

ABSTRACT
Like many bacteria, Bacillus subtilis possesses two DNA translocases that affect chromosome segregation at different steps. Prior to septum closure, nonsegregated DNA is moved into opposite cell halves by SftA, while septum-entrapped DNA is rescued by SpoIIIE. We have used single-molecule fluorescence microscopy and tracking (SMT) experiments to describe the dynamics of the two different DNA translocases, the cell division protein FtsA and the glycolytic enzyme phosphofructokinase (PfkA), in real time. SMT revealed that about 30% of SftA molecules move through the cytosol, while a fraction of 70% is septum bound and static. In contrast, only 35% of FtsA molecules are static at midcell, while SpoIIIE molecules diffuse within the membrane and show no enrichment at the septum. Several lines of evidence suggest that FtsA plays a role in septal recruitment of SftA: an ftsA deletion results in a significant reduction in septal SftA recruitment and a decrease in the average dwell time of SftA molecules. FtsA can recruit SftA to the membrane in a heterologous eukaryotic system, suggesting that SftA may be partially recruited via FtsA. Therefore, SftA is a component of the division machinery, while SpoIIIE is not, and it is otherwise a freely diffusive cytosolic enzyme in vivo. Our developed SMT script is a powerful technique to determine if low-abundance proteins are membrane bound or cytosolic, to detect differences in populations of complex-bound and unbound/diffusive proteins, and to visualize the subcellular localization of slow- and fast-moving molecules in live cells.
IMPORTANCE DNA translocases couple the late events of chromosome segregation to cell division and thereby play an important role in the bacterial cell cycle. The proteins fall into one of two categories, integral membrane translocases or nonintegral translocases. We show that the membrane-bound translocase SpoIIIE moves slowly throughout the cell membrane in B. subtilis and does not show a clear association with the division septum, in agreement with the idea that it binds membrane-bound DNA, which can occur through cell division across nonsegregated chromosomes. In contrast, SftA behaves like a soluble protein and is recruited to the division septum as a component of the division machinery. We show that FtsA contributes to the recruitment of SftA, revealing a dual role of FtsA at the division machinery, but it is not the only factor that binds SftA. Our work represents a detailed in vivo study of DNA translocases at the single-molecule level.
KEYWORDS: Bacillus subtilis, DNA translocase, cell division, chromosome segregation, single-molecule tracking, FtsK, SpoIIIE
INTRODUCTION
Stable transmission of genetic information means that cells must ensure that their DNA is accurately replicated and that each daughter cell receives a completely segregated chromosome. One threat to segregation is that replication of circular chromosomes can lead to the formation of a chromosome dimer, which results from an odd number of recombination events during replication and which needs to be properly resolved. Moreover, a delay in segregation can be detrimental if division occurs prior to complete DNA partitioning. Certain types of stress can damage DNA and disrupt the normal progression of the replication fork. Therefore, cells have evolved pathways to ensure that cell division occurs reliably even when segregation is perturbed (1, 2).
In eukaryotic cells, several checkpoints prevent premature cytokinesis during mitosis (3). In bacteria, however, no such checkpoints separating chromosome segregation and cell division in the absence of DNA damage have been identified. These processes therefore frequently overlap, and cell division can occur even when chromosome segregation is not complete. As a consequence, incompletely segregated chromosomes can be trapped by the invaginating division septum and must be actively transported into the daughter cells to prevent a block in the cell cycle. DNA translocases act in a spatially and temporally defined manner to move DNA away from the closing septum.
In Escherichia coli, FtsK is recruited to transport the trapped DNA into the correct cell in cases when cytokinesis occurs prior to complete chromosome segregation (4). FtsK also recruits DNA recombinases that resolve dimeric and concatenated chromosomes (5). SpoIIIE, a homologue of FtsK in Bacillus subtilis, is primarily involved in chromosome segregation during sporulation. It efficiently transports 70 to 75% of one chromosome during asymmetric septation from the mother cell to the forespore (6, 7). SpoIIIE assembles a coaxially paired channel for each chromosome arm composed of one hexamer in each cell compartment to allow DNA translocation in spite of complete membrane fission (8). SpoIIIE also segregates chromosomes trapped during symmetric cell division (1).
It was recently shown that spherical bacteria also have SpoIIIE/FtsK homologues. In Staphylococcus aureus, for example, SpoIIIE and FtsK operate in independent pathways to ensure correct chromosome management during cell division (9).
