

Abstract
BACKGROUND
Autism spectrum disorder (ASD) has both genetic and environmental origins, including potentially maternal effects. Maternal effects describe the association of one or more maternal phenotypes with liability to ASD in progeny that are independent of maternally transmitted risk alleles. While maternal effects could play an important role, consistent with association to maternal traits such as immune status, no study has estimated maternal, additive genetic, and environmental effects in ASD.
METHODS
Using a population-based sample consisting of all children born in Sweden from 1998 to 2007 and their relatives, we fitted statistical models to family data to estimate the variance in ASD liability originating from maternal, additive genetic, and shared environmental effects. We calculated sibling and cousin family recurrence risk ratio as a direct measure of familial, genetic, and environmental risk factors and repeated the calculations on diagnostic subgroups, specifically autistic disorder (AD) and spectrum disorder (SD), which included Asperger’s syndrome and/or pervasive developmental disorder not otherwise specified.
RESULTS
The sample consisted of 776,212 children of whom 11,231 had a diagnosis of ASD: 4554 with AD, 6677 with SD. We found support for large additive genetic contribution to liability; heritability (95% confidence interval [CI]) was estimated to 84.8% (95% CI: 73.1–87.3) for ASD, 79.6% (95% CI: 61.2–85.1) for AD, and 76.4% (95% CI: 63.0–82.5) for SD.
CONCLUSIONS
There was modest, if any, contribution of maternal effects to liability for ASD, including subtypes AD and SD, and there was no support for shared environmental effects. These results show liability to ASD arises largely from additive genetic variation.
Keywords: Autism, Epidemiology, Genetics, Heritability, Population-based, Psychiatry
Autism spectrum disorder (ASD), a neurodevelopment disorder with significant social, communication, and behavioral challenges (1), has both genetic and environmental origins. Research into its genetic origins has consistently implicated rare and common inherited variation, as well de novo mutation (2–5). Most (6–12) but not all estimates of ASD heritability derive from twin studies, and the remainder derive from direct (2,3) or indirect (13–15) assessments of extended pedigrees. For environmental variation, some of the most consistent findings center on factors of maternal origin or maternal effects. Maternal obesity, hypertension, diabetes, prepregnancy body mass index, polycystic ovary syndrome, hypertensive disorders of pregnancy, and gestational diabetes all have some support for conferring risk for ASD in offspring (16–24). In addition, maternal immune status has been associated with ASD, and altered immune status is quite frequent in studies of mothers and ASD, estimated at >15% for autoimmune diseases and asthma, and 50% for allergies (25).
Here we use “maternal effect” to describe the association of a maternal phenotype with ASD in offspring. That association is assumed to be independent of genetic inheritance of ASD risk alleles from mother to offspring. Quantitative geneticists have long recognized that maternal effects can account for a substantial portion of population variability in an offspring trait (26). Like the offspring trait, the maternal phenotype can itself have both environmental and genetic origins; however, teasing apart these origins requires either carefully designed genetic crosses or extensive and deep pedigree data.
If maternal phenotypes, such as those described above, were a substantial source of liability for ASD, we reasoned that a portion of their impact on liability should be detectable as a maternal effect on ASD. By evaluating full sibling, half-sibling, and cousin relationships, we would predict that maternal effects could account for a substantial portion of the variability of ASD in such a sample. Pawitan et al. (27) proposed a statistical method to estimate the contribution to the variances in liability originating from maternal effects, together with variance due to additive genetic effects and shared environmental effects on binary traits using family data. This method has been applied to preterm birth (28), preeclampsia (29), and schizophrenia and bipolar disorder (30,31). The magnitude of maternal effects for ASD, however, is not yet known, and so far no study has been conducted to separate the maternal effects and additive genetic effects from environmental effects. This is an important gap in our knowledge: if maternal effects were linked to increased ASD risk, investigating their nature is relatively straightforward and such investigations could lead to insights into the etiology of ASD. Moreover, genetically, the presence of maternal effects can bias heritability estimates derived from extended pedigrees because of the (unmodeled) increase of similarity among maternal relatives.
Maternal effects are not estimable from the standard twin design. Monozygotic and dizygotic twins presumably have similar maternal influences on the trait of interest, unless placentation is critical, yet they differ by additive and potentially dominant genetic effects on the trait. Twins also are relatively uncommon in the population and experience disproportionately elevated rates of obstetric complications, such as low birth weight and prematurity. Contrary to twins, contrasts of maternal versus paternal lineages from half-siblings and cousins are informative for maternal effects because additive genetic contributions are predictable and dominant effects should be largely absent, while shared environmental effects should be minimized and are assumed to be zero in our models for cousins. Therefore, differences in recurrence risk between equivalent maternal and paternal lineages should yield legitimate estimates of maternal effects.
In this study, we estimated maternal effects, together with additive genetic and shared environmental effects, for ASD using Swedish family data of nontwin (or singleton) births between the years 1998 to 2007. We also separated ASD into two diagnostic types relevant to that birth cohort, namely DSM-IV autistic disorder (AD), which captures more severely affected individuals, and individuals who have a diagnosis of ASD not as severe as AD, which we term spectrum disorder (SD). AD and SD are known to differ in the distribution of intellectual function, as well as ASD severity; in particular a greater fraction of AD subjects also have comorbid intellectual disability (32,33). Thus, this partition could also be meaningful for maternal effects.
