

Abstract
Intraflagellar transport moves proteins in and out of flagella/cilia and it is essential for the assembly of these organelles. Using whole-genome sequencing, we identified splice site mutations in two IFT genes, IFT81 (fla9) and IFT121 (ift121-2), which lead to flagellar assembly defects in the unicellular green alga Chlamydomonas reinhardtii. The splicing defects in these ift mutants are partially corrected by mutations in two conserved spliceosome proteins, DGR14 and FRA10. We identified a dgr14 deletion mutant, which suppresses the 3′ splice site mutation in IFT81, and a frameshift mutant of FRA10, which suppresses the 5′ splice site mutation in IFT121. Surprisingly, we found dgr14-1 and fra10 mutations suppress both splice site mutations. We suggest these two proteins are involved in facilitating splice site recognition/interaction; in their absence some splice site mutations are tolerated. Nonsense mutations in SMG1, which is involved in nonsense-mediated decay, lead to accumulation of aberrant transcripts and partial restoration of flagellar assembly in the ift mutants. The high density of introns and the conservation of noncore splicing factors, together with the ease of scoring the ift mutant phenotype, make Chlamydomonas an attractive organism to identify new proteins involved in splicing through suppressor screening.
Keywords: IFT, RNA splicing, NMD, whole-genome sequencing, Chlamydomonas
1. Introduction
Pre-messenger RNA splicing, which removes noncoding introns from nascent RNAs to produce functional mRNAs, is an important and precisely controlled process. Some introns (Groups I, II and III), which are found in bacteria, fungi and organelles, are self-spliced. Splicing of most introns found in eukaryotic nuclei is facilitated by the spliceosome, which is a dynamic complex that contains multiple uridine-rich small nuclear ribonucleoproteins (snRNPs) and proteins associated with these snRNPs [1,2]. The major spliceosome, which is composed of U1, U2, U4/U6 and U5 snRNPs and associated proteins, recognizes conserved nucleotide sequences at the 5′ splice donor site (GT), the 3′ splice acceptor site (AG) and the branch site. Mutations in these splice sites usually cause aberrant transcripts and it is estimated that splice site mutations cause approximately 15% of human genetic diseases [3]. The minor spliceosome, which is composed of U11, U12, U4atac, U6atac and U5 and many of the associated proteins found in the major spliceosome, is responsible for the removal of only approximately 0.3% of introns. It recognizes different conserved nucleotide sequences at the 5′ (AT) and 3′ (AC) splice sites. A few human diseases are associated with defects in the minor spliceosome [4]. Biochemical studies identified over 200 proteins associated with the major spliceosome and they can be grouped into different spliceosomal complexes. Some proteins are found to have core functions in the spliceosome while others are considered peripheral, and are present at specific points in the splicing process and are postulated to have noncore functions [2]. In our study described here, we focus on two peripheral proteins, DGCR14 and FRA10AC1 [5].
The DGCR14 (DiGeorge syndrome (DGS) critical region gene 14) gene is located in the minimal DGS critical region on human chromosome 22. DGS (velo-cardio-facial syndrome or 22q11 deletion syndrome) is caused by a deletion of about 46 genes within an approximately 2.5 Mb region and is associated with heart defects, cleft palate, low levels of calcium in the blood, poor immune system and delayed physical and social developments [6]. DGCR14 is a highly conserved protein [6–8] that localizes to the nucleus [7–9]. In Schizosaccharomyces pombe, deletion of the DGCR14 homologue Bis1 affects cell viability during stationary growth but not exponential growth [7]. In Caenorhabditis elegans, while loss of function of ESS-2/DGCR14 in wild-type worms has no obvious defects, a loss of function ess-2 allele in mutants with splice acceptor site mutations affects the stability of both correctly and incorrectly spliced transcripts. DGCR14/ESS-2 was proposed to facilitate splicing [8].
When cells are exposed to partial DNA replication stress, gaps, constrictions or breaks are likely to form at specific sites along the chromosome. Those are considered chromosomal fragile sites [10]. A rare group of chromosomal fragile sites are induced by exposure to folate, and the most frequent folate-sensitive human autosomal fragile site occurs at 10q23 [11]. A CCG expansion in the 5′ UTR of a gene, FRA10AC1 (FRA10A associated CGG repeat 1), is proposed to create the fragile site. The conserved FRA10AC1 (C10orf4) protein [12] was identified as a spliceosomal protein [13,14] and it localizes to the nucleus [15]. Yeast two-hybrid assays revealed that FRA10AC1 interacts with DGCR14 [2]. No functional study of the involvement of FRA10AC1 in pre-mRNA splicing has been reported.
Pre-mRNA splicing defects can lead to accumulation of aberrant transcripts, which can be deleterious to cells [3]. Degradation of these aberrant transcripts, which usually harbour premature termination codon (PTC), is controlled by the nonsense-mediated mRNA decay (NMD) surveillance system. The NMD machinery contains three conserved core components, UPF1, UPF2 and UPF3, which are found in all eukaryotic cells [16]. Phosphorylation of the RNA helicase UPF1, usually performed by the kinase SMG1, regulates NMD in some eukaryotes. In mouse, SMG1 is required for embryogenesis. About 9% of PTC-containing alternatively spliced transcripts show significant increase in SMG1-depleted mouse cells [17]. In C. elegans, transcripts with nonsense mutations accumulate in smg1 mutants [18]. In Drosophila, a likely null smg1 mutant has only a modest effect on NMD efficiency [19]. SMG1 is present in other land plants but not in Arabidopsis thaliana. No SMG1 gene has been identified in either Saccharomyces cerevisiae or S. pombe [20].
Intraflagellar transport (IFT) is a process that moves proteins between the cell body and the cilia/flagella, which are microtubule-based organelles that protrude from the cell body. This bidirectional process is essential for the formation and maintenance of the flagellum. The unicellular green alga Chlamydomonas reinhardtii assembles two flagella that confer the ability to swim in liquid medium. Mutations in IFT genes affect flagellar assembly and the mutant phenotypes are easily detectable due to the inability to oppose gravity by swimming [21–23].
In this study, we used whole-genome sequencing to identify splice site mutations in two IFT genes, IFT81 and IFT121. The missplicing events of IFT81 and IFT121, which include intron retention, exon skipping and adoption of new splice sites, can be corrected by mutations in either DGCR14 or FRA10AC1, but not by mutations in SMG1. Our study provides the first functional study of the involvement of FRA10AC1 in pre-mRNA splicing and suggests that, like C. elegans, the Chlamydomonas DGR14 protein is involved in pre-mRNA splicing regulation. As in other organisms, the Chlamydomonas SMG1 protein is involved in NMD. These ift mutants provide a new resource to identify new players in RNA splicing through suppressor screening, and Chlamydomonas serves as a tractable model system to study RNA splicing.
2. Material and methods
2.1. Strains and culture conditions
Strains were obtained from the Chlamydomonas Resource Center at the University of Minnesota. They include fla9, CC-1918; LMJ.RY0402.144851; and S1D2, CC-2290. The fla9 strain was backcrossed multiple times to wild-type cells to remove any unlinked modifiers. These strains were routinely maintained on Sager and Granick (R) medium agar plates. Ultraviolet mutagenesis to isolate the ift121-2 mutant and to screen for suppressors was performed as previously described [24].
The fla9 cells, when first obtained from the Chlamydomonas Resource Center, displayed a temperature-sensitivity phenotype as reported previously [25]. These cells maintained their flagella and swimming ability at 21°C and became aflagellate when cells were shifted to 32°C. Thus, we were able to analyse flagellar phenotype from fla9 cells, described in figures 1 and 2. Approximately 2 years after these initial studies, we noted the fla9 cells become aflagellate at all temperatures tested (21°C, 25°C and 32°C). To exclude any putative spontaneous mutations, we performed at least five rounds of meiotic crosses and analysed over 300 progeny. The identified IFT81 mutation in fla9 always cosegregates with the aflagellate phenotype (figure 3) and the same splicing pattern of IFT81 persists in the aflagellate cells (figure 2d). The fla9; dgr14-1; DGR14-TG cells, which had short flagella similar to fla9 when first identified (figure 1a), become aflagellate during the same period (figure 3). Missplicing of IFT81 remains the same in fla9; dgr14-1; DGR14-TG (figure 2d). In addition to the fla9 strain we maintained in the laboratory, we acquired fla9 from the Chlamydomonas Resource Center. We tested the flagellar phenotype of these two fla9 strains, fla9; dgr14-1, and fla9; dgr14-1; DGR14-TG in both R and TAP media, with trace elements obtained from the Chlamydomonas Resource Center and a different Chlamydomonas laboratory, at 21°C. The aflagellate phenotype persists in both fla9 strains and in fla9; dgr14-1; DGR14-TG while fla9; dgr14-1 displays more than 80% flagellated cells in the same media. We used EDTA acid to prepare trace elements, which led to no precipitation in the final product, while other trace elements were prepared with sodium EDTA that resulted in precipitation and filtration to obtain the final product [26]. Therefore, difference in trace elements does not contribute to the change of the fla9 phenotype. Given the phenotype observed, we consider the fla9 mutant in our hands has lost its temperature-sensitive phenotype and the aflagellate phenotype at all temperatures is the fla9 mutant phenotype we study onward.