The SpoIIIE/FtsK translocases of rod-shaped bacteria have an N terminus containing transmembrane domains, followed by a linker and two domains that make up the motor (α and β). The motor forms a homohexamer around DNA (10, 11). Three-dimensional (3D)-structured illumination microscopy coupled with scanning number and brightness analysis in B. subtilis have shown that SpoIIIE clusters in sporulation and division septa contain 47 ± 20 molecules, 70% of which are in hexameric state (12). The γ-domain of SpoIIIE/FtsK confers directional DNA translocation by preferentially interacting with 8-nucleotide recognition sequences (SpoIIIE recognition sequences [SRS] and FtsK orienting/polarizing sequences [KOPS]) (13, 14).
In B. subtilis, strains harboring spoIIIE alleles that lack the γ-domain exhibit a considerable decrease in sporulation efficiency. The sporulation defect of spoIIIE Δγ cells can be partly or fully rescued by any of a variety of missense mutations in the linker and motor domains of SpoIIIE Δγ. However, the suppressors do not rescue chromosome translocation defects during vegetative growth (15). This argues for the importance of the γ-subunit during vegetative growth.
SpoIIIE/FtsK pumps chromosomes toward the chromosome dimer resolution site (dif site) located near the terminus region using the polarity of SRS/KOPS sequences, which are recognized by the SpoIIIE/FtsKγ domain and permit the loading of the translocase onto the DNA in one specific orientation (16). SRS/KOPS are distributed over the chromosome and oriented toward the terminus region, where they are found at a high frequency (17). In E. coli, recombination at the dif site happens through the action of the site-specific recombinases XerD and XerC (18, 19), which catalyze the formation and resolution of a Holliday junction intermediate at dif, where each recombinase mediates a strand exchange reaction (20). FtsK arranges the dif sites in close proximity at the division septum and directly activates XerD (21). In Bacillus subtilis, RipX and CodV are the homologues of XerD and XerC, respectively (22, 23). Both RipX and CodV bind to the B. subtilis dif sites and catalyze strand exchange in vitro (5).
B. subtilis has another DNA translocase that acts synergistically with SpoIIIE in the rescue of DNA that might become trapped by the division septum during vegetative growth: the soluble protein SftA. Both SftA and SpoIIIE display strong similarity with the C-terminal domain of FtsK (2). SftA appears to be a component of the divisome. It facilitates dimer resolution by bringing the dif sites into close proximity, most likely using a KOPS-like mode for translocation directionality. SftA translocates DNA during septation, while SpoIIIE is only recruited to rescue septum-entrapped DNA after division is completed (1, 2). SpoIIIE is essential for sporulation but not during vegetative growth. However, SpoIIIE becomes indispensable under conditions where chromosome segregation is impaired (24). In vivo studies revealed that neither SpoIIIE nor SftA is essential for RipX-dependent recombination at a dif site, but they contribute additively to dimer resolution (25). RipX and CodV form a preassembled complex on the chromosome during the cell cycle, probably to allow an immediate initiation of dimer resolution. The sftA spoIIIE double-mutant strain has a more severe phenotype than either of the single mutants, suggesting considerable overlap in their roles (25).
In this work, we analyze the dynamics of SftA and SpoIIIE at the single-molecule level in real time. We find that most SftA molecules are present at the septum, while SpoIIIE molecules are generally mobile within the membrane. We also show evidence for a potential role of FtsA in the recruitment of SftA to the midcell using a heterologous eukaryotic system and an in vivo deletion assay.
RESULTS
The localization pattern of SftA is affected by the absence of FtsA.