METHODS AND MATERIALS
Study Population and Family Structure
Using the Swedish Medical Birth Register (34), we created a cohort consisting of all singletons who were born in Sweden between 1998 and 2007 and lived beyond 2 years of age. Note that children born in 2007 were 8 years old in 2014, the close of follow-up. The Swedish Multi-generation Register (35), which consists of all children born in Sweden since 1932 (and alive in 1961) and their parents, was used to identify parents and grandparents of eligible children and construct seven family types. We first identified cousin pairs: those related through mothers who were sisters were maternal parallel cousins (mPCs); those related through fathers who were brothers were paternal parallel cousins (pPCs); and cousins of other relationships were cross-cousins (CCs). Of the families that remained, the next pairings were families with half-siblings (HSs), of either maternal (mHS) or paternal origin (pHS); note that these families could also contain full siblings. Finally, we formed “nonpaired families,” consisting of full siblings. If there were more than two eligible pairings per family, say mPCs and CCs, then only eligible children of the oldest two siblings formed the paired family; we also limited families to a maximum of six children.
Outcome Variables
ASD, AD, and SD were ascertained using the Swedish National Patient Register. In addition to routine medical and developmental screening, all children had mandatory developmental assessment at 4 years of age, consisting of motor, language, cognitive, and social development. Children with a suspected developmental disorder were referred for further assessment to a specialized team. Diagnostic information was reported to the National Patient Register using the ICD-10: AD (ICD-10: F84.0); SD, (ICD-10: F84.5 “Asperger’s syndrome”), and/or pervasive developmental disorder not otherwise specified (ICD-10: F84.9). See the Supplement for more details regarding diagnosis.
Statistical Methods
All analyses are conducted using R version 3.2.29 (36). To compare ASD risk in families of different relatedness, we calculated the recurrence risk and family recurrence risk ratio (FRR). The two measures are expected to vary across paired families in the presence of a true underlying maternal effect; for instance, first, the FRR should be greater for mPCs than pPCs. Second, we fitted variance component models to estimate the contribution of maternal, additive, and environmental effects, and we repeated those analyses separately for AD and SD. FRR is calculated as the ratio of family/sibling recurrence risk and the population prevalence (37). To estimate the recurrence risk, we identified all index cases and calculated the proportion of individuals diagnosed among their full siblings, half-siblings, and cousins of this proband. Then, for each family type, we defined the recurrence risk as the proportion of affected individuals among all siblings/cousins of those affected and defined FRR as this proportion divided by the prevalence. Confidence intervals were calculated by assuming a binomial distribution (38). For the severity subtypes, we require the proband and relative pairs to be of the same diagnostic subtype.
Liability Modeling
Maternal, additive genetic, and shared environmental effects can be estimated because the strength of genetically induced correlation of a trait varies by degree of relatedness between pairs. A residual term, which is commonly interpreted as unshared environmental effects, can also be estimated, although, in reality, it could be influenced by unmodeled genetic effects as well.
We assume a genetic coefficient of relationship of 0.5 for full siblings, 0.25 for half-siblings, and 0.125 for cousins (27,39). Maternal effect coefficients are assumed to be 1 for full siblings and mHSs, 0.5 for mPCs, and 0 for other relative pairs. Shared environment is unique to maternal families assumed to be living together, specifically full siblings and mHSs (27), and its coefficient is set to 1 for these children and 0 otherwise (Table 1). Note that, without data from cousins, differentiating maternal effect from the shared environmental effect would be impossible (Table 1).
Table 1.
Assumed Genetic and Environmental Correlations Between Relative Pairs
| Variance Components | Relative pair relation | |||||
|---|---|---|---|---|---|---|
| Full Siblings | Half-siblings | Cousins | ||||
| mHSs | pHSs | mPCs | CCs | pPCs | ||
| Additive Genetic Effect | 0.5 | 0.25 | 0.25 | 0.125 | 0.125 | 0.125 |
| Maternal Effect | 1 | 1 | 0 | 0.5 | 0 | 0 |
| Shared Environmental Effect | 1 | 1 | 0 | 0 | 0 | 0 |
| Unshared Environmental Effect | 0 | 0 | 0 | 0 | 0 | 0 |
CCs, cross cousins; mHSs, maternal half-siblings; mPCs, maternal parallel cousins; pHSs, paternal half-siblings; pPCs, paternal parallel cousins.
Following Pawitan et al. (27), the liability model captures random effects for additive genetic Ai, maternal Mi, shared environment Ci, and unshared environment ei (error residuals), for the ith family in the total N paired families. Let , for i = 1, …, N, be the vector of binary outcomes from ni members. All paired families are assumed to be independent. Let xi, …, xN be the corresponding covariate matrices, each of size ni × p. We incorporate two categorical covariates into the model, sex (male vs. female) and birth year (2003–2007 vs. 1998–2002). Conditional on the random effects, we assume yij in to be an independent Bernoulli event with parameter , following a probit model: Φ−1(pi) = xiβ+Ai+ Mi+Ci+ei, where β is a p-vector of intercept and fixed regression parameters (sex and birth year). For each random effect, the random parameter captures the dependencies between members in the family; the design vector shows contribution to the outcome. To complete this specification, we assume , , , and , , where σ2 denotes variance component of each random effect, and R denotes respective correlation matrix. Marginal probabilities are calculated for all family units, with a Monte Carlo algorithm employed to optimize the log-likelihood of the generalized linear mixed model (27). Heritability is estimated by proportion of total variance explained by additive genetic component or effect (27), , and the fraction of variation explained by other variance components computed similarly. The lower and upper bounds of the 95% confidence interval for ith variance component is estimated by using profile likelihood approach (40). The reported probit-link values for fixed parameters are in standard normal quantile scale, which could be transformed to values comparable to general linear mixed model without random components by multiplying by (40).
