

Abstract
The circadian timing system influences the functional properties of most, if not all, physiological processes. Central to the mammalian timing system is the suprachiasmatic nucleus (SCN) of the hypothalamus. The SCN functions as a ‘master clock’ that sets the phasing of ancillary circadian oscillator populations found throughout the body. Further, via an entraining input from the retina, the SCN ensures that the clock oscillators are synchronized to the daily light/dark cycle. A critical component of the SCN timing and entrainment systems is the p44/42 mitogen-activated protein kinase (ERK/MAPK) pathway. Here, we examined the expression and function of phosphoprotein-enriched in astrocytes (PEA-15), an ERK scaffold protein that serves as a key regulator of MAPK signaling. A combination of immunolabeling and Western blotting approaches revealed high levels of PEA-15 within the SCN. PEA-15 expression was enriched in distinct subpopulations of SCN neurons, including arginine vasopressin (AVP)-positive neurons of the SCN shell region. Further, expression profiling detected a significant circadian oscillation in PEA-15 expression within the SCN. Brief photic stimulation during the early subjective night led to a significant increase in PEA-15 phosphorylation, an event that can trigger ERK/PEA-15 dissociation. Consistent with this, co-immunoprecipitation assays revealed that PEA-15 is directly bound to ERK in the SCN, and that photic stimulation leads to their dissociation. Finally, we show that PEA-15 regulates ERK/MAPK-dependent activation of the core clock gene period1. Together, these data raise the prospect that PEA-15 functions as a key regulator of the SCN timing system.
Keywords: C57Bl/6, PEA-15, Circadian, ERK, suprachiasmatic nucleus
Graphical abstract
Here, we report that the ERK scaffold protein PEA-15 is highly expressed in the suprachiasmatic nucleus (SCN). Further, we show that both its expression and phosphorylation state are under the control of the circadian clock. We also report that the PEA-15/ERK complex is regulated by photic input, and that PEA-15 regulates the capacity of the ERK/MAPK pathway to stimulate expression of the core clock gene period1. These data indicate that PEA-15 is ideally positioned to affect SCN clock timing and entrainment.
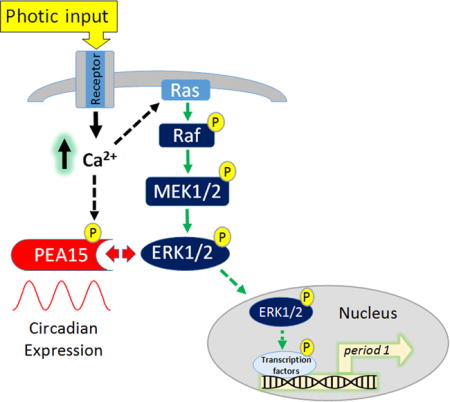
INTRODUCTION
In mammals, the suprachiasmatic nucleus (SCN) of the hypothalamus serves as the master circadian pacemaker (Silver and Moore, 1998; Weaver, 1998; Mohawk et al., 2012). Daily timing cues generated by the SCN set the phasing and amplitude of ancillary clock populations found throughout all organ systems of the body, and the loss of clock output from the SCN leads to the disruption, and/or dysregulation, of a wide array of biochemical, physiological and behavioral rhythms (Stephan and Zucker, 1972; Eastman et al., 1984; Yoo et al., 2004).
At a cellular level, the inherent timing capacity of SCN neurons is generated by a transcriptional/translational/posttranslational feedback loop, which is centered on the rhythmic expression of the core clock genes period1/2 and cryptochrome1/2 (Shearman et al., 2000; Partch et al., 2014). The expression of period and cryptochrome genes is driven by a heterodimeric transcription factor formed by CLOCK/BMAL1, which binds to an E-Box motif within the 5′ regulatory regions of these genes. The rhythmicity of the circadian oscillator is set by a negative feedback circuit in which PERIOD/CRYPTOCHROME protein complexes negatively regulate CLOCK/BMAL1-mediated transcription, and the frequency of the oscillatory period is set by the accumulation and degradation rates of the PERIOD/CRYPTOCHROME protein complexes (Shearman et al., 2000; Meng et al., 2008).
This core clock feedback loop is influenced by an array of activity-regulated kinase and phosphatase signaling pathways (Vanselow and Kramer, 2007; Virshup et al., 2007). In addition, kinase pathways are principal routes through which retinal input sets the phasing of the molecular oscillator (Meijer and Schwartz, 2003; Hirota and Fukada, 2004). Consistent with these ideas, the p44/42 mitogen activated protein kinase (ERK/MAPK) pathway has been shown to play a key role in both the timing and the entrainment of the SCN oscillator (Goldsmith and Bell-Pedersen, 2013). With respect to clock timing, MAPK signaling exhibits a marked rhythm, and the abrogation of MAPK activity leads to a damping of clock gene rhythms and reduction in neuronal firing properties (Obrietan et al., 1998; Akashi et al., 2008). With respect to clock entrainment, photic stimulation during the circadian night triggers a marked increase in ERK/MAPK pathway activation, and the pharmacological suppression of MAPK signaling leads to the disruption of light-evoked clock resetting (Obrietan et al., 1998; Butcher et al., 2002; Coogan and Piggins 2003). Given that MAPK signaling has a role in key aspects of cellular timing and entrainment, several studies have examined intracellular signaling events that affect ERK/MAPK signaling (Shimizu et al., 2003; Cheng et al., 2006; Hainich et al., 2006). Here, we focused on the expression and the function of the ERK (the MAPK pathway effector kinase) scaffold protein PEA-15 (Phosphoprotein Enriched in Astrocytes 15) in the SCN. As the name denotes, PEA-15 was originally shown to be highly enriched in astrocytes (Araujo et al. 1993); however, more recent work found that PEA-15 is also expressed in neurons (Sharif et al., 2004). Much of the interest in PEA-15 has been centered on its functional role as a scaffolding protein for ERK and Fas-associated death domain protein (FADD) (Renganathan et al., 2005; Fiory et al., 2009; Valmiki and Ramos, 2009). With respect to MAPK signaling, PEA-15 has been shown to sequester ERK in the cytoplasm, and thus suppress its ability to translocate to the nucleus and stimulate inducible gene expression (Formstecher et al., 2001; Renganathan et al., 2005). Interestingly, the ability of PEA-15 to function as a scaffold is regulated by the activation state of the cell. Along these lines, the phosphorylation of PEA-15 at Serine-104 and Serine-116 (via protein kinase C, and CaM kinase II, respectively) leads to the dissociation of the ERK-PEA15 complex, and in turn, ERK nuclear translocation and the induction of MAPK-mediated gene expression (Kubes et al., 1998; von Kriegsheim et al., 2009; Sulzmaier et al., 2012). Here we report on the expression and regulation of PEA-15 in the SCN, and provide mechanistic insights into its potential roles in circadian clock timing and entrainment.
MATERIALS AND METHODS
Animals
In total, 166 C57Bl/6 mice (females and males, which were equally distributed across all experimental groups; aged 6 to 14 weeks) were used. Mice were generated from an in-house breeding colony, which was established from founders acquired from Jackson Laboratory (Bar Harbor, ME). Animals were maintained on a standard 12:12 light/dark cycle and were provided ad libitum access to water and food. All studies were approved by the Ohio State University Institutional Animal Care and Use Committee.
Circadian and light pulse experiments
Initially, animals were entrained to a 12:12 light/dark cycle for a minimum of 4-weeks. For the circadian analysis of PEA-15 and Phospho-PEA-15, mice were dark-adapted for 2 days and then sacrificed at 4-hr intervals over a 24 hour period. For light pulse experiments, animals were dark-adapted for 2 days and then exposed to white light (100 lux) for 30 seconds, and then euthanized. Control animals not exposed to white light were handled in a parallel manner with the light-treated animals. For both the circadian and light pulse experiments, mice were killed under dim red light (~ 5 lux) via rapid cervical dislocation. Brains were quickly removed, immersed in ice-cold physiological saline, cut into 500 μm coronal sections using a vibratome, and sections were fixed in paraformaldehyde (4%) for six hours at 4°C. Sections were then cryoprotected in 30% sucrose overnight, and then cut into thin sections (40 μm) with a freezing microtome. Data were collected from 6 animals per time point (or experimental condition).
Immunohistochemical labeling and analysis
Free-floating thin sections containing the SCN were immunolabeled using the following antibodies (all raised in rabbit): PEA-15 (1:9,000 dilution, catalog code: 2780, Cell Signaling Technology, Danvers, MA, USA), phospho-Serine-104 PEA-15 (pSer-104 PEA-15; 1:250, catalog code: 2776, Cell Signaling Technology), phospho-Serine-116 PEA-15 (pSer-116 PEA-15; 1:100 dilution, catalog code: 59216, Abcam, Cambridge, MA), and phospho-Threonine-202/Tyrosine-204 ERK (p-ERK; 1:3,000 dilution, catalog code: 9101, Cell Signaling Technology). Briefly, sections were treated with 0.3% hydrogen peroxide, blocked for 1 hour in 10% normal goat serum (NGS), and then incubated overnight at 4°C with the primary antibody diluted in 5% NGS/PBS containing 0.02% Triton X-100. Sections were then incubated with a biotinylated anti-rabbit secondary (1:1000 dilution, catalog code: BA-1000, Vector Laboratories, San Carlos CA) at room temperature for 2 hours, followed by 45 min incubation with horseradish peroxidase (HRP)/Avidin/Biotin complex (catalog code: PK-4000, Vectastain, Vector Laboratories). Exposure to a nickel-intensified DAB substrate (catalog code: SK-4100, Vector Laboratories) was used to visualize the immunolabeling/HRP complex. Of note, for the PEA-15 labeling, DAB development times were longer for ZT condition (Fig. 1A and 1B) than for CT data sets presented throughout the rest of the manuscript. Sections were mounted on gelatin-coated slides and coverslipped using Permount (Fischer Scientific). Sections were washed 3 times between each labeling step. Whole brain coronal sections were acquired with Nikon SMZ800N microscope (Nikon USA, Melville, NY); SCN images were acquired using a Leica DM IRB microscope (Leica Biosystems, Nussloch, Germany). Immunohistochemical labeling was quantitated using densitometry (0–255 scale) and cell counting; both methods used the ImageJ software program (National Institutes of Health, Bethesda, Maryland, USA). Densitometric analysis was used to examine PEA-15 and phospho-PEA-15 (pSer-104 PEA-15 and pSer- 116 PEA-15) expression over the circadian cycle, and to analyze light-evoked pSer-104 PEA-15, pSer-116 PEA-15 and p-ERK expression.
