detection [30,34,62,93,106,110] can help to determine the effect of specific mutations. These approaches are usually used in research rather than in healthcare practice.
Other advanced research techniques are used for the characterization of new mutations in newly-discovered genes or mutations causing MSMD. These methods for the confirmation of a pathogenic effect of new mutations are beyond the scope of this review, and they have been carefully reviewed elsewhere [117]. Briefly, transfection of different cell lines with wild-type or mutated genes may be useful for the evaluation of a mutated allele in terms of protein expression and function. It is possible to perform expression assays, as disease-causing variations commonly have altered expression. Also, transfection of a wild-type copy of the mutated gene into patient cells (for example, in EBV-B or T-saimiri cells) that restores protein function can reveal a possible loss-of-function mutation [119]. Furthermore, new techniques such as CRISPR/Cas9 open up the possibility of reversing the mutation in patient cells or mutating control cells, especially in the event that no cells are available from the patient, to confirm that the phenotype that is observed in the patient is due to the mutation.
Discussion
Tuberculosis was thought by many to be a hereditary disease until the discovery of the characteristic bacterium by Koch in 1882 [120]. It was not until the middle of the 20th century that infections after BCG vaccination were understood to be related to inborn errors of immunity [16], and only in 1996 did Jouanguy [17] and Newport [18] et al. show for the first time that inheritable single monogenic defects in the IFN-γ circuit conferred susceptibility to mycobacterial infection rather than to a broad range of pathogens.
These findings boosted the concept of atypical PID in which monogenic defects confer selective susceptibility to specific pathogens [121,122]. Twenty years and ten disease-causing genes later, MSMD diagnosis is still a clinical challenge. In the present review, we provide an overview of the different assays available for the study of suspected defects in the IFN-γ circuit that can be performed in diagnostic and research laboratories. Table 5 summarizes their advantages and disadvantages.
Table 5.
Strengths and limitations of the different techniques for MSMD diagnosis.
| Procedure | Test | Strengths | Limitations | MSMD defect detected (published) |
|---|---|---|---|---|
| Culture and cytokine determination | Basal IFN-γ in plasma | Easy and cheap to perform, when established, high levels of IFN-γ are indicative of IFN-γ receptor defect | Differences between kits, normalization of IFN-γ levels after acute phase of the infection | AR IFN-γ receptors defects |
| WB culture with BCG stimulation | Most similar condition to reality | Partial defects can be occult, some MSMD defects have normal results; big variability in healthy controls, use of BCG difficult a lot ISO regulations acceptance Needs to be performed in fresh blood (max. 48h after extraction) |
AR IFN-γ receptors, IL-12Rβ1, IL-12p40, ISG15/TYK2 defects | |
| PBMCs culture with BCG stimulation | Can be performed in cryopreserved cells | Partial defects can be occult, some MSMD defects have normal results; big variability in healthy controls, use of BCG difficult a lot ISO regulations acceptance | ||
| WB culture with mitogen stimulation | NO use of BCG | Mycobacterial-specific immunity is not tested Needs to be performed in fresh blood (max. 48h after extraction) |
AR IFN-γR1, IL-12Rβ1, IL-12p40 defects NEMO deficiency performed in PBMCs |
|
| PBMCs culture with mitogen stimulation | NO use of BCG. Can be performed in cryopreserved cells |
Mycobacterial-specific immunity is not tested | ||
| Cytometry (in primary cells) | IFN-γ binding | Useful to detect defects in IFN-γR1 when it is expressed in the membrane | There are some defects that bind IFN-γ but have no functional IFN-γR1 | IFN-γR1 defects |
| IFN-γR1 determination | Easy to perform, rapid diagnosis of some forms of IFN-γR1deficiency. | Some forms of IFN-γR1 deficiency present a non-functional form of IFN-γR1 in the membrane, so presence of the receptor does not exclude the defect. | IFN-γR1 defects | |
| IFN-γR2 determination | Easy to perform, rapid diagnosis of some forms of IFNGR2 deficiency | Some forms of IFNGR2 present a non-functional form of IFN-γR2 in the membrane, so presence of the receptor does not exclude the defect. | IFN-γR2 defects | |
| STAT1 phosphorylation determination | Rapid test that evaluates the function of both IFN-γR1/IFN-γR2 and can detect some forms of STAT1 deficiency | Partial forms of IFN-γ R1/2 defects can be occult if only high levels of IFN-γ are used, some STAT1 defects present normal STAT1 phosphorylation after IFN-γ stimulation | IFN-γR1/2 defects, STAT1, TYK2 (help) | |
| IL-12Rβ1 determination | Rapid/easy test; all but one mutation in IL12Rβ1 present a lack of IL-12Rβ1 in the membrane. | There is a form of IL-12Rβ1 defect that present IL-12Rβ1 in the membrane of act lymphocytes, so it’s present does not exclude the defect | IL-12Rβ1 defects | |
| STAT4 determination | Evaluates both IL-12Rβ1 and Tyk2 function. | Long technique; no STAT4 antibody to detect total STAT4. | IL-12Rβ1, TYK2 defects | |
| Genetics | Sanger | When the number of candidate genes is small, it can be easy, rapid and cheap. Easy to analyze and interpret | Slow and expensive if there is not a clear orientation of the defect. Not useful for the discovery of new genes. | All known defects |
| Next generation sequencing | Indicated when there is no clear candidate gene, study of all known defects at once | Expensive, difficult to analyze and interpret results due to the huge amount of generated data. Especially with whole genome sequencing, gene panels are more affordable and easy to interpret. | All known defects, discovery of new genes or new manifestations of known genes | |
| Useful for new genes discovery; detection of mutations in non-coding/regulatory regions with whole genome sequencing (not whole exome sequencing) |
PBMC: peripheral mononuclear cells; WB: whole blood.