Figure 1.
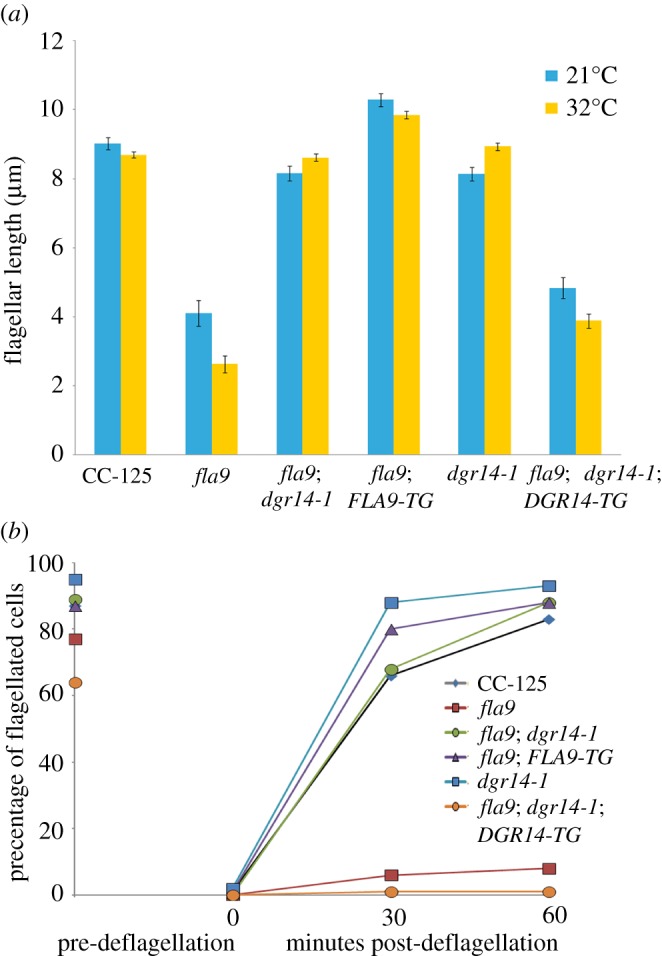
Flagellar assembly and regeneration defects in fla9 can be rescued by both wild-type IFT81 gene and a suppressor mutation, dgr14-1. (a) Measurement of flagellar length (n = 100) in individual strains at both 21°C (blue) and 32°C (yellow). Bars indicate the standard deviation of the mean. (b) Percentage of flagellated cells (n = 100) in each strain before and after flagellar amputation by pH shock. Cells were kept at 32°C during flagellar regeneration.
Figure 2.

A splice acceptor site mutation affects splicing of IFT81. (a) Gene structure of IFT81 and position of the fla9 mutation. Green box, 5′ UTR; orange boxes, exons; black solid lines, introns; purple box, 3′ UTR; blue arrow, position of the fla9 mutation. The single-nucleotide change from A to G is indicated in red. (b) Alternative splicing of IFT81 in cells grown at 21°C. (i) Representation of multiple IFT81 transcripts between exons 6 and 9. (ii) RT-PCR products of IFT81 between exons 1–4 (top), exons 6–9 (middle), and exons 9–11 (bottom). Individual bands are labelled according to their compositions. (c) Immunoblot of IFT81. Ten micrograms of flagellar proteins isolated from cells grown at 21°C were used in each lane. The same membrane was later probed with an anti-α-tubulin (TUA1) antibody to serve as a loading control. (d) RT-PCR of IFT81 exons 6–9 in various strains grown at 25°C. (e) RT-PCR of DGR14 at both 5′ and 3′ ends of the gene. Amplification of the ribosomal protein gene CRY1 serves as a loading control.
Figure 3.

Flagellar assembly in splice site ift mutants is restored by mutations in splicing factors. The percentage of flagellated cells was determined by counting 100 cells in triplicates in each strain. Error bars represent the standard deviation of the mean.
2.2. Meiotic mapping of fla9 and whole-genome sequencing
A cross between fla9 and wild-type (CC-124) showed 2 : 2 segregation of the aflagellate phenotype at 32°C in 87 tetrads. It suggests that the fla9 mutant contains either a single mutation or multiple tightly linked mutations. fla9 was mated to a highly polymorphic strain S1D2 (CC-2290) [27] and in 235 meiotic progeny fla9 maps between 3.255 and 3.780 Mb on chromosome 17 in Phytozome v. 5.5 of the Chlamydomonas genome. Chlamydomonas genomic DNA for whole-genome sequencing was prepared as previously described [24,28]. Three micrograms of DNA were submitted to the Genome Technology Access Core (Washington University) for library construction, Illumina sequencing and initial data analysis. SNP calling and subtraction of irrelevant SNPs/short indels were performed as previously described [28]. Large indels were identified by SoftSearch [29]. Around 12 000 breakpoints were found in strain 4c that was identified as an extragenic suppressor and compared to those found in the pf27 strain [30] and in fla11-2 [31] to identify indels unique to the 4c strain.
2.3. cDNA preparation, TA cloning and sequencing
For RNA isolation, cells from two R medium agar plates grown for 5 days were resuspended in 40 ml nitrogen-free medium (M-N/5) for 2 h at room temperature to allow flagellar assembly. The cells were then collected and RNA extraction was performed with the RNeasy Mini Kit (Qiagen) according to the manufacturer's recommendation. Two micrograms of RNA was used in a reverse transcription reaction with SuperScript III (Invitrogen) with random primers as previously described [32]. Gel-purified reversed transcribed cDNA products generated by Phusion (New England Biolabs) at the annealing temperature of 64°C were either subjected to direct Sanger sequencing (GeneWiz) or subjected to TA cloning. Primer sequences used in this study are listed in the electronic supplementary material, table S1. For TA-cloning, poly(A) tails were added to the gel-purified PCR products with TAQ polymerase and the fragments were later cloned into the pCR4-TOPO vector (Invitrogen). Plasmid DNA for Sanger sequencing was prepared by FastPlasmid Mini Kit (5 Prime) and sequenced with both T3 and T7 primers at GeneWiz.
2.4. BAC DNA preparation and Chlamydomonas transformation
Chlamydomonas BAC DNA was prepared using a QIAGEN Midiprep kit as previously described [33]. For rescue of fla9, two micrograms of isolated BAC DNA was transformed into fla9 cells by electroporation [32]. Cells were separated into 96 tubes each containing 20 ml liquid rich medium at 32°C. Swimming cells in these tubes were enriched by transferring the top 5 ml liquid into fresh 20 ml liquid rich medium every two days. After five rounds of transfer, crude DNA preparation, PCR and enzyme digestion to identify both mutant and transformed IFT81 genes were performed from all transformants. For rescue of 4c, the 40B10 BAC DNA was digested with HindIII and SbfI to obtain a 7.5 kb fragment, which includes approximately 3.5 kb upstream of the start codon of DGR14. The fragment was then cloned into HindIII and PstI sites of a pBlueScript SK vector (Stratagene). For rescue of fra10, the 3.1 kb genomic DNA fragment was amplified by FRA10-1F and FRA10-1R (electronic supplementary material, table S1) using Phusion DNA polymerase followed by the addition of poly(A) with Taq DNA polymerase for 10 min at 72°C. The amplified fragment was then cloned into the pCR4-TOPO vector (Invitrogen).
2.5. Flagellar length measurement
To measure Chlamydomonas flagellar length, Chlamydomonas cells were resuspended in liquid M-N/5 medium for 4 h and treated with autolysin for 30 min at room temperature. Cells were then resuspended in microtubule stabilization buffer (MTSB) [34] at room temperature. Multi-well slides (ThermoFisher) were coated with 0.1% poly-l-lysine (Sigma-Aldrich) for 5 min before being washed with MilliQ water once and allowed to dry completely. Cells were applied to wells on the slides and left in the dark for 2 min at room temperature. Excess cell suspension was removed by pipetting. Lysis buffer (MTSB + 1% Nonidet P-40) was added to individual wells and cells were lysed for 2 min at room temperature. Excess cell suspension was removed by pipetting. To wash off the lysis buffer, MTSB was added to individual wells and removed by pipetting. Samples were fixed with MTSB + 4% paraformaldehyde for 30 min at room temperature. Slides were then submerged in cold methanol (−20°C) for 2 × 5 min and left to dry at room temperature. Nucleo-flagellar apparatuses [35] are attached to individual wells and are visible under a phase-contrast microscope. The samples were blocked by 100% blocking buffer (BB, 5% BSA and 1% fish gelatin in PBS) for 1 h at room temperature. The samples were stained with a primary antibody (anti-acetylated α-tubulin, Sigma) at 1 : 500 dilution with 20% BB at 4°C overnight. The samples were washed six times with 20% BB, followed by 1-h inoculation at room temperature with a secondary antibody (Alexa-594-conjugated goat anti-mouse, Invitrogen) at 1 : 1000 dilution with 20% BB. The samples were washed six times with 20% BB and mounted in Fluoromount-G (SouthernBiotech). The images were captured with an UltraVIEW VoX laser spinning disk confocal microscope (PerkinElmer) and acquired by Volocity software (PerkinElmer). ImageJ was used to measure 100 flagella from 50 cells from each strain.