Figure 1. PEA-15 expression in the SCN.
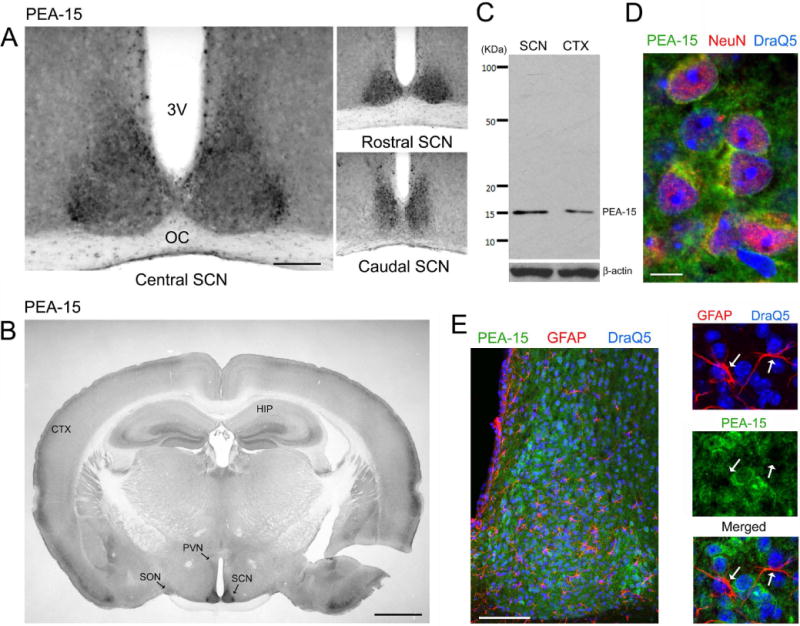
A) Representative PEA-15 immunolabeling within the central, rostral and caudal SCN. 3V, Third ventricle; OC, Optic chiasm Bar: 200 microns. B) Low magnification (4×) image of PEA-15. Marked expression of PEA-15 was observed in the SCN, with a lower level of expression observed in the paraventricular nucleus (PVN) and supraoptic nucleus (SON). HIP: hippocampus; CTX: cortex. Bar = 1 mm. C) Western blotting of cortical (CTX) and SCN lysates for PEA-15. As a loading control, the membrane was re-probed for β-actin. D) High magnification confocal image of PEA-15 (green), the neuronal nuclear marker NeuN (red) and the nuclear stain DraQ5 (blue) in the central SCN. PEA-15 was detected within the neuronal cytoplasm and perinuclear regions. Bar: 10 microns. E) Left panel: Low magnification image of the SCN immunolabeled for PEA-15 (green), the astrocytic marker, GFAP (red) and DraQ5 (blue). Right panels: magnified region from the central SCN: the three channels were overlaid in different combinations to aid in analyzing astrocytic PEA-15 expression. Arrows denote the locations of two astrocytes, as assessed using GFAP labeling. Of note, limited PEA-15 was detected in astrocytes. Bar = 100 microns.
Immunofluorescent labeling and analysis
Initially, thin-sectioned tissue was blocked with 10% NGS in PBS containing 0.02% Triton-X 100 for 1 hour, and then incubated overnight at 4°C with one, or a combination, of the following primary antibodies: mouse PEA-15 (1:500 dilution, catalog code: 166678, Santa Cruz Biotechnologies, Dallas, TX), rabbit PEA-15 (1:250 dilution, catalog code: #2780, Cell Signaling Technology), rabbit NeuN (1:3,000 dilution, catalog code: ABN78, Milipore), rabbit VIP (1:200 dilution, catalog code: SC-20727, Santa Cruz Biotechnologies), guinea pig AVP (1:1000 dilution, catalog code: 4562, Peninsula Laboratories International), rabbit pERK (1:3000 dilution, catalog code: 9101, Cell Signaling Technologies), mouse GFAP (1:1000 dilution, catalog code: 131-17719, Invitrogen), rabbit pSer-104 PEA-15 (1:200 dilution, catalog code: 2776, Cell Signaling Technologies), and Alexa Fluor conjugated pERK (1:100, catalog code: 4344, Cell Signaling Technologies). Of note, for the analysis of light-evoked pSer-104 PEA-15 and pERK (Fig. 5E), tissue was sequentially labeled with the rabbit pSer-104 PEA-15 primary antibody (1:200 dilution, catalog code: 2776, Cell Signaling Technologies), and then labeled with Alexa Fluor 594- conjugated anti-rabbit, followed by labeling with the Alexa Fluor 488-conjugated pERK (1:100, catalog code: 4344, Cell Signaling Technologies). Sequential labeling was required to ensure that cross labeling of the Alexa Fluor secondary used to detect pSer-104-PEA-15 did not occur because both primary antibodies were generated in rabbits. Next, tissue was labeled with the nucleic acid stain DRAQ5 (1:5,000 dilution, catalog code: 4084, Cell Signaling Technologies) and then mounted on glass slides and coverslipped with Fluoromount-G (catalog code: 0100-01, SouthernBiotech, Birmingham, AL). Sections were washed 3 times between each labeling step. Fluorescence images were acquired using a Leica SP8 confocal microscope; acquisition parameters (e.g., contrast settings, pinhole) for each fluorescent channel were held constant for each experimental group.
Figure 5. Light regulates PEA-15 phosphorylation in the SCN.
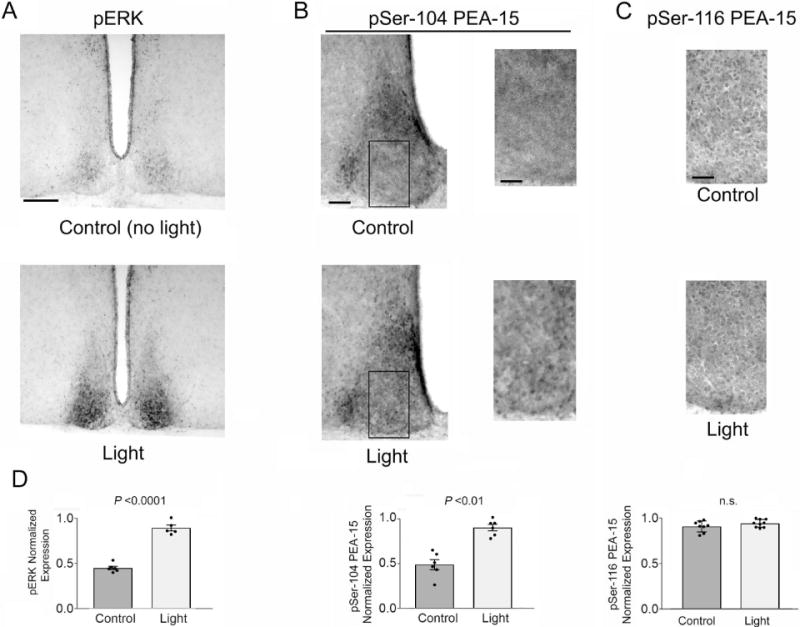
Mice were dark-adapted two days prior to a 30-second light pulse at CT 15, and then immediately sacrificed. A) IHC labeling was used to detect a light-evoked increase in ERK phospho-activation, relative to the control, no light, condition. Bar = 225 microns. B) Left panels: Representative 20× images of the SCN immunolabeled for the expression of pSer-104 PEA-15. Boxed regions are magnified (right panels), and used to highlight the increase in Ser104 phosphorylation in response to light. Bar = 100 microns. C) Representative high magnification images of pSer-116 PEA-15 in the SCN core region following the 30 sec light-pulse paradigm described in A. Note the consistent number of Ser-116 phosphorylated cells in the control and light-pulsed animal. Bar = 30 microns. D) Quantification of pERK-expression (left panel), pSer-104 PEA-15- positive cells within the SCN core (middle panel) and pSer-116 PEA-15-positive cells within the SCN core (right panel) in control and light-pulsed mice. Analysis was performed on 5–9 mice/group. Data were analyzed via the Student’s t-test. n.s.: not significant.
For the assessment of PEA-15 expression in AVP and VIP cells, confocal micrographs from double labeled SCN tissue (PEA-15/AVP and PEA-15/VIP) were initially thresholded to remove background/non-specific labeling. Next, immunopositive cells within the AVP (dorsal SCN) and VIP (central SCN) channels were digitally marked, and then the immunopositive cells within the PEA-15 channel were marked. The channels were then merged and the percentages of AVPergic and VIPergic cells that were positive for PEA-15 were determined.
Circadian expression analysis
Circadian expression analysis of PEA-15, was performed on the whole SCN, as well as the SCN core and shell regions (Fig. 3B); pSer-104 PEA-15 and pSer-116 PEA-15 analysis was performed on the whole SCN (Fig. 4). Regions were manually defined based on anatomical landmarks and our experience with the functional subdivisions of the SCN using ImageJ software. An example of our manual outline method is shown in figure 2A. Profiling was also performed on the hypothalamus; for this, measurements were made within a defined digital circle (400 microns in diameter) which was placed within the hypothalamus, just lateral to the SCN. The densitometry value from the SCN (and the noted subregions) was divided by the adjacent hypothalamic region to generate a normalized expression ratio. MATLAB R2015b (The MathWorks, Inc., Natick, MA, USA) was used to generate best fit cosine curves for the data represented in figure 3B and 4D.
Figure 3. Profiling PEA-15 expression across the circadian cycle.