Kaimer et al. (2) showed that SftA was recruited as an early component to the cell division machinery before DivIB, DivIC, FtsL, or penicillin-binding 2B (Pbp2B). SftA no longer localized to midcell in an FtsZ depletion strain but to distinct foci on the lateral membrane, which suggests that SftA has an intrinsic membrane affinity, binds to a membrane-associated protein that is recruited to midcell by FtsZ, or binds to FtsZ directly. To identify additional recruitment factors, we localized SftA-YFP (YFP, yellow fluorescent protein) in mutants lacking early accessory division proteins EzrA, ZapA, and SepF or in a strain in which FtsA is deleted. The results are shown in Fig. 1. In an ezrA deletion background, SftA was still recruited to aberrantly formed division septa: 21.6% of the cells (n = 166) formed double septa, and 7% of cells had an additional septum close to the old pole. The cells were also longer than the wild type by 20%, as was previously published (26). Seventy-eight percent of ezrA-null cells (n = 210) contained midcell SftA-YFP signals, compared with 76% of wild-type cells (n = 240). In a zapA deletion mutant, which does not have any observable phenotype under laboratory growth conditions, SftA-YFP also localized at midcell in 86% of the cells indistinguishable from wild-type cells (n = 150). The same findings apply to a ΔsepF mutant strain where SftA localized at midcell in 79% of the 120 cells counted. Cells were slightly elongated, which is in accordance with previously published data (27). SepF becomes essential in cells where FtsA is deleted, suggesting that SepF can complement the function of FtsA in cell division (26). We also tested DivIVA as potential interaction partner for SftA but did not find a pronounced lack of formation of SftA rings or foci in divIVA mutant cells (Fig. 1E). In fact, SftA localized normally in 75% of the cells (n = 125). Deletion of divIVA leads to filamentation and polar divisions that in turn cause a minicell phenotype (28).
FIG 1.

Localization of SftA-YFP in strains that carry mutations of cell division proteins. White arrowheads show the septal localization of SftA. (A) SftA-YFP in ΔezrA mutant; white lines show double-division septa. (B) SftA-YFP in ΔdivIVA mutant; white line shows aberrantly formed septum close to the cell pole. (C) SftA-YFP in ΔzapA mutant. (D) SftA-YFP in ΔsepF; white line points at a septum formed near the pole. (E) SftA-YFP in ΔezrA ΔftsA mutant; white lines show abnormal septa. (F) SftA-YFP in ΔftsH mutant. Images are overlays of SftA-YFP signals (green) and membranes stained with FM 4-64 (red). White bars = 2 μm.
An ftsA-null mutant has problems dividing and forms long filamentous cells. However, when cloned into a ΔezrA background, the resulting double mutant grows well and does not show a strong cell division defect (26). Therefore, a strain was created which expresses SftA-YFP from the original locus and harbors ΔezrA ΔftsA-null mutations. The strain formed notably elongated cells, but a cell length closer to that of wild-type cells was restored when cells were grown in 0.01 mM isopropyl-β-d-thiogalactopyranoside (IPTG; driving the expression of the ftsZ gene lying downstream of ftsA). Interestingly, SftA-YFP localized in this strain in only 42% of the cells (n = 200), i.e., in considerably fewer cells than in the wild type (Fig. 1E), although the cells were slightly longer than wild-type cells.
In order to assess the role of FtsH in SftA localization, SftA-YFP was cloned into a ΔftsH mutant strain. The deletion had no effect on the midcell localization of SftA (Fig. 1F), which excludes FtsH as a recruiting factor for SftA. However, SftA-YFP midcell bands/foci were much fainter in the absence of FtsH than in wild-type cells (Fig. 1F).
Because SftA shows a localization defect in the absence of FtsA, we decided to focus our efforts on FtsA and FtsZ given the fact that they are the earliest division proteins and the core behind the recruitment of the rest of the septal proteins to the division site. It could very well be that several factors contribute together to the septal recruitment of SftA, and shedding light on at least one of them could help our future understanding of the interplay between the division proteins.
Coexpression of SftA and FtsA in S2 Schneider cells.
To study the putative interaction of SftA and FtsA, we employed a heterologous expression system. S2 Schneider cells are derived from Drosophila flies and are unlikely to contain specific binding partners that interact with bacterial proteins. In order to investigate a possible interaction with FtsA or FtsZ, S2 Schneider cells were cotransfected with SftA-YFP and untagged FtsA or FtsZ. As a control, cells were transfected either with SftA-YFP alone or with FtsA-YFP or FtsZ-YFP alone. Expression of all recombinant YFP-tagged proteins in this experiment was verified by Western blot with anti-green fluorescent protein (anti-GFP) antibodies (Fig. 2G).
FIG 2.

(A to F) S2 Schneider cell cotransfection experiments. Cell were transfected with plasmids expressing the corresponding proteins as shown in the panels. White arrowheads point to membrane assemblies of SftA. Images were taken through the middle of the cells in panels A to C, and circles with bars indicate the focal plane in panels D to F. (G) Western blot analysis with anti-GFP antibodies of the expressed recombinant proteins in the S2 Schneider cell experiment. The letters in parentheses correspond to cells harvested from the respective experiments in images. White bars = 5 μm.