2.6. Flagellar regeneration
Flagellar amputation of Chlamydomonas cells was performed by pH shock [36]. After pH neutralization, cells were pelleted by centrifugation (1000 × g, 2 min) and resuspended in fresh R medium. The cells were kept at 32°C and a small portion of cells was fixed in 0.2% glutaraldehyde at each time point for visualization and cell count.
2.7. Immunoblot
Chlamydomonas flagellar isolation was performed as previously described after dibucaine amputation [37]. Ten micrograms of flagellar proteins were used in each strain. Immunoblots were performed as previously described [38]. The primary antibodies used include IFT81.1 (a gift from Dr. Doug Cole, 1 : 350 dilution) and anti-α-tubulin (DM1A, Sigma-Aldrich, 1 : 5000 dilution). The secondary antibody used was HRP-conjugated goat anti-mouse antibody (Bio-Rad, 1 : 5000 dilution).
2.8. Protein sequence alignment and prediction of protein structures
Protein sequences were obtained from NCBI and they were aligned by MUSCLE [39]. Colour-coded alignment of protein sequences was obtained using COLORFY [32]. Coiled-coil domains were predicted using COILS [40] and α-helices were predicted by YASPIN [41].
2.9. Analysis of orthologues of DGR14 and FRA10 in eukaryotes
Orthologues of DGR14 and FRA10 were obtained from the EggNog database [42]. To ensure that absence of an orthologue is not due to incomplete genome assembly, we required species used in the analysis to contain at least four out of five core splicing proteins, U2AF, PRP8, PRP17, PRP19 and SLU7. The absence of an orthologue in each species is also verified by BLAST against proteins in the NCBI database. Intron density was acquired from Rogozin et al. [43] and a median density is reported when there are multiple species in a given class.
3. Results
3.1. The fla9 mutant contains a splice site mutation in IFT81
The fla9 mutant was isolated as a temperature-sensitive mutant in an N-methyl-nitro N-nitrosoguanidine mutagenesis screen [25]. The cells grow flagella at 21°C and maintain their flagella at 32°C for 6 h. However, the cells fail to regenerate flagella at 32°C and thus become aflagellate once they go through cell division at 32°C. When we first obtained the fla9 strain from the Chlamydomonas Resource Center, they displayed the mutant phenotype as expected (figure 1). The fla9 cells had short flagella (approx. 4 µm) when compared to wild-type (CC-125) cells (approx. 9 µm) at 21°C. While the wild-type cells maintained their flagellar length 4 h after they were switched to 32°C, the fla9 cells had even shorter flagella (approx. 2.6 µm) (figure 1a). Prolonged (overnight) inoculation at 32°C eventually resulted in aflagellate cells. Only approximately 5% of fla9 cells were capable of regenerating flagella 1 h after pH shock [36] at 32°C, compared to approximately 80% of wild-type (CC-125) cells (figure 1b).
The fla9 allele was previously mapped to chromosome 17 [44] and we mapped it to a region between 3.255 and 3.780 Mb. Whole-genome sequencing of fla9 identified only one change (AG to GG) in the region (table 1) and it affects the 3′ splice site of intron 7 in the IFT81 gene (figure 2a) [38]. We designed a PCR-based assay to detect this change (electronic supplementary material, table S1) and it cosegregated with the flagellar defect in 50 meiotic fla9 progeny. Thus, it is tightly linked to the fla9 mutant.
Table 1.
Summary of mutants identified in this study.
| mutant | affected gene | mutation | position |
|---|---|---|---|
| fla9 | IFT81 | c. 823-2A > G | chromosome_17: 3 365 104 |
| dgr14-1 | FAP208, FAL13, Cre11.g482101, Cre11.g482150, FBB9, Cre11.g482250 | 33 kb deletion | chromosome_11: 3 603 615–3 636 297 |
| ift121-2 | IFT121 | c. 2754 +1G > A | chromosome_11: 2 411 434 |
| ift121-2 rev26 | IFT121 | c.2748G > A, p. K916 K | chromosome_11: 2 411 441 |
| ift121-2 rev28 | IFT121 | c. 2820 + 1G > A | chromosome_11: 2 411 221 |
| fra10 | Cre07.g336250 | c. 600delC, p. K201fs | chromosome_7: 3 538 004 |
| smg1-1 | Cre13.g572050 | c. 2263C > T, p. Q755X | chromosome_13: 1 413 437 |
| smg1-2 | Cre13.g572050 | c. 5587G > T, p. E1863X | chromosome_13: 1 417 593 |
| smg1-3 | Cre13.g572050 | c. 6787A > T, p. K2263X | chromosome_13: 1 419 322 |
| smg1-4 | Cre13.g572050 | c. 14452G > T, p. E4818X | chromosome_13: 1 430 695 |
| smg1-5 | Cre13.g572050 | c. 14494G > T, p. E4832X | chromosome_13: 1 430 737 |
We transformed 1E18 BAC (chromosome 17, 3 318 187–3 378 344) DNA into the fla9 mutant [45,46]. Ninety-six independent transformants were recovered after enriching for cells that regain the ability to swim at 32°C. Eighty-one of them had both mutant and wild-type alleles based on the PCR-based assay. The remaining 15 transformants, which still have the mutant allele, may carry suppressor mutations occurring elsewhere, but were not studied further. Backcrosses of nine randomly selected rescued transformants showed the rescue event is extragenic. One of such rescued strain (fla9; FLA9-TG) was randomly selected for further analysis. This rescued strain has approximately 10 µm flagella at both 21°C and 32°C (figure 1a) and it regenerates flagella at 32°C, similar to what we observe in wild-type cells (figure 1b).
3.2. The splice site mutation in fla9 leads to alternative splicing of IFT81
We expect the change at the 3′ splice site in fla9 to affect splicing of the IFT81 gene. PCR fragments from exons 1–4 and exons 9–11 are identical in length and intensity between wild-type and fla9 strains (figure 2b), which shows that the stability of the IFT81 mRNA is not affected in the fla9 strain. By contrast, PCR fragments from exons 6–9 show differences in wild-type and fla9 cells. In wild-type CC-125 cells, a single band, that corresponds to the predicted length of exons 6–9, is observed (figure 2b, band A). In fla9 cells, three bands are amplified (figure 2b, bands B, D and C). Sanger sequencing indicates that the predominant band B includes intron 7, and that exon 8 is skipped in band C (figure 2b). The middle band (D) contains a mix of wild-type and misspliced products. This band was subcloned and nine single colonies were subjected to Sanger sequencing. Two different splicing products are found: four different single colonies contain the wild-type cDNA and the other five colonies have a deletion of the first nine nucleotides of exon 8. The deletion is predicted to remove three amino acids (V275N276E277) in IFT81 protein. The glutamic acid is conserved in all IFT81 proteins examined (electronic supplementary material, figure S1). Nevertheless, an immunoblot using anti-IFT81 monoclonal antibody [47] shows that the IFT81 protein is absent in the fla9 mutant at both 21°C (figure 2c) and 32°C (electronic supplementary material, figure S2). In the fla9 strain rescued with wild-type FLA9 (fla9; FLA9-TG), the wild-type cDNA (band A) becomes the predominant species but the rescued strain still shows low levels of intron inclusion and exon skipping fragments (bands B and C) (figure 2b and electronic supplementary material, figure S2). Correspondingly, the IFT81 protein is expressed in the fla9; FLA9-TG strain at a lower level than found in wild-type cells (figure 2c and electronic supplementary material, figure S2).
3.3. Mutations in the DGCR14 gene suppress the flagellar defect in fla9
A fla9 strain with a spontaneous mutation (4c) shows flagellar assembly and regeneration at 32°C (figure 1, fla9; dgr14-1). By PCR and enzyme digestion, the fla9 splice site mutation is still present in 4c. Similar to the fla9; FLA9-TG strain, the major RT-PCR band amplified by the IFT81 exon 6–9 primers in the 4c strain is the wild-type product at both 21°C and 32°C (figure 2b and electronic supplementary material, figure S2). We detected the IFT81 protein in 4c cells (fla9; dgr14-1) but the protein abundance is lower than that found in the fla9; FLA9-TG strain at both temperatures (figure 2c and electronic supplementary material, figure S2).