Sensitivity and Complementary Analyses
We split additive genetic effect into maternal and paternal contributions (27). We assumed this additive genetic effect of maternal contributions to be independent of the maternal effect. A sensitivity analysis excluding half-siblings was conducted to avoid potential bias introduced by the assumption that children live with their mother.
When classifying families, we restricted the family size at six and took the first child born into a family between years 1998 and 2007 as the proband. A hierarchy was applied in the pedigree by prioritizing cousins over half-siblings. Also, in our evaluation of diagnostic subtypes, we restricted siblings of probands to match on subtype. Any of these restrictions could introduce some bias into the FRR calculations. For this reason, we also took a different approach to estimating the FRR for ASD and its subtypes AD and SD by degree of relatedness. To determine the FRR, we defined an index group as either all cases or a sample of control subjects of the same size as the number of cases. For each of the index individuals, we then determined the fraction of relatives of a certain type with a case diagnosis and the FRR (see the Supplement for details). Because control index subjects are randomly chosen, we repeat this process 100 times to get average estimates. Based on these estimates, we also derive rough estimates of genetic effects and heritability, as described in the Supplement.
RESULTS
From the cohort of children born between 1998 and 2007, we obtained 272,869 families in total, of which 55,117 (168,054 children) were mPCs, 104,400 (316,489 children) were CCs, and 54,776 (166,454 children) were pPCs; among those children who do not fit in any of the cousin paired families, 5185 (11,780 children) were pHSs and 6658 (14,865 children) were mHSs (Figure 1). In total, there were 776,212 children in our primary analysis, 51.52% male and 48.48% female. Of these, there were 11,231 (14.47 per 1000) individuals diagnosed with ASD, specifically 4554 with AD and 6677 with SD (Table 2). Sex ratios were 2.80 for ASD, 2.98 for AD, and 2.67 for SD. The rates of ASD in half-siblings were substantially higher than in cousins or full siblings.
Figure 1.
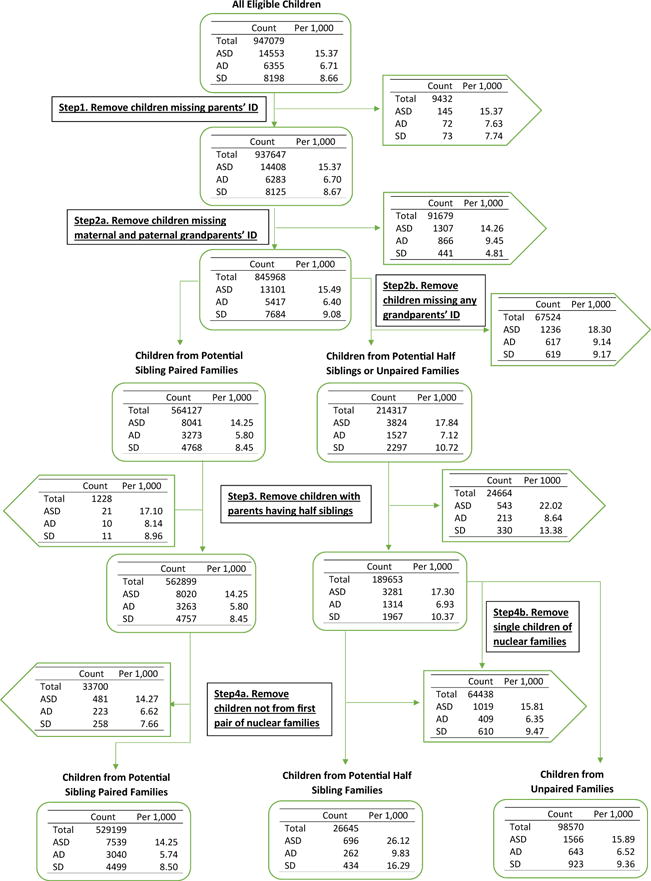
Flow chart describing the study population. AD, autistic disorder; ASD, autism spectrum disorder; ID, identification; SD, Asperger and pervasive development disorders not otherwise specified combined.
Table 2.
Outcome and Characteristics Across Families of Different Relatedness and Counts of Subjects Diagnosed With ASD, AD, and SD
| Outcome | Family types | ||||||
|---|---|---|---|---|---|---|---|
| mPCs | CCs | pPCs | pHSs | mHSs | Unpaired | Total | |
| Number of Families | 55,117 | 104,400 | 54,776 | 5185 | 6658 | 46,733 | 272,869 |
| Number of Children | 168,054 | 316,489 | 166,454 | 11,780 | 14,865 | 98,570 | 776,212 |
| Male, n (%) | 86,272 (51.34) | 163,214 (51.57) | 85,720 (51.50) | 6007 (50.99) | 7753 (52.16) | 50,927 (51.67) | 399,893 (51.52) |
| Female, n (%) | 81,782 (48.66) | 153,275 (48.43) | 80,734 (48.50) | 5773 (49.01) | 7112 (47.84) | 47,643 (48.33) | 376,319 (48.48) |
| Number of Cases | |||||||
| ASD | 2317 | 4397 | 2255 | 275 | 421 | 1566 | 11,231 |
| AD | 962 | 1798 | 889 | 94 | 168 | 643 | 4554 |
| SD | 1355 | 2599 | 1366 | 181 | 253 | 923 | 6677 |
| Outcome Rate (Cases per 1,000 Persons) | |||||||
| ASD | 13.79 | 13.89 | 13.55 | 23.34 | 28.32 | 15.89 | 14.47 |
| AD | 5.72 | 5.68 | 5.34 | 7.98 | 11.30 | 6.52 | 5.87 |
| SD | 8.06 | 8.21 | 8.21 | 15.37 | 17.02 | 9.36 | 8.60 |
AD, autistic disorder; ASD, autism spectrum disorder; CCs, cross cousins; mHSs, maternal half siblings; mPCs, maternal parallel cousins; pHSs, paternal half siblings; pPCs, paternal parallel cousins; SD, Asperger and pervasive development disorders not otherwise specified combined; Unpaired, full siblings not paired to cousins or half-siblings.
