

ABSTRACT
Growing bacteria have a high concentration of ribosomes to ensure sufficient protein synthesis, which is necessary for genome replication and cellular division. To elucidate whether metabolic activity of soil microorganisms is coupled with growth, we investigated the relationship between rRNA and DNA synthesis in a soil bacterial community using quantitative stable isotope probing (qSIP) with H218O. Most soil bacterial taxa were metabolically active and grew, and there was no significant difference between the isotopic composition of DNA and RNA extracted from soil incubated with H218O. The positive correlation between 18O content of DNA and rRNA of taxa, with a slope statistically indistinguishable from 1 (slope = 0.96; 95% confidence interval [CI], 0.90 to 1.02), indicated that few taxa made new rRNA without synthesizing new DNA. There was no correlation between rRNA-to-DNA ratios obtained from sequencing libraries and the atom percent excess (APE) 18O values of DNA or rRNA, suggesting that the ratio of rRNA to DNA is a poor indicator of microbial growth or rRNA synthesis. Our results support the notion that metabolic activity is strongly coupled to cellular division and suggest that nondividing taxa do not dominate soil metabolic activity.
IMPORTANCE Using quantitative stable isotope probing of microbial RNA and DNA with H218O, we show that most soil taxa are metabolically active and grow because their nucleic acids are significantly labeled with 18O. A majority of the populations that make new rRNA also grow, which argues against the common paradigm that most soil taxa are dormant. Additionally, our results indicate that relative sequence abundance-based RNA-to-DNA ratios, which are frequently used for identifying active microbial populations in the environment, underestimate the number of metabolically active taxa within soil microbial communities.
KEYWORDS: rRNA, DNA, quantitative stable isotope probing, qSIP, atom percent excess 18O values of nucleic acids, APE 18O, density shift, rRNA-to-DNA ratio, relative sequence abundance, microbial activity, microbial growth, soil, atom percent excess, environmental microbiology, soil microbiology
INTRODUCTION
Growing bacteria require many proteins to replicate their DNA and divide and therefore must also have a large pool of ribosomes to synthesize these proteins. Ribosome concentrations in pure cultures correlated positively with microbial growth rates: fast-growing bacteria contained relatively more ribosomes, measured as rRNA-to-DNA ratios, than slowly growing bacteria (1–5). rRNA-to-DNA ratios have been extended to environmental studies of multispecies bacterial communities, in which the ratios are used as indicators of bacterial metabolic activity (6–8). By comparing relative abundances in sequencing libraries from RNA and DNA directly extracted from the environment, ostensibly active and growing microbial populations, with high rRNA-to-DNA ratios, and inactive microbial populations, with low rRNA-to-DNA ratios, can be identified (9).
Growth of microbial taxa can be characterized by DNA stable isotope probing (DNA-SIP) with H218O (10). This technique identifies organisms that assimilate and grow on isotopically labeled substrates (11), and detection of DNA with high 18O content provides direct evidence for microbial growth because oxygen from water is incorporated into bacterial genomes during replication (10). H218O-DNA-SIP has been applied to study microbial growth in a wide range of ecosystems, including terrestrial (12–15) and aquatic (16) habitats.
Whereas DNA-SIP investigates DNA replication, a measure of cell division, RNA-SIP characterizes RNA synthesis, an indicator of microbial metabolic activity (17–20). Thus, stable isotope probing of rRNA may detect metabolically active populations that are not growing, and comparison of RNA- and DNA-SIP results can characterize the relationship between metabolic activity and microbial population growth.
Few studies have simultaneously compared DNA- and RNA-SIP results of microbial communities. Most used 13C-containing substrates (18, 20, 21) and generally found that incorporation of the isotope into microbial RNA (mRNA or rRNA) was faster than incorporation into DNA. There has been only one study that investigated microbial growth and activity in soil using DNA- and RNA-SIP with H218O (22). This study found that, after incubation of the soil with H218O, the bacterial communities recovered in heavy RNA-SIP fractions were similar to communities characterized through bulk RNA analysis. The community represented in the heavy fractions of DNA was also similar to the community in nonfractionated DNA, leading the investigators to propose that most soil microorganisms were active and grew during the incubation with H218O, but their study did not quantitatively assess the isotopic enrichment of individual taxa.
In the present study, we used quantitative stable isotope probing (qSIP) (23) with H218O to investigate rRNA synthesis and growth of microbial taxa in soil. Following incubation of the soil with H218O, we calculated shifts in rRNA and DNA densities of bacterial taxa using all SIP fractions and used the density shift to calculate the 18O composition of nucleic acids. This approach not only captures a greater number of taxa, including taxa present in fractions of intermediate densities, but also quantifies the atom percent excess 18O values (APE 18O) of rRNA and DNA for all members of the community and is unaffected by taxon-specific GC content. In addition to qSIP analysis, we compared ratios of rRNA to DNA in sequencing libraries to the isotopic composition of rRNA or DNA for all taxa. Our objective was to elucidate whether metabolic activity, assessed by measuring rRNA synthesis, and growth, assessed by measuring DNA synthesis, were coupled for all soil taxa. We hypothesized that growing populations will also be metabolically active. Additionally, we were interested to find if nongrowing taxa synthetized new rRNA of if microorganisms were able to grow with minimal rRNA synthesis.
RESULTS
Labeling of microbial RNA and DNA.
Incubation of soil with H218O resulted in incorporation of 18O into microbial RNA and DNA (Fig. 1). The presence of 18O within nucleic acids increased their densities so that substantial weighted average density (WAD) shifts were observed after isopycnic centrifugation (Fig. 1, P < 0.05, on all days). There was no significant interaction between the density shift of RNA or DNA and time (F1.012, 4.048 = 0.383, P = 0.572, Greenhouse-Geisser epsilon = 0.506), showing that incorporation of 18O into RNA was similar to incorporation of 18O into DNA on each day. However, there was a significant main effect for time (F1.012, 4.048 = 15.620, P = 0.016, Greenhouse-Geisser epsilon = 0.506), with an increase in nucleic acid density over time. Specifically, RNA density shifts significantly increased from 0.0067 ± 0.0018 g/ml on day 1 to 0.0165 ± 0.0056 g/ml on day 8 (F2, 4 = 15.404, P = 0.013); mean changes in DNA density shifts were similar to those observed for RNA but were not statistically significant (F1, 2 = 5.501, P = 0.144). 18O isotope ratio mass spectrometry (IRMS) analysis of total RNA and DNA samples showed similar trends over time. The isotopic enrichment, expressed as delta (δ) 18O values, is provided in Table S1 in the supplemental material.