A meiotic cross between 4c and the wild-type strain shows that 4c carries an extragenic mutation that is unlinked to the fla9 mutation (n = 23). The suppressor mutant itself (dgr14-1) has no flagellar assembly or regeneration defect (figure 1). To identify the causative mutation in 4c, we subjected one of the suppressed meiotic progeny to whole-genome sequencing. With 157× coverage of the genome, we did not identify a causative SNP or short insertion/deletion (indel) (electronic supplementary material, table S2) [28]. Instead, we identified a 32 682-bp sequencing gap on chromosome 11: 3 603 615–3 636 297 by SoftSearch [29] and manual examination of aligned reads. Within this region, six genes, FAP208, FAL13, Cre11.g482101, Cre11.g482150, FBB9 and Cre11.g482250, are either missing or disrupted. Both FAP208 and FBB9 were identified as flagellar proteins [48,49]. Cre11.g482101, Cre11.g482150 and Cre11.g482250 are novel genes. The FAL13 gene contains five exons and encodes a protein of 699 amino acids and shares 30% protein sequence identity (3 × 10−12) with the human DGCR14 protein. Sequence alignment indicates that it shares sequence similarity to DGCR14 homologues in S. pombe, Arabidopsis, Drosophila, C. elegans, zebrafish, mouse and human (electronic supplementary material, figure S3). Owing to its sequence similarity and its putative function in mRNA splicing, we renamed the FAL13 gene as DGR14.
Given that DGCR14 has been implicated in mRNA splicing in C. elegans and the splicing pattern of IFT81 is altered in the 4c strain, we expect introduction of the wild-type DGR14 gene into the fla9; dgr14-1 double mutant leads to short flagella and missplicing of IFT81 as observed in fla9. An approximately 7.5 kb DNA fragment, which contains the full-length DGR14 gene and approximately 3.5 kb upstream of DGR14 (part of Cre11.g482101), was transformed to the fla9; dgr14-1 double mutant [50]. Four transformants with short or no flagella were identified. One of these transformants does not contain a wild-type DGR14 gene. It suggests that the flagellar phenotype is likely due to a random insertion event instead of DGR14 rescue, which is known to occur [51]. The other three transformants carry the transformed wild-type DGR14 gene. We randomly picked one of these transformants for RT-PCR and Sanger sequencing of IFT81 splicing products from exons 6 to 9 (figure 2b, fla9; dgr14-1; DGR14-TG). They are identical to those found in fla9 (figure 2b). Similar to fla9, the fla9; dgr14-1; DGR14-TG cells have short flagella (approx. 4 µm) at both 21°C and 32°C (figure 1a) and they fail to regenerate flagella after amputation at 32°C (figure 1b). No IFT81 protein is detected in fla9; dgr14-1; DGR14 -TG cells (figure 2c and electronic supplementary material, figure S2). In contrast, the dgr14-1 mutant has wild-type IFT81 transcript (figure 2b and electronic supplementary material, figure S2) and wild-type IFT81 protein (figure 2c and electronic supplementary material, figure S2). Therefore, we conclude that the dgr14-1 deletion suppresses the fla9 mutant phenotype by modifying the misspliced IFT81 transcripts.
In addition to dgr14-1, we obtained a Chlamydomonas strain LMJ.RY0402.211897, which has an insertion in intron 5 of DGR14, from the Chlamydomonas Library Project (CLiP) [52]. The insertion leads to reduced level of the DGR14 transcript at the 3′ UTR (figure 2e). This insertional allele is renamed dgr14-2. It was crossed to the 4c strain and all 22 independent fla9 progeny that contain the dgr14-2 allele show normal flagellar assembly (figure 3, fla9; dgr14-2). Examination of IFT81 exons 6–9 in one of these progeny indicates that while mRNA with intron retention is observed, the wild-type transcript is restored (figure 2d). Therefore, both a deletion (dgr14-1) and an insertion (dgr14-2) in DGR14 act as suppressors of the fla9 mutant.
3.4. A splice site mutation in IFT121 causes alternative splicing of IFT121
In a mutant screen for aflagellate mutants, we isolated a new mutant strain, db35, that fails to assemble flagella (figure 3, ift121-2). Whole-genome sequencing indicates that it contains a 5′ splice site (donor) mutation, GT to AT, in the intron between exons 21 and 22 of IFT121 (table 1, figure 4a). This mutant also contained a second mutation in a gene that is unlinked to IFT121 (electronic supplementary material, table S2). We designed PCR based assays to detect both SNPs (electronic supplementary material, table S1) and selected a progeny (db35-1) that contains only the IFT121 SNP to study further. A backcross of db35-1 showed cosegregation of the IFT121 SNP and the aflagellate mutant phenotype (n = 20). Thus, this SNP is tightly linked to the mutant phenotype.
Figure 4.

A splice donor site mutation affects splicing of IFT121. (a) Gene structure of IFT121 and positions of multiple mutations. Green box, 5′ UTR; orange boxes, exons; black solid lines, introns; purple box, 3′ UTR; blue arrows, positions of ift121 mutations. The single-nucleotide changes in individual mutants are indicated in red. (b) Alternative splicing of IFT121 between exons 20–23. (i) Representation of multiple IFT121 transcripts amplified within the region. (ii) RT-PCR products of IFT121. (c) RT-PCR of FRA10 across the gene. Amplification of the ribosomal protein gene CRY1 serves as a loading control.
By RT-PCR, a single band from IFT121 exons 20–23 is amplified in wild-type cells (figure 4b, band A), while two bands are amplified in the db35-1 mutant (figure 4b, ift121-2). Sanger sequencing indicates the larger band (band B) contains intron 21 (electronic supplementary material, figure S4A) and the smaller band (band C) contains a truncated exon 21, in addition to exons 20, 22 and 23. This transcript is generated by adoption of an alternative splice donor site, which is 31 nucleotides upstream of the original site, within exon 21 (electronic supplementary material, figure S4B).
To provide further evidence that the splice site mutation in IFT121 is the causative mutation in this aflagellate mutant, we performed UV mutagenesis on the db35-1 mutant and identified revertants of the aflagellate phenotype of ift121. Two new strains (rev26 and rev28) show restored flagellar assembly (figure 3) and produce no aflagellate progeny when backcrossed to a wild-type strain. Sanger sequencing revealed that both mutants carry changes in the IFT121 gene. In rev26, there is a synonymous mutation of K916 (AAG to AAA) in exon 21 that is 7 nucleotides upstream of the original mutation (figure 4a, electronic supplementary material, figure S4C). RT-PCR of IFT121 in rev26 (figure 4b) reveals correct splicing of exons 20–23 with the silent mutation (Band A′), in addition to the alternatively spliced IFT121 transcripts found in ift121-2 (Bands B and C). Rev28 has a donor site mutation (GT to AT) at the beginning of intron 22 (figure 4a, electronic supplementary material, figure S4D), at position 2 411 221 on chromosome 11. It generates a complex set of IFT121 transcripts (figure 4b, electronic supplementary material, figure S4D) that includes band D (exon 20, deletion of 31 nucleotides from exon 21, exon 22, intron 22 and exon 23) and band C′ (deletion of 31 nucleotides from exon 21, exon 22, inclusion of 7 nucleotides of intron 22 and exon 23). The resulting band C’ now restores an in-frame IFT121 transcript. This transcript replaces 32 amino acids (aa 909–940) from the IFT121 protein sequence (black box, electronic supplementary material, figure S5) with 24 different amino acids. This change affects a few amino acids that are conserved across different species (electronic supplementary material, figure S5). The secondary structure predicted by YASPIN [41] indicates the C-terminus half of the IFT121 protein contains several predicted α-helices (electronic supplementary material, figure S5, magenta blocks). Three small α-helices (aa 899–915; aa 919–929; aa 932–950) are predicted within the region of replacement. Instead, now one large α-helix (aa 900–942) is predicted in the rev28 strain. Therefore, even though amino acid composition is changed around this region, the preserved secondary structure appears to be sufficient to restore flagellar assembly and motility in the ift121-2 rev28 strain. Based on the swimming phenotype and IFT121 splicing events found in these two intragenic revertants, we conclude the splice donor site mutation of IFT121 is the causative mutation and renamed the db35-1 strain ift121-2.
3.5. A frameshift mutation of FRA10 suppresses the ift121-2 mutation
In addition to the intragenic revertants, we isolated two extragenic suppressors of the ift121-2 mutant. One suppressor, sup15, splices IFT121 correctly across exons 20–23 (figure 4b, ift121-2; fra10). Thus, it is likely to be a suppressor that affects splicing. The other suppressor, sup25, retains alternative IFT121 splicing fragments. It is likely to be a suppressor that affects flagellar motility/assembly or mRNA stability but not splicing. Whole-genome sequencing of sup15 (electronic supplementary material, table S1) indicates a single nucleotide deletion (TG to T, table 1), which leads to a frameshift, in the Cre07.g336250 gene. The sup15 strain was backcrossed to wild-type. Fourteen meiotic progeny that contain the ift121-2 mutation but show wild-type flagellar assembly cosegregate with the single nucleotide deletion in Cre07.g336250. This gene encodes a protein that shares 54% identity and 69% similarity (3 × 10−55) to the human folate-sensitive fragile site protein FRA10AC1 (electronic supplementary material, figure S6). We named it FRA10 in Chlamydomonas.