Family Recurrence Risk Ratio
For families accessed through an ASD proband, compared with those without, FRRs were largest for full siblings, intermediate for half-siblings, and smallest for cousins (Figure 2). Families accessed through an AD proband tended to have the largest FRR. The FRRs for cousin pairs were 2.68, 2.23, and 2.14 for mPCs, CCs, and pPCs, respectively, and their confidence intervals strongly overlap (Figure 2), suggesting limited support for a maternal effect. Similar patterns for FRR were seen for families accessed through AD and SD probands (Figure 2).
Figure 2.
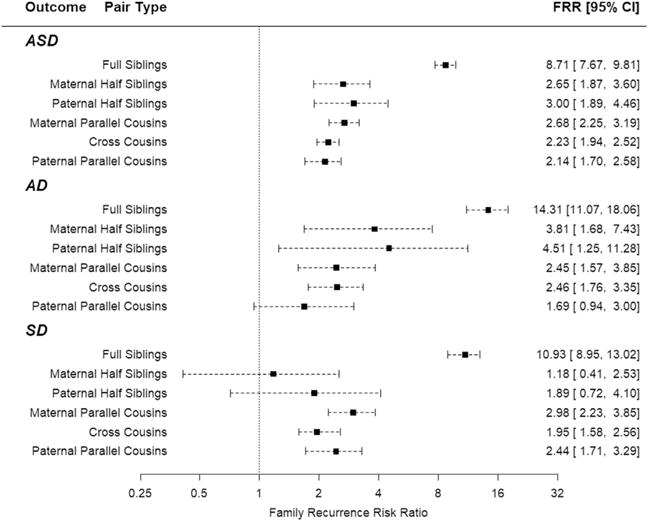
Family recurrence risk ratio (FRR) with 95% confidence intervals (CIs), by outcome and type of sibling/cousin relations. AD, autistic disorder; ASD, autism spectrum disorder; SD, Asperger and pervasive development disorders not otherwise specified combined.
Liability Model for an Outcome of ASD or ASD Subtypes
As expected (Table 3), there was higher risk of ASD and ASD subtypes for male subjects compared with female subjects. Moreover, due to shorter length of follow-up, lower risks were observed in the later birth cohort (years 2003–2007); the effect of birth cohort was smallest for AD, which could reflect either more consistent diagnosis of more severely affected individuals and/or a tendency toward later age of diagnosis among those less severely affected.
Table 3.
Estimated Fraction of Variation Explained for Liability of ASD, AD, and SD, and Estimated Coefficient for Fixed Parameters
| Estimatesa (95% CI) | Outcome | ||
|---|---|---|---|
| ASD | AD | SD | |
| Estimated Variance for Random Components (Fractions of Variation Explained) | |||
| Maternal effect | 0.004 (0, 0.052) | 0.001 (0, 0.074) | 0.007 (0.002, 0.068) |
| Additive genetic effect | 0.848 (0.731, 0.873) | 0.796 (0.612, 0.851) | 0.764 (0.630, 0.825) |
| Shared environmental effect | 0.002 (0, 0.037) | 0.007 (0, 0.097) | 0.002 (0, 0.047) |
| Unshared environmental effect | 0.147 (0.120, 0.201) | 0.195 (0.135, 0.274) | 0.227 (0.163, 0.293) |
| Estimated Coefficient for Fixed Parametersb | |||
| Gender, male | 0.395 (0.385, 0.401) | 0.372 (0.362, 0.387) | 0.356 (0.347, 0.367) |
| Birth cohort, 2003–2007 | −0.246 (−0.259, −0.237) | −0.093 (−0.104, − 0.077) | −0.324 (−0.338, − 0.309) |
AD, autistic disorder; ASD, autism spectrum disorder; CI, confidence interval; SD, Asperger and pervasive development disorders not otherwise specified combined.
The mixed model used a probit link. Random effects used in the liability model included maternal effect, direct genetic effect, shared environmental effect, and unshared environmental effect, and sex (1 = male, 0 = female) and birth cohort (1 = 2003–2007 cohort, 0 = 1998–2002 cohort) as fixed parameters. Coefficient for fixed effects indicates outcome risk associated with certain variable, while adjusted for other parameters.
The reported probit-link values for fixed parameters are in standard normal quantile scale.
Additive genetic effects accounted for the largest effect on liability, with little evidence for maternal and no evidence for shared environmental effects (Table 3, Supplemental Table S1). Similar results were obtained for severity subtypes. Estimates of heritability ranged from 85% for ASD to 76% for SD, with overlapping confidence intervals.
Sensitivity and Complementary Analyses
Sensitivity
Estimating separate maternal and paternal additive genetic effects, there were no statistically significant differences for ASD or subtypes, indicating an equal additive genetic effect contribution from the parents. The contribution tended to be somewhat larger for the maternal than paternal lineage for ASD (Supplemental Table S2). When half-siblings were excluded from the analysis, similar estimates were obtained for ASD and AD (see Table 3 vs. Supplemental Table S3); for SD, however, the estimates of maternal effects were notably larger when half-siblings were excluded versus included, although neither estimate of maternal effects was significantly different than zero.