Some issues in MSMD diagnosis need to be resolved. First, there is a need for awareness about MSMD, so that physicians taking care of children or adults can suspect this disorder. Knowledge of the specific warning signs is of utmost importance, as well as knowledge of other conditions that can lead to susceptibility to mycobacterial diseases and that must be included in the differential diagnosis: patients, especially children, with BCG-itis or BCG-osis, EM infections, or severe tuberculosis, alone or in combination with other intracellular infections, are to be suspected of having MSMD. Global frequency of MSMD has been estimated to be at least 1/50,000, although it was previously thought to be rare.
Second, there is a need to facilitate the diagnosis of MSMD, once suspected. Indeed, the detection of the genetic defect is necessary to offer the patient the best treatment options and genetic counseling, and therefore to decrease mortality. This is exemplified by complete deficiency of IFN-γR where the only curative treatment attempted is HSCT; most other forms of MSMD will benefit from prolonged antibiotics to which exogenous hrIFN-γ therapy can be added - even partial defects of IFN-γR respond to exogenous hrIFN-γ therapy [2,29,123]. When a member of a family is diagnosed with MSMD, BCG vaccination in family members should be avoided until a genetic defect has been ruled out. It is also important to consider that some MSMD etiologies have incomplete penetrance, meaning that not all the individuals presenting with the mutation will present the clinical phenotype [2,20,22]. For example, in IL-12Rβ1 deficiency, it is estimated that 21% of the individuals with MSMD genotype do not show the phenotype at 20 years [2,20]. Genetic counseling in these patients is thus challenging.
Functional tests for MSMD diagnosis are also challenging: in this review we have described a broad array of available tests; however, some of these techniques are limited by the timing and the requirement of qualified staff, making the full diagnosis of MSMD usually only possible in specialized immunology laboratories. Genetic approaches are gaining ground and could overcome these limitations. Nevertheless, genetic results usually need a functional confirmation of the identified mutation.
Conclusions
We describe the currently-available techniques to study patients with suspected MSMD defects in diagnostic and research laboratories. MSMD should be considered in patients with significant infection (severe, disseminated, or recurrent) after BCG vaccination and infection by mycobacteria, particularly EM, especially in combination with Salmonella, Candida or virus. When suspected, acquired causes of immunodeficiency, T cell defects and chronic granulomatous disease first need to be ruled out. Then, MSMD-specific evaluation should be started. The tests performed, and their order, may depend on laboratory facilities, technical staff, and clinical orientation. Genetic studies may be performed after functional studies have suggested a specific defect or may be performed upfront and be followed by functional confirmation. Given the number of different genetic etiologies causing MSMD, NGS technologies may be especially suitable to help in the identification of new disease-causing genes, because almost 60% of patients with suspected MSMD today have no identified genetic cause.
Acknowledgments
Funding All phases of this work were supported by the Plan Nacional de I+D+I and co-financed by the ISCIII – Subdirección General de Evaluación y Fomento de la Investigación Sanitaria – and the Fondo Europeo de Desarrollo Regional (FEDER) under the grants PI12/01990 and PI15/01094 to LA and PI13/00676 to MJ, PI13/01456 and PI16/00759 to CRG. The Laboratory of Human Genetics of Infectious Diseases is supported by grants from the National Institute of Allergy and Infectious Diseases (under grants 5R01AI089970 and 5R37AI095983), the National Center for Research Resources and the National Center for Advancing Sciences of the National Institutes of Health (grant numbers 8UL1TR000043 for J.L.C.), The Rockefeller University, the St. Giles Foundation, Institut National de la Santé et de la Recherche Médicale (INSERM), Paris Descartes University, the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID), and the French National Research Agency (ANR) under the “Investments for the Future” program (grant number ANR-10-IAHU-01), ANR-GENMSMD (for J.B.), SRC2017 (support of Clinical research, Fondation du Souffle et du Fonds de Recherche en Santé Respiratoire, for J.B.). RMB was supported by the European Molecular Biology Organization (EMBO). COQ was supported by ANR-14-CE15-006-01.
Abbreviations
- AD
autosomal dominant
- APC
antigen presenting-cell
- AR
autosomal recessive
- BCG
bacille Calmette-Guérin
- EBV-B cells
Epstein-Barr virus-transformed B cells
- ELISA
enzyme-linked immunosorbent assay
- EM
environmental mycobacteria
- EMSA
electrophoretic mobility shift assay
- GAF
γ-activated factor
- GAS
γ interferon-activated site
- hrIFN-γ
human recombinant IFN-γ
- HSCT
hematopoietic stem cell transplantation
- IFN
interferon
- IFN-γR
IFN-γ receptor
- IL
interleukin
- IRF
interferon regulatory factor
- ISG
interferon-stimulated gene
- IUIS
International Union of Immunology Societies
- JAK2
Janus-associated kinase 2
- LOF
loss of function
- LPS
lipopolysaccharide
- MFI
mean fluorescence intensity
- MSMD
Mendelian susceptibility to mycobacterial disease
- NEMO
nuclear factor-kappa B essential modulator
- NGS
next generation sequencing
- NK
natural killer
- PBMCs
peripheral blood mononuclear cells
- PHA
phytohemagglutinin
- PID
primary immunodeficiency
- PPD
purified protein derivative
- RORC
RAR related orphan receptor C
- ROS
reactive oxygen species
- STAT
signal transducer and activator of transcription
- TB
tuberculosis
- Th
T helper
- TLR
Toll-like receptor
- TNF
tumor necrosis factor
- T-saimiri cells
herpes virus saimiri-transformed T cells
- TYK2
tyrosine kinase 2
- WB
whole blood
- WES
whole exome sequencing
- WGS
whole genome sequencing
- XR-EDA-ID
anhidrotic ectodermal dysplasia with immunodeficiency
Footnotes
Approximate costs of the different techniques are calculated for each sample processed to which the cost of healthy (normal) control/s sample/s must be added; only reagent-derived costs are included. It is important to take into account that prices are approximate and that they may vary depending on the supplier/country or type of kit used. Moreover, when evaluating the costs of implementing these techniques, other costs need to be considered, such as sample preservation, including frozen PBMCs and plasma, DNA extraction and preservation, and general materials such as phosphate buffer saline, plastic materials and culture media. Because all laboratories may not have a flow cytometer, they may need to use flow cytometry facilities, which likely charge the users for the use of the cytometers and for technical assistance. This is an important variable to consider in all flow cytometry techniques as it may significantly increase the final cost.