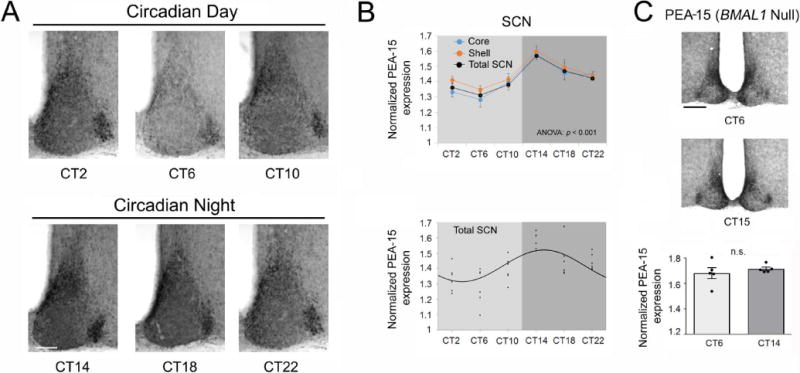
A) Representative IHC labeling for PEA-15; tissue was profiled at 4-hr intervals over a 24-hr period. Bar = 100 microns. B) Top graph: Densitometric analysis of PEA-15 IHC labeling over the circadian cycle in the SCN; profiling was also performed on core and shell subregions (n = 6 animals/time point). Significant time-of-day expression was observed in the SCN and within the noted subregions. Data were analyzed by one-way ANOVA (p < 0.001 for the SCN and for regional analyses). Bottom graph: Scatter-plot of the normalized PEA-15 data for the SCN. Each data point represents the mean value from an individual mouse. Data were fitted to a cosine curve, as described in the Methods section. C) Top: Representative images of PEA-15 expression in BMAL1 null mice. Animals were sacrificed at circadian time 6 (CT 6) and CT 15 (n = 5 animals/time point/genotype). Bar = 175 microns. Bottom: Densitometric analysis (both mean values and scatterplot data from individual mice) of PEA-15 IHC labeling in the SCN of BMAL1 null mice. PEA-15 expression was not significantly different at the two noted circadian times: Data were analyzed by Student’s t-test. n.s.: not significant (p >0.05).
Figure 4. Profiling phosphorylated PEA-15 expression across the circadian cycle.
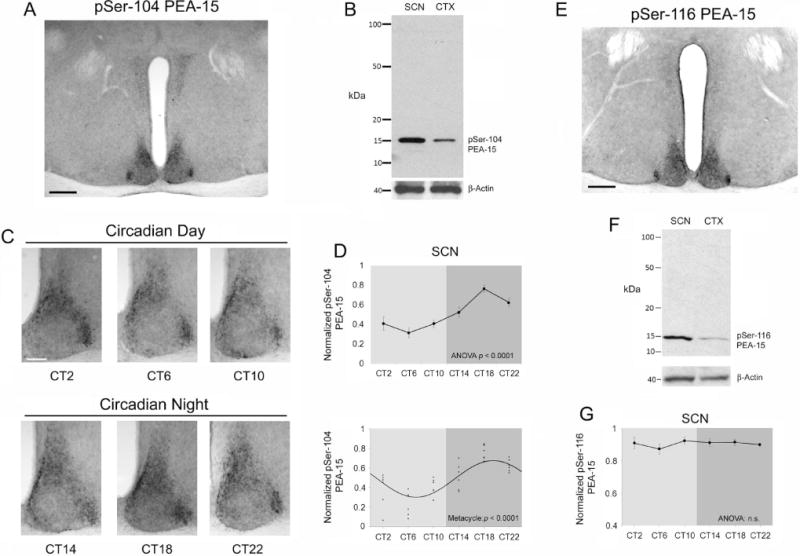
A) Representative 4× image of phospho-Serine-104 PEA-15 (pSer-104 PEA-15). Bar = 300 microns. B) Western blot of SCN and cortical (CTX) lysates for pSer-104 PEA-15. As a loading control, the membrane was re-probed for β- actin. C) Circadian profiling of pSer-104 PEA-15 at 4-hr intervals over a 24-hr period. Representative SCN images reveal an increase in Ser104 PEA-15 phosphorylation during the subjective nighttime. Bar = 100 microns. D) Top graph: Quantitative analysis of pSer-104 PEA-15 expression over the circadian cycle in the SCN (top) (n = 6 animals/time point) and analyzed via one-way ANOVA (p < 0.0001). Bottom panel: Scatter-plot of the normalized pSer-104 PEA-15 data. Data were fitted to a cosine curve, as described in the Methods section. E) Representative 4× image of phospho- Serine-116-PEA-15 (pSer-116 PEA-15) expression in the SCN. Bar = 300 microns. F) Western blot of SCN and cortical (CTX) lysates probed for pSer-116 PEA-15. As a loading control, the membrane was re-probed for β-actin (shown at the bottom). G) Quantitative analysis of pSer-116 PEA-15 expression over the circadian cycle in the SCN (n = 5 animals/time point). One-way ANOVA did not detect a time-of-day change in pSer-116 PEA-15 in the SCN. n.s.: not significant (p>0.05).
Figure 2. Cellular phenotype-specific expression of PEA-15 in the SCN.
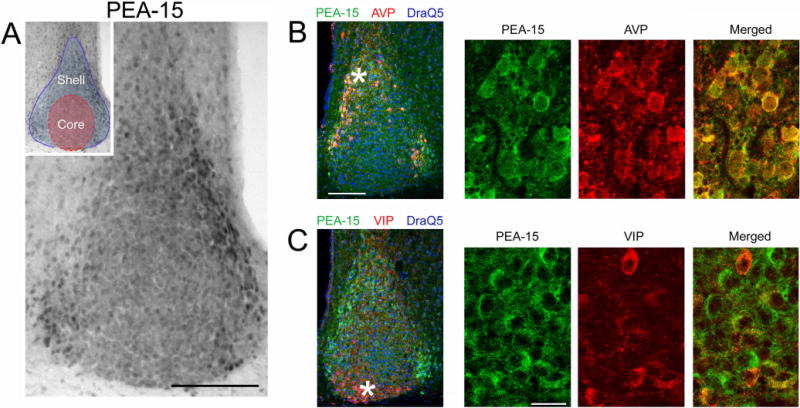
A) Immunohistochemical (IHC) labeling for PEA-15 in the SCN. Enriched PEA-15 was detected in the lateral and dorsal regions of the SCN. Inset: two functionally- and phenotypically-defined regions of the SCN are noted. Red shading corresponds to the core region; the blue outline and shading defines the shell region. Bar = 100 microns. B) Left panel: Fluorescent immunolabeling for PEA-15 (green channel), AVP (red channel) and DraQ5 (blue channel) in the SCN. Asterisk denotes the approximate location of the high magnification images shown in the rightward panels. Bar = 100 microns. C) Left panel: Fluorescent immunolabeling for PEA-15 (green), VIP (red) and DraQ5 (blue) in the SCN. Asterisk denotes the approximate location of the high magnification images shown in the rightward panels. Bar = 20 microns.
Western blots
Samples containing the SCN (and a minimal amount of surrounding hypothalamic tissue) and piriform cortex were isolated from 500 μm coronal sections. Samples were sonicated in 50 μl of RIPA buffer, supplemented with a cocktail of protease inhibitors (Complete Mini Tablet, Roche Diagnostics). Total protein was quantified using a BCA assay (Pierce BCA Assay Kit, catalog code: 23225, Thermo Scientific) and 10 μg of protein was mixed with an equivalent volume of 4X sample buffer, heated to 90°C for 10 min, and electrophoresed through a 10% SDS-PAGE gel using standard procedures. Protein samples were then transblotted onto PVDF membranes (Immobilon P: Millipore) and then blocked with 10% (wt/vol) powdered milk diluted in TBST (20mM Tris base, 150mM NaCl, 0.1% Tween 20, pH7.6) for 1 hr. Next, membranes were probed with antibodies against PEA-15 (1:1000 dilution, catalog code: 166678, Santa Cruz Biotechnologies), pSer-104 PEA-15 (1:1000 dilution, catalog code: 2776, Cell Signaling Technologies) or pSer-116 PEA15 (1:1000 dilution, catalog code: 59216, Abcam, Cambridge, MA). Membranes were then labeled with a goat anti-rabbit IgG secondary antibody conjugated to horseradish peroxidase (HRP, 1:2,000 dilution PerkinElmer, Boston, MA). HRP was detected using the Western-Lightning Plus-ECL kit (PerkinElmer). Membranes were then probed for β-actin (1:100,000 dilution, catalog code: 125-ACT, Phosphosolutions), and antigenicity was detected using the noted HRP labeling method. Membranes were washed three times (5 min/wash) in TBST between each antibody treatment.
Co-immunoprecipitation
Initially, mice were dark-adapted as described above, and then pulsed with light (100 lux, 30 sec) at CT15. Animals were immediately sacrificed, and 500 μm sections through the SCN were prepared as described above. Microdissected SCN were immediately frozen until further processing. Control animals were treated in the same manner, but were not pulsed with light. SCN tissue was pooled from 8 animals per condition in order to have sufficient protein to run a co-IP. Protein was lysed in a non-denaturing buffer (20 mM Tris HCL, 137 mM NaCl, 2 mM EDTA and 1% NP-40) and quantified as described for Western blotting. 300 μg of protein was then pre-cleared by the addition of 0.5 μg IgG antibody for 30 min, followed by 100 μL sepharose bead incubation for 1 hr at 4°C. Next, ERK-antibody bound beads (10 μl/sample; 1:200 dilution, catalog code: #5736, Cell Signaling Technologies) were added to the cleared lysates and incubated overnight at 4°C. Beads were pelleted by centrifugation and the supernatant was removed and incubated (4°C for 3-hours) a second time with 10 μL of ERK- bound beads. Beads were washed (3 times) after each incubation and then boiled in 4× sample buffer for 5 minutes at 95°C to both elute and denature proteins from the antibody complex. Samples were then loaded onto 15% acrylamide gels and processed as described above for Western analysis. PVDF membranes were probed with PEA-15 (1:1,000 dilution, catalog code: 166678, Santa Cruz Biotechnologies) and ERK (1:4,000, catalog code: 4696, Cell Signaling Technology) antibodies. Experiments were repeated 4-times and densitometric analysis of band intensities were performed with ImageJ (https://imagej.nih.gov/ij/). Immunoprecipitated ERK band intensities were normalized to 1% input ERK bands and co-immunoprecipitated PEA-15 band intensities were normalized to 1% PEA-15 bands. The normalized values obtained were then presented as a ratio of PEA-15 to ERK. Ratiometric values from light pulse and control conditions were compared with a two-tailed Student’s t- test.