Complementary Analyses
Next, we explored whether the limitations inherent in the data or the restrictions we put on the data could substantially alter results. Using alternative methods, we computed estimates of additive genetic and maternal effects that fell within the confidence intervals provided by the model (Supplemental Figure S1). Thus, our explorations supported additive genetic effects as the dominant contributor for ASD liability, as well as its subtypes (Supplement). Important features of the data, however, emerged from the alternative analyses, and they are described here.
Our approach to the liability model required grandparental information. As highlighted in Figure 1, this criterion eliminated a different proportion of AD versus SD subjects: the odds of removing AD subjects was 2.82 times larger than it was for SD (Fisher’s exact test, p < 2 × 10−16). Curiously, in contrast to full siblings and cousin pairs, point estimates for both pHSs and mHSs showed higher risk of ASD when the proband was unaffected (Supplemental Tables S5 and S6). For other pairs of relatives, the risk of ASD was no different than the prevalence; however, the rate was >1.5-fold larger for half-siblings, which was highly significant (pHSs, p < 3.6 × 10−7; mHSs, p < 1.9 × 10−5; one-sample Student’s t tests, df = 99), and rate was somewhat larger for SD than AD.
In addition to altering the ratio of AD to SD counts in the population, the emphasis of our liability model on cousin relationships forced a lower count of half-sibling pairs. For these and other reasons, we used a different approach to estimating FRR that obviates these drawbacks for some family relationships (see Methods and Materials and the Supplement). Several results emerged from these analyses. First, the risk for ASD and subtypes showed patterns close to that expected under an additive genetic model (Supplemental Table S7). Yet, when expressed as the FRR, the half-sibling’s recurrence risks were deflated by the higher prevalence of ASD in half-siblings compared with full siblings, and the same phenomenon occurred for the severity subtypes (Supplemental Tables S8 and S9). This feature could account for the lack-of-fit for full siblings in the liability model and provides justification for our emphasis on cousin pairs rather than half-siblings. Second, whether we kept the restriction from the liability model that probands and relative pairs for a severity subtype must match, or relaxed the restriction to allow the relatives to be of any ASD subtype, the patterns of FRR (Supplemental Tables S8 and S9) were similar to that seen in Figure 2, emphasizing the predominant contribution of additive genetic effects and minor, if any, contribution from maternal and shared environmental effects. Indeed, fitting these results to a simple method of moments model yielded point estimates for heritability consistent with terms in the liability model, although with a somewhat higher estimate of maternal effect for SD (0.14 vs. 0.007) (Supplemental Tables S10–S12, Supplemental Figure S1).
DISCUSSION
Many studies have evaluated the heritability of ASD (2,8,11,12,15,41), most but not all using twins, and no study has estimated the maternal effect of ASD. The majority of these studies conclude that ASD is a substantially heritable trait. Our results support this conclusion: in this Swedish population sample, we estimated heritability as 84.8%. The results agree quite well with the most recent meta-analysis of twin studies (12), which also estimated heritability in the range of 64% to 91%.
The results for AD and SD were similar to those for ASD (Table 3, Supplemental Figure S1). Based on the bulk of the analyses, we believe these subtypes are consistent with an additive genetic model for liability. Specifically, first- and second-degree relatives of AD probands who are diagnosed with the more severe form of ASD tend to have greater risk and recurrence risk than relatives of SD probands, and this tendency holds across various alternative analyses (Table 3, Supplemental Tables S9 and S10). These results suggest that severity maps onto load of liability alleles. If that were true, we reasoned that first-degree relatives of AD probands would more likely to be diagnosed with AD, and conversely first-degree relatives of SD probands would more likely to be diagnosed with SD, than the alternative level of severity. This is indeed the case; the same diagnosis for proband and full siblings is roughly twofold more likely than is different severity subtypes (AD proband and sibling, p < 2.2 × 10−16; SD pro-band and sibling, p = 8.7 × 10−6; Fisher exact test) (Supplemental Table S13). Also consistent with our “genetic load” hypothesis, recurrence risk for SD for a half-sibling of an AD proband is greater than that for an SD proband. For cousins, however, no clear pattern emerges, suggesting that our hypothesis requires additional evaluation, especially using a multiple threshold model for AD and SD in the same population.
Curiously, the requirement of three-generation pedigrees generated significantly greater loss of AD and SD subjects (Figure 1). This pattern could be related to our hypothesized greater genetic load for AD versus SD families, and it could relate also to other psychopathology (42–44). This explanation is one of many, however. Another observation without obvious explanation is the significantly increased rate of ASD in half-siblings, compared with population prevalence. This pattern could be due to increased ASD genetic load carried by parents who tend to have multiple partners; it could be due to parental experience with ASD offspring; or it could also be due to parental age, which we expect to be greater, on average, for multipartner parents. Parental age is now an established risk factor (45–49).
Our interest was broader than heritability of ASD per se. One of the foci for its risk involves factors of maternal origin, such as maternal obesity, polycystic ovary syndrome, hypertensive disorders of pregnancy, and gestational diabetes. If these maternal phenotypes were a substantial source of liability for ASD in offspring, we reasoned their impact could be detected as a maternal effect. The maternal effect would be independent of the substantial additive genetic effect described above. It is important to note, however, that maternal effects per se can be influenced by both genetic and environmental factors. Our general linear mixed model covers the genetic part explicitly (Figure 3, Tables 1 and 4), whereas the FRR should reflect whichever of these components are relevant (Figure 2, Table 3). Because recent results suggest that certain maternal phenotypes influence ASD status of offspring, we wondered how much of the population-level variation in ASD diagnosis could be ascribed to maternal effects.
Figure 3.
