

Abstract
C-CBL (CBL) encodes a multifunctional protein engaged in the regulation of intracellular signaling pathways.1,2 It was first identified as a cellular counterpart of the viral oncogene, v-CBL, that causes murine lymphoma.3,4 Although no genetic evidence existed suggesting its role in human carcinogenesis, the recent discovery of c-CBL mutations in myeloid cancers has unveiled a unique oncogenic mechanism mediated by gain-of-function of a mutated tumor suppressor, closely associated with allelic conversion of 11q arms.5-9 In this review, we summarize our current knowledge about c-CBL mutations and discuss the molecular mechanisms of their gain-of-function.
Keywords: c-CBL, 11qUPD, myeloproliferative neoplasms, gain-of-function, MDS/MPN, tyrosine kinases
Myeloproliferative Neoplasms and Related Disorders
Myeloproliferative neoplasms (MPNs) are a heterogeneous group of blood cancers, characterized by clonal hematopoiesis that causes excessive production of one or more components of mature blood cells with hypercellular bone marrow and extramedullary hematopoiesis.10 Some patients also show abnormalities in cell morphology and differentiation with dysplastic bone marrow, and are classified into myelo-dysplastic/myeloproliferative neoplasms (MDS/MPN) in the World Health Organization (WHO) classification.11 A genetic hallmark of MPN and MDS/MPN is frequent mutations of genes on signal transduction pathways, which have been causally linked to hypersensitivity of neoplastic progenitors to growth factors and cytokines.10 A notable example is JAK2 V617F mutations found in most cases of polycythemia vera (PV), a form of MPNs that is characterized by overproduction of mature erythrocytes together with other blood components.12-14
JAK2 Mutations in MPNs
These mutants encode constitutive active kinases that transmit signals from erythropoietin receptor, and induce a hypersensitive proliferative response to erythropoietin.12 Of particular interest about JAK2 mutations in PV is the presence of one or more subclones showing acquired uniparental disomy (aUPD) involving the 9p arm that leads to homozygous JAK2 mutations (JAK2mut/mut) by allelic conversion (Fig. 1).15 One of the initial discoveries of JAK2 mutations relied on the detailed mapping of loss of heterozygosity (LOH) caused by aUPD in 9p.13 The consequence of 9p-aUPD is loss of wild type JAK2 and duplication of mutated JAK2, but the latter seems to be more important for the clonal selection of UPD clones, because mutated JAK2 is duplicated without loss of wild-type allele in 9p trisomy in some cases.16 Similarly gain-of-function mutations of cMPL are frequently found in primary myelofibrosis in close association with 1p-aUPD.17 Thus, aUPD, or copy number neutral LOH, is associated not only with biallelic loss-of-function of classical tumor suppressor genes in the Knudson’s paradigm,18 but also with gain-of-function of proto-oncogenes. Moreover, genome-wide analysis of genetic imbalances in a variety of myeloid neoplasms revealed that aUPD is another genetic feature of MPNs, where 42% of chronic myelomonocytic leukemia (CMML) cases had one or more regions of aUPD and were grouped into several discrete clusters, which may or may not harbor mutations of known cancer related genes.9 Among these one of the most prominent is the cluster that is defined by 11q-aUPD, from which mutated c-CBL proto-oncogene was identified.9
Figure 1.
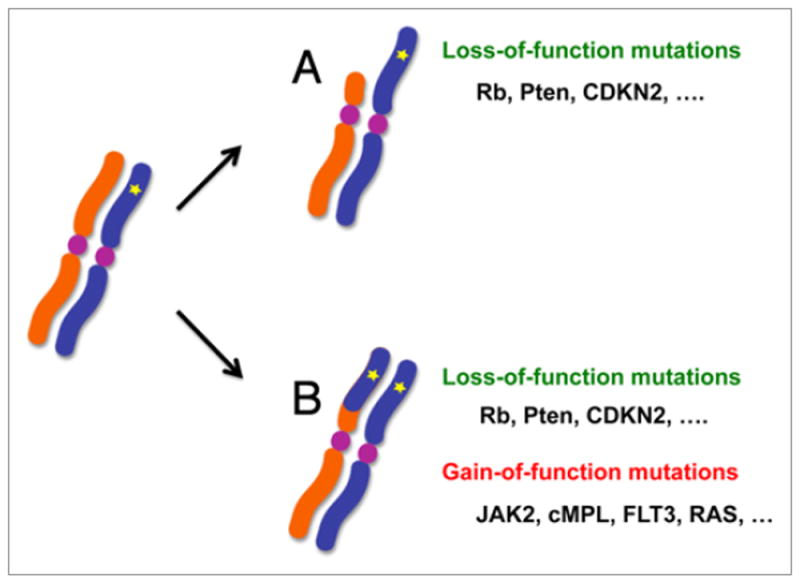
In cancer cells, LOH is frequently associated with a mutated tumor suppressor locus, in which a normal copy of the tumor suppressor is lost by simple allelic deletion (A), or replaced by the mutated copy through allelic conversion that leads to copy number neutral LOH or aUPD (B). In either case, the common consequence is biallelic loss-of-function of the tumor suppressor. In addition, LOH caused by aUPD is also implicated in the common mechanism of homozygous mutations of proto-oncogenes. A number of gain-of-function oncogenic mutations found in aUPD regions have been shown to exist in a homozygous state, including mutations of JAK2 (9pUPD), MPL (1pUPD), NRAS (1pUPD), KRAS (12pUPD), BRAF and FLT3 (13qUPD). The clonal outgrowth of aUPD-positive clones indicates that two copies of mutations confer a growth advantage to aUPD positive cells through their gain-of-function.