Reporter gene assay
COS-7 cells were seeded into 24-well plates and maintained in 10% FBS DMEM with 1% penicillin/streptomycin. At 75% confluency, cells were transfected using lipofectamine-2000 (Invitrogen; per the manufacturer’s instructions) with a mouse period1-luciferase reporter construct (Butcher et al., 2005), and with either a PEA-15 expression vector or an empty expression vector control. The PEA-15 expression vector (VectorBuilder, Cyagen Biosciences, Santa Clara, CA) was engineered to express PEA-15 and GFP from separate minimal CMV promoters. To assess the concentration-dependent effects of transgenic PEA-15 on period1-luciferase expression, cells were lysed 48 hours after transfection, and total luminescence was measured in a plate-reader using luciferin as a substrate, per the manufacturer’s instructions (Luciferase Assay System, catalog code: E1500, Promega, Madison,WI). To examine the effects of PEA-15 and the MAPK pathway on stimulus-evoked period1-luciferase expression, 24 hours after transfection, cells were serum starved for 16-hours, and then pre-treated for 30 minutes with U0126 (10 μM, catalog code: 9903S, Cell Signaling Technology) or DMSO (control). Cells were then stimulated with 20% serum for 8-hours, followed by lysis and luminescence analysis, as described above. Luminescence values were determined by subtracting background counts from non-transfected control lysates, and then normalizing the counts to total protein levels. One-way ANOVA, followed by Bonferroni multiple comparison post-hoc test was used to analyze for statistical differences between conditions, p <0.05 was used to ascribe significance.
Statistical Analyses
For the quantitative analysis of ERK activation in relation to PEA-15 expression in the shell and core (Fig. 6A), 20× confocal images of the SCN were analyzed using ImageJ software. To this end, the SCN was digitally demarcated into core and shell regions. Next, mean PEA-15 and pERK expression levels (0–255 scale) were calculated for each region. Values for pERK and PEA-15 were divided by expression levels from an adjacent lateral hypothalamic region (as described above), to generate normalized fluorescence intensity. For the light-pulse assays, mean values were generated by averaging values from three central SCN sections per animal. These values were used to generate a mean ± the standard error of the mean for treatment groups. Regional quantification was performed by an individual that was blinded to the experimental conditions.
Figure 6. pERK and PEA-15 expression patterns and functional interactions.
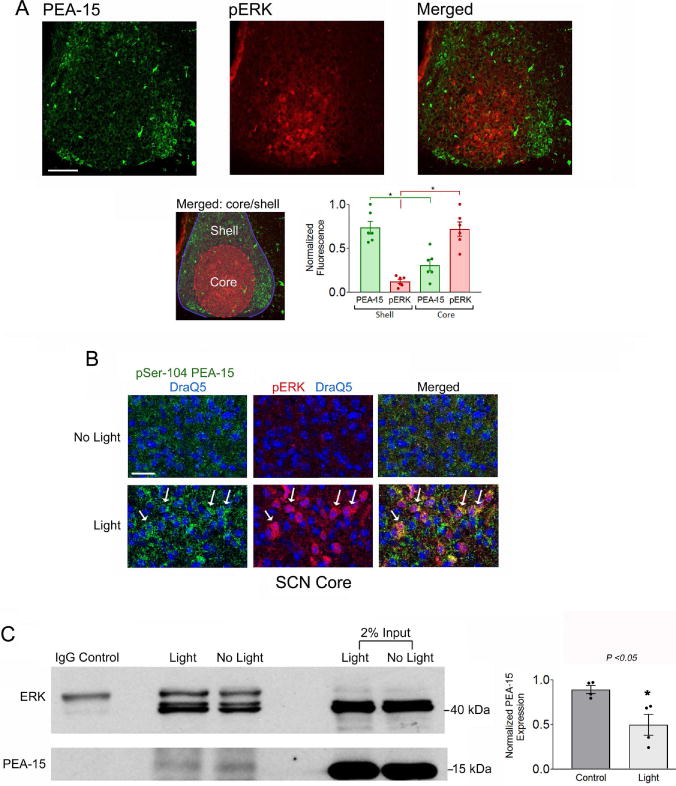
A) Top panels: Mice were dark-adapted for two days, pulsed with light (30 seconds, 100 lux) at CT15 and the SCN tissue was immunolabeled for PEA-15 and phospho-ERK (pERK). Light trigged marked ERK activation within the SCN core, a region that exhibited low levels of PEA-15, relative to the shell region. Bar = 75 microns. Bottom left panel: the merged image was color-coded to delineate the two noted regions; Red shading corresponds to the core region; the blue outline and shading defines the shell region. Bottom right panel: Densitometric analysis of PEA-15 and pERK expression in the core and shell regions of the SCN: data were analyzed by Student’s t-test. *: p < 0.0001. B) Representative confocal images of pSer-104 PEA-15 (green channel), pERK (red channel) and DraQ5 (blue channel) from the SCN core region. Relative to the no light condition, light triggered a marked increase in PEA-15 phosphorylation and ERK activation. Arrows denote cells with relatively high levels of pSer-104 PEA-15 and pERK. Bar = 40 microns. C) Left panel: Co-immunoprecipitation of SCN tissue from control and light-treated animals sacrificed at CT15. Total ERK was immunoprecipitated and probed via Western blotting for ERK and PEA-15. Relative to the control condition, the amount of immunoprecipitated PEA- 15 was reduced following light treatment. Tissue was pooled from 8 SCN samples for each condition, and the experimental results were performed using four separate trials. Right panel: Quantitative analysis of PEA-15 expression in the immunoprecipitated lysates from control and light-treated mice. Data were averaged from quadruplicate determinations and presented as normalized values and were analyzed by Student’s t-test *: p < 0.05.
Statistical analysis was performed with GraphPad Prism 3.0 software, and all data are represented as the mean ± the standard error of the mean (SEM). For all experiments, statistical significance was set at p < 0.05, as denoted in the text/figures. Statistical comparisons between two groups were conducted using Student’s t-tests (two-tailed). Comparisons of more than two groups were made using a one-way ANOVA. When significance was obtained from ANOVA tests, multiple comparison Bonferroni post-hoc tests were conducted in PRISM 3.0 to determine significant differences between any two groups.
RESULTS
As a starting point, C57Bl/6 mice were sacrificed during the middle of the day, zeitgeber time 6 (ZT 6), and coronal sections through the SCN were processed via immunohistochemistry (IHC) for the expression of PEA-15. Representative data in figure 1A revealed that PEA-15 was enriched throughout the rostro- caudal axis of the SCN. Further, low magnification whole-brain coronal imaging (Fig. 1B) showed elevated PEA-15 expression in the SCN relative to forebrain regions, including the hippocampus and cortex. Likewise, the high level of PEA-15 expression in the SCN contrasts with the modest expression levels within surrounding dorsal and lateral regions of the hypothalamus, as well as within well-demarcated hypothalamic nuclei, including the paraventricular nucleus and supraoptic nucleus (Fig. 1B). The specificity of the PEA-15 antibody used for IHC was tested via Western blotting; tissue lysates from both the SCN and the cortex identified a single 15 KDa band (Fig 1C), which is consistent with the molecular weight of PEA-15, thus supporting the specificity of the IHC expression pattern.
As the name denotes, PEA-15 has been reported to be enriched in astrocytic cell populations (Araujo et al. 1993). However, the expression pattern that we described here suggests that PEA-15 is also expressed in neuronal cells within the SCN. To test this idea, SCN tissue was colabeled for PEA-15 and for the neuronal-nuclear-specific marker, NeuN. Confocal analysis revealed a cellular level colocalization between PEA-15 (within the cytoplasm) and NeuN (within the nucleus) (Fig 1D). Further, colabeling for PEA-15 and the glial marker GFAP detected modest levels of colocalized expression (Fig 1E). Together, these data indicated that PEA-15 is enriched in neuronal cell populations of the SCN.
Although PEA-15 is expressed broadly within the SCN, high magnification images detected enriched expression within well-described SCN subregions. Notably, compared to ventro-medial SCN (also referred to as the SCN ‘core’), relatively high levels of PEA-15 were detected in the dorsomedial and lateral regions (also referred to as the SCN ‘shell’), (Fig 2A). These high-expressing regions roughly correspond with the subpopulation of arginine vasopressin (AVP)-positive SCN shell neurons, and indeed, co-immunolabeling detected marked PEA-15 expression in AVPergic neurons (Fig. 2B). PEA-15 expression was detected in ~ 93% of AVP-positive neurons (350 neurons analyzed: 50/per mouse; n= 7 mice). PEA-15 was also detected in a subset of SCN core neurons, as assessed by double immunolabeling for PEA-15 and vasoactive intestinal peptide (VIP; a marker of SCN core neurons: Fig. 2C). PEA-15 expression was detected in ~ 73% of VIP-positive neurons (490 neurons analyzed: 70/per mouse; n= 7 mice).
Next we examined whether the expression of PEA-15 is modulated over the circadian cycle in the SCN. To this end, mice were dark-adapted for 2 days, and tissue was profiled via IHC for PEA-15 at 4-hr intervals (Fig. 3A). Quantitative analysis of the SCN detected a significant time-of-day difference in PEA-15 expression, with a peak occurring during the early subjective night, and nadir occurring during the middle of the subjective day, as assessed by a one-way ANOVA for the total SCN (Fig. 3B; F(5,30) = 6.475, p = 0.0003). Of note, significant time-of-day changes in PEA-15 expression were also observed in both the core and the shell subregions of the SCN (Fig. 3B; Core; F(5, 30) = 7.580, p = 0.0001, and shell; F(5,30) = 6.688; p = 0.0003; one-way ANOVA), while circadian changes in PEA-15 expression were not detected in lateral hypothalamic regions surrounding the SCN (F(5, 28) = 0.6916, p = 0.6340; one-way ANOVA). To test whether the SCN rhythm in PEA-15 expression is gated by the circadian clock, PEA-15 expression was profiled in tissue from BMAL1 null mice. BMAL1 is a key component of the molecular circadian clock timing system, and its deletion results in a loss of circadian timekeeping capacity (Bunger et al., 2000). Interestingly, the significant time-of-day difference in the peak and trough levels of PEA-15 expression were not detected in BMAL1 null mice (Fig. 3C; t(6) = 0.5798, p = 0.5832; Student’s t-test). Together, these data indicate that PEA-15 is a clock-controlled gene in the SCN.