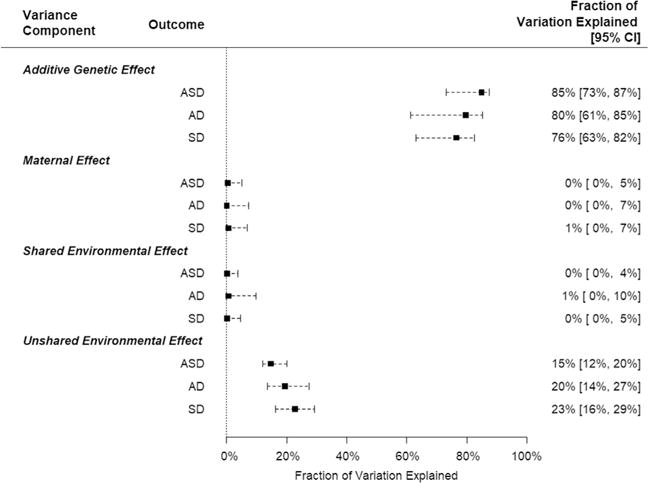
Fraction of total variation explained by each variance component with 95% confidence interval (CI), by outcome. AD, autistic disorder; ASD, autism spectrum disorder; SD, Asperger and pervasive development disorders not otherwise specified combined.
When we estimate a maternal effect, however, its impact on population-level variability in diagnosis is limited or nonexistent. This critical finding paves the way toward insights into causal mechanisms that could mediate the effects of maternal factors. While many maternal traits associated with ASD risk are heritable, their impact on ASD risk could be sporadic, thereby limiting detectability of maternal effects through shared maternal ancestry. There are other explanations, however. Consider the association between maternal immune status and ASD risk. This too could be too sporadic to detect with our design. Alternatively, if genetic factors increased risk for both altered maternal immune status and offspring ASD, there would be no evidence for maternal effects. Under such a model, it is not that maternal immune status contributes to ASD risk but rather that there is a shared genetic risk that contributes to altered maternal immune status and ASD risk in the child. In this scenario, animal models that perturb immune pathways in pregnant dams would have limited construct validity as ASD models.
While the current data cannot address this fully, it seems likely that maternal risk factors contribute only modestly to liability for ASD, possibly adding to existing genetic vulnerability of the child. If this hypothesis were true, it parallels the prevailing genetic model in which inherited variation accounts for the bulk of liability, but de novo variation also plays some role (3,43,50). This hypothesis can be evaluated by population-based studies, such as this one, but it must also integrate maternal risk factors relevant to each birth and the genetics of the children. Then, using mixed-effects models, one can partition variability in liability into sources such as additive and de novo genetic, maternal, and potentially other environmental exposures.
There could also be other factors partly explaining our results. Because maternal metabolic conditions associated with ASD have well-known effects on fertility, the maternal effect could to some extent be masked by a decrease in fertility. It is also reasonable to sound this note of caution: while we can be confident that inherited genetic variation accounts for the bulk of liability to ASD and maternal genetic contributions must be far smaller, we cannot conclude the maternal genetic contribution is zero for several reasons. First, although our population sample is large, even larger samples could detect modest effects that we did not. Second, we fit a relatively simple quantitative genetics model to the data, and geneticists will recognize it is only a model of reality, not reality itself. Moreover, three-generation pedigrees make only simple genetic models estimable in most settings. Finally, not all of our results are consistent with zero maternal contribution. In fitting the data for severity subtypes, some evidence emerged for maternal effects on liability, although they were never large.
Regardless of the caveats, our general conclusions—large additive genetic contribution to liability for ASD and modest, if any, maternal genetic contribution—are strongly supported by the data. We show that these conclusions are robust to most modeling assumptions and how risk and recurrence risk are estimated.
Supplementary Material
Acknowledgments
This study was supported by grants from the National Institutes of Health (Grant Nos. HD073978 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institute of Environmental Health Sciences, and National Institute of Neurological Disorders and Stroke to AR); Grant No. MH097849 from the National Institute of Mental Health (NIMH) to JDB; by the Beatrice and Samuel A. Seaver Foundation; NIMH Grant No. R37MH057881 (to KR and BD); by the Mindich Child Health and Development Institute (to DEG, JB, AR, SS, BM), and the Friedman Brain Institute, Icahn School of Medicine at Mount Sinai.
The sponsors of the study had no role in study design, data collection, data analysis, data interpretation, writing of the report, or in the decision to submit the paper for publication. The corresponding author had full access to all data in the study and had final responsibility for the decision to submit for publication.
Footnotes
DISCLOSURES
The authors report no biomedical financial interests or potential conflicts of interest.
Supplementary material cited in this article is available online at https://doi.org/10.1016/j.biopsych.2017.09.007.