c-CBL Mutations in MDS/MPNs
Although c-CBL mutations have been reported in a variety of myeloid neoplasms including acute myeloid leukemia, myelodysplastic syndromes, as well as classical myeloproliferative disorders, the majority of c-CBL-mutated cases are MDS/MPN, including CMML (~15%), juvenile myelomonocytic leukemia (JMML) (~17%), and atypical chronic myeloid leukemia (~5%).5-9,19,20 In most cases, c-CBL mutations are associated with 11q-aUPD involving c-CBL locus, which converts these mutations into a homozygous state. Loss of wild-type c-CBL is rarely caused by chromosomal deletion.6,7,9 c-CBL mutations exclusively occur independent of RAS and PTPN11 in CMML and JMML.8,9 Notably, c-CBL mutations have a germline origin in some JMML cases.8 Approximately half of the c-C-BL mutations in JMML cases involve Y371, while mutations are widely distributed within linker/RING finger domain in other neoplasms. c-CBL mutants strongly transform fibroblasts and enhance proliferation of hematopoietic progenitors in methylcellulose culture.9 These genetic and functional observations indicate that mutant c-CBL may have some gain-of-function, which promotes clonal evolution, especially of aUPD-positive clones carrying two copies of the mutations.
c-CBL as a Tumor Suppressor Gene
c-CBL proto-oncogene is a cellular homologue of a viral oncogene, v-CBL, isolated from the Casitas-NS-lymphoma virus that induces murine lymphoma.3,4 Together with other two homologues CBL-b and CBL-c, it comprises the CBL family of proteins. All c-CBL proteins have an N-terminal domain for binding to phosphorylated tyrosine kinases (TKB domain) connected through a linker sequence to the RING finger, but CBL-c lacks most of the C-terminal domains shared by c-CBL and CBL-b (Fig. 2A). While c-CBL has multivalent molecular functions in signal transduction and cytoskeletal regulation, the most intensively studied-function is its role in negative regulation of receptor tyrosine kinase (RTK) signalings, which depends on the E3 ubquitin ligase activity of this molecule.1,21 After RTKs are phosphorylated on cytokine stimulation, c-CBL binds to the phosphorylated RTKs through the TKB domain, and mono-ubiquitinates these RTKs at multiple sites in concert with the E2 conjugating enzyme, which is followed by internalization and degradation/recycling of the phosphorylated RTKs.21 Thus, c-CBL prevents excessive RTK signaling after cytokine/growth factor stimulation and potentially acts as a tumor suppressor. c-CBL-/- mice have an enlarged thymus, splenomegaly with extramedullary hematopoiesis.22,23 In these mice, hematopoietic progenitor pools are expanded,9,24 and their hematopoietic progenitors exhibit hypersensitive proliferative responses to cytokine stimulations. When introduced into BCR/ABL transgenic mice, a c-CBL-/- allele accelerates blastic crisis.9 Moreover, c-CBL-/- mice developed invasive cancer spontaneously (in preparation), further supporting that c-CBL has tumor suppressor functions.
Figure 2.