Much of the functionality of Pea-15 is regulated by changes in its phosphorylation state. In particular, the phosphorylation of Ser-104 and Ser-116 (via PKC and CaMKII, respectively) has been shown to affect its interactions with a number of proteins, including ERK and Fas-associated death domain protein (reviewed in Fiory et al., 2009). Given the key role that phosphorylation plays in the function of PEA- 15, we examined its phosphorylation state over the circadian cycle and in response to photic input.
Initially, we analyzed the expression of the Ser-104 phosphorylated form of PEA-15 (pSer-104 PEA-15). At ZT 6, the IHC expression pattern of pSer-104 PEA-15 within the SCN largely paralleled the expression pattern of the total protein; thus, high levels of pSer-104 PEA-15 were detected within the shell regions of the SCN, with more modest expression within the core (Fig. 4A). Western analysis using the same pSer-104 PEA-15-specific antibody used for IHC detected a single band at ~ 15 kDa, which is consistent with the molecular weight of PEA-15 (Fig. 4B). Next, circadian profiling, using the dark-adaptation and temporal profiling approaches described above for PEA-15, revealed a significant oscillation in pSer-104 PEA-15, (Fig. 4D; F(5,39) = 11.26, p < 0.0001, one-way ANOVA); the total SCN data were also presented in a scatter-plot format, and fitted to a cosine curve. As with the total protein profile, pSer104 PEA-15 expression levels did not change over the circadian cycle in lateral hypothalamic regions (F(5,39) = 1.348, p = 0.2528, one-way ANOVA).
Next, we examined PEA-15 phosphorylation at Ser-116 (pSer-116 PEA-15) using a Ser-116 phosphorylation-specific PEA-15 antibody. IHC labeling detected an expression pattern that paralleled the pattern of total PEA-15; hence marked labeling was found throughout the SCN, with the highest levels observed in the shell regions (Fig. 4E). IHC analysis was complemented with Western blotting (using the same antibody), which confirmed the selectively of the antibody and confirmed the relatively high level of pSer-116 PEA-15 expression in the SCN (Fig. 4F). Interestingly, circadian profiling of pSer-116 PEA-15 in the SCN did not indicate an oscillation (Fig. 4G; F(5,24) = 0.4958, p = 0.7762, one-way ANOVA). Together these data reveal complex, clock-gated, regulation of PEA-15 phosphorylation in the SCN.
Several reports have shown that PKC and CaMKII (again, two kinases that target PEA-15) are activated following photic stimulation in the SCN (Yokota et al., 2001; Agostino et al., 2004; Lee et al., 2007; Bonsall and Lall, 2013). This, coupled with the noted functional relevance of PEA-15 phosphorylation led us to examine photic regulation of PEA-15 phosphorylation. As a starting point, mice were dark-adapted for 2 days, and then exposed to a 30 second light (100 lux) pulse at CT15, and then immediately sacrificed. Analysis was focused on rapid changes in the phosphorylation state of PEA-15, given that at the 30 second photic stimulation time point, strong ERK activation was observed, as assessed via the IHC labeling for the Threonine and Tyrosine phosphorylated forms of ERK1 and ERK2 (a marker of MAPK pathway activation) (Fig. 5A and 5D left panel; t(9) = 12.27, p < 0.0001, Student’s t-test). IHC profiling revealed that 30 seconds of photic stimulation led to a significant increase in the Ser-104 phosphorylated form of PEA-15 (Fig. 5B and 5D middle panel; t(10) = 6.766, p < 0.0001, Student’s t-test). The representative image shows that this light-evoked increase in phosphorylation was detected specifically within the core region of the SCN. However, using the 30 second light pulse paradigm, an increase in Ser-116 phosphorylation was not detected (Fig. 5C and 5D right panel; t(15) = 1.391, p = 0.1845, Student’s t-test). Further, a longer, 15 min, light stimulus did not lead to an increase in the phosphorylation state of Ser-116 (or Ser-104), relative to the control condition (data not shown), suggesting that the functional effects of light on PEA-15 phosphorylation are rapid and transient.
We then turned our attention to an analysis of the spatial and temporal relationships between ERK activation and PEA-15 in the SCN. To this end, SCN tissue from light-treated (30 sec, 100 lux) mice was double immunolabeled for PEA-15 and activated ERK (Fig. 6A). Interestingly, light-evoked ERK activation was largely limited to SCN core neurons that express relatively low levels of PEA-15; thus, shell neurons that express relatively high PEA-15 formed a well-demarcated boundary between ERK responsive and non- responsive cells in the SCN (Fig. 6A, lower left panel). Quantitative analysis of the noted SCN sub-regional expression pattern of PEA-15 and pERK is provided in figure 6A (lower right panel; PEA-15 core versus shell; t(10) = 2.646, p = 0.0245 and pERK core versus shell; t(10) = 7.059, p < 0.0001, Student’s t- test).
As noted, the functional interaction between PEA-15 and ERK is dynamically regulated by PEA-15 phosphorylation. Given that photic stimulation triggers both ERK activation and PEA-15 phosphorylation (Fig. 5), we analyzed the cellular-level colocalization of these two events, and whether light triggers the dissociation of the PEA-15/ERK complex. Initially, double immunofluorescent labeling was used to detect a colocalized, cellular-level, increase in ERK phosphorylation and PEA-15 phosphorylation at Ser-104 following light treatment (Fig. 6B), thus indicating that photic stimulation triggers a rapid and coordinated cellular-level regulation of the MAPK pathway and PEA-15. Next, to test for an interaction between PEA-15 and ERK, we performed co-immunoprecipitation assays on SCN lysates from control and light-treated animals at CT15. To this end, an antibody bead conjugate was used to pull down total ERK, and the immunoprecipitate was profiled via Western blotting for ERK and PEA-15. Under the control, no light condition, PEA-15 co-immunoprecipitated with ERK, thus indicating that ERK and PEA- 15 are complexed within the SCN (Fig. 6C). Interestingly, this interaction was affected by light exposure; light treatment led to a significant reduction in PEA-15 bound to ERK (Fig. 6C; t(6) = 3.163, p = 0.0195, Student’s t-test), thus supporting the idea that photic stimulation triggers the rapid dissociation of ERK from cytoplasmic PEA-15.
Finally, several studies have shown that MAPK signaling can alter both the amplitude and the phasing of the oscillator cells (Butcher et al., 2005; Nomura et al., 2006). Key to this effect would appear to be the ability of the MAPK cascade to drive the expression of the core clock gene period1. Given that PEA-15 scaffolds ERK in the cytoplasm, which could affect the capacity of MAPK signaling to regulate gene transactivation, we examined the effects of PEA-15 on period1 transcription. For these studies, Cos7 cells were cotransfected with a period1-luciferase reporter construct and a PEA-15 construct that also expresses GFP (Fig. 7A). Interestingly, transgenic expression of PEA-15 led to a concentration-dependent decrease in period1 transcription (Fig. 7B; F(9,29) = 42.67, p < 0.0001, one-way ANOVA). Post- hoc analysis revealed that a striking reduction in period1 transcription occurred with the transfection of just 0.1 μg of PEA-15 (t(7) = 7.566, p < 0.001, Bonferroni multiple comparison). Further, pretreatment with U0126 (10 μM), an inhibitor of the MAPK pathway, also reduced period1 transcription (t(7) = 7.886, p < 0.001, Bonferroni multiple comparison); however, there was no additive suppressive effect on period1 transcription when U0126 was administered in combination with transgenic PEA-15 (t(7) = 1.283, p > 0.05, Bonferroni multiple comparison), thus indicating that the effects of PEA-15 on period1 transcription are mediated by the suppression of MAPK signaling. Further, serum (a potent MAPK pathway activator) substantially evoked period1 transcription, (Fig 7C; F(7,23) = 63.72, p < 0.0001, one-way ANOVA) and this induction was blocked by overexpression of PEA-15 (Fig. 7C; t(7) = 2.26, p > 0.05, Bonferroni multiple comparison). Importantly, the addition of U0126 did not augment the inhibitory effects of transgenic PEA-15 on period1 transcription (Fig 7C; t(7) = 2.671, p > 0.05, Bonferroni multiple comparison). Thus, via its role as a regulator of MAPK signaling, these data raise the prospect that PEA-15 may influence both the timing and entrainment of the SCN circadian clock.
Figure 7. PEA-15 regulates period1 expression.