References
- 1.Lauritsen MB. Autism spectrum disorders. Eur Child Adolesc Psych. 2013;22:S37–S42. doi: 10.1007/s00787-012-0359-5. [DOI] [PubMed] [Google Scholar]
- 2.Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46:881–885. doi: 10.1038/ng.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klei L, Sanders SJ, Murtha MT, Hus V, Lowe JK, Willsey AJ, et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol Autism. 2012;3:9. doi: 10.1186/2040-2392-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kosmicki JA, Samocha KE, Howrigan DP, Sanders SJ, Slowikowski K, Lek M, et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat Genet. 2017;49:504–510. doi: 10.1038/ng.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanders SJ, Xin H, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87:1215–1233. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson GM. Twin studies in autism: What might they say about genetic and environmental influences. J Autism Dev Disord. 2012;42:1526–1527. doi: 10.1007/s10803-012-1552-6. [DOI] [PubMed] [Google Scholar]
- 7.Deng W, Zou X, Deng H, Li J, Tang C, Wang X, et al. The relationship among genetic heritability, environmental effects, and autism spectrum disorders: 37 pairs of ascertained twin study. J Child Neurol. 2015;30:1794–1799. doi: 10.1177/0883073815580645. [DOI] [PubMed] [Google Scholar]
- 8.Frazier TW, Thompson L, Youngstrom EA, Law P, Hardan AY, Eng C, et al. A twin study of heritable and shared environmental contributions to autism. J Autism Dev Disord. 2014;44:2013–2025. doi: 10.1007/s10803-014-2081-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68:1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hallmayer J, Glasson EJ, Bower C, Petterson B, Croen L, Grether J, et al. On the twin risk in autism. Am J Hum Genet. 2002;71:941–946. doi: 10.1086/342990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taniai H, Nishiyama T, Miyachi T, Imaeda M, Sumi S. Genetic influences on the broad spectrum of autism: Study of proband-ascertained twins. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:844–849. doi: 10.1002/ajmg.b.30740. [DOI] [PubMed] [Google Scholar]
- 12.Tick B, Bolton P, Happe F, Rutter M, Rijsdijk F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J Child Psychol Psychiatry. 2016;57:585–595. doi: 10.1111/jcpp.12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gronborg TK, Schendel DE, Parner ET. Recurrence of autism spectrum disorders in full- and half-siblings and trends over time: A population-based cohort study. JAMA Pediatr. 2013;167:947–953. doi: 10.1001/jamapediatrics.2013.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ozonoff S, Young GS, Carter A, Messinger D, Yirmiya N, Zwaigenbaum L, et al. Recurrence risk for autism spectrum disorders: A Baby Siblings Research Consortium Study. Pediatrics. 2011;128:E488–E495. doi: 10.1542/peds.2010-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, Reichenberg A. The familial risk of autism. JAMA. 2014;311:1770–1777. doi: 10.1001/jama.2014.4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Casas M, Chatzi L, Carsin AE, Amiano P, Guxens M, Kogevinas M, et al. Maternal pre-pregnancy overweight and obesity, and child neuropsychological development: Two Southern European birth cohort studies. Int J Epidemiol. 2013;42:506–517. doi: 10.1093/ije/dyt002. [DOI] [PubMed] [Google Scholar]
- 17.Gardner RM, Lee BK, Magnusson C, Rai D, Frisell T, Karlsson H, et al. Maternal body mass index during early pregnancy, gestational weight gain, and risk of autism spectrum disorders: Results from a Swedish total population and discordant sibling study. Int J Epidemiol. 2015;44:870–883. doi: 10.1093/ije/dyv081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kosidou K, Dalman C, Widman L, Arver S, Lee BK, Magnusson C, et al. Maternal polycystic ovary syndrome and the risk of autism spectrum disorders in the offspring: a population-based nationwide study in Sweden. Mol Psychiatry. 2016;21:1441–1448. doi: 10.1038/mp.2015.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mehta SH, Kerver JM, Sokol RJ, Keating DP, Paneth N. The association between maternal obesity and neurodevelopmental outcomes of offspring. J Pediatr. 2014;165:891–896. doi: 10.1016/j.jpeds.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 20.Rivera HM, Christiansen KJ, Sullivan EL. The role of maternal obesity in the risk of neuropsychiatric disorders. Front Neurosci. 2015;9:194. doi: 10.3389/fnins.2015.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez A. Maternal pre-pregnancy obesity and risk for inattention and negative emotionality in children. J Child Psychol Psychiatry. 2010;51:134–143. doi: 10.1111/j.1469-7610.2009.02133.x. [DOI] [PubMed] [Google Scholar]
- 22.Silverman BL, Rizzo T, Green OC, Cho NH, Winter RJ, Ogata ES, et al. Long-term prospective evaluation of offspring of diabetic mothers. Diabetes. 1991;40:121–125. doi: 10.2337/diab.40.2.s121. [DOI] [PubMed] [Google Scholar]
- 23.Tanda R, Salsberry PJ, Reagan PB, Fang MZ. The impact of prepregnancy obesity on children’s cognitive test scores. Matern Child Health J. 2013;17:222–229. doi: 10.1007/s10995-012-0964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tuovinen S, Raikkonen K, Kajantie E, Leskinen JT, Henriksson M, Pesonen AK, et al. Hypertensive disorders in pregnancy and intellectual abilities in the offspring in young adulthood: The Helsinki Birth Cohort Study. Ann Med. 2012;44:394–403. doi: 10.3109/07853890.2011.573497. [DOI] [PubMed] [Google Scholar]