FIG 1.
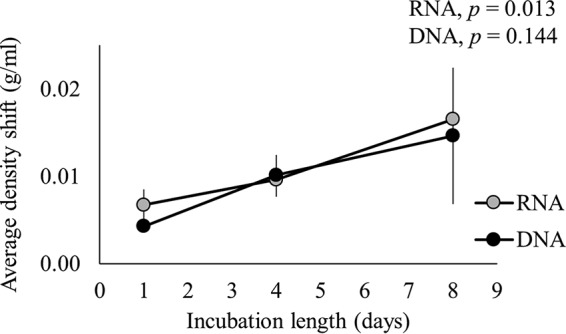
Density shifts of RNA or DNA extracted from soil incubated with H218O for 1, 4, or 8 days. Bars show the means ± standard deviations (n = 3). P values indicate statistical significance for a main effect of time.
Correlation between labeled RNA and DNA for taxa.
There was a strong positive correlation between the overall atom percent excess (APE) 18O of rRNA and DNA [ρ(345 simultaneously detected taxa) = 0.859, P < 0.005] (Fig. 2). Positive values of APE 18O indicate isotopic enrichment of nucleic acids above natural abundance levels of 18O (approximately 0.2 atom%), which resulted from assimilation of H218O followed by incorporation of the isotope during nucleic acid synthesis. For all time points, the incorporation of 18O into rRNA and DNA was described by the following equation: APE 18O of DNA = 0.96 × APE 18O of rRNA − 0.001, with the 95% confidence interval (CI) around the slope from 0.90 to 1.02 (model II regression, major axis [MA] analysis), indicating that, for a given taxon, the amount of 18O assimilated into rRNA was not different from the amount of 18O assimilated into DNA. The number of taxa simultaneously detected in DNA and RNA qSIP analyses was 83 on day 1, 126 on day 4, and 136 in day 8 (Table 1). Taxa unique to each library type could not be included in the correlations. There were 106, 52, and 73 unique taxa in the rRNA libraries on days 1, 4, and 8, respectively, and 28, 74, and 51 unique taxa in the DNA libraries on days 1, 4, and 8, respectively (Table 1).
FIG 2.
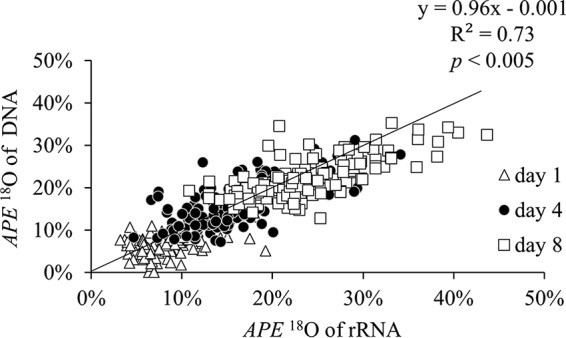
Correlation between atom percent excess (APE) 18O values of DNA and rRNA among soil taxa on days 1, 4, and 8. The equation shown is for the overall regression, which includes all time points. The black line represents a 1:1 ratio, where the APE 18O of DNA and rRNA are equal.
TABLE 1.
Number of taxa detected in rRNA or DNA libraries at each time point
| Day | No. of taxa present in RNA libraries | No. of taxa present in DNA libraries | No. of taxa unique to RNA libraries | No. of taxa unique to DNA libraries | No. of taxa shared between RNA and DNA libraries | No. of taxa present in RNA or DNA libraries |
|---|---|---|---|---|---|---|
| 1 | 189 | 111 | 106 | 28 | 83 | 217 |
| 4 | 178 | 200 | 52 | 74 | 126 | 252 |
| 8 | 209 | 187 | 73 | 51 | 136 | 260 |
Correlation between labeled RNA and DNA for phyla.
APE 18O of rRNA and DNA were also correlated for individual soil phyla (Fig. 3). All correlations were positive, and the APE 18O of the nucleic acids increased over time similarly to the overall correlation shown in Fig. 2. The confidence intervals around the regression slopes overlapped 1 for all phyla except Acidobacteria, which had a slope significantly less than 1 (slope = 0.72; 95% CI around the slope, 0.61 to 0.86). Some groups, for instance, Thaumarchaeota, Chloroflexi, or Verrucomicrobia, had lower APE 18O of their nucleic acids than other groups, including Actinobacteria or Proteobacteria (Fig. 3). The regressions also demonstrated that, within each phylum, taxa varied in 18O compositions of their DNA and rRNA. For example, among the Actinobacteria, Streptomycetaceae contained highly 18O-labeled rRNA (43.72% ± 7.41%) and DNA (32.51% ± 9.44%), whereas Rubrobacter had much lower 18O content of rRNA (16.61% ± 4.78%) and DNA (16.34% ± 6.76%) even after 8 days of incubation with H218O. Similar differences were observed among taxa from other phyla (Fig. 3).
FIG 3.

Correlations between APE 18O of DNA and rRNA of major soil phyla at incubation times of 1, 4, and 8 days. For each phylum, the equation represents the overall relationship, comprising all time points, between the two variables. Symbols represent taxa within the phylum, and error bars represent 95% confidence intervals of the APE 18O values.
Correlation between RNA-to-DNA ratio and nucleic acid labeling.
Across taxa, ratios of rRNA to DNA in sequencing libraries were not correlated with their APE 18O of DNA or rRNA [ρ(335) = 0.025, P = 0.649 for DNA, and ρ(574) = −0.082, P = 0.051 for rRNA] (Fig. 4A and B), indicating that the ratios were not a sound indicator of metabolic activity. Ratios of relative abundances of RNA to DNA are commonly used to assess microbial activity in nature. Active taxa are characterized by high ratios (or at least ratios of >1), and inactive taxa are characterized low ratios (ratios of <1). We therefore expected that taxa with high ratios would also be highly labeled with 18O due to substantial rRNA synthesis or growth, but this was not observed. The ratios were greater than 1 for only 35 (44%) taxa on day 1, 70 (56%) taxa on day 4, and 57 (44%) taxa on day 8. These proportions, which supposedly indicate active taxa, were considerably lower than proportions of active taxa identified through qSIP.
FIG 4.