We transformed the ift121-2; fra10 double mutant with a 3.1 kb DNA fragment that includes full-length wild-type FRA10 gene and approximately 0.9 kb upstream DNA. It is expected that rescue of the fra10 mutant results in aflagellate cells as observed in ift121-2. We obtained 10 aflagellate transformants. Five of them contain the wild-type FRA10 gene (ift121-2; fra10; FRA10-TG) while the other five may generate the aflagellate phenotype through random insertion [51]. The IFT121 transcript profiles in all five FRA10-TG transformants, obtained from four independent transformations, were analysed. The transcript products found in all ift121-2; fra10; FRA10-TG transformants and in ift121-2 are similar (figure 4b and electronic supplementary material, figure S8). Therefore, the frameshift mutation of FRA10 acts as a suppressor to restore the wild-type splicing pattern in ift121-2. The abundance of FRA10 transcripts in both ift121-2; fra10 and ift121-2; fra10; FRA10-TG is similar to the levels in wild-type (CC-125) (figure 4c). Given the single-nucleotide deletion, which is predicted to cause a frameshift, is found in the last exon (exon 4) of the gene, the mutant transcript is unlikely to be subjected to NMD [53].
3.6. DGR14 and FRA10 mutations can suppress both splice donor and acceptor site mutations
The dgr14 mutations suppress the splice acceptor site in fla9 and the fra10 mutation suppresses the splice donor site in ift121-2. Since both DGR14 and FRA10 were identified as spliceosomal C complex proteins [13,14] and they show protein–protein interaction [2], we asked whether mutations in these two spliceosomal proteins suppress both splice donor and acceptor site mutations. Flagellar assembly is restored in both fla9; fra10 and ift121-2; dgr14-1 strains (figure 3). Correspondingly, the wild-type IFT81 transcript is restored in the fla9; fra10 double mutant (figure 2d) and the wild-type IFT121 transcript is restored in the ift121-2; dgr14-1 mutant (figure 4b). Cells are aflagellate in fla9; fra10; FRA1-TG and in ift121-2; dgr14-1; DGR14-TG strains (figure 3). We conclude that a mutation in either DGR14 or FRA10 is sufficient to suppress both splice donor and acceptor site mutations in these IFT genes.
3.7. Nonsense mutations in the SMG1 gene stabilize the misspliced transcripts in the ift mutants
In an independent screen for suppressors of a paralyzed flagella mutant, we identified five nonsense mutants in the SMG1 gene (Cre13.g572050) (table 1; figure 5e) by whole-genome sequencing. Chlamydomonas SMG1 protein shares 32% identity and 46% similarity to its human homologue (1 × 10−110) (electronic supplementary material, figure S7). Since one of the important roles of NMD is to remove transcripts harbouring a premature terminated codon (PTC), we asked whether the smg1 mutations affect the misspliced IFT81 transcripts in fla9 and the misspliced IFT121 transcripts in ift121-2.
Figure 5.

Nonsense mutations in smg1 affect flagellar assembly in splice site ift mutants. (a) Distribution of flagellar length in wild-type (CC-124) and various fla9 mutants. Lengths of 100 flagella (represented by open circles) from 50 cells were measured in each strain. Horizontal bar represents the median flagellar length in each strain. (b) Alternative splicing of IFT81 between exons 6–9. (i) Representation of wild-type and intron retention transcripts. The red stop sign represents premature termination codon. (ii) RT-PCR products of IFT81 amplified from the same cells used in (a). (c) Distribution of flagellar length in wild-type (CC-124) and various ift121-2 mutants. Lengths of 100 flagella (represented by open circles) from 50 cells were measured in each strain. Horizontal bar represents the median flagellar length in each strain. (d) Alternative splicing of IFT121 between exons 20 to 23. (i) Representation of multiple IFT121 transcripts amplified within the region. (ii) RT-PCR products of IFT121 amplified from the same cells used in (c). (e) Gene structure of SMG1 and positions of multiple nonsense mutations. Green box, 5′ UTR; orange boxes, exons; black solid lines, introns; purple box, 3′ UTR; vertical black lines, positions of smg1 nonsense mutations. The positions of individual mutations in the SMG1 protein are indicated. Numbers of exons are indicated below the orange boxes. Owing to limitation of space, only odd numbers are included. The blue horizontal bars indicate regions amplified by RT-PCR in (f). (f) RT-PCR products of SMG1 in multiple exons. Amplification of the ribosomal protein gene CRY1 serves as a loading control.
We generated three double mutant strains (fla9; smg1-2, fla9; smg1-5 and ift121-2; smg1-2) and two triple mutants (fla9; dgr14-1; smg1-2 and ift121-2; fra10; smg1-2). While both fla9 and ift121-2 single mutant strains have very short or no flagella, both mutants show various flagellar lengths in the smg1 background (figure 5a,c). However, these cells display no motility. The triple mutant cells, similar to the fla9; dgr14-1 or ift121; fra10 double mutants, are motile and have normal flagellar length.
Semi-quantitative RT-PCR of IFT81 (figure 5b) followed by Sanger sequencing revealed that in the fla9; smg1-2 double mutant, the abundance of the intron inclusion transcript (band B), which bears a PTC, is approximately fivefold greater than that in the fla9 mutant. In the fla9; smg1-5 double mutant, the abundance is approximately fourfold greater. No wild-type transcript (band A) is observed in either double mutant. This suggests that IFT81 splicing is not altered in the smg1 mutant background. In the triple mutants fla9; dgr14-1; smg1-2 and fla9; dgr14-1; fra10, both the wild-type and intron inclusion transcripts are detected. The abundance of these transcripts is not significantly increased.
In the ift121-2; smg1-2 double mutant, the abundance of the truncated transcript (band C), which contains a PTC, increases approximately ninefold (figure 5d). Interestingly, no accumulation of the intron inclusion transcript (band B), which also harbours a PTC, is observed in the double mutant. A close examination of the transcript sequence reveals that it contains four in-frame AUG codons within 200 nucleotides downstream of the PTC, a widespread mechanism used by human genes to escape NMD surveillance [54]. No wild-type IFT121 transcript (band A) is observed in the double mutant. In the ift121-2; fra10; smg1-2 triple mutant, we detected all three transcripts and there is about fourfold accumulation of the truncated transcript (band C) but not in the intron inclusion transcript (band B).
The smg1-2 mutation does not affect the abundance of the mutant fra10 transcript in the ift121-2; fra10; smg1-2 mutant (figure 4c). This is consistent with our hypothesis that the frameshift in the last exon of the FRA10 transcript is not subjected to NMD. In contrast, the nonsense SMG1 mutant transcripts accumulate in both the smg1-2 and smg1-5 mutant strains (figure 5f). It indicates that the NMD pathway is likely to be compromised in the smg1 mutants.
4. Discussion
4.1. Misregulation of RNA splicing via mutations in cis-acting RNA sequences
RNA splicing is necessary to produce mature RNA for almost all genes in vertebrates. Misregulation of RNA splicing, by both cis-acting RNA sequences and trans-acting RNA splicing factors, are linked to cancers and other human diseases [3,55].
In this study, we report mutations in the splice sites of two Chlamydomonas IFT genes that lead to aberrant splicing of their transcripts and defects in flagellar assembly. The isolation of two intragenic revertants of ift121-2 shows novel ways that cells can rescue a splice site defect. In ift121-2, the mutation in the donor splice site in intron 21 leads to an alternative donor site 31 nucleotides upstream (electronic supplementary material, figure S4A). Exonic splicing enhancers (ESEs) are short oliognucleotide sequences in exons around the splice sites that bind to splicing factors and facilitate splicing [56]. It is estimated that approximately 4% of synonymous changes are deleterious to splicing by affecting ESE sequences [57]. We used the RESCUE program for human ESE prediction [56], and it reveals nine ESEs within 12 nucleotides upstream of the canonical splice donor site of intron 21. In the ift121-2 rev26 mutant, a synonymous change that is 7 nucleotides upstream of the original mutation partially restores wild-type splicing (electronic supplementary material, figure S4B). The single nucleotide change in rev26 is predicted to change the sequences of eight ESEs and to add a new ESE. These ESEs may have higher affinity for splicing factors around the noncanonical splice site (AT in ift121-2) and this recruitment could facilitate splicing.
A genome-wide analysis of alternative spliced transcripts in Chlamydomonas indicated that both constitutive splicing and alternative splicing events use GT as the consensus splice donor site [58]. However, in the ift121-2 rev28 mutant, the splicing machinery opts for a noncanonical splice donor site (GC). It is unclear why this site is chosen, but it results in an in-frame reading frame. While this choice changes the primary protein sequence, it is unlikely to change the secondary structure based on structure predictions [41].
4.2. Correction of RNA splicing mistakes via mutations in spliceosomal proteins
Both DGCR14 and FRA10AC1 were identified in proteomic studies of human spliceosomes but are absent in yeast spliceosomes [2]. While yeast and human spliceosomes share common core structures and proteins, human spliceosomes contain more spliceosomal proteins that play regulatory roles [59]. Analysis of yeast postcatalytic spliceosome structure [60–62] revealed formation of non-Watson–Crick base pairings between G (+1) of the 5′ splice site and G (−1) of the 3′ splice site, and between A (−2) of the 3′ splice site and the conserved adenine at the branch point, which is linked to G (+1) of the 5′ splice site. While the structure of human postcatalytic spliceosome has not been revealed yet, we expect it uses similar base pairing mechanisms but contains more spliceosomal proteins that may include DGCR14 and FRA10AC1. In our study, the splice site mutations disrupt G (+1) of the 5′ splice site and A (−2) of the 3′ splice site. These mutations in dgr14 and fra10 mutants show increased wild-type splicing. Therefore, DGR14 and FRA10 are likely to be involved in facilitating recognition/interaction among G (+1), A (−2) and A at the branch point. However, it is unclear whether the involvement is direct or indirect and it will require additional assays to address their functions.