- 25.Lyall K, Ashwood P, Van de Water J, Hertz-Picciotto I. Maternal immune-mediated conditions, autism spectrum disorders, and developmental delay. J Autism Dev Disord. 2014;44:1546–1555. doi: 10.1007/s10803-013-2017-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Falconer DS. Introduction to Quantitative Genetics. London, UK: Longman; 1981. [Google Scholar]
- 27.Pawitan Y, Reilly M, Nilsson E, Cnattingius S, Lichtenstein P. Estimation of genetic and environmental factors for binary traits using family data. Stat Med. 2004;23:449–465. doi: 10.1002/sim.1603. [DOI] [PubMed] [Google Scholar]
- 28.Svensson AC, Sandin S, Cnattingius S, Reilly M, Pawitan Y, Hultman CM, et al. Maternal effects for preterm birth: A genetic epidemiologic study of 630,000 families. Am J Epidemiol. 2009;170:1365–1372. doi: 10.1093/aje/kwp328. [DOI] [PubMed] [Google Scholar]
- 29.Cnattingius S, Reilly M, Pawitan Y, Lichtenstein P. Maternal and fetal genetic factors account for most of familial aggregation of pre-eclampsia: A population-based Swedish cohort study. Am J Med Genet A. 2004;130A:365–371. doi: 10.1002/ajmg.a.30257. [DOI] [PubMed] [Google Scholar]
- 30.Lichtenstein P, Yip BH, Bjork C, Pawitan Y, Cannon TD, Sullivan PF, et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: A population-based study. Lancet. 2009;373:234–239. doi: 10.1016/S0140-6736(09)60072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yip BH, Bjork C, Lichtenstein P, Hultman CM, Pawitan Y. Covariance component models for multivariate binary traits in family data analysis. Stat Med. 2008;27:1086–1105. doi: 10.1002/sim.2996. [DOI] [PubMed] [Google Scholar]
- 32.Miles JH, Takahashi TN, Bagby S, Sahota PK, Vaslow DF, Wang CH, et al. Essential versus complex autism: Definition of fundamental prognostic subtypes. Am J Med Genet A. 2005;135A:171–180. doi: 10.1002/ajmg.a.30590. [DOI] [PubMed] [Google Scholar]
- 33.Spiker D, Lotspeich LJ, Dimiceli S, Myers RM, Risch N. Behavioral phenotypic variation in autism multiplex families: Evidence for a continuous severity gradient. Am J Med Genet. 2002;114:129–136. doi: 10.1002/ajmg.10188. [DOI] [PubMed] [Google Scholar]
- 34.Cnattingius S, Ericson A, Gunnarskog J, Kallen B. A quality study of a medical birth registry. Scand J Soc Med. 1990;18:143–148. doi: 10.1177/140349489001800209. [DOI] [PubMed] [Google Scholar]
- 35.Ekbom A. The Swedish Multi-Generation Register. In: Dillner J, editor. Methods in Biobanking. Totowa, NJ: Humana Press Inc; 2011. pp. 215–220. [DOI] [PubMed] [Google Scholar]
- 36.R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: 2015. [Google Scholar]
- 37.Olson JM, Cordell HJ. Ascertainment bias in the estimation of sibling genetic risk parameters. Genet Epidemiol. 2000;18:217–235. doi: 10.1002/(SICI)1098-2272(200003)18:3<217::AID-GEPI3>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 38.Risch N, Hoffmann TJ, Anderson M, Croen LA, Grether JK, Windham GC. Familial recurrence of autism spectrum disorder: Evaluating genetic and environmental contributions. Am J Psychiatry. 2014;171:1206–1213. doi: 10.1176/appi.ajp.2014.13101359. [DOI] [PubMed] [Google Scholar]
- 39.Rijsdijk FV, Sham PC. Analytic approaches to twin data using structural equation models. Brief Bioinform. 2002;3:119–133. doi: 10.1093/bib/3.2.119. [DOI] [PubMed] [Google Scholar]
- 40.Noh M, Yip B, Lee Y, Pawitan Y. Multicomponent variance estimation for binary traits in family-based studies. Genet Epidemiol. 2006;30:37–47. doi: 10.1002/gepi.20099. [DOI] [PubMed] [Google Scholar]
- 41.Hoekstra RA, Bartels M, Verweij CJH, Boomsma DI. Heritability of autistic traits in the general population. Arch Pediatr Adolesc Med. 2007;161:372–377. doi: 10.1001/archpedi.161.4.372. [DOI] [PubMed] [Google Scholar]
- 42.Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–994. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson EB, Samocha KE, Kosmicki JA, McGrath L, Neale BM, Perlis RH, et al. Autism spectrum disorder severity reflects the average contribution of de novo and familial influences. Proc Natl Acad Sci U S A. 2014;111:15161–15165. doi: 10.1073/pnas.1409204111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson EB, St Pourcain B, Anttila V, Kosmicki JA, Bulik-Sullivan B, Grove J, et al. Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nat Genet. 2016;48:552–555. doi: 10.1038/ng.3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Croen LA, Najjar DV, Fireman B, Grether JK. Maternal and paternal age and risk of autism spectrum disorders. Arch Pediatr Adolesc Med. 2007;161:334–340. doi: 10.1001/archpedi.161.4.334. [DOI] [PubMed] [Google Scholar]
- 46.Grether JK, Anderson MC, Croen LA, Smith D, Windham GC. Risk of autism and increasing maternal and paternal age in a large North American population. Am J Epidemiol. 2009;170:1118–1126. doi: 10.1093/aje/kwp247. [DOI] [PubMed] [Google Scholar]
- 47.Hultman CM, Sandin S, Levine SZ, Lichtenstein P, Reichenberg A. Advancing paternal age and risk of autism: New evidence from a population-based study and a meta-analysis of epidemiological studies. Mol Psychiatr. 2011;16:1203–1212. doi: 10.1038/mp.2010.121. [DOI] [PubMed] [Google Scholar]
- 48.Lampi KM, Hinkka-Yli-Salomaki S, Lehti V, Helenius H, Gissler M, Brown AS, et al. Parental age and risk of autism spectrum disorders in a Finnish national birth cohort. J Autism Dev Disord. 2013;43:2526–2535. doi: 10.1007/s10803-013-1801-3. [DOI] [PubMed] [Google Scholar]
- 49.Sandin S, Schendel D, Magnusson P, Hultman C, Suren P, Susser E, et al. Autism risk associated with parental age and with increasing difference in age between the parents. Mol Psychiatr. 2016;21:693–700. doi: 10.1038/mp.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.