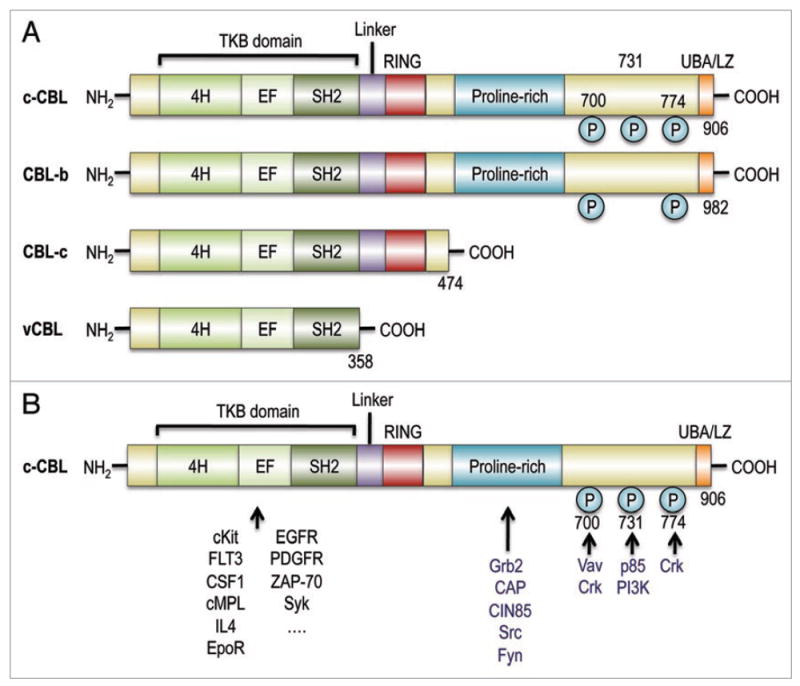
(A) Structure of CBL family proteins. CBL family proteins in mammals have highly conserved domains, where an N-terminal TKB domain, consisting of a four-helix bundle (4H), a Ca2+-binding EF (EF), and a src-homology (SH2) domains, is connected to a RING finger domain via a linker. c-CBL and CBL-b, but not CBL-c, have a proline-rich and other C-terminal components that end with a ubiquitin-associated and leucine zipper (UBA/LZ) domain. Their viral form, v-CBL, is truncated just after its SH2 domain. (B) CBL family proteins interact with a number of signal transducing molecules. Through their TKB domain, CBL family proteins target phosphrylated tyrosine kinases, including growth factor receptors and cytokine receptors, as well as, non-receptor tyrosine kinases. Ubiquitin conjugating enzymes have contact with CBL proteins via the linker/RING finger domain, which is central to the E3 ubiquitin ligase activity. The proline-rich domain provides a binding site for SH3 domains of Grab2, CAP and Src-family kinases. The C-terminal portion contains three tyrosine residues, Y700, Y731 and Y774, which are the major phosphorylated tyrosines, and which bind to the p85 subunit of PI3 kinase (Y731), Vav (Y700) and Crk proteins (Y700 and Y774).
Gain of Function of CBL Mutants
How can we reconcile with the tumor suppressor functions of c-CBL on the one hand, and the oncogenic properties of c-CBL mutants on the other? A simple explanation would be an inhibition of tumor suppressor function of wild type c-CBL by mutant c-CBL. Most c-CBL mutations in MPNs occur within the linker/RING finger domains, through which c-CBL binds E2 conjugating enzymes, and thus are expected to compromise the E3 ligase activity of the molecule. In fact, when expressed in fibroblasts, tumor-derived linker and RING finger mutants show severely compromised E3 ubiquitin ligase activity.9,20 Moreover, two linker mutants (Q367P and Y371S) have been shown to inhibit the activity of wild-type c-CBL protein, although they do not make direct contact with E2 enzymes but with the TKB domain.25 As expected from the inhibitory action of these mutants with regard to E3 ubiquitin ligase activity, transduction of the latter mutants into NIH3T3 or hematopoietic cells lead to prolonged activation of tyrosine kinases after stimulation with a variety of cytokines and growth factors, including epidermal growth factor, stem cell factor (SCF), Interleukin 3 (IL3), thrombopoietin, and FLT3 ligand.9,20 Given the diverse spectrum of kinase targets of CBL, the enhanced sensitivity of these cells to a variety of cytokines is well expected.