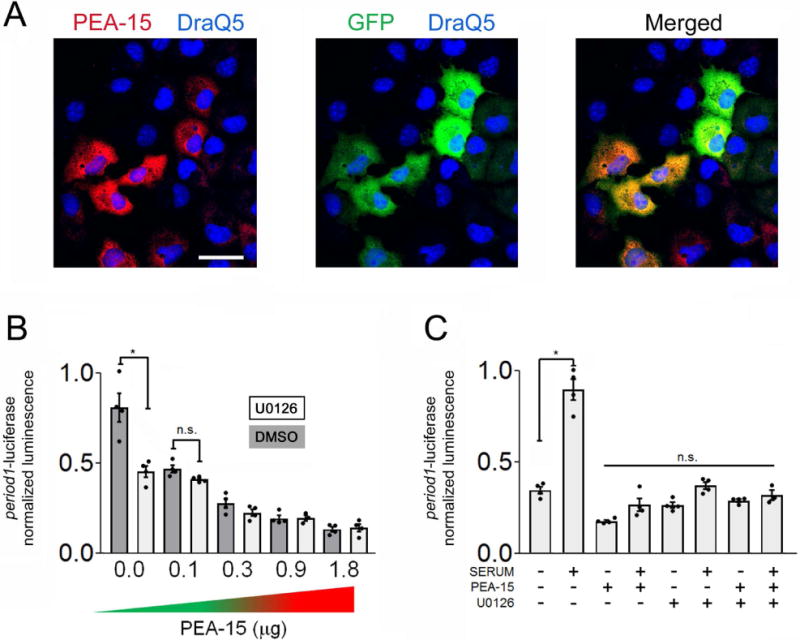
A) Cos7 cells were transfected with a PEA-15 reporter construct that also expresses the transfection marker GFP. Representative images of PEA-15 (red channel), GFP (green channel) and DraQ5 labeling (blue channel). Bar = 20 microns. B) The period1-luciferase reporter was cotransfected with increasing concentrations of the PEA-15 expression vector, or an empty expression vector. Note the concentration dependent reduction in period1-dependent transcription. Further, pretreatment with U0126 (10 μM) reduced period1-dependent transcription, but it did not markedly affect the suppressive effects of transgenic PEA-15. C) Serum-evoked period1 transcription was suppressed by transfection with PEA-15 (1.0 μg). Pretreatment with U0126 (10 μM) reduced serum-induced period1-dependent transcription, but it did not have additive suppressive effects with transgenic PEA- 15. For all experiments, data were averaged from triplicate determinations and are representative of 3 independent trials analyzed by a one-way ANOVA followed by Bonferroni multiple comparison tests *: p < 0.001; n.s: not significant (p > 0.05).
DISCUSSION
Here, we report on PEA-15 expression and function in the SCN. The central findings of this study reveal that 1) PEA-15 is enriched in SCN neurons, 2) PEA-15 expression follows the circadian cycle, 3) PEA-15 phosphorylation is modulated as a function of clock phase and photic input, 4) PEA-15 functions as a light-sensitive ERK scaffold, and 5) PEA-15 regulates MAPK-dependent activation of the core clock gene period1. Together, these findings reveal a hitherto uncharacterized level of intracellular signaling that could contribute to key aspects of SCN clock physiology.
The high level of PEA-15 expression that we detected in the SCN was a striking initial finding. Interestingly, PEA-15 exhibited enriched expression within the AVP-positive shell neurons of the SCN. The AVPergic cell population functions as the main circadian pacemaker cells of the SCN, and work in coordination with the VIPergic cells of the SCN core to generate a precisely phased and robust intra-SCN pacemaker network (Vosko et al., 2007; Li et al., 2009; Mieda et al., 2015; Ono et al., 2016). Further, AVPergic neurons have also been shown to function as an output from the SCN, conferring clock timing cues to a number of brain regions (Li et al., 2009; Kalsbeek et al., 2010). The reasons for elevated PEA-15 expression in AVPergic shell neurons are not known. However, it is notable that high levels of clock-gated ERK activity have been reported within the AVP cell population during the transition period between the subjective day and subjective night (Obrietan et al., 1998); given this, it is reasonable to speculate that the high levels of PEA-15 within the shell may be required to ensure the functional fidelity of ERK/MAPK signaling within AVPergic neurons.
PEA-15 levels followed a circadian expression pattern within the SCN, with a single peak detected during the subjective night and a trough during the mid-subjective day. Expression with a 24-hr period is a strong indicator that a gene is under control (either directly or indirectly) of the core clock molecular oscillator; this interpretation is supported by the observation that the peak in the PEA-15 rhythm was abrogated in BMAL1 null mice. However, it should be noted that the analysis of PEA-15 in BMAL1 null mice was limited to two time-points, CT6 and CT15, and thus a shift in the phasing of a PEA-15 rhythm could have evaded detection. With respect to the underlying transcriptional pathways that drive rhythmic PEA-15 expression, enhancer analysis did not identify an E-box motif within the 5′ regulatory region of PEA-15. Hence, rhythms in PEA-15 are likely not mediated by a direct CLOCK/BMAL1-driven transcriptional mechanism. However, a CRE/AP-1-like motif has been reported approximately 100 bases upstream of the PEA-15 transcription start site (Ungaro et al., 2012). This is notable, given that the CREB/CRE pathway serves as an ancillary, circadian-gated, transcriptional pathway within the SCN (Obrietan et al., 1999; O’Neill et al., 2008). Hence, CREB/CRE-mediated transcription may drive rhythmic PEA-15 expression in the SCN.
With respect to the regulation of MAPK signaling, much of the functionality of PEA-15 is regulated by phosphorylation states of Ser-104 and Ser-116 (reviewed by Fiory et al., 2009). Although the details of the functional interactions between ERK and PEA-15 are not well understood, and may be subject to cell- type, or context-specific secondary events, work to date indicates that these two phosphorylation events allow for ERK dissociation, which in turn would allow for the activated form of ERK to translocate to the nucleus. Consistent with this, phospho-mimetic mutagenesis of Ser-104 and Ser-116 results in a robust and lasting accumulation of ERK in the nucleus; in turn, nuclear ERK accumulation is a key event in the induction of MAPK-dependent gene expression (Formstecher et al., 2001; Krueger et al., 2005; Hunter et al., 2011).
The phosphorylation of PEA-15 at Ser-104 and Ser-116 are mediated by PKC and CaMKII, respectively (Araujo et al., 1993; Estellés et al., 1996; Kubes et al., 1998). As noted, both PKC and CaMKII are expressed in the SCN and have been reported to contribute to clock timing and to clock entrainment. Along these lines, the disruption of PKC (via PKC isoform-specific gene deletion and pharmacological approaches) has been shown to suppress light-induced clock entrainment (Jakubcakova et al., 2007; Lee et al., 2007). Further, CaMKII signaling has been reported to couple glutamatergic signaling to entrainment of the circadian clock (Fukushima et al., 1997). In addition, CaMKII was shown to play a role in SCN neuronal synchronization (Kon et al., 2014).
Given the role of PEA-15 as a regulator of ERK localization, coupled with work showing MAPK signaling plays a key role in clock entrainment, we examined whether photic stimulation alters the phosphorylation state of PEA-15. We found that brief light exposure during the night time domain triggered an increase in PEA-15 phosphorylation at Ser-104. Interestingly, the phosphorylation of PEA- 15 was quite rapid, with a significant increase detected following 30 seconds of light treatment. This rapid activation is noteworthy, given that activation of the MAPK pathway, as assessed by ERK phosphorylation, was also detected at the 30 second post-stimulus time point. Hence, if a principal function of PEA-15 in the SCN is to regulate ERK subcellular localization, then a rapid light-evoked increase in PEA-15 phosphorylation would be expected to occur. Further, at a mechanistic level, a light evoked increase in PEA-15 phosphorylation would be expected to trigger the dissociation of ERK, and indeed, co-IP analysis revealed that photic stimulation led to the release of ERK from PEA-15. This rapid dissociation of ERK from PEA-15 is supported by structure/function x-ray crystallography studies (Mace et al., 2013). Interestingly, these studies also showed that PEA-15 not only functions as an ERK scaffold, but that it also regulates the functional capacity of complexed ERK, and further, PEA-15 protects activated ERK from dephosphorylation. Thus, one could envision PEA-15 serving as an intracellular gate on MAPK signaling in the SCN, limiting the full functional capacity of activated ERK, until a coordinated set of parallel signaling events triggers the phosphorylation of PEA-15 at Ser-104. Here it is worth noting that photic stimulation was not found to increase the Ser-116 phosphorylated form of PEA-15. Given the noted functional roles for CaMKII in the SCN, the reason for the lack of an effect is not clear. However, the high basal level of Ser-116 phosphorylation may be suggestive of constitutive phosphorylation, which would preclude further phosphorylation by light.
As a state variable of the clock, the rapid light-evoked expression of period genes has been implicated in clock resetting (Albrecht et al., 1997; Shearman et al., 1997; Shigeyoshi et al., 1997), and given that MAPK signaling is a potent regulator of period1 expression (Akiyama et al., 1999; Travnickova-Bendova et al., 2002; Tischkau et al., 2003; Cao et al., 2013), we examined the potential effects that PEA-15 has on period1 gene expression. Here we show that both the basal and inducible expression of a period1- luciferase reporter construct was suppressed by PEA-15 overexpression. Importantly, the effects of PEA- 15 were not additive with the effects of the MAPK pathway inhibitor U0126, thus indicating that PEA15 reduces period1 transcription by suppressing ERK signaling. In many respects, these findings are consistent with studies showing that PEA-15 exerts very potent effects on MAPK-dependent transcription. Along these lines, overexpression of PEA-15 in culture has been shown to dramatically reduce ERK-dependent gene transcription, which can be rescued when PEA-15 is constitutively phosphorylated at Ser-104 (Krueger et al., 2005; Vaidyanathan et al., 2007). Further, stimulus-evoked ERK-mediated transcription is enhanced in thymocytes isolated from PEA-15 null mice (Pastorino et al., 2010). These studies, combined with the work reported here, provide putative mechanisms by which PEA-15 modulates light-evoked MAPK signaling, and in turn the clock resetting mechanism.
In total, these studies reveal PEA-15 to be neuronally-enriched, clock-gated and light responsive in the SCN. Given its high expression in the SCN, coupled with its role as a critical regulator of MAPK signaling, PEA-15 may be well positioned to contribute to the functional fidelity of signaling networks that regulate SCN clock timing and entrainment. Current work is focused on the examination of clock-timing in PEA-15 null mice.
Acknowledgments
National Institutes of Health; Grant code: MH103361, NS066345, NS091302 National Science Foundation; Grant code: 1354612
ABBREVIATIONS
- AP-1
Activator protein 1
- AVP
Arginine vasopressin
- CAMKII
Ca2+/calmodulin-dependent protein kinase II
- CT
Circadian Time
- CRE
cAMP response element
- CREB
cAMP response element-binding protein
- DAB
Diaminobenzidine
- ERK
Extracellular signaling related kinase
- FADD
Fas-associated death domain
- GFAP
Glial fibrillary acidic protein
- IHC
Immunohistochemistry
- PEA-15
Phosphoprotein enhanced in astrocytes
- PKC
Protein kinase C
- MAPK
Mitogen-activated protein kinase
- VIP
Vasoactive intestinal peptide
- ZT
Zeitgeber time
Footnotes
DR. KARL OBRIETAN (Orcid ID : 0000-0002-7525-7121)
CONFLICT OF INTEREST
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
K.W., K.H., J.O. and K.O. designed the experiments. K.W., L.S.Q, and K.S performed the experiments. K.W., L.S.Q., and S.A. analyzed the data. K.W., S.A., K.H., and K.O. wrote the manuscript.