Hands-on time is an estimation of the time needed to perform the technique; however, the response time (turn-around time) can vary depending on different factors including i) the need to batch patient samples, ii) the number of patient samples, and iii) the time required for analysis (from receipt of specimen to reporting the result).
Disclosure statements:
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- 1.Casanova JLL, Abel L. Genetic dissection of immunity to mycobacteria: the human model. Annu Rev Immunol. 2002;20:581–620. doi: 10.1146/annurev.immunol.20.081501.125851. [DOI] [PubMed] [Google Scholar]
- 2.Bustamante J, Boisson-Dupuis SSS, Abel L, et al. Mendelian susceptibility to mycobacterial disease: Genetic, immunological, and clinical features of inborn errors of IFNgamma immunity. Semin Immunol. 2014;26:454–470. doi: 10.1016/j.smim.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Picard C, Bobby Gaspar H, Al-Herz W, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J Clin Immunol. 2018;38:96–128. doi: 10.1007/s10875-017-0464-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mishra A, Akhtar S, Jagannath C, et al. Pattern recognition receptors and coordinated cellular pathways involved in tuberculosis immunopathogenesis: Emerging concepts and perspectives. Mol Immunol. 2017;87:240–248. doi: 10.1016/j.molimm.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Alsina L, Israelsson E, Altman MC, et al. A narrow repertoire of transcriptional modules responsive to pyogenic bacteria is impaired in patients carrying loss-of-function mutations in MYD88 or IRAK4. Nat Immunol. 2014;15:1134–1142. doi: 10.1038/ni.3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Cunha J, Ramanujam S, Wagner RJ, et al. In vitro and in vivo secretion of human ISG15, an IFN-induced immunomodulatory cytokine. J Immunol. 1996;157:4100–4108. [PubMed] [Google Scholar]
- 7.Bogunovic D, Byun M, Durfee LA, et al. Mycobacterial Disease and Impaired IFN- Immunity in Humans with Inherited ISG15 Deficiency. Science. 2012;337:1684–1688. doi: 10.1126/science.1224026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cottle LE. Mendelian susceptibility to mycobacterial disease. Clin Genet. 2011;79:17–22. doi: 10.1111/j.1399-0004.2010.01510.x. [DOI] [PubMed] [Google Scholar]
- 9.Ramirez-Alejo N, Santos-Argumedo L. Innate Defects of the IL-12/IFN-γ Axis in Susceptibility to Infections by Mycobacteria and Salmonella. J Interf Cytokine Res. 2014;34:307–317. doi: 10.1089/jir.2013.0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Torrado E, Cooper AM. Cytokines in the Balance of Protection and Pathology During Mycobacterial Infections. In: Divangahi M, editor. The New Paradigm of Immunity to Tuberculosis. Springer New York; New York, NY: 2013. pp. 121–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zundler S, Neurath MF. Interleukin-12: Functional activities and implications for disease. Cytokine Growth Factor Rev. 2015;26:559–568. doi: 10.1016/j.cytogfr.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Watford WT, Hissong BD, Bream JH, et al. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol Rev. 2004;202:139–156. doi: 10.1111/j.0105-2896.2004.00211.x. [DOI] [PubMed] [Google Scholar]
- 13.Bogunovic D, Boisson-Dupuis S, Casanova J-L. ISG15: leading a double life as a secreted molecule. Exp Mol Med. 2013;45:e18–e18. doi: 10.1038/emm.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Majoros A, Platanitis E, Kernbauer-Hölzl E, et al. Canonical and Non-Canonical Aspects of JAK–STAT Signaling: Lessons from Interferons for Cytokine Responses. Front Immunol. 2017;8:29. doi: 10.3389/fimmu.2017.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stark GR. How cells respond to interferons revisited: From early history to current complexity. Cytokine Growth Factor Rev. 2007;18:419–423. doi: 10.1016/j.cytogfr.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mimouni J. Our experiences in three years of BCG vaccination at the center of the O.P.H.S. at Constantine; study of observed cases (25 cases of complications from BCG vaccination) Alger Medicale. 1951;55:1138–1147. [PubMed] [Google Scholar]
- 17.Jouanguy E, Altare F, Lamhamedi S, et al. Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette-Guerin infection. N Engl J Med. 1996;335:1956–1961. doi: 10.1056/NEJM199612263352604. [DOI] [PubMed] [Google Scholar]
- 18.Newport MJ, Huxley CM, Huston S, et al. A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N Engl J Med. 1996;335:1941–1949. doi: 10.1056/NEJM199612263352602. [DOI] [PubMed] [Google Scholar]
- 19.Feinberg J, Fieschi C, Doffinger R, et al. Bacillus Calmette Guérin triggers the IL-12/IFN-γ axis by an IRAK-4- and NEMO-dependent, non-cognate interaction between monocytes, NK, and T lymphocytes. Eur J Immunol. 2004;34:3276–3284. doi: 10.1002/eji.200425221. [DOI] [PubMed] [Google Scholar]