Although these experimental data support a dominant negative mechanism of mutant c-CBL, a simple dominant negative model is defied by an experiment, in which mutant c-CBL was transduced into c-CBL-/- hematopoietic progenitors. Lin-Sca1+cKit+ (LSK) hematopoietic progenitors from c-CBL-/- mice showed enhanced survival or proliferative responses after stimulation with a variety of cytokines, including SCF, IL3, or thrombopoietin, as compared to those from c-CBL+/+ mice. However, transduction of mutant c-CBL into c-CBL-/- progenitors dramatically augmented the responses to these cytokines and also to FLT3 ligand, while the effect of mutant c-CBL-transduction into c-CBL+/+ progenitors was unremarkable even as compared to mock-transduced CBL-/- progenitors.9 The augmented sensitivity to these cytokines in c-CBL-/- cells was nothing to do with the inhibition of c-CBL functions, and thus is considered to represent a true gain-of-function of the mutant c-CBL. The gain-of-function nature of c-CBL mutations is also predicted from the fact that in myeloid neoplasms, 11qLOH is caused by aUPD in most cases and rarely accompanies 11q deletion, although in this case the target gene has tumor suppressor functions. Interestingly, the effect of the gain-of-function effect largely disappears by introducing wild type c-CBL or in the presence of the wild-type c-CBL allele,9 which might explain the observation that the wild type c-CBL allele was lost in most MDS/MPN cases with c-CBL mutations as a result of allelic conversion or aUPD.
Origin of the Gain-of-Function of Mutant CBL
The exact mechanism through which mutant c-CBL acquires oncogenic functions even in c-CBL-/- cells is still elusive. Because the gain-of-function of mutant c-CBL is largely neutralized by the presence of wild type c-CBL, one possibility is that it could be mediated by the inhibition of some ‘CBL-like’ activity still present in c-CBL-/- cells, most likely CBL-b. Both c-CBL and CBL-b are expressed in immature hematopoietic progenitors, and c-CBL mutant inhibits E3 ubiquitin ligase activity of both c-CBL proteins.9,26 Although c-CBL/CBL-b-double knockout mice are embryonic lethal, conditional double knockout in T cells shows hypersensitive to anti-CD3 stimulations and prolonged TCR-signaling, as compared to c-CBL or CBL-b single null T cells.27 This reminds us of the gain-of-function of mutated TP53, which explains the difference in the phenotypes between TP53-/- and TP53mut/- mice. TP53-/- mice develop tumors at a high frequency, but they are mostly sarcomas or lymphomas and development of carcinoma is very rare, whereas TP53mut/- mice also develop carcinoma in various organs. Thus, TP53 mutant has more than null functions, which are thought to be mediated by the inhibition of its homologues, TP63 and TP73, expressed in epithelial tissues (Fig. 3).28,29 Like c-CBL, TP53 tumor suppressor gene was first identified as an oncogene through its mutated, oncogenic forms in cancer cells. On the other hand, the model of gain-of-function mediated through CBL-b inhibition fails to explain why CBL-b mutations are extremely rare in CMML. According to this model, essentially no difference would be expected between the mutations of c-CBL and CBL-b, as long as in either case, compromised E3 ubiquitin ligase activity would result. The linker-RING finger mutants of c-CBL would be expected also to be able to inhibit E3 ubiquitin ligase activity of the wild-type c-CBL.
Figure 3.
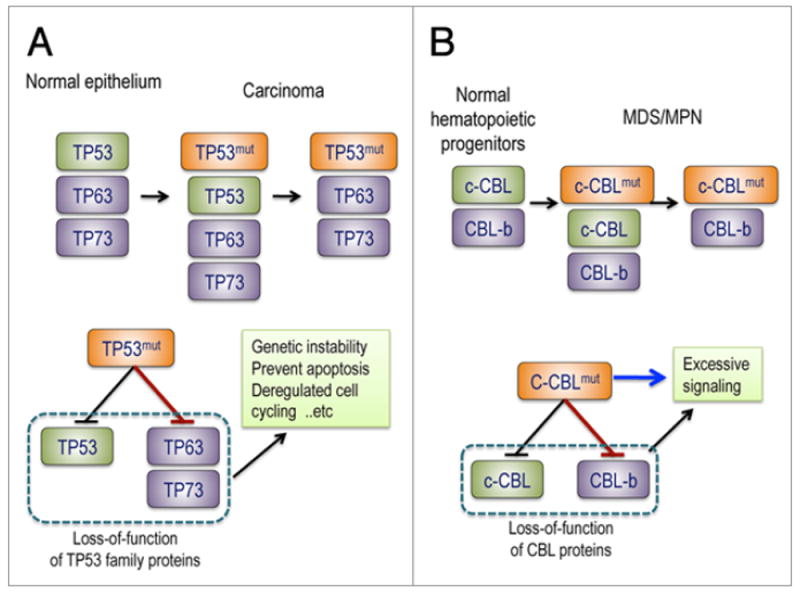
Possible mechanisms of gain-of-function of mutated TP53 and c-CBL. The gain-of-function of TP53 mutants is associated with their potential to induce carcinoma in mice as well as in human, which is considered to be mediated by inhibition of TP63 and TP73. TP53-deficient mice frequently develop sarcomas and lymphomas but only rarely carcinomas, which are thought to be suppressed by TP53 homologues, TP63 and TP73, in epithelial tissues, in the face of loss of TP53. Mutant TP53 inhibits tumor suppressor functions of TP63 and TP73, and compromises TP53-like activity. Similarly, the gain-of-function of CBL mutants found in MDS/MPN may be explained by the inhibition of CBL-b (red arrow), which would result in more profound defects in negative regulation of tyrosine kinase signaling compared to simple loss of c-CBL. On the other hand, c-CBL is thought to have positive regulatory functions that are not directly related to the E3 ubiquitin ligase activity and could be the source of the gain-of-function of c-CBL mutants (blue arrow).