Expression of SftA-YFP alone in S2 Schneider cells resulted in a diffuse fluorescence pattern, arguing against the ability of SftA to target the general phospholipids within the membrane, which are similar between bacteria and eukaryotes (Fig. 2A). While FtsZ-YFP formed multiple assemblies across the cell (Fig. 2B), FtsA-YFP largely assembled at the membrane (Fig. 2D), in agreement with its possession of an amphipathic helix (29). This localization pattern of FtsA is similar to the localization of YFP-MreB or of B. subtilis bactofilins, which were shown to be recruited to the membrane by an amphipathic helix and intrinsic membrane affinity, respectively (30). Interestingly, when coexpressed, FtsA caused the recruitment of SftA-YFP to the membrane, where clear assemblies were observed, hinting toward a direct interaction between the two proteins (Fig. 2E).
However, when SftA-YFP was coexpressed with FtsZ, we observed diffuse localization that is reminiscent of SftA localization but no indication of a specific interaction of SftA and FtsZ (Fig. 2C). Cotransfection of FtsZ-YFP and of FtsA resulted in the recruitment of FtsZ to the membrane, as can be seen in Fig. 2F.
Single-molecule microscopy of SftA, SpoIIIE, and PfkA reveals different localization patterns.
SftA can be purified as a soluble enzyme (2). However, inspection of its N-terminal sequence has suggested the presence of a membrane-targeting helix (31). During the depletion of FtsZ, SftA signals can still be observed at the membrane (2). We wished to gain insight into the question of whether SftA is a membrane-associated protein or if it is a truly cytosolic enzyme that becomes recruited to the division site. SftA is not highly abundant, so a possible freely diffusing fraction in the membrane or in the cytosol is not detectable by conventional epifluorescence or by total internal-reflection fluorescence (TIRF) microscopy. Nevertheless, using highly sensitive single-molecule fluorescence microscopy and tracking (SMT), we are able to observe diffusive molecules. By illuminating an area of about 10 μm in diameter with parallel light from the center of a laser beam, which results in an even and intense illumination, we can follow the real-time movement of single molecules (see Fig. S1A and C in the supplemental material) using fast stream acquisitions, once all except for the last few molecules have been bleached by the laser. Single molecules can be identified from single-step bleaching (Fig. S1B and C). We are using the U-track software (32) for automated spot detection and tracking, which is based on Gaussian fitting of fluorescent spots, resulting in a subpixel resolution, and MicrobeTracker (33) for automated cell detection. Our custom-developed single-molecule analysis software (Matlab based) integrates the positional coordinates of the molecules into the coordinate system of the cell and quantitatively analyzes its diffusive behavior and localization.
Besides SftA, we imaged and tracked SpoIIIE and phosphofructokinase (PfkA) during exponential growth in minimal medium, both expressed from their respective original promoters and each fused to monomeric YFP. PfkA is a glycolytic enzyme, which we used as a control for a freely diffusive enzyme, and SpoIIIE served as an indicator of a membrane-associated DNA translocase. Examples of movies for SftA are Movies S1 (real time), S2 (dynamic molecule, slow motion), and S3 (static molecule, slow motion), for SpoIIIE in Movies S4 (real time) and S5 (mobile molecule, slow motion), and for PfkA in Movies S6 (real time) and S7 (single dynamic molecule, slow motion), all in the supplemental material. The three proteins showed a characteristic localization pattern when we summed up all the pixel intensities from consecutive images of an acquired stream (Fig. 3A). Additionally, we projected the positions of the molecules from all cells and all movies into a normalized bacterial cell (dimensions, 3 by 1 μm) and plotted these positions in a density plot (Fig. 3B). The density of PfkA-YFP shows a clear localization in the cytosol, consistent with it being a freely diffusive enzyme (Fig. 3B). On the other hand, the normalized localization of SftA-YFP showed that there is a higher density of SftA-YFP spots in positions corresponding to midcell and the cell poles. This was not observed for PfkA-YFP, where localization was more equally distributed to the center of the cell (Fig. 3B). In contrast to SftA and PfkA, SpoIIIE preferentially localized at the membrane, where it showed a homogeneous distribution. Note that during stream acquisition, the focal plane is not always exactly at the central plane of the cell; if the focus moves up or down, molecules that travel along the y axis are also observed. Therefore, tracks that appear to be in the cytosol are also observed for SpoIIIE.