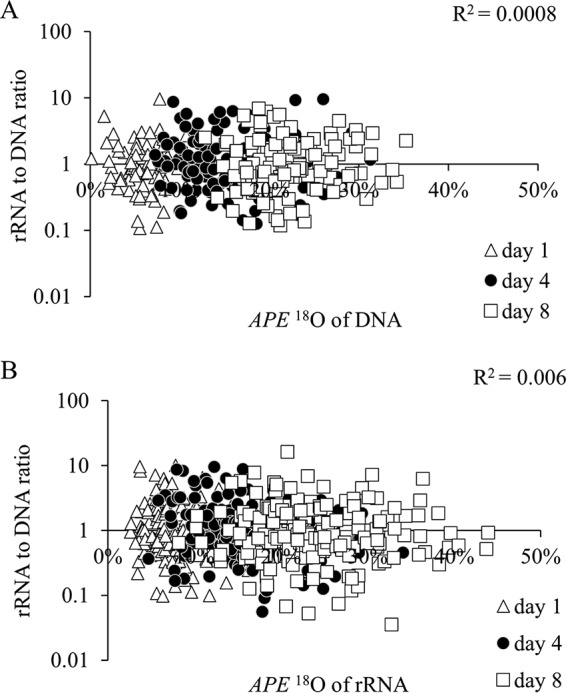
Log scale relationship between taxon-specific ratios of rRNA to DNA in sequencing libraries and the APE 18O of DNA (A) or rRNA (B) at days 1, 4, and 8.
DISCUSSION
We expected bacterial RNA to become significantly more labeled with 18O than DNA because RNA turns over faster than DNA (24–27) and because bacteria can be metabolically active without growing (28, 29). We hypothesized that, compared to RNA, a small fraction of the DNA extracted from soil incubated with H218O would be new (i.e., labeled with 18O) because DNA is present in dormant or dead cells (30), whereas high RNA concentrations are associated with cellular activity (31–33). Additionally, we hypothesized that all RNA extracted from soil incubated with H218O would be new and labeled with 18O after 8 days of incubation. Assuming that half of the oxygen atoms in RNA come from water (34), the APE 18O of new RNA should be at least 50%. However, only ∼23% of O atoms in RNA were 18O, indicating that approximately half of the ribonucleotides must have been made prior to H218O addition and persisted throughout the incubation.
Although we found evidence for old rRNA in soil, we also found that most soil taxa were synthesizing new rRNA and were growing. Overall, rates of rRNA and DNA synthesis were statistically indistinguishable from each other in this soil bacterial community, a result that contrasts sharply with the expectation that RNA synthesis could occur without DNA replication or that metabolism in soil is largely independent of microbial growth. A number of labeled taxa were detected only in the rRNA or DNA libraries. This suggested that extremely rare taxa can also be metabolically active and can grow.
The degree to which taxa incorporated 18O from water into their nucleic acids varied, indicating differences in growth and metabolic rates among bacterial populations in soil. Taxa with low APE 18O of rRNA and DNA likely simply had lower growth or metabolic rates than taxa with high APE 18O of nucleic acids. For instance, Chloroflexi or Planctomycetes express oligotrophic characteristics (35, 36), thus possibly explaining the low APE 18O of their nucleic acids. In contrast, Bacteroidetes are known copiotrophs, and Proteobacteria comprise many copiotrophic genera (36, 37), which can explain the higher growth and metabolic activity of these groups in a rewetted soil. Alternatively, variation in the APE 18O of nucleic acids among populations may reflect the presence of old rRNA and DNA synthetized prior to H218O addition. The larger the amount of older nucleic acids that persist through the incubation, the lower is the measured 18O composition of the nucleic acid extracts.
Unlike 13C substrates, which require de novo nucleotide synthesis to become incorporated into nucleic acids (20), 18O from H218O can become incorporated into new rRNA and new DNA during polymerization of salvaged nucleotides (38, 39). Salvaged nucleotides were previously present in nucleic acids while de novo nucleotides were newly synthesized. In an environment with high concentrations of H218O, de novo nucleotides will contain more 18O than salvaged nucleotides because 18O may be incorporated into the ribose sugar or the base during nucleotide synthesis. In contrast, nucleic acids generated with salvaged ribonucleotides incorporate 18O isotopes only as branch oxygen atoms in phosphodiester bonds that link nucleotides together (40). Consequently, the isotopic composition of RNA or DNA in soil microorganisms incubated with H218O is impacted by the source of nucleotides used in their synthesis. Microorganisms that heavily salvage nucleotides will have lower 18O compositions of nucleic acids even during high rRNA or DNA synthesis than microorganisms that newly synthesize nucleotides (12). It is possible that 13C-RNA-SIP studies could fail to capture organisms that salvage ribonucleotides in early stages of an experiment, while H218O-based SIP studies would find these populations to contain RNA with only moderate 18O content.
Within a cell, newly synthetized RNA and DNA molecules should have similar 18O isotopic compositions because both nucleic acids are made from rapidly interchanging nucleotide pools. Ribonucleotide reductase catalyzes the conversion of ribonucleotides to deoxyribonucleotides (41, 42), so that it is likely that both ribonucleotides and deoxyribonucleotides, which can be salvaged or synthetized de novo, have the same 18O composition. It is only when a cell synthesizes RNA but not DNA that we would expect to find differences in 18O composition between the nucleic acids.
The isotopic composition of rRNA and DNA can be related quantitatively to microbial activity (rRNA synthesis) and growth (DNA synthesis) if degradation rates of old and new, 18O-enriched, nucleic acids are similar. The degree of 18O incorporation then reflects the level of activity or growth for each taxon. Currently, ratios of relative abundance of rRNA to DNA in sequencing libraries are often used to assess microbial activity in soil (see, for example, references 7 and 8), and in aquatic environments (see, for example, references 43, 44, and 45). Taxa with ratios below 1 are considered inactive (9, 46) because they are represented by fewer rRNA than DNA sequences (47). This may occur when other populations have very high rRNA-to-DNA ratios or when a substantial fraction of DNA, originally derived from taxa with low ratios, is released extracellularly and becomes adsorbed to the soil matrix (30). The extant population could be highly active but misclassified as inactive because of the extracellular (old) DNA. Sequencing DNA does not distinguish between old and new nucleic acids, but it is possible to do so using stable isotope probing.