Another, but not necessarily exclusive, explanation of the gain-of-function of mutant c-CBL would be related to positive roles of c-CBL as a signal transducer rather than an attenuator (Figs. 3A and 4). c-CBL not only binds to a number of phosphorylated tyrosine kinases through its TKB domain, which is indispensable for the negative regulation of these kinases, but also interacts with more than 150 different proteins through a number of C-terminal domains and residues, and acts as a multi-domain adaptor protein, involved in signal transduction (Fig. 2B).2 When recruited to phosphorylated tyrosine kinases, c-CBL is also phosphorylated at multiple tyrosine residues, and provides docking sites for the SH2 domains of Vav (pY700),30 CrkL (pY700 and pY774)31-34 and the p85 subunit of PI3 kinase (Y731).35-37 c-CBL also binds to Grab2,38-40 CAP,41 and Src family tyrosine kinases36 through the proline-rich domain. Several lines of evidence suggest that c-CBL positively transmits signals through these interactions. For example, c-CBL promotes cell survival and proliferation, depending on the PI3 kinase pathway,42,43 and also enhances activation of MAP kinases after stimulation of Met tyrosine kinase.44 c-CBL is also a key substrate/effector of Src kinase, which plays a central role in bone resorption and osteoclast migration.45,46 It also is involved in cytoskeletal rearrangements through activation of Rac1, Cdc42, and R-Ras.47,48 Normally, mediated by its E3 ligase activity, kinase-bound phosphorylated c-CBL rapidly undergoes degradation,26 by which positive signaling should be terminated. Thus, once linker/RING finger mutations abolish the E3 ligase activity of c-CBL, the consequence would be prolonged signaling due not only to loss of negative regulation of tyrosine kinase, but also to enhanced positive regulatory functions, which should appear as gain-of-function (Fig. 4).
Figure 4.
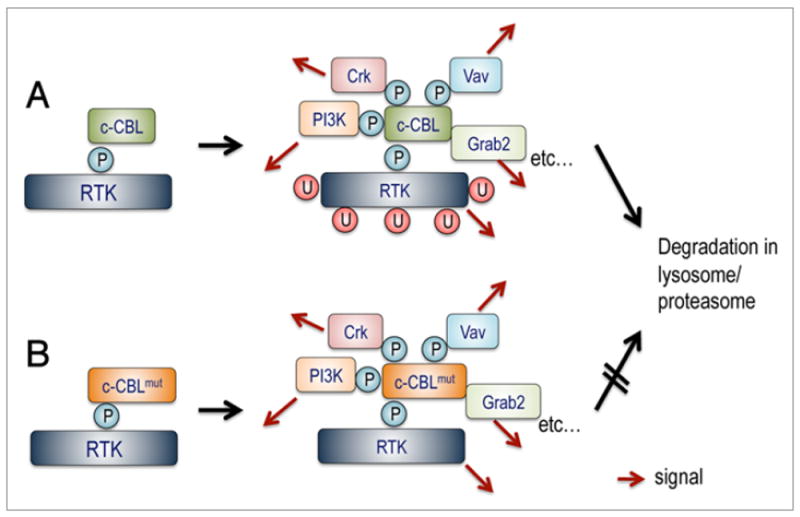
Positive regulation of signal transduction by c-CBL. (A) Having E3 ubiquitin ligase activity for negative regulation of signaling, c-CBL also works as an adaptor protein for multiple signal transduction molecules. When bound to phosphorylated tyrosine kinases, c-CBL is rapidly phosphorylated at multiple tyrosine residues, which in turn provide binding sites for a number of signal transduction molecules. Several lines of evidence suggest that binding to these molecules plays important roles in positive regulation of signal transduction (red arrows). Normally, phosphorylated c-CBL undergoes degradation, which is mediated by its E3 ubiquitin ligase activity. Thus, degradation of mutated c-CBL could be retarded, leading to prolonged transmission of positive signals (B).