- 20.van de Vosse E, Haverkamp MH, Ramirez-Alejo N, et al. IL-12Rβ1 Deficiency: Mutation Update and Description of the IL12RB1 Variation Database. Hum Mutat. 2013;34:1329–1339. doi: 10.1002/humu.22380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Beaucoudrey L, Samarina A, Bustamante J, et al. Revisiting human IL-12Rβ1 deficiency: a survey of 141 patients from 30 countries. Medicine (Baltimore) 2010;89:381–402. doi: 10.1097/MD.0b013e3181fdd832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Altare F, Lammas D, Revy P, et al. Rapid Publication Inherited Interleukin 12 Deficiency in a Child with Bacille Calmette-Guérin and Salmonella enteritidis Disseminated Infection. J Clin Invest. 1998;102:2035–2040. doi: 10.1172/JCI4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prando C, Samarina A, Bustamante J, et al. Inherited IL-12p40 Deficiency. Medicine (Baltimore) 2013;92:109–122. doi: 10.1097/MD.0b013e31828a01f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, Bogunovic D, Payelle-Brogard B, et al. Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature. 2015;517:89–93. doi: 10.1038/nature13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bustamante J, Picard C, Boisson-Dupuis S, et al. Genetic lessons learned from X-linked Mendelian susceptibility to mycobacterial diseases. Ann N Y Acad Sci. 2011;1246:92–101. doi: 10.1111/j.1749-6632.2011.06273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hambleton S, Salem S, Bustamante J, et al. IRF8 Mutations and Human Dendritic-Cell Immunodeficiency. N Engl J Med. 2011;365:127–138. doi: 10.1056/NEJMoa1100066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kreins AY, Ciancanelli MJ, Okada S, et al. Human TYK2 deficiency: Mycobacterial and viral infections without hyper-IgE syndrome. J Exp Med. 2015;212:1641–1662. doi: 10.1084/jem.20140280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jouanguy E, Dupuis S, Pallier A, et al. In a novel form of IFN-γ receptor 1 deficiency, cell surface receptors fail to bind IFN-γ. J Clin Invest. 2000;105:1429–1436. doi: 10.1172/JCI9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dorman SE, Picard C, Lammas D, et al. Clinical features of dominant and recessive interferon γ receptor 1 deficiencies. Lancet. 2004;364:2113–2121. doi: 10.1016/S0140-6736(04)17552-1. [DOI] [PubMed] [Google Scholar]
- 30.Sologuren I, Boisson-Dupuis S, Pestano J, et al. Partial recessive IFN-γR1 deficiency: genetic, immunological and clinical features of 14 patients from 11 kindreds. Hum Mol Genet. 2011;20:1509–1523. doi: 10.1093/hmg/ddr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jouanguy E, Lamhamedi-Cherradi S, Lammas D, et al. A human IFNGR1 small deletion hotspot associated with dominant susceptibility to mycobacterial infection. Nat Genet. 1999;21:370–378. doi: 10.1038/7701. [DOI] [PubMed] [Google Scholar]
- 32.Rosenzweig SD, Schwartz OM, Brown MR, et al. Characterization of a Dipeptide Motif Regulating IFN- Receptor 2 Plasma Membrane Accumulation and IFN- Responsiveness. J Immunol. 2004;173:3991–3999. doi: 10.4049/jimmunol.173.6.3991. [DOI] [PubMed] [Google Scholar]
- 33.Moncada-Velez M, Martinez-Barricarte R, Bogunovic D, et al. Partial IFN- R2 deficiency is due to protein misfolding and can be rescued by inhibitors of glycosylation. Blood. 2013;122:2390–2401. doi: 10.1182/blood-2013-01-480814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chapgier A, Boisson-Dupuis S, Jouanguy E, et al. Novel STAT1 Alleles in Otherwise Healthy Patients with Mycobacterial Disease. PLoS Genet. 2006;2:e131. doi: 10.1371/journal.pgen.0020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dupuis S. Impairment of Mycobacterial But Not Viral Immunity by a Germline Human STAT1 Mutation. Science. 2001;293:300–303. doi: 10.1126/science.1061154. [DOI] [PubMed] [Google Scholar]
- 36.Hirata O, Okada S, Tsumura M, et al. Heterozygosity for the Y701C STAT1 mutation in a multiplex kindred with multifocal osteomyelitis. Haematologica. 2013;98:1641–1649. doi: 10.3324/haematol.2013.083741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bustamante J, Arias Aa, Vogt G, et al. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol. 2011;12:213–221. doi: 10.1038/ni.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bustamante J, Picard C, Fieschi C, et al. A novel X-linked recessive form of Mendelian susceptibility to mycobaterial disease. J Med Genet. 2006;44:e65–e65. doi: 10.1136/jmg.2006.043406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Filipe-Santos O, Bustamante J, Chapgier A, et al. Inborn errors of IL-12/23- and IFN-γ-mediated immunity: molecular, cellular, and clinical features. Semin Immunol. 2006;18:347–361. doi: 10.1016/j.smim.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 40.Olbrich P, Martínez-Saavedra MT, Perez-Hurtado JM, et al. Diagnostic and therapeutic challenges in a child with complete Interferon-γ Receptor 1 deficiency. Pediatr Blood Cancer. 2015;62:2036–2039. doi: 10.1002/pbc.25625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zwerling A, Behr MA, Verma A, et al. The BCG World Atlas: A Database of Global BCG Vaccination Policies and Practices. PLoS Med. 2011;8:e1001012. doi: 10.1371/journal.pmed.1001012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boisson-Dupuis S, Bustamante J, El-Baghdadi J, et al. Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. Immunol Rev. 2015;264:103–120. doi: 10.1111/imr.12272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boisson-Dupuis S, El Baghdadi J, Parvaneh N, et al. IL-12Rbeta1 deficiency in two of fifty children with severe tuberculosis from Iran, Morocco, and Turkey. PLoS One. 2011;6:e18524. doi: 10.1371/journal.pone.0018524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caragol I, Raspall M, Fieschi C, et al. Clinical Tuberculosis in 2 of 3 Siblings with Interleukin-12 Receptor 1 Deficiency. Clin Infect Dis. 2003;37:302–306. doi: 10.1086/375587. [DOI] [PubMed] [Google Scholar]