DATA ACCESSIBILITY STATEMENT
All data is available from the corresponding author on request.
References
- Agostino PV, Ferreyra GA, Murad AD, Watanabe Y, Golombek DA. Diurnal, circadian and photic regulation of calcium/calmodulin-dependent kinase II and neuronal nitric oxide synthase in the hamster suprachiasmatic nuclei. Neurochem Int. 2004;44:617–625. doi: 10.1016/j.neuint.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Akashi M, Hayasaka N, Yamazaki S, Node K. Mitogen-activated protein kinase is a functional component of the autonomous circadian system in the suprachiasmatic nucleus. J Neurosci. 2008;28:4619–4623. doi: 10.1523/JNEUROSCI.3410-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama M, Kouzu Y, Takahashi S, Wakamatsu H, Moriya T, Maetani M, Watanabe S, Tei H, Sakaki Y, Shibata S. Inhibition of light- or glutamate-induced mPer1 expression represses the phase shifts into the mouse circadian locomotor and suprachiasmatic firing rhythms. J Neurosci. 1999;19:1115–1121. doi: 10.1523/JNEUROSCI.19-03-01115.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht U, Sun ZS, Eichele G, Lee CC. A differential response of two putative mammalian circadian regulators, mper1 and mper2, to light. Cell. 1997;91:1055–1064. doi: 10.1016/s0092-8674(00)80495-x. [DOI] [PubMed] [Google Scholar]
- Araujo H, Danziger N, Cordier J, Glowinski J, Chneiweiss H. Characterization of PEA-15, a major substrate for protein kinase C in astrocytes. J Biol Chem. 1993;268:5911–5920. [PubMed] [Google Scholar]
- Bonsall DR, Lall GS. Protein kinase C differentially regulates entrainment of the mammalian circadian clock. Chronobiol Int. 2013;30:460–469. doi: 10.3109/07420528.2012.741170. [DOI] [PubMed] [Google Scholar]
- Butcher GQ, Doner J, Dziema H, Collamore M, Burgoon PW, Obrietan K. The p42/44 mitogen-activated protein kinase pathway couples photic input to circadian clock entrainment. J Biol Chem. 2002;277:29519–29525. doi: 10.1074/jbc.M203301200. [DOI] [PubMed] [Google Scholar]
- Butcher GQ, Lee B, Cheng HYM, Obrietan K. Light stimulates MSK1 activation in the suprachiasmatic nucleus via a PACAP-ERK/MAP kinase-dependent mechanism. J Neurosci. 2005;25:5305–5313. doi: 10.1523/JNEUROSCI.4361-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Butcher GQ, Karelina K, Arthur JSC, Obrietan K. Mitogen and stress-activated protein kinase 1 (MSK1) modulates photic entrainment of the suprachiasmatic circadian clock. Eur J Neurosci. 2013;37:130–140. doi: 10.1111/ejn.12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HYM, Dziema H, Papp J, Mathur DP, Koletar M, Ralph MR, Penninger JM, Obrietan K. The molecular gatekeeper Dexras1 sculpts the photic responsiveness of the mammalian circadian clock. J Neurosci. 2006;26:12984–12995. doi: 10.1523/JNEUROSCI.4253-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coogan AN, Piggins HD. Circadian and photic regulation of phosphorylation of ERK1/2 and Elk-1 in the suprachiasmatic nuclei of the Syrian hamster. J Neurosci. 2003;23:3085–3093. doi: 10.1523/JNEUROSCI.23-07-03085.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman CI, Mistlberger RE, Rechtschaffen A. Suprachiasmatic nuclei lesions eliminate circadian temperature and sleep rhythms in the rat. Physiol Behav. 1984;32:357–368. doi: 10.1016/0031-9384(84)90248-8. [DOI] [PubMed] [Google Scholar]
- Estellés A, Yokoyama M, Nothias F, Vincent JD, Glowinski J, Vernier P, Chneiweiss H. The major astrocytic phosphoprotein PEA-15 is encoded by two mRNAs conserved on their full length in mouse and human. J Biol Chem. 1996;271:14800–14806. doi: 10.1074/jbc.271.25.14800. [DOI] [PubMed] [Google Scholar]
- Fiory F, Formisano P, Perruolo G, Beguinot F. Frontiers: PED/PEA-15, a multifunctional protein controlling cell survival and glucose metabolism. Am J Physiol - Endocrinol Metab. 2009;297:E592–E601. doi: 10.1152/ajpendo.00228.2009. [DOI] [PubMed] [Google Scholar]
- Formstecher E, Ramos JW, Fauquet M, Calderwood DA, Hsieh JC, Canton B, Nguyen XT, Barnier JV, Camonis J, Ginsberg MH, Chneiweiss H. PEA-15 Mediates Cytoplasmic Sequestration of ERK MAP Kinase. Dev Cell. 2001;1:239–250. doi: 10.1016/s1534-5807(01)00035-1. [DOI] [PubMed] [Google Scholar]
- Fukushima T, Shimazoe T, Shibata S, Watanabe A, Ono M, Hamada T, Watanabe S. The involvement of calmodulin and Ca2+/calmodulin-dependent protein kinase II in the circadian rhythms controlled by the suprachiasmatic nucleus. Neurosci Lett. 1997;227:45–48. doi: 10.1016/s0304-3940(97)00297-8. [DOI] [PubMed] [Google Scholar]
- Goldsmith CS, Bell-Pedersen D. Diverse roles for MAPK signaling in circadian clocks. Adv Genet. 2013;84:1–39. doi: 10.1016/B978-0-12-407703-4.00001-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainich EC, Pizzio GA, Golombek DA. Constitutive activation of the ERK-MAPK pathway in the suprachiasmatic nuclei inhibits circadian resetting. FEBS Lett. 2006;580:6665–6668. doi: 10.1016/j.febslet.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Hatori M, Gill S, Mure LS, Goulding M, O’Leary DDM, Panda S. Lhx1 maintains synchrony among circadian oscillator neurons of the SCN. eLife. 2014;3:e03357. doi: 10.7554/eLife.03357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota T, Fukada Y. Resetting Mechanism of Central and Peripheral Circadian Clocks in Mammals. Zoolog Sci. 2004;21:359–368. doi: 10.2108/zsj.21.359. [DOI] [PubMed] [Google Scholar]
- Hirota T, Lee JW, Lewis WG, Zhang EE, Breton G, Liu X, Garcia M, Peters EC, Etchegaray JP, Traver D, Schultz PG, Kay SA. High-Throughput Chemical Screen Identifies a Novel Potent Modulator of Cellular Circadian Rhythms and Reveals CKIα as a Clock Regulatory Kinase. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter I, Mascall KS, Ramos JW, Nixon GF. A phospholipase Cγ1-activated pathway regulates transcription in human vascular smooth muscle cells. Cardiovasc Res. 2011;90:557–564. doi: 10.1093/cvr/cvr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubcakova V, Oster H, Tamanini F, Cadenas C, Leitges M, van der Horst GTJ, Eichele G. Light entrainment of the mammalian circadian clock by a PRKCA-dependent posttranslational mechanism. Neuron. 2007;54:831–843. doi: 10.1016/j.neuron.2007.04.031. [DOI] [PubMed] [Google Scholar]
- Kalsbeek A, Fliers E, Hofman MA, Swaab DF, Buijs RM. Vasopressin and the output of the hypothalamic biological clock. J Neuroendocrinol. 2010;22:362–372. doi: 10.1111/j.1365-2826.2010.01956.x. [DOI] [PubMed] [Google Scholar]
- Kon N, Yoshikawa T, Honma S, Yamagata Y, Yoshitane H, Shimizu K, Sugiyama Y, Hara C, Kameshita I, Honma K, Fukada Y. CaMKII is essential for the cellular clock and coupling between morning and evening behavioral rhythms. Genes Dev. 2014;28:1101–1110. doi: 10.1101/gad.237511.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger J, Chou FL, Glading A, Schaefer E, Ginsberg MH. Phosphorylation of Phosphoprotein Enriched in Astrocytes (PEA-15) Regulates Extracellular Signal-regulated Kinase-dependent Transcription and Cell Proliferation. Mol Biol Cell. 2005;16:3552–3561. doi: 10.1091/mbc.E04-11-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubes M, Cordier J, Glowinski J, Girault JA, Chneiweiss H. Endothelin induces a calcium-dependent phosphorylation of PEA-15 in intact astrocytes: identification of Ser104 and Ser116 phosphorylated, respectively, by protein kinase C and calcium/calmodulin kinase II in vitro. J Neurochem. 1998;71:1307–1314. doi: 10.1046/j.1471-4159.1998.71031307.x. [DOI] [PubMed] [Google Scholar]
- Lee B, Almad A, Butcher GQ, Obrietan K. Protein kinase C modulates the phase-delaying effects of light in the mammalian circadian clock. Eur J Neurosci. 2007;26:451–462. doi: 10.1111/j.1460-9568.2007.05664.x. [DOI] [PubMed] [Google Scholar]
- Li JD, Burton KJ, Zhang C, Hu SB, Zhou QY. Vasopressin receptor V1a regulates circadian rhythms of locomotor activity and expression of clock-controlled genes in the suprachiasmatic nuclei. Am J Physiol Regul Integr Comp Physiol. 2009;296:R824–830. doi: 10.1152/ajpregu.90463.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace PD, Wallez Y, Egger MF, Dobaczewska MK, Robinson H, Pasquale EB, Riedl SJ. Structure of ERK2 bound to PEA-15 reveals a mechanism for rapid release of activated MAPK. Nat Commun. 2013;4:1681. doi: 10.1038/ncomms2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer JH, Schwartz WJ. In Search of the Pathways for Light-Induced Pacemaker Resetting in the Suprachiasmatic Nucleus. J Biol Rhythms. 2003;18:235–249. doi: 10.1177/0748730403018003006. [DOI] [PubMed] [Google Scholar]