In contrast to the CBL-b inhibition model, the uni-laterality of c-CBL mutations could be more easily explained, because c-CBL and CBL-b have distinct biological functions, as clearly shown by the phenotypes of c-CBL-/- and CBL-b-/- mice.22,23,49,50 For example, CBL-b lacks one of the major phosphorylated tyrosines, Y731, that provides a docking site for the p85 subunit of PI3 kinase. Although the exact molecular basis for the distinct functions between both CBL proteins remains to be elucidated, c-CBL-specific positive regulatory function in immature hematopoietic progenitors may be important for the pathogenesis of myeloid neoplasms.
Conclusion
Allelic conversion leading to aUPD is an important genetic mechanism of clonal evolution in the pathogenesis of MPN, and associated not only with loss-of-function of tumor suppressor genes, but also with gain-of-function mutations of proto-oncogenes. Homozygous c-CBL mutations that characterize a subset of MDS/MPD carrying 11q-aUPD, represent a unique example of gain-of-function mutations of tumor suppressor/proto-oncogene. These linker/RING finger mutations convert c-CBL, which otherwise act as a tumor suppressor, to a gain-of-function oncogenic protein. Although its exact molecular mechanism is still unknown, the gain-of-function of oncogenic c-CBL mutants seems to be related to disintegration of negative and positive regulatory machineries of normal c-CBL protein. Detailed analysis of the oncogenic mechanisms of c-CBL mutants is warranted, which should shed light on a novel aspect of physiological function of c-CBL. Considering their expression and functions in a broad spectrum of tissues, CBL family genes may be mutated in other human cancers.
References
- 1.Thien CB, Langdon WY. Cbl: many adaptations to regulate protein tyrosine kinases. Nat Rev Mol Cell Biol. 2001;2:294–307. doi: 10.1038/35067100. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt MH, Dikic I. The Cbl interactome and its functions. Nat Rev Mol Cell Biol. 2005;6:907–18. doi: 10.1038/nrm1762. [DOI] [PubMed] [Google Scholar]
- 3.Langdon WY, Hartley JW, Klinken SP, Ruscetti SK, Morse HC., 3rd v-cbl, an oncogene from a dual-recombinant murine retrovirus that induces early B-lineage lymphomas. Proc Natl Acad Sci USA. 1989;86:1168–72. doi: 10.1073/pnas.86.4.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blake TJ, Shapiro M, Morse HC, 3rd, Langdon WY. The sequences of the human and mouse c-cbl proto-oncogenes show v-cbl was generated by a large truncation encompassing a proline-rich domain and a leucine zipper-like motif. Oncogene. 1991;6:653–7. [PubMed] [Google Scholar]
- 5.Abbas S, Rotmans G, Lowenberg B, Valk PJ. Exon 8 splice site mutations in the gene encoding the E3-ligase CBL are associated with core binding factor acute myeloid leukemias. Haematologica. 2008;93:1595–7. doi: 10.3324/haematol.13187. [DOI] [PubMed] [Google Scholar]
- 6.Dunbar AJ, Gondek LP, O’Keefe CL, Makishima H, Rataul MS, Szpurka H, et al. 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res. 2008;68:10349–57. doi: 10.1158/0008-5472.CAN-08-2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grand FH, Hidalgo-Curtis CE, Ernst T, Zoi K, Zoi C, McGuire C, et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myelo-proliferative neoplasms. Blood. 2009;113:6182–92. doi: 10.1182/blood-2008-12-194548. [DOI] [PubMed] [Google Scholar]
- 8.Loh ML, Sakai DS, Flotho C, Kang M, Fliegauf M, Archambeault S, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood. 2009;114:1859–63. doi: 10.1182/blood-2009-01-198416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S, et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature. 2009;460:904–8. doi: 10.1038/nature08240. [DOI] [PubMed] [Google Scholar]
- 10.Van Etten RA, Shannon KM. Focus on myeloproliferative diseases and myelodysplastic syndromes. Cancer Cell. 2004;6:547–52. doi: 10.1016/j.ccr.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 11.Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–51. doi: 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- 12.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 13.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 14.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–97. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 15.Levine RL, Pardanani A, Tefferi A, Gilliland DG. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer. 2007;7:673–83. doi: 10.1038/nrc2210. [DOI] [PubMed] [Google Scholar]