- 45.Jouanguy E, Lamhamedi-Cherradi S, Altare F, et al. Partial interferon-gamma receptor 1 deficiency in a child with tuberculoid bacillus Calmette-Guérin infection and a sibling with clinical tuberculosis. J Clin Invest. 1997;100:2658–2664. doi: 10.1172/JCI119810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parvaneh N, Barlogis V, Alborzi A, et al. Visceral leishmaniasis in two patients with IL-12p40 and IL-12Rβ1 deficiencies. Pediatr Blood Cancer. 2017;64:e26362. doi: 10.1002/pbc.26362. [DOI] [PubMed] [Google Scholar]
- 47.Obinata K, Lee T, Niizuma T, et al. Two cases of partial dominant interferon-γ receptor 1 deficiency that presented with different clinical courses of bacille Calmette-Guérin multiple osteomyelitis. J Infect Chemother. 2013;19:757–760. doi: 10.1007/s10156-012-0493-5. [DOI] [PubMed] [Google Scholar]
- 48.Villella A, Picard C, Jouanguy E, et al. Recurrent Mycobacterium avium osteomyelitis associated with a novel dominant interferon gamma receptor mutation. Pediatrics. 2001;107:E47. doi: 10.1542/peds.107.4.e47. [DOI] [PubMed] [Google Scholar]
- 49.Jirapongsananuruk O, Luangwedchakarn V, Niemela JE, et al. Cryptococcal osteomyelitis in a child with a novel compound mutation of the IL12RB1 gene. Asian Pacific J allergy Immunol. 2012;30:79–82. [PubMed] [Google Scholar]
- 50.Boudjemaa S, Dainese L, Héritier S, et al. Disseminated BCG osteomyelitis related to STAT 1 gene deficiency mimicking a metastatic neuroblastoma. Pediatr Dev Pathol. 2017;3:255–261. doi: 10.1177/1093526616686255. [DOI] [PubMed] [Google Scholar]
- 51.Shamriz O, Engelhard D, Rajs AP, et al. Mycobacterium szulgai Chronic Multifocal Osteomyelitis in an Adolescent With Inherited STAT1 Deficiency. Pediatr Infect Dis J. 2013;32:1345–1347. doi: 10.1097/01.inf.0000437067.43859.4c. [DOI] [PubMed] [Google Scholar]
- 52.Sasaki Y, Nomura A, Kusuhara K, et al. Genetic basis of patients with bacille Calmette-Guerin osteomyelitis in Japan: identification of dominant partial interferon-gamma receptor 1 deficiency as a predominant type. J Infect Dis. 2002;185:706–709. doi: 10.1086/339011. [DOI] [PubMed] [Google Scholar]
- 53.Arend SM, Janssen R, Gosen JJ, et al. Multifocal osteomyelitis caused by nontuberculous mycobacteria in patients with a genetic defect of the interferon-gamma receptor. Neth J Med. 2001;59:140–151. doi: 10.1016/s0300-2977(01)00152-8. [DOI] [PubMed] [Google Scholar]
- 54.Conti F, Lugo-Reyes SO, Blancas Galicia L, et al. Mycobacterial disease in patients with chronic granulomatous disease: A retrospective analysis of 71 cases. J Allergy Clin Immunol. 2016;138:241–248.e3. doi: 10.1016/j.jaci.2015.11.041. [DOI] [PubMed] [Google Scholar]
- 55.Picard C, Casanova JL. Inherited disorders of cytokines. Curr Opin Pediatr. 2004;16:648–658. doi: 10.1097/01.mop.0000145919.92477.5f. [DOI] [PubMed] [Google Scholar]
- 56.Orange JS, Levy O, Brodeur SR, et al. Human nuclear factor kappa B essential modulator mutation can result in immunodeficiency without ectodermal dysplasia. J Allergy Clin Immunol. 2004;114:650–656. doi: 10.1016/j.jaci.2004.06.052. [DOI] [PubMed] [Google Scholar]
- 57.Orange JS, Brodeur SR, Jain A, et al. Deficient natural killer cell cytotoxicity in patients with IKK-γ/NEMO mutations. J Clin Invest. 2002;109:1501–1509. doi: 10.1172/JCI14858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spinner MA, Sanchez LA, Hsu AP, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123:809–821. doi: 10.1182/blood-2013-07-515528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hsu AP, Sampaio EP, Khan J, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118:2653–2655. doi: 10.1182/blood-2011-05-356352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mace EM, Hsu AP, Monaco-Shawver L, et al. Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56bright subset. Blood. 2013;121:2669–2677. doi: 10.1182/blood-2012-09-453969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vairo D, Tassone L, Tabellini G, et al. Severe impairment of IFN-gamma and IFN-alpha responses in cells of a patient with a novel STAT1 splicing mutation. Blood. 2011;118:1806–1817. doi: 10.1182/blood-2011-01-330571. [DOI] [PubMed] [Google Scholar]