We observed no obvious defects in viability, mating efficiency and motility in the single mutants of dgr14 and fra10 or the dgr14-1; fra10 double mutant. This is consistent with the observation that deletion of Bis1, the DGCR14 homologue found in fission yeast, does not affect viability during exponential growth [7]. We performed transcriptome analysis of dgr14, fra10 and the double mutant but find very few changes in RNA splicing and abundance (M Pandey, G Stormo and SK Dutcher 2018, unpublished work). Similarly, no phenotypic or splicing defect has been report in the ess-2 mutant in C. elegans [8].
Given that both DGCR14 and FRA10AC1 are not core splicing proteins [2], we asked whether these two proteins are present in different species across multiple eukaryotic kingdoms (figure 6). The presence/absence of orthologues of each protein in individual species was extracted from the EggNOG database [42] and NCBI BLAST. In Saccharomyces cerevisiae, approximately 5% of genes contain introns and its spliceosome contains fewer than 100 proteins [63,64]. The intron density per 1 kb of coding sequence is approximately 0.1 [43]. In contrast, approximately 43% of Schizosaccharomyces pombe genes contain introns and its intron density is ten-fold higher [43,64]. The DGCR14 homologue Bis1, which is proposed to be a stress response protein [7], is present in four different species of the fission yeasts [64], while both DGCR14 and FRA10AC1 homologues are absent in the Saccharomycetes lineage (n = 17). In addition to Saccharomycetes, DGCR14 is absent in the oomycetes Phytophthora ramorum (Pram) and Phytophthora capsici (Pcap). However, the genome of Phytophthora sojae contains DGCR14. Therefore, the absence of DGCR14 in Pram and Pcap may arise from incomplete genome assembly. Even though DGCR14 is not essential to RNA splicing, it is present in most species analysed (figure 6). FRA10AC1 is absent in Ascomycota, a subgroup of fungi. It is also absent in Ustilaginomycotina, which have very low intron density (approx. 0.4) when compared to other organisms in Basidiomycota [43]. The absence of FRA10AC1 correlates with low intron density and we suggest that these two observations may be related. An exception is observed in the unicellular green algae Ostreococcus, in which the intron density is low (approx. 0.6) but both FRA10AC1 and DGCR14 are present. In general, higher intron density (greater than 3) may require additional noncore spliceosomal proteins to recognize intron boundaries and promote splicing, therefore the presence of both proteins is observed in these species. In Chlamydomonas, 88% of the genes contain introns and an intron density of approximately 6.3, which is similar to the vertebrates with an intron density of 6.9 [43,58].
Figure 6.

Distribution of orthologues of DGCR14 and FRA10AC1 in multiple eukaryotic kingdoms. The median intron density (per 1 kb coding region) in each class is calculated from Rogozin et al. [43]. Open circles, protein absent from the class; half closed circles, protein absent from some species within the class; closed circles, protein present in the class. The lengths of branches do not represent evolutionary distance. The numbers of species analysed in each class are indicated on the right.
4.3. The ift mutants provide new insights about intraflagellar transport
Mutations of IFT81 in humans lead to multiple symptoms that include polydactyly, nephronophthisis [65], asphyxiating thoracic dystrophy, short rib polydactyly [66] and retinal dystrophy [65,67]. In the fla9 mutant strain reported here, the most prevalent transcript shows retention of IFT81 intron 7 and produces no protein that is detected by the monoclonal anti-IFT81 antibody [47], which recognizes the C-terminus of IFT81 (D Cole 2014, personal communication, figure 2c and electronic supplementary material, figure S2). In the ift81-1 mutant strain, which was generated by an insertion in exon 7 of IFT81, no IFT81 protein was detected by immunoblot with the same antibody [23]. We propose that the fla9 mutant transcript produces a truncated IFT81 protein that contains the first approximately 270 amino acids, not detected by the antibody. It has been reported that the N-termini of IFT81 and IFT74 are crucial to flagellar assembly and may dimerize to form a binding module for tubulin [23,68]. In the fla9; smg1 double mutant, accumulation of this transcript produces enough truncated IFT81 proteins to interact with the N-terminus of IFT74 and to allow variable flagellar assembly (figure 5a). It is worth noting that the flagella remain immotile in fla9; smg1 cells, which suggests the C-terminus of IFT81 is required for cargo needed for flagellar motility.
Mutations in IFT121/WDR35 result in cranioectodermal dysplasia [69–73], short rib polydactyly syndrome [74], Ellis–van Creveld syndrome [75] and respiratory dysfunction [72]. Studies using truncation mutants indicated the N-terminus of IFT121 (aa 1–640) is important for interactions with IFT122, Arl13b and INPP5E while the C-terminus (aa 641–1181) is important for its ciliary localization [76]. In the ift121-2; smg1-2 mutant, the accumulated misspliced transcript (Band C in figure 5d) is expected to encode a truncated IFT121 protein that contains the first 908 amino acids with an additional 56 novel amino acids. We propose that accumulation of this truncated IFT121 protein is responsible for flagellar assembly. An insertional mutant of IFT121 (ift121-1) has been previously reported. The exact location of gene disruption is unknown but it is within the last one-third of the gene [77]. Similar to our ift121-2 mutant, the ift121-1 mutant is aflagellate and it is unclear whether ift121-1 can assemble flagella in a smg1 background.
Hypoxia has been reported to partially restore flagellar assembly in truncated IFT46 and IFT74 mutants [78,79]. Removal of the first 196 amino acids from IFT74 in the ift74-1 mutant leads to a slight accumulation of the truncated protein and the mutant cells assemble immotile flagella when cells are not aerated [79]. When stressed, a truncated IFT46 protein, which lacks the N-terminal 100 amino acids, accumulates and the mutant cells assemble flagella with various lengths [78]. Our study on fla9; smg1 and ift121-1; smg1 mutants provides additional evidence that over-accumulation of truncated IFT proteins can partially rescue flagellar assembly defects. To further understand the detailed mechanism, over-expression of truncated constructs of IFT81 and IFT121, and their interactions with other IFT proteins, will be necessary.
Supplementary Material
Acknowledgements
We offer special thanks to Dr. Gianni Piperno for early characterization of the 4c suppressor strain. We thank Dr. Gary Stormo and Manishi Pandey for comments on the manuscript and Dr. Douglas Cole for the IFT81 antibody.
Data accessibility
The sequence data for the strains in the electronic supplementary material, table S2 are deposited at BioProject ID PRJNA407207 at the National Center for Bioinformatics Information (NCBI). All other data are presented in the electronic supplementary material, tables and figures.
Authors' contributions
H.L. carried out molecular laboratory work, participated in data analysis and the design of the study, carried out sequence alignments and drafted the manuscript; Z.Z. carried out molecular laboratory work; C.I. isolated the original suppressor mutation and S.K.D. conceived the study, designed the study, carried out genetic laboratory work, coordinated the study and helped draft the manuscript. All authors gave final approval for publication.
Competing interests
We declare we do not have competing interest.
Funding
This work was supported by grants from the Children's Discovery Institute (PD-II-2014-379), the National Heart Lung, and Blood Institute (R01 HL128370), and the National Institute of General Medical Sciences (R01 GM32843) to S.K.D. We thank the Genome Technology Access Center (GTAC) in the Department of Genetics at Washington University School of Medicine, which is partially supported by National Cancer Institute Cancer Center Support grant #P30 CA91842 to the Siteman Cancer Center, and by the Institute for Clinical and Translational Sciences (ICTS)/Clinical and Translational Science Award (CTSA) grant UL1RR024992 from the National Center for Research Resources (NCRR), a component of the NIH and NIH Roadmap for Medical Research.