SIP analysis of bacterial nucleic acids showed that 99.5% of taxa were synthesizing new rRNA while 100% of taxa had grown at the end of the incubation. In contrast, rRNA-to-DNA ratios indicated that only 44% of taxa were active. Many studies documented the presence of inactive taxa in the environment (27, 33, 48) through measuring the relative abundance ratios in sequencing libraries. This study finds that taxa with low rRNA-to-DNA ratios contain newly synthesized nucleic acids, suggesting that relative abundance ratios may not be appropriate for identifying active populations in soil. We find that rRNA-to-DNA ratios underestimated the number of active taxa. Recently, Steven and colleagues (49) have also found that rRNA-to-DNA ratios tend to misclassify active populations as dormant, with a rate of false detection as high as 47% when the simulated communities comprised populations that differed in the rRNA amplification (amount of ribosomes per cell) associated with a specific growth rate (49).
qSIP reflects the uptake of an isotope by entire populations, not by single microorganisms. It is therefore impossible to determine what fractions (if any) of a microbial population are more or less active relative to each other. It is likely that individual cells have different metabolic rates and thus assimilate different amounts of 18O, but our approach detects only the isotopic enrichment of the whole group. Identifying regions of DNA and RNA capable of differentiating ecotypes and using these as targets for qSIP analysis could provide finer resolution. Still, despite this shortcoming, we were able to draw interesting conclusions about microbial taxa, certainly applicable to important questions in microbial ecology. Adding water to dry soils does not provide an energy or a carbon source, but it still likely stimulates the growth and activity of some populations, some of which otherwise may have remained inactive and nongrowing. The qSIP approach using 18O-labeled water could therefore potentially overestimate growth and activity of some populations. Nevertheless, addition of 200 mg of water certainly had less impact on microbial growth and activity than, for example, addition of an equivalent amount of carbon. Furthermore, we were able to detect significant differences in the APE 18O of nucleic acids among taxa as some populations did not assimilate significant amounts of 18O, indicating that the technique is able to distinguish differential rates of growth and activity among taxa in complex communities. Last, this study focused on only one ecosystem; other ecosystems may show different patterns of microbial activity and growth when explored through qSIP. Further research is therefore needed to explore microbial community dynamics in different ecosystems and to assess whether qSIP consistently provides evidence for high metabolic activity and growth of microbial taxa in the environment.
Conclusion.
Contrary to our expectation, we found a strong correlation between the 18O composition of a microbial population's rRNA and DNA with a slope approximating 1. This indicates that it is rare for a cell to synthesize new rRNA without synthesizing DNA. We observed some taxa that had less enrichment in rRNA than DNA, possibly indicating that they contained rRNA prior to H218O addition. We also found that the nucleotides of most dominant soil taxa were enriched in 18O, suggesting that a large fraction of the soil microbial community appears active rather than dormant. Last, our study suggests that rRNA-to-DNA ratios may underestimate the number of active taxa in natural ecosystems.
MATERIALS AND METHODS
Sample collection.
Soil samples were obtained from the A-horizon (∼20 cm deep) at three locations (n = 3) (34°57′21.879″N, 111°45′14.859″W; 34°57′21.289″N, 111°45′14.189″W; and 34°57′21.591″N, 111°45′14.683″W) near Sedona, AZ, USA, in April 2014. This soil was characterized as sandy loam with an average pH of 6.95 ± 0.42, with a soil moisture content of 4.11% ± 0.24% at the time of sampling. The organic matter content was 13.7% ± 1.7%, and concentrations of NO3-N and P were 13.5 ± 7.0 and 14.3 ± 0.6 ppm, respectively. The samples were immediately transported at room temperature to Northern Arizona University. Following sieving through a 2-mm-pore-size sieve, the soils were air dried overnight on clean metal trays.
Sample processing.
Two grams of soil was incubated with 400 μl of 95 atom% H218O or with 400 μl of sterile, natural-abundance [18O]water in 15-ml Falcon tubes for 1, 4, or 8 days in the dark. Total RNA and DNA were extracted from 1 gram of soil using an RNA PowerSoil Total RNA isolation kit and DNA elution accessory kit (Mo Bio Laboratories, Carlsbad, CA) immediately at the end of each incubation. All extracts were visualized on 1% agarose E-gels (Life Technologies, Grand Island, NY) and quantified with a Qubit 2.0 Fluorimeter (Life Technologies) and Qubit RNA assay kit or double-stranded DNA (dsDNA) assay kit (Life Technologies). Total RNA was digested with DNA-free DNase treatment removal reagents (Ambion, Life Technologies, Grand Island, NY) and visualized on E-gels, and the concentration of the treated RNA was determined with the Qubit.
Isopycnic ultracentrifugation.
Nucleic acids were ultracentrifuged in 3.3 ml OptiSeal polyallomer tubes (Beckman Coulter) using an Optima MAX benchtop ultracentrifuge (Beckman Coulter, Fullerton, CA). For RNA samples, 2.585 ml of 1.99 g/ml cesium trifluoroacetate (CsTFA) solution (GE Healthcare, Piscataway, NJ), 110 μl of deionized formamide (Sigma-Aldrich, St. Louis, MO), and 505 μl of RNase-free water (Life Technologies, Grand Island, NY) were combined. For DNA samples, a cesium chloride (CsCl) solution consisting of 2.7 ml of saturated CsCl and 400 μl of gradient buffer (200 mM Tris, pH 8.0, 200 mM KCl, and 2 mM EDTA) was prepared. For ultracentrifugation of RNA, 3.2 ml of the CsTFA solution and 1 μg of total RNA were added to each tube and centrifuged in a TLN-100 rotor at 60,000 rpm (127,000 × g) at 18°C for 72 h. For centrifugation of DNA, 3.1 ml of the CsCl solution and 1 μg of DNA were added to each tube. DNA was centrifuged identically to RNA. CsTFA and CsCl gradients were separated into 100-μl or 150-μl fractions using a manual fraction recovery system (Beckman Coulter) for RNA samples or an automated fractionation system (Brandel, Gaithersburg, MD) for DNA samples. All parts of the manual system were cleaned with ethanol (EtOH), and all parts of the automated Brandel system were thoroughly cleaned with ethanol and air between each sample. Additionally, samples incubated with natural-abundance [18O]water were processed separately from samples incubated with H218O to minimize potential carryover of the label. The density of each fraction was measured with a digital refractometer (Reichert Ophthalmic Instruments, Depew, NJ). RNA fractions were purified by adding 200 μl of ice-cold isopropanol and 100 ng of ultrapure glycogen (Affymetrix, Santa Clara, CA) to each 1.5-ml tube. Tubes were vortexed and incubated at −20°C overnight. Purification was done as described by Whiteley et al. (50). DNA fractions were purified by combining two volumes of water, one volume of isopropanol, and 100 ng of ultrapure glycogen (Affymetrix, Santa Clara, CA). Tubes were also vortexed and stored at room temperature overnight. DNA was further purified by centrifuging the fractions for 15 to 30 min at 13,400 × g, discarding the supernatant, adding 500 μl of 70% EtOH, centrifuging for 5 min at the same speed, discarding the supernatant again, centrifuging the tubes one more time at 13,400 × g for 30 s, and finally removing traces of the supernatant. Pellets were air dried for 10 min and resuspended in 20 μl of 1× Tris-EDTA (TE) buffer. Final concentrations of nucleic acids in each fraction were measured using a Qubit fluorimeter (Life Technologies).
cDNA was obtained from RNA samples, including fractionated and nonfractionated samples, using a Maxima H-Minus First Strand cDNA synthesis kit (Thermo Fisher Scientific, Waltham, MA) and random pentadecamer primers (Eurofins MWG Operon, Huntsville, AL). Each 20-μl reaction mixture contained 5 μl of RNA template, 5.0 μM primer, 0.5 mM concentrations of the deoxynucleotide triphosphates (dNTPs), 1× reverse transcriptase (RT) buffer, and sterile water.