FIG 3.
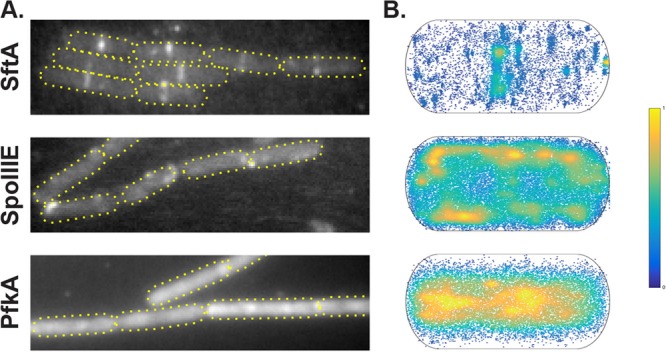
(A) Z-projection of all frames from a selected movie of SftA-YFP, SpoIIIE-YFP, and PfkA-YFP. (B) Heat map of single-molecule localizations of SftA-YFP, SpoIIIE-YFP, and PfkA-YFP plotted into a standardized cell. High abundance is indicated in yellow and low abundance in dark blue.
SftA divides into two fractions, a static (likely septum bound) and a mobile fraction.
As a next step in our evaluation of SMT data, we analyzed the diffusive properties of individual molecules, which yields information on the diffusion rates and on the percentage of molecules in an immobile (or very slowly moving) and mobile state. To this end, we focused on the distribution of distances that molecules move between consecutive image acquisitions. For a molecule following simple Brownian diffusion, this probability density function (PDF) takes the form of a Gaussian, for which the variance is proportional to the diffusion constant D (see Materials and Methods). The experimentally determined PDF of SftA shows, however, that it cannot be explained by a single Gaussian fit (Fig. 4A, black dotted line). Instead, it is more likely the superposition of two Gaussians (Fig. 4A, green solid line), corresponding to one fraction of mobile and another fraction of immobile molecules. From fitting the variances and respective areas under the two Gaussians, we then determined the diffusion constants and relative fractions of molecules in the mobile and immobile states (Table 1). Our results show that the majority (73%) of SftA displacement steps correspond to a static state of the molecule with a diffusion constant of 0.046 μm2/s, which is close to what we expect from the localization error of our system (20 nm). The remaining 27% of the SftA displacement steps were mobile, with D = 0.46 μm2/s (Fig. 4A). These data suggest that about two-thirds of SftA molecules are septum bound, and one-third of the molecules diffuse through the cytosol. Compared with determined diffusion rates of membrane proteins (34), the mobile fraction of SftA moves at a much higher rate, in agreement with SftA being a cytosolic protein. Indeed, for the freely diffusing enzyme PfkA-YFP, we found that 95% move with a diffusion constant of 1.2 μm2/s (Fig. 4C and Table 1), which is 2.5 times faster than the mobile fraction of StfA. It is possible that the mobile fraction and the diffusion constants for PfkA-YFP are underestimated because of the long exposure time of about 30 ms. In other words, some very fast PfkA molecules may only be detectable with faster acquisition times. However, given that the same acquisition settings were used, PfkA serves as a good control for the diffusive behavior of SftA, whose PDF curve is much narrower than that of PfkA (Fig. 4A and 3C).
FIG 4.

Single-molecule microscopy of SftA-YFP and SpoIIIE-YFP. (A) Probability density function (PDF) of displacements obtained from SftA-YFP tracked in wild-type and ftsA ezrA double-mutant cells. Histograms were simultaneously fitted to a Gaussian mixture model assuming 2 different types of diffusive behavior (green line). Indeed, the multivariate fit matched the data better than assuming a single Gaussian distribution (outer dotted line). The dashed line and the inner dotted line correspond to the distributions of the larger and smaller frame-to-frame displacements representing fast and slowly diffusing molecules, respectively. (B) PDF of SpoIIIE-YFP in untreated cells and cells treated with 50 ng/ml mitomycin C (MMC). (C) PDF of PfkA-YFP. (D) Bubble plot showing diffusion coefficients of the diffusive subfractions seen for SftA-YFP, SpoIIIE-YFP, FtsA-YFP, and PfkA-YFP.
TABLE 1.