- 16.Najfeld V, Montella L, Scalise A, Fruchtman S. Exploring polycythaemia vera with fluorescence in situ hybridization: additional cryptic 9p is the most frequent abnormality detected. Br J Haematol. 2002;119:558–66. doi: 10.1046/j.1365-2141.2002.03763.x. [DOI] [PubMed] [Google Scholar]
- 17.Kawamata N, Ogawa S, Yamamoto G, Lehmann S, Levine RL, Pikman Y, et al. Genetic profiling of myeloproliferative disorders by single-nucleotide polymorphism oligonucleotide microarray. Exp Hematol. 2008;36:1471–9. doi: 10.1016/j.exphem.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1:157–62. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- 19.Caligiuri MA, Briesewitz R, Yu J, Wang L, Wei M, Arnoczky KJ, et al. Novel c-CBL and CBL-b ubiquitin ligase mutations in human acute myeloid leukemia. Blood. 2007;110:1022–4. doi: 10.1182/blood-2006-12-061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sargin B, Choudhary C, Crosetto N, Schmidt MH, Grundler R, Rensinghoff M, et al. Flt3-dependent transformation by inactivating c-Cbl mutations in AML. Blood. 2007;110:1004–12. doi: 10.1182/blood-2007-01-066076. [DOI] [PubMed] [Google Scholar]
- 21.Joazeiro CA, Wing SS, Huang H, Leverson JD, Hunter T, Liu YC. The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science. 1999;286:309–12. doi: 10.1126/science.286.5438.309. [DOI] [PubMed] [Google Scholar]
- 22.Murphy MA, Schnall RG, Venter DJ, Barnett L, Bertoncello I, Thien CB, et al. Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. Mol Cell Biol. 1998;18:4872–82. doi: 10.1128/mcb.18.8.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naramura M, Kole HK, Hu RJ, Gu H. Altered thymic positive selection and intracellular signals in Cbl-deficient mice. Proc Natl Acad Sci USA. 1998;95:15547–52. doi: 10.1073/pnas.95.26.15547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rathinam C, Thien CB, Langdon WY, Gu H, Flavell RA. The E3 ubiquitin ligase c-Cbl restricts development and functions of hematopoietic stem cells. Genes Dev. 2008;22:992–7. doi: 10.1101/gad.1651408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng N, Wang P, Jeffrey PD, Pavletich NP. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell. 2000;102:533–9. doi: 10.1016/s0092-8674(00)00057-x. [DOI] [PubMed] [Google Scholar]
- 26.Zeng S, Xu Z, Lipkowitz S, Longley JB. Regulation of stem cell factor receptor signaling by Cbl family proteins (Cbl-b/c-Cbl) Blood. 2005;105:226–32. doi: 10.1182/blood-2004-05-1768. [DOI] [PubMed] [Google Scholar]
- 27.Naramura M, Jang IK, Kole H, Huang F, Haines D, Gu H. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat Immunol. 2002;3:1192–9. doi: 10.1038/ni855. [DOI] [PubMed] [Google Scholar]
- 28.Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–72. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 29.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–60. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 30.Marengere LE, Mirtsos C, Kozieradzki I, Veillette A, Mak TW, Penninger JM. Proto-oncoprotein Vav interacts with c-Cbl in activated thymocytes and peripheral T cells. J Immunol. 1997;159:70–6. [PubMed] [Google Scholar]
- 31.de Jong R, ten Hoeve J, Heisterkamp N, Groffen J. Crkl is complexed with tyrosine-phosphorylated Cbl in Ph-positive leukemia. J Biol Chem. 1995;270:21468–71. doi: 10.1074/jbc.270.37.21468. [DOI] [PubMed] [Google Scholar]
- 32.Buday L, Khwaja A, Sipeki S, Farago A, Downward J. Interactions of Cbl with two adapter proteins, Grb2 and Crk, upon T cell activation. J Biol Chem. 1996;271:6159–63. doi: 10.1074/jbc.271.11.6159. [DOI] [PubMed] [Google Scholar]
- 33.Sattler M, Salgia R, Okuda K, Uemura N, Durstin MA, Pisick E, et al. The proto-oncogene product p120CBL and the adaptor proteins CRKL and c-CRK link c-ABL, p190BCR/ABL and p210BCR/ABL to the phosphatidylinositol-3’ kinase pathway. Oncogene. 1996;12:839–46. [PubMed] [Google Scholar]