Sequencing and data processing.
Libraries were prepared from fractionated and nonfractionated cDNA or DNA by preamplifying the 16S rRNA gene with the PCR primers 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACVSGGGTATCTAAT-3′) (Eurofins MWG Operon, Huntsville, AL). PCR was carried out in 8-μl reaction mixtures containing 1 μl of template (DNA or cDNA), 4 μl (2×) Phusion Mastermix (water, 10× RedJuice [40% 1 M Tris, pH 8.5, phenol red, 60% glycerol]) with 5× HF buffer (10 mM dNTPs, Phusion HSII polymerase), 2.4 μl nuclease-free water, 0.25 μl (50 M) MgCl2, and 0.06 μl (25.0 mM) of each primer. Preamplification was done in triplicate using a Tetrad PCR thermal cycler (Bio-Rad, Hercules, CA). Cycling conditions started with a 2-min denaturation step at 95°C, followed by 15 cycles of 30 s at 95°C, 30 s at 55°C, 4 min at 60°C, and a final hold at 10°C. The three PCR replicates were pooled, visualized (1% agarose gel), and diluted 10-fold with Tris-Cl (pH 8.0). One microliter of the diluted PCR products was amplified with 515F and 806R indexed primers (51). Carboxyl-modified (0.1%) Sera-Mag Magnetic Speed-beads (Thermo Fisher Scientific, Freemont, CA) in 18% polyethylene glycol (PEG) were used to purify the indexed amplicons, which were then quantified with a Quant-it PicoGreen double-stranded DNA assay kit (Life Technologies). A PerkinElmer automatic liquid handler (Waltham, MA) was used to pool the amplicons, with final concentrations between 2 and 3.5 nM. Pools were subsequently bead purified with the Sera-Mag beads and quantified with a Kapa SYBR Fast qPCR kit (KAPA Biosystems, Inc., Wilmington, MA). DNA and cDNA libraries were sequenced in separate runs on an Illumina MiSeq platform. All runs used the 2-by-150 paired-end read chemistry and followed identical protocols. Sequencing was done at the Environmental Genetics and Genomics Facility (EnGGen) at Northern Arizona University.
An in-house chained workflow (52), which is based on QIIME, version 1.7 (51), was used to process the sequencing data. Information about the workflow can be found at GitHub (https://github.com/alk224/akutils-v1.2). First, sequences were screened and filtered for PhiX contamination (5 to 6%). Next, reads and index files were joined and demultiplexed. The chained workflow was used for chimera filtering, operational taxonomic unit (OTU) picking (swarm algorithm, open reference, 97% similarity), sequence alignment (Greengenes, version 13_5 database) (53), and taxonomy assignment (Ribosomal Data Project classifier) (54). Singletons were removed. OTU tables were filtered and summarized. FastTree (55) was used to generate a phylogenetic tree, and PyNAST (56) was used to produce a filtered sequence alignment file. Normalized and nonnormalized core diversity analyses were run using a rarefaction depth of 5,000 sequences per sample for cDNA and 2,500 sequences per sample for DNA libraries to maximize the number of retained samples in each library type. The resulting L7 (species-level) nonnormalized OTU tables, which comprised 591 cDNA libraries and 521 DNA libraries and contained 665 and 930 taxa, respectively, were used in subsequent analyses.
IRMS analysis.
Nonfractionated RNA and DNA samples were quantified with a Quant-iT RiboGreen RNA assay kit (Invitrogen, Carlsbad, CA) or a Quant-iT PicoGreen dsDNA assay kit (Invitrogen) and diluted with UltraPure salmon sperm DNA solution (Invitrogen, Carlsbad, CA) to 200 μg of oxygen. The diluted samples were pipetted into silver capsules (Costech Analytical Technologies, Inc., Valencia, CA), and the capsules were placed in a 96-well tray, set on a heat block at 50°C until water evaporated, closed, weighted, and sent to the Stable Isotope Facility at the University of California, Davis, for isotopic analysis on an Elementar PyroCube (Elementar Analysensysteme GmbH, Hanau, Germany) interfaced to a Isoprime VisION (Isoprime Ltd., Stockport, United Kingdom, a unit of Elementar Analysensysteme GmbH, Hanau, Germany).
Data preparation for qSIP analysis.
For qSIP analysis, we retained sequences of taxa that were present in at least 40% of fractions from one sample or present in at least two of the three replicate field samples. This filtering removed ∼470 rare taxa from the cDNA libraries (here referred to as rRNA libraries) and ∼750 taxa from the DNA libraries. These taxa were rare and comprised less than 5.5% of the rRNA and less than 10.7% of the DNA libraries. We conserved as many taxa as possible for the analysis for which we had reliable and robust data. A taxon was therefore omitted only if we could not reliably calculate the weighted average density (WAD) for its DNA or RNA. Because reliably calculated WADs could indicate no isotope labeling, there was no inherent bias toward growing organisms in this analysis.
Weighted average densities were calculated for both types of nucleic acids. The sample-specific WADs was obtained as follows: the concentration of nucleic acids (RNA or DNA) in each fraction was multiplied by the density of the fraction. The products were summed across density fractions (the numerator in the equation below) and divided by the sum of the concentrations of nucleic acids across all fractions (the denominator in the equation below):
| (1) |
where WADs is the weighted average density of a nucleic acid sample (s) (i.e., RNA or DNA), k is the fraction of a nucleic acid sample, K is the total number of fractions from a nucleic acid sample, [nucleic acid] is the concentration of a nucleic acid in each fraction (either RNA or DNA), and density is the density of a nucleic acid in each fraction.