- 62.Kong X-F, Ciancanelli M, Al-Hajjar S, et al. A novel form of human STAT1 deficiency impairing early but not late responses to interferons. Blood. 2010;116:5895–5906. doi: 10.1182/blood-2010-04-280586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chapgier A, Kong XF, Boisson-Dupuis S, et al. A partial form of recessive STAT1 deficiency in humans. J Clin Invest. 2009;119:1502–1514. doi: 10.1172/JCI37083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chapgier A, Wynn RF, Jouanguy E, et al. Human complete Stat-1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo. J Immunol. 2006;176:5078–5083. doi: 10.4049/jimmunol.176.8.5078. [DOI] [PubMed] [Google Scholar]
- 65.Dupuis S, Jouanguy E, Al-Hajjar S, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003;33:388–391. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- 66.Moens L, Frans G, Bosch B, et al. Successful hematopoietic stem cell transplantation for myelofibrosis in an adult with warts-hypogammaglobulinemia-immunodeficiency-myelokathexis syndrome. J Allergy Clin Immunol. 2016;138:1485–1489.e2. doi: 10.1016/j.jaci.2016.04.057. [DOI] [PubMed] [Google Scholar]
- 67.Hambleton S, Goodbourn S, Young DF, et al. STAT2 deficiency and susceptibility to viral illness in humans. Proc Natl Acad Sci. 2013;110:3053–3058. doi: 10.1073/pnas.1220098110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eletto D, Burns SO, Angulo I, et al. Biallelic JAK1 mutations in immunodeficient patient with mycobacterial infection. Nat Commun. 2016;7:13992. doi: 10.1038/ncomms13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Okada S, Markle JG, Deenick EK, et al. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science. 2015;349:606–613. doi: 10.1126/science.aaa4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brode SK, Jamieson FB, Ng R, et al. Increased risk of mycobacterial infections associated with anti-rheumatic medications. Thorax. 2015;70:677–682. doi: 10.1136/thoraxjnl-2014-206470. [DOI] [PubMed] [Google Scholar]
- 71.Lake MA, Ambrose LR, Lipman MCI, et al. “Why me, why now?” Using clinical immunology and epidemiology to explain who gets nontuberculous mycobacterial infection. BMC Med. 2016;14:54. doi: 10.1186/s12916-016-0606-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cisneros JR, Murray KM. Corticosteroids in Tuberculosis. Ann Pharmacother. 1996;30:1298–1303. doi: 10.1177/106002809603001115. [DOI] [PubMed] [Google Scholar]
- 73.Orme IM, Ordway DJ. Host Response to Nontuberculous Mycobacterial Infections of Current Clinical Importance. Infect Immun. 2014;82:3516–3522. doi: 10.1128/IAI.01606-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thaker H, Neilly IJ, Saunders PG, et al. Remember mycobacterial disease in hairy cell leukaemia (HCL) J Infect. 2001;42:213–214. doi: 10.1053/jinf.2001.0816. [DOI] [PubMed] [Google Scholar]
- 75.Kraut EH. Clinical manifestations and infectious complications of hairy-cell leukaemia. Best Pract Res Clin Haematol. 2003;16:33–40. doi: 10.1016/s1521-6926(02)00085-3. [DOI] [PubMed] [Google Scholar]
- 76.Netea MG, Hoitink O, Kullberg BJ, et al. Defective interferon-gamma production in patients with hairy cell leukaemia. Neth J Med. 2008;66:340–343. [PubMed] [Google Scholar]
- 77.Arlotta A, Cefalo MG, Maurizi P, et al. Critical pulmonary infection due to nontuberculous mycobacterium in pediatric leukemia: report of a difficult diagnosis and review of pediatric series. J Pediatr Hematol Oncol. 2014;36:66–70. doi: 10.1097/MPH.0b013e3182841737. [DOI] [PubMed] [Google Scholar]
- 78.Kampmann B, Hemingway C, Stephens A, et al. Acquired predisposition to mycobacterial disease due to autoantibodies to IFN-γ. J Clin Invest. 2005;115:2480–2488. doi: 10.1172/JCI19316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee W-I, Huang J-L, Wu T-S, et al. Patients with inhibitory and neutralizing auto-antibodies to interferon-γ resemble the sporadic adult-onset phenotype of Mendelian Susceptibility to Mycobacterial Disease (MSMD) lacking Bacille Calmette–Guerin (BCG)-induced diseases. Immunobiology. 2013;218:762–771. doi: 10.1016/j.imbio.2012.08.281. [DOI] [PubMed] [Google Scholar]
- 80.Hanitsch LG, Löbel M, Müller-Redetzky H, et al. Late-Onset Disseminated Mycobacterium avium intracellulare Complex Infection (MAC), Cerebral Toxoplasmosis and Salmonella Sepsis in a German Caucasian Patient with Unusual Anti-Interferon-Gamma IgG1 Autoantibodies. J Clin Immunol. 2015;35:361–365. doi: 10.1007/s10875-015-0161-5. [DOI] [PubMed] [Google Scholar]
- 81.Wu U-I, Chuang Y-C, Sheng W-H, et al. Use of QuantiFERON-TB Gold Intube assay in screening for neutralizing anti-interferon-γ autoantibodies in patients with disseminated nontuberculous mycobacterial infection. Clin Microbiol Infect. 2018;24:159–165. doi: 10.1016/j.cmi.2017.06.029. [DOI] [PubMed] [Google Scholar]
- 82.Fieschi C, Dupuis S, Picard C, et al. High levels of interferon gamma in the plasma of children with complete interferon gamma receptor deficiency. Pediatrics. 2001;107:E48. doi: 10.1542/peds.107.4.e48. [DOI] [PubMed] [Google Scholar]