References
- 1.Roy S, Gilbert W. 2006. The evolution of spliceosomal introns: patterns, puzzles and progress. Nat. Rev. Genet. 7, 211 (doi:10.1038/nrg1807) [DOI] [PubMed] [Google Scholar]
- 2.Hegele A. et al 2012. Dynamic protein–protein interaction wiring of the human spliceosome. Mol. Cell 45, 567–580. (doi:10.1016/j.molcel.2011.12.034) [DOI] [PubMed] [Google Scholar]
- 3.Scotti MM, Swanson MS. 2016. RNA mis-splicing in disease. Nat. Rev. Genet. 17, 19–32. (doi:10.1038/nrg.2015.3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turunen JJ, Niemelä EH, Verma B, Frilander MJ. 2013. The significant other: splicing by the minor spliceosome. RNA 4, 61–76. (doi:10.1002/wrna.1141) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wahl MC, Will CL, Lührmann R. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718. (doi:10.1016/j.cell.2009.02.009) [DOI] [PubMed] [Google Scholar]
- 6.Guna A, Butcher N, Bassett A. 2015. Comparative mapping of the 22q11.2 deletion region and the potential of simple model organisms. J. Neurodevelop. Disord. 7, 1–16. (doi:10.1186/s11689-015-9113-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taricani L, Tejada ML, Young PG. 2002. The fission yeast ES2 homologue, Bis1, interacts with the Ish1 stress-responsive nuclear envelope protein. J. Biol. Chem. 277, 10 562–10 572. (doi:10.1074/jbc.M110686200) [DOI] [PubMed] [Google Scholar]
- 8.Noma K, Goncharov A, Jin Y. 2014. Systematic analyses of rpm-1 suppressors reveal roles for ESS-2 in mRNA splicing in Caenorhabditis elegans. Genetics 198, 1101–1115. (doi:10.1534/genetics.114.167841) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindsay EA, Harvey EL, Scambler PJ, Baldini A. 1998. ES2, a gene deleted in DiGeorge syndrome, encodes a nuclear protein and is expressed during early mouse development, where it shares an expression domain with a goosecoid-like gene. Hum. Mol. Genet. 7, 629–635. (doi:10.1093/hmg/7.4.629) [DOI] [PubMed] [Google Scholar]
- 10.Dillon LW, Burrow AA, Wang Y-H. 2010. DNA instability at chromosomal fragile sites in cancer. Curr. Genomics 11, 326–337. (doi:10.2174/138920210791616699) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petit P, Fryns J, Berghe H, Hecht F. 1986. Population cytogenetics of autosomal fragile sites. Clin. Genet. 29, 96–100. (doi:10.1111/j.1399-0004.1987.tb02768.x) [DOI] [PubMed] [Google Scholar]
- 12.Vlachakis D, Pavlopoulou A, Kazazi D, Kossida S. 2013. Unraveling microalgal molecular interactions using evolutionary and structural bioinformatics. Gene 528, 109–119. (doi:10.1016/j.gene.2013.07.039) [DOI] [PubMed] [Google Scholar]
- 13.Bessonov S, Anokhina M, Will CL, Urlaub H, Luhrmann R. 2008. Isolation of an active step I spliceosome and composition of its RNP core. Nature 452, 846–850. (doi:10.1038/nature06842) [DOI] [PubMed] [Google Scholar]
- 14.Schmidt C, Grønborg M, Deckert J, Bessonov S, Conrad T, Lührmann R, Urlaub H. 2014. Mass spectrometry-based relative quantification of proteins in precatalytic and catalytically active spliceosomes by metabolic labeling (SILAC), chemical labeling (iTRAQ), and label-free spectral count. RNA 20, 406–420. (doi:10.1261/rna.041244.113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarafidou T, et al. 2004. Folate-sensitive fragile site FRA10A is due to an expansion of a CGG repeat in a novel gene, FRA10AC1, encoding a nuclear protein. Genomics 84, 69–81. (doi:10.1016/j.ygeno.2003.12.017) [DOI] [PubMed] [Google Scholar]
- 16.He F, Jacobson A. 2015. Nonsense-mediated mRNA decay: degradation of defective transcripts is only part of the story. Annu. Rev. Genet. 49, 339–366. (doi:10.1146/annurev-genet-112414-054639) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McIlwain DR, Pan Q, Reilly PT, Elia AJ, McCracken S, Wakeham AC, Itie-Youten A, Blencowe BJ, Mak TW. 2010. Smg1 is required for embryogenesis and regulates diverse genes via alternative splicing coupled to nonsense-mediated mRNA decay. Proc. Natl Acad. Sci. USA 107, 12 186–12 191. (doi:10.1073/pnas.1007336107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pulak R, Anderson P. 1993. mRNA surveillance by the Caenorhabditis elegans smg genes. Genes Dev. 7, 1885–1897. (doi:10.1101/gad.7.10.1885) [DOI] [PubMed] [Google Scholar]
- 19.Metzstein MM, Krasnow MA. 2006. Functions of the nonsense-mediated mRNA decay pathway in Drosophila development. PLoS Genet. 2, e180 (doi:10.1371/journal.pgen.0020180) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lloyd JPB, Davies B. 2013. SMG1 is an ancient nonsense-mediated mRNA decay effector. Plant J. 76, 800–810. (doi:10.1111/tpj.12329) [DOI] [PubMed] [Google Scholar]
- 21.Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, Cole DG. 2000. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene Tg737, are required for assembly of cilia and flagella. J. Cell Biol. 151, 709–718. (doi:10.1083/jcb.151.3.709) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iomini C, Babaev-Khaimov V, Sassaroli M, Piperno G. 2001. Protein particles in Chlamydomonas flagella undergo a transport cycle consisting of four phases. J. Cell Biol. 153, 13–24. (doi:10.1083/jcb.153.1.13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kubo T, Brown JM, Bellve K, Craige B, Craft JM, Fogarty K, Lechtreck KF, Witman GB. 2016. Together, the IFT81 and IFT74 N-termini form the main module for intraflagellar transport of tubulin. J. Cell Sci. 129, 2106–2119. (doi:10.1242/jcs.187120) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dutcher SK, et al. 2012. Whole-genome sequencing to identify mutants and polymorphisms in Chlamydomonas reinhardtii. G3 2, 15–22. (doi:10.1038/srep3423210.1534/g3.111.000919) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adams GM, Huang B, Luck DJ. 1982. Temperature-sensitive, assembly-defective flagella mutants of Chlamydomonas reinhardtii. Genetics 100, 579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harris EH. 1989. The Chlamydomonas sourcebook: A comprehensive guide to biology and laboratory use. Cambridge, MA: Academic Press. [DOI] [PubMed] [Google Scholar]
- 27.Gross CH, Ranum LP, Lefebvre PA. 1988. Extensive restriction fragment length polymorphisms in a new isolate of Chlamydomonas reinhardtii. Curr. Genet. 13, 503–508. (doi:10.1007/BF02427756) [DOI] [PubMed] [Google Scholar]
- 28.Lin H, Dutcher SK. 2015. Chapter 18 - Genetic and genomic approaches to identify genes involved in flagellar assembly in Chlamydomonas reinhardtii. In Methods in cell biology (eds Renata B, Wallace FM), pp. 349–386. Cambridge, MA: Academic Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hart SN, Sarangi V, Moore R, Baheti S, Bhavsar JD, Couch FJ, Kocher J-PA, Tang H. 2013. SoftSearch: integration of multiple sequence features to identify breakpoints of structural variations. PLoS ONE 8, e83356 (doi:10.1371/journal.pone.0083356) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alford LM, Mattheyses AL, Hunter EL, Lin H, Dutcher SK, Sale WS. 2013. The Chlamydomonas mutant pf27 reveals novel features of ciliary radial spoke assembly. Cytoskeleton 70, 804–818. (doi:10.1002/cm.21144) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dutcher, et al. In preparation. Intraflagellar transport (IFT) is need for assembling the ciliary necklace.