The WADs of nucleic acids (rRNA or DNA) were also obtained for individual taxa (t) (WADt). The calculations were identical to those for whole nucleic acid samples described above, except that the concentration of rRNA was multiplied by taxon-specific relative abundance in the rRNA libraries, and the concentration of DNA was multiplied by taxon-specific relative abundance in the DNA libraries.
| (2) |
where WADt is the weighted average density of a nucleic acid of a taxon (t), k is the fraction of a nucleic acid sample, K is the total number of fractions from a nucleic acid sample, RelAbt is the relative abundance of a taxon (t) in the sequencing library (i.e., rRNA or DNA library), [nucleic acid] is the concentration of a nucleic acid in each fraction, and density is the density of a nucleic acid in each fraction.
A correction, which was based on the 18O isotopic composition of each nucleic acid type, was applied to WADs and WADt. The isotopic composition of RNA and DNA was obtained by IRMS analysis (see Table S1 in the supplemental material) and was expressed as atom percent excess 18O (APE 18O) of RNA or DNA using the fractional abundance (F) and isotope mixing models. First, the δ18O values were converted to atom percent values as follows: F = [(δ/1,000 + 1)Rst]/[(δ/1,000 + 1)Rst +1], where F is fractional abundance (atom percent), δ is the sample-specific delta 18O value obtained by IRMS analysis, Rst is the ratio of molar abundance of heavy to light isotopes in the Vienna Standard Mean Ocean Water (VSMOW). The 18O/16O ratio is 2005.20 ppm.
Next, the atom percent 18O of each sample was determined using the isotope mixing model:
| (3) |
| (4) |
where pA is the proportion of RNA or DNA sample, pB is the proportion of salmon sperm DNA required for dilution of RNA or DNA samples for IRMS analyses, At%A is the atom percent 18O of the RNA or DNA sample, At%B is the atom percent 18O of salmon sperm DNA, and At% mix is the atom percent 18O of the mixture of the RNA or DNA sample diluted with salmon sperm DNA.
To obtain the isotopic enrichment of each labeled RNA or DNA sample relative to the natural abundance of the isotope, we calculated its atom percent excess 18O (APE 18O). This was done by subtracting the APE 18O of a nonlabeled sample from the APE 18O of a corresponding labeled sample. The APE 18O of each RNA or DNA sample was then converted to an expected WADs using the equations below:
| (5) |
| (6) |
The first equation was developed using the known molecular weight of the rRNA molecule at different 18O isotopic compositions (natural abundance and 100% 18O content), verified by empirical measurements of the density of RNA at natural abundance of 18O. Details about the second equation, for DNA, were provided by Hungate et al. (23). All calculated WADs values, here referred to as measured WADs, were adjusted using the expected WADs:
| (7) |
| (8) |
The WADs were adjusted individually for each sample and corresponding fractions. The statistical software package R (57) was used to determine all taxon-specific shifts in nucleic acid density as well as all taxon-specific atom percent excess 18O (APE 18O) values of rRNA and DNA as described by Hungate et al. (23). The code was adapted for RNA analysis, which included setting the GC content to 50%, molecular weight to 339.49, and number of oxygen atoms to 7. All computer code can be found at https://bitbucket.org/QuantitativeSIP/qsip_repo.
Ratio calculations.
Relative rRNA-to-DNA ratios were obtained by dividing a taxon-specific relative abundance in nonfractionated 16S rRNA libraries by its relative abundance in the nonfractionated DNA (i.e., 16S rRNA gene-based) libraries.
Statistical analyses.
Analysis of APE 18O of rRNA and DNA was performed in R (57). Other statistical analyses were performed in SPSS (58), with statistical significance set to an alpha (α) of 0.05. The Shapiro-Wilk test, Levene's test, and Mauchly's test were used to test for assumption of normality, homogeneity of variances, and sphericity, respectively. Additionally, we used a t test to determine if shifts in nucleic acid densities significantly differed from zero. A two-way mixed analysis of variance (ANOVA) was used to test for a significant interaction between density shift or APE 18O of nucleic acids of phyla and time. Last, we used a Spearman's rank order correlation test and a model II regression analysis in R to assess the strength and significance of the correlation between taxon-specific APE 18O of rRNA and DNA and between the ratio of relative abundance in rRNA to DNA libraries and the APE 18O of DNA or rRNA of taxa. Correlations and regressions were generated only with taxa that were present in both the DNA and RNA libraries. These taxa were referred to as the shared taxa. All other analyses included taxa that were present in only one library type, either DNA or RNA. These taxa were referred to as the unique taxa.
Accession number(s).
All sequences have been deposited in the NCBI Sequence Read Archive (SRA) under BioSample accession numbers SAMN07960499 to SAMN07960874, SAMN07965143 to SAMN07965605, and SAMN07968111 to SAMN07968486. Data can directly be accessed at https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP123236.
Data availability.
Workflow used for sequencing data processing is available at https://github.com/alk224/akutils-v1.2. R code used for calculating taxon-specific APE 18O of nucleic acids is available at https://bitbucket.org/QuantitativeSIP/qsip_repo.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by award 1142096 from the National Science Foundation, Division Of Solar Programs, the Department of Energy's Biological Systems Science Division, Program in Genomic Science (DE-SC0010579 and DE-SC0016207), and by the IGERT Fellowship. The funding agencies were involved neither in designing the study, collecting and interpreting the data, nor in deciding to submit the work for publication.
We declare that we have no conflicts of interest.
Additionally, we thank Paul Dijkstra, Matthew Bowker, Rebecca Mau, Michaela Hayer, Ben Koch, and Lela Andrews from Northern Arizona University and Joshua Sackett from the Desert Research Institute.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02441-17.