- 83.Martínez-Barricarte R, Megged O, Stepensky P, et al. Mycobacterium simiae Infection in Two Unrelated Patients with Different Forms of Inherited IFN-γR2 Deficiency. J Clin Immunol. 2014;34:904–909. doi: 10.1007/s10875-014-0085-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rottman M, Soudais C, Vogt G, et al. IFN-γ Mediates the Rejection of Haematopoietic Stem Cells in IFN-γR1-Deficient Hosts. PLoS Med. 2008;5:e26. doi: 10.1371/journal.pmed.0050026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dorman SE, Holland SM. Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest. 1998;101:2364–2369. doi: 10.1172/JCI2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Puel A, Reichenbach J, Bustamante J, et al. The NEMO mutation creating the most-upstream premature stop codon is hypomorphic because of a reinitiation of translation. Am J Hum Genet. 2006;78:691–701. doi: 10.1086/501532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vogt G, Chapgier A, Yang K, et al. Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nat Genet. 2005;37:692–700. doi: 10.1038/ng1581. [DOI] [PubMed] [Google Scholar]
- 88.Rosenzweig SD, Dorman SE, Uzel G, et al. A novel mutation in IFN-gamma receptor 2 with dominant negative activity: biological consequences of homozygous and heterozygous states. J Immunol. 2004;173:4000–4008. doi: 10.4049/jimmunol.173.6.4000. [DOI] [PubMed] [Google Scholar]
- 89.Holland SM, Dorman SE, Kwon A, et al. Abnormal regulation of interferon-gamma, interleukin-12, and tumor necrosis factor-alpha in human interferon-gamma receptor 1 deficiency. J Infect Dis. 1998;178:1095–1104. doi: 10.1086/515670. [DOI] [PubMed] [Google Scholar]
- 90.Noordzij JG, Hartwig NG, Verreck FA, et al. Two patients with complete defects in interferon gamma receptor-dependent signaling. J Clin Immunol. 2007;27:490–496. doi: 10.1007/s10875-007-9097-8. [DOI] [PubMed] [Google Scholar]
- 91.Prando C, Boisson-Dupuis S, Grant AV, et al. Paternal uniparental isodisomy of chromosome 6 causing a complex syndrome including complete IFN-γ receptor 1 deficiency. Am J Med Genet Part A. 2010;152A:622–629. doi: 10.1002/ajmg.a.33291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kong X-FF, Vogt G, Itan Y, et al. Haploinsufficiency at the human IFNGR2 locus contributes to mycobacterial disease. Hum Mol Genet. 2013;22:769–781. doi: 10.1093/hmg/dds484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tsumura M, Okada S, Sakai H, et al. Dominant-negative STAT1 SH2 domain mutations in unrelated patients with mendelian susceptibility to mycobacterial disease. Hum Mutat. 2012;33:1377–1387. doi: 10.1002/humu.22113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Janssen R, Van Wengen A, Verhard E, et al. Divergent role for TNF-alpha in IFN-gamma-induced killing of Toxoplasma gondii and Salmonella typhimurium contributes to selective susceptibility of patients with partial IFN-gamma receptor 1 deficiency. J Immunol. 2002;169:3900–3907. doi: 10.4049/jimmunol.169.7.3900. [DOI] [PubMed] [Google Scholar]
- 95.Okada S, Ishikawa N, Shirao K, et al. The novel IFNGR1 mutation 774del4 produces a truncated form of interferon-gamma receptor 1 and has a dominant-negative effect on interferon-gamma signal transduction. J Med Genet. 2007;44:485–491. doi: 10.1136/jmg.2007.049635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Takeda K, Kawai T, Nakazawa Y, et al. Augmentation of antitubercular therapy with IFNγ in a patient with dominant partial IFNγ receptor 1 deficiency. Clin Immunol. 2014;151:25–28. doi: 10.1016/j.clim.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 97.Allende LM, Lopez-Goyanes A, Paz-Artal E, et al. A point mutation in a domain of gamma interferon receptor 1 provokes severe immunodeficiency. Clin Diagn Lab Immunol. 2001;8:133–137. doi: 10.1128/CDLI.8.1.133-137.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kilic SS, van Wengen A, de Paus Ra, et al. Severe disseminated mycobacterial infection in a boy with a novel mutation leading to IFN-γR2 deficiency. J Infect. 2012;65:568–572. doi: 10.1016/j.jinf.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 99.Filipe-Santos O, Bustamante J, Haverkamp MH, et al. X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production. J Exp Med. 2006;203:1745–1759. doi: 10.1084/jem.20060085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pedraza-Sánchez S, Herrera-Barrios MT, Aldana-Vergara R, et al. Bacille Calmette–Guérin infection and disease with fatal outcome associated with a point mutation in the interleukin-12/interleukin-23 receptor beta-1 chain in two Mexican families. Int J Infect Dis. 2010;14:e256–e260. doi: 10.1016/j.ijid.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 101.Ehlayel M, de Beaucoudrey L, Fike F, et al. Simultaneous presentation of 2 rare hereditary immunodeficiencies: IL-12 receptor beta1 deficiency and ataxia-telangiectasia. J Allergy Clin Immunol. 2008;122:1217–1219. doi: 10.1016/j.jaci.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 102.Fieschi C. A novel form of complete IL-12/IL-23 receptor 1 deficiency with cell surface-expressed nonfunctional receptors. Blood. 2004;104:2095–2101. doi: 10.1182/blood-2004-02-0584. [DOI] [PubMed] [Google Scholar]