- 32.Lin H, Kwan AL, Dutcher SK. 2010. Synthesizing and salvaging NAD+: lessons learned from Chlamydomonas reinhardtii. PLoS Genet. 6, e1001105 (doi:10.1371/journal.pgen.1001105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dutcher SK, Morrissette NS, Preble AM, Rackley C, Stanga J. 2002. Epsilon-tubulin is an essential component of the centriole. Mol. Biol. Cell. 13, 3859–3869. (doi:10.1038/srep3423210.1091/mbc.E02-04-0205) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Awata J, Takada S, Standley C, Lechtreck KF, Bellvé KD, Pazour GJ, Fogarty KE, Witman GB. 2014. NPHP4 controls ciliary trafficking of membrane proteins and large soluble proteins at the transition zone. J. Cell Sci. 127, 4714–4727. (doi:10.1242/jcs.155275) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wright RL, Salisbury J, Jarvik JW. 1985. A nucleus–basal body connector in Chlamydomonas reinhardtii that may function in basal body localization or segregation. J. Cell Biol. 101, 1903–1912. (doi:10.1083/jcb.101.5.1903) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Albee AJ, Kwan AL, Lin H, Granas D, Stormo GD, Dutcher SK. 2013. Identification of cilia genes that affect cell-cycle progression using whole-genome transcriptome analysis in Chlamydomonas reinhardtti. G3 3, 979–991. (doi:10.1534/g3.113.006338) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Craige B, Brown JM, Witman GB. 2001. Isolation of Chlamydomonas flagella. In Current protocols in cell biology, vol. 59, 3.41 Hoboken, NJ: John Wiley & Sons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin H, Miller ML, Granas DM, Dutcher SK. 2013. Whole genome sequencing identifies a deletion in protein phosphatase 2A that affects its stability and localization in Chlamydomonas reinhardtii. PLoS Genet. 9, e1003841 (doi:10.1371/journal.pgen.1003841) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. (doi:10.1093/nar/gkh340) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lupas A, Van Dyke M, Stock J. 1991. Predicting coiled coils from protein sequences. Science 252, 1162–1164. (doi:10.1126/science.252.5009.1162) [DOI] [PubMed] [Google Scholar]
- 41.Lin K, Simossis VA, Taylor WR, Heringa J. 2005. A simple and fast secondary structure prediction method using hidden neural networks. Bioinformatics 21, 152–159. (doi:10.1093/bioinformatics/bth487) [DOI] [PubMed] [Google Scholar]
- 42.Huerta-Cepas J, et al. 2016. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44(D1), D286–DD93. (doi:10.1093/nar/gkv1248) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rogozin IB, Carmel L, Csuros M, Koonin EV. 2012. Origin and evolution of spliceosomal introns. Biol. Dir. 7, 11 (doi:10.1186/1745-6150-7-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang B, Ramanis Z, Dutcher SK, Luck DJL. 1982. Uniflagellar mutants of Chlamydomonas: evidence for the role of basal bodies in transmission of positional information. Cell 29, 745–753. (doi:10.1016/0092-8674(82)90436-6) [DOI] [PubMed] [Google Scholar]
- 45.Dutcher SK, Trabuco EC. 1998. The UNI3 gene is required for assembly of basal bodies of Chlamydomonas and encodes delta-tubulin, a new member of the tubulin superfamily. Mol. Biol. Cell. 9, 1293–1308. (doi:10.1091/mbc.9.6.1293) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Myster SH, Knott JA, Wysocki KM, O'Toole E, Porter ME. 1999. Domains in the 1α dynein heavy chain required for inner arm assembly and flagellar motility in Chlamydomonas. J. Cell Biol. 146, 801–818. (doi:10.1083/jcb.146.4.801) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, Rosenbaum JL. 1998. Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J. Cell Biol. 141, 993–1008. (doi:10.1083/jcb.141.4.993) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pazour GJ, Agrin N, Leszyk J, Witman GB. 2005. Proteomic analysis of a eukaryotic cilium. J. Cell Biol. 170, 103–113. (doi:10.1083/jcb.200504008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li JB, et al. 2004. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell 117, 541–552. (doi:10.1016/S0092-8674(04)00450-7) [DOI] [PubMed] [Google Scholar]
- 50.Sizova I, Fuhrmann M, Hegemann P. 2001. A Streptomyces rimosus aphVIII gene coding for a new type phosphotransferase provides stable antibiotic resistance to Chlamydomonas reinhardtii. Gene 277, 221–229. (doi:10.1016/S0378-1119(01)00616-3) [DOI] [PubMed] [Google Scholar]
- 51.Tam LW, Lefebvre PA. 1993. Cloning of flagellar genes in Chlamydomonas reinhardtii by DNA insertional mutagenesis. Genetics 135, 375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li X, et al. 2016. An indexed, mapped mutant library enables reverse genetics studies of biological processes in Chlamydomonas reinhardtii. Plant Cell 28, 367–387. (doi:10.1105/tpc.15.00465) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thermann R, et al. 1998. Binary specification of nonsense codons by splicing and cytoplasmic translation. EMBO J. 17, 3484–3494. (doi:10.1093/emboj/17.12.3484) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lindeboom RGH, Supek F, Lehner B. 2016. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet. 48, 1112–1118. (doi:10.1038/ng.3664) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dvinge H, Kim E, Abdel-Wahab O, Bradley RK. 2016. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer. 16, 413–430. (doi:10.1038/nrc.2016.51) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fairbrother WG, Yeh R-F, Sharp PA, Burge CB. 2002. Predictive identification of exonic splicing enhancers in human genes. Science 297, 1007–1013. (doi:10.1126/science.1073774) [DOI] [PubMed] [Google Scholar]
- 57.Cáceres EF, Hurst LD. 2013. The evolution, impact and properties of exonic splice enhancers. Genome Biol. 14, R143 (doi:10.1186/gb-2013-14-12-r143) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Labadorf A, Link A, Rogers MF, Thomas J, Reddy AS, Ben-Hur A. 2010. Genome-wide analysis of alternative splicing in Chlamydomonas reinhardtii. BMC Genomics 11, 114 (doi:10.1186/1471-2164-11-114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhan X, Yan C, Zhang X, Lei J, Shi Y. 2018. Structure of a human catalytic step I spliceosome. Science 359, 537–545. (doi:10.1126/science.aar6401) [DOI] [PubMed] [Google Scholar]
- 60.Bai R, Yan C, Wan R, Lei J, Shi Y. 2017. Structure of the post-catalytic spliceosome from Saccharomyces cerevisiae. Cell 171, 1589–1598. (doi:10.1016/j.cell.2017.10.038) [DOI] [PubMed] [Google Scholar]
- 61.Wilkinson ME, Fica SM, Galej WP, Norman CM, Newman AJ, Nagai K. 2017. Postcatalytic spliceosome structure reveals mechanism of 3′-splice site selection. Science 358, 1283–1288. (doi:10.1126/science.aar3729) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu S, et al. 2017. Structure of the yeast spliceosomal postcatalytic P complex. Science 358, 1278–1283. (doi:10.1126/science.aar3462) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fabrizio P, et al. 2009. The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome. Mol. Cell 36, 593–608. (doi:10.1016/j.molcel.2009.09.040) [DOI] [PubMed] [Google Scholar]
- 64.Rhind N, et al. 2011. Comparative functional genomics of the fission yeasts. Science 332, 930–936. (doi:10.1126/science.1203357) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Perrault I, et al. 2015IFT81, encoding an IFT-B core protein, as a very rare cause of a ciliopathy phenotype. J. Med. Genet. 52, 657–665. (doi:10.1136/jmedgenet-2014-102838) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Duran I, et al. 2016. Destabilization of the IFT-B cilia core complex due to mutations in IFT81 causes a spectrum of short-rib polydactyly syndrome. Sci. Rep. 6, 34232 (doi:10.1038/srep34232) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dharmat R, et al. 2017. IFT81 as a candidate gene for nonsyndromic retinal degeneration. Invest. Ophthalmol. Vis. Sci. 58, 2483–2490. (doi:10.1167/iovs.16-19133) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bhogaraju S, et al. 2013. Molecular basis of tubulin transport within the cilium by IFT74 and IFT81. Science 341, 1009–1012. (doi:10.1126/science.1240985) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gilissen C, et al. 2010. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am. J. Hum. Genet. 87, 418–423. (doi:10.1016/j.ajhg.2010.08.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bacino CA, Dhar SU, Brunetti-Pierri N, Lee B, Bonnen PE. 2012. WDR35 mutation in siblings with Sensenbrenner syndrome: a ciliopathy with variable phenotype. Am. J. Med. Genet. Part A 158A, 2917–2924. (doi:10.1002/ajmg.a.35608) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hoffer JL, Fryssira H, Konstantinidou AE, Ropers HH, Tzschach A. 2013. Novel WDR35 mutations in patients with cranioectodermal dysplasia (Sensenbrenner syndrome). Clin. Genet. 83, 92–95. (doi:10.1111/j.1399-0004.2012.01880.x) [DOI] [PubMed] [Google Scholar]
- 72.Li Y, et al. 2015. Respiratory motile cilia dysfunction in a patient with cranioectodermal dysplasia. Am. J. Med. Genetic. Part A 167, 2188–2196. (doi:10.1002/ajmg.a.37133) [DOI] [PubMed] [Google Scholar]
- 73.Smith C, Lamont RE, Wade A, Bernier FP, Parboosingh JS, Innes AM. 2016. A relatively mild skeletal ciliopathy phenotype consistent with cranioectodermal dysplasia is associated with a homozygous nonsynonymous mutation in WDR35. Am. J. Med. Genet. Part A. 170, 760–765. (doi:10.1002/ajmg.a.37514) [DOI] [PubMed] [Google Scholar]
- 74.Mill P, et al. 2011. Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am. J. Hum. Genet. 88, 508–515. (doi:10.1016/j.ajhg.2011.03.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Caparrós-Martín JA, et al. 2015. Specific variants in WDR35 cause a distinctive form of Ellis-van Creveld syndrome by disrupting the recruitment of the EvC complex and SMO into the cilium. Hum. Mol. Genet. 24, 4126–4137. (doi:10.1093/hmg/ddv152) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fu W, Wang L, Kim S, Li J, Dynlacht BD. 2016. Role for the IFT-A complex in selective transport to the primary cilium. Cell Rep. 17, 1505–1517. (doi:10.1016/j.celrep.2016.10.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Behal RH, Miller MS, Qin H, Lucker BF, Jones A, Cole DG. 2012. Subunit interactions and organization of the Chlamydomonas reinhardtii intraflagellar transport complex A proteins. J. Biol. Chem. 287, 11 689–11 703. (doi:10.1074/jbc.M111.287102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hou Y, Witman GB. 2017. The N-terminus of IFT46 mediates intraflagellar transport of outer arm dynein and its cargo-adaptor ODA16. Mol. Biol. Cell. 28, 2420–2433. (doi:10.1091/mbc.E17-03-0172) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brown JM, Cochran DA, Craige B, Kubo T, Witman GB. 2015. Assembly of IFT trains at the ciliary base depends on IFT74. Curr. Biol. 25, 1583–1593. (doi:10.1016/j.cub.2015.04.060) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequence data for the strains in the electronic supplementary material, table S2 are deposited at BioProject ID PRJNA407207 at the National Center for Bioinformatics Information (NCBI). All other data are presented in the electronic supplementary material, tables and figures.