REFERENCES
- 1.Rosset R, Julien J, Monier R. 1966. Ribonucleic acid composition of bacteria as a function of growth rate. J Mol Biol 18:308–320. doi: 10.1016/S0022-2836(66)80248-6. [DOI] [PubMed] [Google Scholar]
- 2.Dortch Q, Roberts T, Clayton J, Ahmed S. 1983. RNA/DNA ratios and DNA concentrations as indicators of growth rate and biomass in planktonic marine organisms. Mar Ecol Prog Ser 13:61–71. doi: 10.3354/meps013061. [DOI] [Google Scholar]
- 3.Kemp PF, Lee S, Laroche J. 1993. Estimating the growth-rate of slowly growing marine-bacteria from RNA content. Appl Environ Microbiol 59:2594–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muttray AF, Mohn WW. 1998. RNA/DNA ratio as an indicator of metabolic activity in resin acid-degrading bacteria. Water Sci Technol 37:89–93. [Google Scholar]
- 5.Muttray AF, Yu Z, Mohn WW. 2001. Population dynamics and metabolic activity of Pseudomonas abietaniphila BKME-9 within pulp mill wastewater microbial communities assayed by competitive PCR and RT-PCR. FEMS Microbiol Ecol 38:21–31. doi: 10.1111/j.1574-6941.2001.tb00878.x. [DOI] [Google Scholar]
- 6.Rodríguez-Blanco A, Ghiglione JF, Catala P, Casamayor EO, Lebaron P. 2009. Spatial comparison of total vs. active bacterial populations by coupling genetic fingerprinting and clone library analyses in the NW Mediterranean Sea. FEMS Microbiol Ecol 67:30–42. doi: 10.1111/j.1574-6941.2008.00591.x. [DOI] [PubMed] [Google Scholar]
- 7.Baldrian P, Kolařík M, Štursová M, Kopecký J, Valášková V, Větrovský T, Žifčáková L, Šnajdr J, Rídl J, Vlček Č, Voříšková J. 2012. Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J 6:248–258. doi: 10.1038/ismej.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foesel BU, Nägele V, Naether A, Wüst PK, Weinert J, Bonkowski M, Lohaus G, Polle A, Alt F, Oelmann Y, Fischer M, Friedrich MW, Overmann J. 2014. Determinants of Acidobacteria activity inferred from the relative abundances of 16S rRNA transcripts in German grassland and forest soils. Environ Microbiol 16:658–675. doi: 10.1111/1462-2920.12162. [DOI] [PubMed] [Google Scholar]
- 9.Roszak DB, Colwell RR. 1987. Survival strategies of bacteria in the natural environment. Microbiol Rev 51:365–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwartz E. 2007. Characterization of growing microorganisms in soil by stable isotope probing with H218O. Appl Environ Microbiol 73:2541–2546. doi: 10.1128/AEM.02021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neufeld JD, Wagner M, Murrell JC. 2007. Who eats what, where and when? Isotope-labelling experiments are coming of age. ISME J 1:103–110. [DOI] [PubMed] [Google Scholar]
- 12.Aanderud ZT, Lennon JT. 2011. Validation of heavy-water stable isotope probing for the characterization of rapidly responding soil bacteria. Appl Environ Microbiol 77:4589–4596. doi: 10.1128/AEM.02735-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woods A, Watwood M, Schwartz E. 2011. Identification of a toluene-degrading bacterium from a soil sample through H218O DNA stable isotope probing. Appl Environ Microbiol 77:5995–5999. doi: 10.1128/AEM.05689-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwartz E, Van Horn DJ, Buelow HN, Okie JG, Gooseff MN, Barrett JE, Takacs-Vesbach CD. 2014. Characterization of growing bacterial populations in McMurdo dry valley soils through stable isotope probing with 18O-water. FEMS Microbiol Ecol 89:415–425. doi: 10.1111/1574-6941.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mau RL, Liu CM, Aziz M, Schwartz E, Dijkstra P, Marks JC, Price LB, Keim P, Hungate BA. 2015. Linking soil bacterial biodiversity and soil carbon stability. ISME J 9:1477–1480. doi: 10.1038/ismej.2014.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayer M, Schwartz E, Marks JC, Koch BJ, Morrissey EM, Schuettenberg AA, Hungate BA. 2016. Identification of growing bacteria during litter decomposition in freshwater through H218O quantitative stable isotope probing. Environ Microbiol Rep 8:975–982. doi: 10.1111/1758-2229.12475. [DOI] [PubMed] [Google Scholar]
- 17.Manefield M, Whiteley AS, Griffiths RI, Bailey MJ. 2002. RNA stable isotope probing, a novel means of linking microbial community function to phylogeny. Appl Environ Microbiol 68:5367–5373. doi: 10.1128/AEM.68.11.5367-5373.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lueders T, Manefield M, Friedrich MW. 2004. Enhanced sensitivity of DNA- and rRNA-based stable isotope probing by fractionation and quantitative analysis of isopycnic centrifugation gradients. Environ Microbiol 6:73–78. doi: 10.1046/j.1462-2920.2003.00536.x. [DOI] [PubMed] [Google Scholar]
- 19.Whiteley AS, Manefield M, Lueders T. 2006. Unlocking the “microbial black box” using RNA-based stable isotope probing technologies. Curr Opin Biotechnol 17:67–71. doi: 10.1016/j.copbio.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Dumont MG, Pommerenke B, Casper P, Conrad R. 2011. DNA-, rRNA- and mRNA-based stable isotope probing of aerobic methanotrophs in lake sediment. Environ Microbiol 13:1153–1167. doi: 10.1111/j.1462-2920.2010.02415.x. [DOI] [PubMed] [Google Scholar]
- 21.Manefield M, Griffiths R, McNamara NP, Sleep D, Ostle N, Whiteley A. 2007. Insights into the fate of a 13C labelled phenol pulse for stable isotope probing (SIP) experiments. J Microbiol Methods 69:340–344. doi: 10.1016/j.mimet.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 22.Rettedal EA, Brozel VS. 2015. Characterizing the diversity of active bacteria in soil by comprehensive stable isotope probing of DNA and RNA with H218O. Microbiologyopen 4:208–219. doi: 10.1002/mbo3.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hungate BA, Mau RL, Schwartz E, Caporaso JG, Dijkstra P, van Gestel N, Koch BJ, Liu CM, McHugh TA, Marks JC, Morrissey EM, Price LB. 2015. Quantitative microbial ecology through stable isotope probing. Appl Environ Microbiol 81:7570–7581. doi: 10.1128/AEM.02280-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mandelstam J. 1960. The intracellular turnover of protein and Nucleic acids and its role in biochemical differentiation. Bacteriol Rev 24:289–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manefield M, Whiteley AS, Ostle N, Ineson P, Bailey MJ. 2002. Technical considerations for RNA-based stable isotope probing: An approach to associating microbial diversity with microbial community function. Rapid Commun Mass Spectrom 16:2179–2183. doi: 10.1002/rcm.782. [DOI] [PubMed] [Google Scholar]
- 26.Wellington EM, Berry A, Krsek M. 2003. Resolving functional diversity in relation to microbial community structure in soil: exploiting genomics and stable isotope probing. Curr Opin Microbiol 6:295–301. doi: 10.1016/S1369-5274(03)00066-3. [DOI] [PubMed] [Google Scholar]