- Meng QJ, Logunova L, Maywood ES, Gallego M, Lebiecki J, Brown TM, Sládek M, Semikhodskii AS, Glossop NRJ, Piggins HD, Chesham JE, Bechtold DA, Yoo SH, Takahashi JS, Virshup DM, Boot-Handford RP, Hastings MH, Loudon ASI. Setting clock speed in mammals: the CK1ε tau mutation in mice accelerates the circadian pacemaker by selectively destabilizing PERIOD proteins. Neuron. 2008;58:78–88. doi: 10.1016/j.neuron.2008.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieda M, Ono D, Hasegawa E, Okamoto H, Honma KI, Honma S, Sakurai T. Cellular clocks in AVP neurons of the SCN are critical for interneuronal coupling regulating circadian behavior rhythm. Neuron. 2015;85:1103–1116. doi: 10.1016/j.neuron.2015.02.005. [DOI] [PubMed] [Google Scholar]
- Mohawk JA, Green CB, Takahashi JS. Central and Peripheral Circadian Clocks in Mammals. Annu Rev Neurosci. 2012;35:445–462. doi: 10.1146/annurev-neuro-060909-153128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura K, Takeuchi Y, Fukunaga K. MAP kinase additively activates the mouse Per1 gene promoter with CaM kinase II. Brain Res. 2006;1118:25–33. doi: 10.1016/j.brainres.2006.08.087. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Impey S, Smith D, Athos J, Storm DR. Circadian Regulation of cAMP Response Element-mediated Gene Expression in the Suprachiasmatic Nuclei. J Biol Chem. 1999;274:17748–17756. doi: 10.1074/jbc.274.25.17748. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Impey S, Storm DR. Light and circadian rhythmicity regulate MAP kinase activation in the suprachiasmatic nuclei. Nat Neurosci. 1998;1:693–700. doi: 10.1038/3695. [DOI] [PubMed] [Google Scholar]
- O’Neill JS, Maywood ES, Chesham JE, Takahashi JS, Hastings MH. cAMP-dependent signaling as a core component of the mammalian circadian pacemaker. Science. 2008;320:949–953. doi: 10.1126/science.1152506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono D, Honma S, Honma KI. Differential roles of AVP and VIP signaling in the postnatal changes of neural networks for coherent circadian rhythms in the SCN. Sci Adv. 2016;2:e1600960. doi: 10.1126/sciadv.1600960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partch CL, Green CB, Takahashi JS. Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 2014;24:90–99. doi: 10.1016/j.tcb.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino S, Renganathan H, Caliva MJ, Filbert EL, Opoku-Ansah J, Sulzmaier FJ, Gawecka JE, Werlen G, Shaw AS, Ramos JW. The death effector domain protein PEA-15 negatively regulates T-cell receptor signaling. FASEB J. 2010;24:2818–2828. doi: 10.1096/fj.09-144295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renganathan H, Vaidyanathan H, Knapinska A, Ramos JW. Phosphorylation of PEA-15 switches its binding specificity from ERK/MAPK to FADD. Biochem J. 2005;390:729–735. doi: 10.1042/BJ20050378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangoram AM, Saez L, Antoch MP, Gekakis N, Staknis D, Whiteley A, Fruechte EM, Vitaterna MH, Shimomura K, King DP, Young MW, Weitz CJ, Takahashi JS. Mammalian Circadian Autoregulatory Loop. Neuron. 1998;21:1101–1113. doi: 10.1016/s0896-6273(00)80627-3. [DOI] [PubMed] [Google Scholar]
- Serchov T, Heumann R. Ras Activity Tunes the Period and Modulates the Entrainment of the Suprachiasmatic Clock. Front Neurol. 2017;8:264. doi: 10.3389/fneur.2017.00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif A, Renault F, Beuvon F, Castellanos R, Canton B, Barbeito L, Junier MP, Chneiweiss H. The expression of PEA-15 (phosphoprotein enriched in astrocytes of 15 kDa) defines subpopulations of astrocytes and neurons throughout the adult mouse brain. Neuroscience. 2004;126:263–275. doi: 10.1016/j.neuroscience.2004.02.039. [DOI] [PubMed] [Google Scholar]
- Shearman LP, Sriram S, Weaver DR, Maywood ES, Chaves I, Zheng B, Kume K, Lee CC, Der GTJ, van Horst Hastings MH, Reppert SM. Interacting Molecular Loops in the Mammalian Circadian Clock. Science. 2000;288:1013–1019. doi: 10.1126/science.288.5468.1013. [DOI] [PubMed] [Google Scholar]
- Shearman LP, Zylka MJ, Weaver DR, Kolakowski LF, Reppert SM. Two period homologs: circadian expression and photic regulation in the suprachiasmatic nuclei. Neuron. 1997;19:1261–1269. doi: 10.1016/s0896-6273(00)80417-1. [DOI] [PubMed] [Google Scholar]
- Shigeyoshi Y, Taguchi K, Yamamoto S, Takekida S, Yan L, Tei H, Moriya T, Shibata S, Loros JJ, Dunlap JC, Okamura H. Light-induced resetting of a mammalian circadian clock is associated with rapid induction of the mPer1 transcript. Cell. 1997;91:1043–1053. doi: 10.1016/s0092-8674(00)80494-8. [DOI] [PubMed] [Google Scholar]
- Shimizu K, Okada M, Nagai K, Fukada Y. Suprachiasmatic nucleus circadian oscillatory protein, a novel binding partner of K-Ras in the membrane rafts, negatively regulates MAPK pathway. J Biol Chem. 2003;278:14920–14925. doi: 10.1074/jbc.M213214200. [DOI] [PubMed] [Google Scholar]
- Silver R, Moore RY. The suprachiasmatic nucleus and circadian function: an introduction. Chronobiol Int. 1998;15:vii–x. doi: 10.3109/07420529808998698. [DOI] [PubMed] [Google Scholar]
- Stephan FK, Zucker I. Circadian rhythms in drinking behavior and locomotor activity of rats are eliminated by hypothalamic lesions. Proc Natl Acad Sci USA. 1972;69:1583–1586. doi: 10.1073/pnas.69.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzmaier FJ, Opoku-Ansah J, Ramos JW. Phosphorylation is the switch that turns PEA-15 from tumor suppressor to tumor promoter. Small GTPases. 2012;3:173–177. doi: 10.4161/sgtp.20021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischkau SA, Mitchell JW, Tyan SH, Buchanan GF, Gillette MU. Ca2+/cAMP Response Element-binding Protein (CREB)-dependent Activation of Per1 Is Required for Light-induced Signaling in the Suprachiasmatic Nucleus Circadian Clock. J Biol Chem. 2003;278:718–723. doi: 10.1074/jbc.M209241200. [DOI] [PubMed] [Google Scholar]
- Travnickova-Bendova Z, Cermakian N, Reppert SM, Sassone-Corsi P. Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMAL1 activity. Proc Natl Acad Sci U S A. 2002;99:7728–7733. doi: 10.1073/pnas.102075599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungaro P, Mirra P, Oriente F, Nigro C, Ciccarelli M, Vastolo V, Longo M, Perruolo G, Spinelli R, Formisano P, Miele C, Beguinot F. Peroxisome proliferator-activated receptor-γ activation enhances insulin-stimulated glucose disposal by reducing ped/pea-15 gene expression in skeletal muscle cells: evidence for involvement of activator protein-1. J Biol Chem. 2012;287:42951–42961. doi: 10.1074/jbc.M112.406637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan H, Opoku-Ansah J, Pastorino S, Renganathan H, Matter ML, Ramos JW. ERK MAP kinase is targeted to RSK2 by the phosphoprotein PEA-15. Proc Natl Acad Sci USA. 2007;104:19837–19842. doi: 10.1073/pnas.0704514104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valmiki MG, Ramos JW. Death effector domain-containing proteins. Cell Mol Life Sci. 2009;66:814–830. doi: 10.1007/s00018-008-8489-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanselow K, Kramer A. Role of Phosphorylation in the Mammalian Circadian Clock. Cold Spring Harb Symp Quant Biol. 2007;72:167–176. doi: 10.1101/sqb.2007.72.036. [DOI] [PubMed] [Google Scholar]
- Virshup DM, Eide EJ, Forger DB, Gallego M, Harnish EV. Reversible Protein Phosphorylation Regulates Circadian Rhythms. Cold Spring Harb Symp Quant Biol. 2007;72:413–420. doi: 10.1101/sqb.2007.72.048. [DOI] [PubMed] [Google Scholar]
- von Kriegsheim A, Baiocchi D, Birtwistle M, Sumpton D, Bienvenut W, Morrice N, Yamada K, Lamond A, Kalna G, Orton R, Gilbert D, Kolch W. Cell fate decisions are specified by the dynamic ERK interactome. Nat Cell Biol. 2009;11:1458–1464. doi: 10.1038/ncb1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosko AM, Schroeder A, Loh DH, Colwell CS. Vasoactive intestinal peptide and the mammalian circadian system. Gen Comp Endocrinol. 2007;152:165–175. doi: 10.1016/j.ygcen.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver DR. The Suprachiasmatic Nucleus: A 25-Year Retrospective. J Biol Rhythms. 1998;13:100–112. doi: 10.1177/074873098128999952. [DOI] [PubMed] [Google Scholar]
- Yokota S, Yamamoto M, Moriya T, Akiyama M, Fukunaga K, Miyamoto E, Shibata S. Involvement of calcium-calmodulin protein kinase but not mitogen-activated protein kinase in light-induced phase delays and Per gene expression in the suprachiasmatic nucleus of the hamster. J Neurochem. 2001;77:618–627. doi: 10.1046/j.1471-4159.2001.00270.x. [DOI] [PubMed] [Google Scholar]
- Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, Menaker M, Takahashi JS. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci U S A. 2004;101:5339–5346. doi: 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]