Diffusion constants and percentages of static and mobile molecule fractions
| Protein or strain | No. of cells | No. of tracks | Cell length (mean ± SD) (μm) | Avg life time (s) | D (μm2 · s−1)a | D1 (μm2 · s−1)b | F1 (%)c | D2 (μm2 · s−1)d | F2 (%)e |
|---|---|---|---|---|---|---|---|---|---|
| SftA-YFP | |||||||||
| WTf | 87 | 881 | 2.95 ± 0.91 | 0.325 | 0.086 | 0.046g | 73 | 0.41g | 27 |
| ΔftsA ΔezrA mutant | 98 | 789 | 3.11 ± 1.17 | 0.341 | 0.11 | 54 | 46 | ||
| SpoIIIE-YFP | 65 | 833 | 3.09 ± 0.69 | 0.349 | 0.14 | 0.096 | 68 | 0.34 | 32 |
| PfkA-YFP | 43 | 4,093 | 3.04 ± 0.74 | 0.361 | 0.99 | 0.16 | 9 | 1.2 | 91 |
| FtsA-YFP | 60 | 3,031 | 3.43 ± 1.0 | 0.246 | 0.5 | 0.084 | 35 | 1.2 | 65 |
D, average diffusion constant of all molecules.
D1, diffusion constant of static fraction.
F1, percentage of static molecules.
D2, diffusion constant of mobile fraction.
F2, percentage of mobile molecules.
WT, wild type.
The same diffusion rate was used to be able to compare fractions F1 and F2 between wild-type and ftsA ezrA deletion strains.
Tracking analysis of SftA-YFP in an ftsA mutant strain.
In order to assess the influence of FtsA on the localization of SftA and its behavior at the single-molecule level, SftA-YFP was tracked in strain NEJ14 in which FtsA is absent. This is possible when also EzrA is absent, a protein that destabilizes FtsZ rings (35). Note that by itself, the absence of EzrA has no effect on SftA localization (Fig. 1). Midcell SftA-YFP assemblies were observed but much less frequently than in wild-type cells (Fig. 1A). FtsA therefore has an influence on SftA localization. At the single-molecule level, the simultaneous fit of SftA molecules tracked in the wild type and in the ftsA deletion strain showed that in the absence of FtsA, 54% of the steps were static, as opposed to 73% of the steps in wild-type cells (P = 0.005) (Fig. 4A and Table 1). Thus, the percentage of static molecules dropped in a statistically significant manner, revealing that SftA becomes more dynamic in the absence of FtsA. The loss of static molecules and the increase in dynamic ones are best seen in Fig. 4A by the broader distribution of displacements in the FtsA deletion strain.
We also scored the number of SftA molecules in wild-type and ftsA-null cells that are immobile for a certain number of consecutive acquisition times. In wild-type cells, SftA molecules dwelled on average for 106 ms in a radius of 120 nm (which is considered a static event), while in ftsA-null cells, the molecules dwelled on average for 60 ms (Fig. 5A, left, and Table 2), which is significantly shorter, as tested by a two-sample Kolmogorov-Smirnov test. Please note that the y axis is on a log scale. Therefore, the higher number of stopping SftA molecules for shorter times (on the left of the x axis) in wild-type cells has a much larger weight than the higher number of stopping events for longer time intervals in the mutant cells (for which we have no good explanation at the moment). Also note that due to molecule bleaching, these data are an underestimate of the true dwell times. However, the average lifetime (bleaching time) for SftA-YFP was 325 ms (Table 1), which is much higher than the determined average dwell time, showing that our numbers are a good estimate of actual dwell times in vivo. Note that the y axis is on a log scale, such that the weight of the short times is much greater than that of the longer dwell times. Even though the molecules showed shorter stop times in cells lacking FtsA, the effect was lower than we expected if FtsA were the sole factor recruiting SftA to the septum.
FIG 5.

Dwell time density distributions of SftA-YFP and SpoIIIE-YFP. The dwell time represents the time that a molecule stays within a radius of defined size (here, 120 nm). (A) PDF and cumulative density function (CDF) of dwell times determined for SftA in wild-type and in FtsA-depleted cells. (B) PDF and CDF of dwell times calculated for SpoIIIE-YFP in presence and absence of MMC.
TABLE 2.
Trapping times
| Protein | Description | Trapping time (ms) |
|---|---|---|
| SftA | WT | 106 ± 2.8 |
| ΔftsA ΔezrA mutant | 60 ± 1 | |
| SpoIIIE | Untreated | 166 ± 1.7 |
| Treated with MMC | 154 ± 2.1 |
A majority of FtsA molecules are freely diffusing.