- 27.Lillis L, Doyle E, Clipson N. 2009. Comparison of DNA- and RNA-based bacterial community structures in soil exposed to 2,4-dichlorophenol. J Appl Microbiol 107:1883–1893. doi: 10.1111/j.1365-2672.2009.04369.x. [DOI] [PubMed] [Google Scholar]
- 28.Novitsky JA. 1987. Microbial growth rates and biomass production in a marine sediment: evidence for a very active but mostly nongrowing community. Appl Environ Microbiol 53:2368–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nold SC, Ward DM. 1996. Photosynthate partitioning and fermentation in hot spring microbial mat communities. Appl Environ Microbiol 62:4598–4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dlott G, Maul JE, Buyer J, Yarwood S. 2015. Microbial rRNA:rRNA gene ratios may be unexpectedly low due to extracellular DNA preservation in soils. J Microbiol Methods 115:112–120. doi: 10.1016/j.mimet.2015.05.027. [DOI] [PubMed] [Google Scholar]
- 31.Bremer H, Dennis PP. 1996. Modulation of chemical composition and other parameters of the cell by growth rate, p 1553–1569. In Neidhardt FC, Curtiss R III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE (ed), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed ASM Press, Washington, DC. [Google Scholar]
- 32.Ramos C, Mølbak L, Molin S. 2000. Bacterial activity in the rhizosphere analyzed at the single-cell level by monitoring ribosome contents and synthesis rates. Appl Environ Microbiol 66:801–809. doi: 10.1128/AEM.66.2.801-809.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeAngelis KM, Silver WL, Thompson AW, Firestone MK. 2010. Microbial communities acclimate to recurring changes in soil redox potential status. Environ Microbiol 12:3137–3149. doi: 10.1111/j.1462-2920.2010.02286.x. [DOI] [PubMed] [Google Scholar]
- 34.Chaney SG, Duffy JJ, Boyer PD. 1972. Patterns of oxygen interchange between water, substrates, and phosphate compounds of Escherichia coli and Bacillus subtilis. J Biol Chem 247:2145–2150. [PubMed] [Google Scholar]
- 35.Davis KER, Sangwan P, Janssen PH. 2011. Acidobacteria, Rubrobacteridae and Chloroflexi are abundant among very slow-growing and mini-colony-forming soil bacteria. Environ Microbiol 13:798–805. doi: 10.1111/j.1462-2920.2010.02384.x. [DOI] [PubMed] [Google Scholar]
- 36.Leff JW, Jones SE, Prober SM, Barberán A, Borer ET, Firn JL. 2015. Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe. Proc Natl Acad Sci U S A 112:10967–10972. doi: 10.1073/pnas.1508382112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fierer N, Bradford MA, Jackson RB. 2007. Toward an ecological classification of soil bacteria. Ecology 88:1354–1364. doi: 10.1890/05-1839. [DOI] [PubMed] [Google Scholar]
- 38.Ebbole DJ, Zalkin H. 1987. Cloning and characterization of a 12-gene cluster from Bacillus subtilis encoding nine enzymes for de novo purine nucleotide synthesis. J Biol Chem 262:8274–8287. [PubMed] [Google Scholar]
- 39.Berg JM, Tymoczko JL, Stryer L. 2002. Biochemistry 5th ed. W. H. Freeman, New York, NY. [Google Scholar]
- 40.Richards OC, Boyer PD. 1966. 18O Labeling of deoxyribonucleic acid during synthesis and stability of the label during replication. J Mol Biol 19:109–119. doi: 10.1016/S0022-2836(66)80053-0. [DOI] [PubMed] [Google Scholar]
- 41.Reichard P. 1993. From RNA to DNA, why so many ribonucleotide reductases? Science 260:1773–1777. doi: 10.1126/science.8511586. [DOI] [PubMed] [Google Scholar]
- 42.Herrick J, Sclavi B. 2007. Ribonucleotide reductase and the regulation of DNA replication: an old story and an ancient heritage. Mol Microbiol 63:22–34. doi: 10.1111/j.1365-2958.2006.05493.x. [DOI] [PubMed] [Google Scholar]
- 43.Brettar I, Christen R, Höfle MG. 2012. Analysis of bacterial core communities in the central Baltic by comparative RNA-DNA-based fingerprinting provides links to structure-function relationships. ISME J 6:195–212. doi: 10.1038/ismej.2011.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Campbell BJ, Kirchman DL. 2013. Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J 7:210–220. doi: 10.1038/ismej.2012.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hunt DE, Lin Y, Church MJ, Karl DM, Tringe SG, Izzo LK, Johnson ZI. 2013. Relationship between abundance and specific activity of bacterioplankton in open ocean surface waters. Appl Environ Microbiol 79:177–184. doi: 10.1128/AEM.02155-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schimel JP, Gulledge J. 1998. Microbial community structure and global trace gases. Glob Chang Biol 4:745–758. doi: 10.1046/j.1365-2486.1998.00195.x. [DOI] [Google Scholar]
- 47.Reid NM, Addison SL, Macdonald LJ, Lloyd-Jones G. 2011. Biodiversity of active and inactive bacteria in the gut flora of wood-feeding huhu beetle larvae (Prionoplus reticularis). Appl Environ Microbiol 77:7000–7006. doi: 10.1128/AEM.05609-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Portillo MC, Gonzalez JM, Saiz-Jimenez C. 2008. Metabolically active microbial communities of yellow and grey colonizations on the walls of Altamira Cave, Spain. J Appl Microbiol 104:681–691. doi: 10.1111/j.1365-2672.2007.03594.x. [DOI] [PubMed] [Google Scholar]
- 49.Steven B, Hesse C, Soghigian J, Gallegos-Graves LV, Dunbar J. 2017. Simulated ribosomal RNA/DNA ratios show potential to misclassify active populations as dormant. Appl Environ Microbiol 83:e00696-17. doi: 10.1128/AEM.00696-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whiteley AS, Thomson B, Lueders T, Manefield M. 2007. RNA stable-isotope probing. Nat Protoc 2:838–844. doi: 10.1038/nprot.2007.115. [DOI] [PubMed] [Google Scholar]
- 51.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krohn A. 2016. akutils-v12: facilitating analyses of microbial communities through QIIME Zenodo 10:5281. [Google Scholar]
- 53.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Price MN, Dehal PS, Arkin AP. 2010. FastTree—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caporaso JG, Bittinger K, Bushman FD, Desantis TZ, Andersen GL, Knight R. 2010. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.R Core Team. 2014. R: a language and environment for statistical computing (R version 3.2.1). The R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 58.IBM Corp. 2016. IBM SPSS Statistics for Windows, version 24.0. IBM Corp., Armonk, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Workflow used for sequencing data processing is available at https://github.com/alk224/akutils-v1.2. R code used for calculating taxon-specific APE 18O of nucleic acids is available at https://bitbucket.org/QuantitativeSIP/qsip_repo.