- 34.Ribon V, Hubbell S, Herrera R, Saltiel AR. The product of the cbl oncogene forms stable complexes in vivo with endogenous Crk in a tyrosine phosphorylation-dependent manner. Mol Cell Biol. 1996;16:45–52. doi: 10.1128/mcb.16.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, et al. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–78. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 36.Hunter S, Burton EA, Wu SC, Anderson SM. Fyn associates with Cbl and phosphorylates tyrosine 731 in Cbl, a binding site for phosphatidylinositol 3-kinase. J Biol Chem. 1999;274:2097–106. doi: 10.1074/jbc.274.4.2097. [DOI] [PubMed] [Google Scholar]
- 37.Hartley D, Corvera S. Formation of c-Cbl.phosphatidylinositol 3-kinase complexes on lymphocyte membranes by a p56lck-independent mechanism. J Biol Chem. 1996;271:21939–43. doi: 10.1074/jbc.271.36.21939. [DOI] [PubMed] [Google Scholar]
- 38.Fukazawa T, Miyake S, Band V, Band H. Tyrosine phosphorylation of Cbl upon epidermal growth factor (EGF) stimulation and its association with EGF receptor and downstream signaling proteins. J Biol Chem. 1996;271:14554–9. doi: 10.1074/jbc.271.24.14554. [DOI] [PubMed] [Google Scholar]
- 39.Jiang X, Huang F, Marusyk A, Sorkin A. Grb2 regulates internalization of EGF receptors through clathrin-coated pits. Mol Biol Cell. 2003;14:858–70. doi: 10.1091/mbc.E02-08-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meisner H, Czech MP. Coupling of the proto-oncogene product c-Cbl to the epidermal growth factor receptor. J Biol Chem. 1995;270:25332–5. doi: 10.1074/jbc.270.43.25332. [DOI] [PubMed] [Google Scholar]
- 41.Baumann CA, Ribon V, Kanzaki M, Thurmond DC, Mora S, Shigematsu S, et al. CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature. 2000;407:202–7. doi: 10.1038/35025089. [DOI] [PubMed] [Google Scholar]
- 42.Ueno H, Sasaki K, Honda H, Nakamoto T, Yamagata T, Miyagawa K, et al. c-Cbl is tyrosine-phosphorylated by interleukin-4 and enhances mitogenic and survival signals of interleukin-4 receptor by linking with the phosphatidylinositol 3’-kinase pathway. Blood. 1998;91:46–53. [PubMed] [Google Scholar]
- 43.Grishin A, Sinha S, Roginskaya V, Boyer MJ, Gomez-Cambronero J, Zuo S, et al. Involvement of Shc and Cbl-PI 3-kinase in Lyn-dependent proliferative signaling pathways for G-CSF. Oncogene. 2000;19:97–105. doi: 10.1038/sj.onc.1203254. [DOI] [PubMed] [Google Scholar]
- 44.Garcia-Guzman M, Larsen E, Vuori K. The proto-oncogene c-Cbl is a positive regulator of Met-induced MAP kinase activation: a role for the adaptor protein Crk. Oncogene. 2000;19:4058–65. doi: 10.1038/sj.onc.1203750. [DOI] [PubMed] [Google Scholar]
- 45.Tanaka S, Amling M, Neff L, Peyman A, Uhlmann E, Levy JB, et al. c-Cbl is downstream of c-Src in a signalling pathway necessary for bone resorption. Nature. 1996;383:528–31. doi: 10.1038/383528a0. [DOI] [PubMed] [Google Scholar]
- 46.Meng F, Lowell CA. A beta1 integrin signaling pathway involving Src-family kinases, Cbl and PI-3 kinase is required for macrophage spreading and migration. EMBO J. 1998;17:4391–403. doi: 10.1093/emboj/17.15.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scaife RM, Job D, Langdon WY. Rapid microtubule-dependent induction of neurite-like extensions in NIH 3T3 fibroblasts by inhibition of ROCK and Cbl. Mol Biol Cell. 2003;14:4605–17. doi: 10.1091/mbc.E02-11-0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gotoh T, Niino Y, Tokuda M, Hatase O, Nakamura S, Matsuda M, et al. Activation of R-Ras by Rasguanine nucleotide-releasing factor. J Biol Chem. 1997;272:18602–7. doi: 10.1074/jbc.272.30.18602. [DOI] [PubMed] [Google Scholar]
- 49.Bachmaier K, Krawczyk C, Kozieradzki I, Kong YY, Sasaki T, Oliveira-dos-Santos A, et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 2000;403:211–6. doi: 10.1038/35003228. [DOI] [PubMed] [Google Scholar]
- 50.Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, et al. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403:216–20. doi: 10.1038/35003235. [DOI] [PubMed] [Google Scholar]