- 103.de Souza TL, Fernandes RCdSC, da Silva JA, et al. Microbial disease spectrum linked to a novel IL-12Rβ1 N-terminal signal peptide stop-gain homozygous mutation with paradoxical receptor cell-surface expression. Front Microbiol. 2017;8:1–10. doi: 10.3389/fmicb.2017.00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity. 2012;36:515–528. doi: 10.1016/j.immuni.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Qu H-Q, Fisher-Hoch SP, McCormick JB. Molecular immunity to mycobacteria: knowledge from the mutation and phenotype spectrum analysis of Mendelian susceptibility to mycobacterial diseases. Int J Infect Dis. 2011;15:e305–e313. doi: 10.1016/j.ijid.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sampaio EP, Bax HI, Hsu AP, et al. A novel STAT1 mutation associated with disseminated mycobacterial disease. J Clin Immunol. 2012;32:681–689. doi: 10.1007/s10875-012-9659-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kong X-FF, Vogt G, Chapgier A, et al. A novel form of cell type-specific partial IFN-gammaR1 deficiency caused by a germ line mutation of the IFNGR1 initiation codon. Hum Mol Genet. 2010;19:434–444. doi: 10.1093/hmg/ddp507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Storgaard M, Varming K, Herlin T, et al. Novel mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infections. Scand J Immunol. 2006;64:137–139. doi: 10.1111/j.1365-3083.2006.01775.x. [DOI] [PubMed] [Google Scholar]
- 109.de Paus RA, Kilic SS, van Dissel JT, et al. Effect of amino acid substitutions in the human IFN-γR2 on IFN-γ responsiveness. Genes Immun. 2011;12:136–144. doi: 10.1038/gene.2010.74. [DOI] [PubMed] [Google Scholar]
- 110.Kagawa R, Fujiki R, Tsumura M, et al. Alanine-scanning mutagenesis of human signal transducer and activator of transcription 1 to estimate loss- or gain-of-function variants. J Allergy Clin Immunol. 2017;140:232–241. doi: 10.1016/j.jaci.2016.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Uzel G, Frucht DM, Fleisher TA, et al. Detection of intracellular phosphorylated STAT-4 by flow cytometry. Clin Immunol. 2001;100:270–276. doi: 10.1006/clim.2001.5078. [DOI] [PubMed] [Google Scholar]
- 112.Chi CY, Chu CC, Liu JP, et al. Anti-IFN-gamma autoantibodies in adults with disseminated nontuberculous mycobacterial infections are associated with HLA-DRB1*16:02 and HLA-DQB1*05:02 and the reactivation of latent varicella-zoster virus infection. Blood. 2013;121:1357–1366. doi: 10.1182/blood-2012-08-452482. [DOI] [PubMed] [Google Scholar]
- 113.Patel SY, Ding L, Brown MR, et al. Anti-IFN-gamma autoantibodies in disseminated nontuberculous mycobacterial infections. J Immunol. 2005;175:4769–4776. doi: 10.4049/jimmunol.175.7.4769. [DOI] [PubMed] [Google Scholar]
- 114.Browne SK, Burbelo PD, Chetchotisakd P, et al. Adult-onset immunodeficiency in Thailand and Taiwan. N Engl J Med. 2012;367:725–734. doi: 10.1056/NEJMoa1111160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Conti F, Aragão Filho WC, Prando C, et al. Phagocyte nicotinamide adenine dinucleotide phosphate oxidase activity in patients with inherited IFN-γR1 or IFN-γR2 deficiency. J Allergy Clin Immunol. 2015;135:1393–1395.e1. doi: 10.1016/j.jaci.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Meyts I, Bosch B, Bolze A, et al. Exome and genome sequencing for inborn errors of immunity. J Allergy Clin Immunol. 2016;138:957–969. doi: 10.1016/j.jaci.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Casanova J-L, Conley ME, Seligman SJ, et al. Guidelines for genetic studies in single patients: lessons from primary immunodeficiencies. J Exp Med. 2014;211:2137–2149. doi: 10.1084/jem.20140520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Döffinger R, Jouanguy E, Dupuis S, et al. Partial interferon-gamma receptor signaling chain deficiency in a patient with bacille Calmette-Guerin and Mycobacterium abscessus infection. J Infect Dis. 2000;181:379–384. doi: 10.1086/315197. [DOI] [PubMed] [Google Scholar]
- 119.Martínez-Barricarte R, de Jong SJ, Markle J, et al. Current Protocols in Immunology. John Wiley & Sons, Inc; Hoboken, NJ, USA: 2016. Transduction of Herpesvirus saimiri -Transformed T Cells with Exogenous Genes of Interest; pp. 7.21C.1–7.21C.12. [DOI] [PubMed] [Google Scholar]
- 120.Smith T. Parasitism and Disease. Princeton: Princeton University Press; 1934. [Google Scholar]
- 121.Casanova J-L. Human genetic basis of interindividual variability in the course of infection. Proc Natl Acad Sci. 2015;112:7118–7127. doi: 10.1073/pnas.1521644112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Casanova J-L. Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc Natl Acad Sci U S A. 2015;112:7128–7137. doi: 10.1073/pnas.1521651112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Holland SM. Treatment of infections in the patient with Mendelian susceptibility to mycobacterial infection. Microbes Infect. 2000;2:1579–1590. doi: 10.1016/s1286-4579(00)01314-9. [DOI] [PubMed] [Google Scholar]