In contrast to the predominantly septum-bound SftA, 65% of FtsA-YFP molecules were mobile and 35% were static (Table 1). Therefore, a majority of FtsA molecules are not septum bound, in contrast to SftA. Like PfkA, mobile FtsA-YFP had a diffusion coefficient of 1.2 μm2/s, showing that our acquisition settings are not limited in determining diffusion rates for SftA or for SpoIIIE. The static fraction of FtsA-YFP had a diffusion coefficient of 0.08 μm2/s, somewhat faster than that of SftA but in a similar range, indicating that immobile SftA and FtsA fractions behave similarly when bound to the Z-ring. Note that the static fractions move faster than deduced from our localization error (about 20 nm), indicating that septum-bound FtsA and SftA move on a long time scale, i.e., are not entirely static. The “static” PfkA fraction moved with a speed of 0.16 μm2/s (Table 1), showing that these are not truly bound molecules but rather slow-diffusing events (note that even freely diffusing molecules will stochastically stop for short periods of time).
Tracking analysis of SpoIIIE in cells treated and not treated with MMC.
Since SftA and SpoIIIE act synergistically in B. subtilis, we were interested in observing the behavior of SpoIIIE-YFP at the single-molecule level. SpoIIIE-YFP was imaged under normal conditions and under conditions where double-strand breaks were induced by the use of 50 ng/μl mitomycin C (MMC). The microscopy setup used and the exposure time were exactly the same as in the experiments described for SftA.
SpoIIIE assembles at the division (or sporulation) septum to rescue (or pump into the forespore) septum-entrapped DNA after division is completed and is only indispensable for viability under conditions in which chromosome segregation is impaired (1, 2). There has been considerable discussion about the localization of SpoIIIE during vegetative growth, whether it is monomeric and randomly distributed throughout the membrane, with its assembly being triggered upon DNA binding at the sporulation septum (36), or whether it is part of the division machinery. Through photoactivation localization microscopy (PALM) imaging, it has been shown that SpoIIIE is present in division septa in more than 30% of exponentially growing cells, localized to the FtsZ ring, and in 55% of cells during membrane invagination, partially assembled into hexameric motors (12). SpoIIIE either formed dynamic clusters with no defined localization or static clusters that localized to the future septation sites. The dynamic clusters were large, around 100 nm in size, and contained highly dynamic SpoIIIE molecules (12). This study only investigated SpoIIIE inside these clusters and not the single SpoIIIE molecules in the membrane. On the other hand, Kaimer et al. (2) stated that SpoIIIE forms foci in only 1.6% of the cells under normal conditions, which increases to 5.6% after MMC addition. Biller and Burkholder (1) found SpoIIIE-YFP foci in just 4% of growing bacteria. None of the previous studies investigated the dynamics of all, mobile and static, SpoIIIE molecules.
Under our experimental conditions, a majority of SpoIIIE molecules were slowly moving with and without MMC treatment. Under both conditions, two populations of steps were detected: a large slowly diffusing population (68%, 0.1 μm2/s, twice as fast as the static SftA fraction) and a small more quickly moving population (32% with 0.34 μm2/s, Table 1 and Fig. 4B). We interpret these data as indicative of a majority of SpoIIIE molecules moving as hexamers and a minority as monomers. After the addition of MMC, our analysis revealed no change in the dynamic behavior of SpoIIIE (Fig. 3B and D). Interestingly, although the number of SpoIIIE molecules at the septum increases after the induction of DNA damage (MMC) in a subset of cells, this is not reflected in longer dwell times of SpoIIIE. A comparison of the cumulative density functions (CDF) shows very little difference between nontreated (Fig. 5B, bottom, blue curve) and MMC-treated cells (Fig. 5B, bottom, red curve). Therefore, even though SpoIIIE visibly accumulates at the division septum in 5% of the cells upon MMC treatment (Kaimer et al. [2]), there is not a significant change in the (low) diffusion rates of the protein. The finding that SpoIIIE is largely diffusive (with a slow and a faster population) and not enriched at midcell reinforces the idea that it is not a static component of the cell division machinery.
Visualization of the subcellular position of slow- and fast-moving molecules.
We wished to not only determine the most likely position of the proteins (Fig. 3) or the relative fractions of mobile and immobile molecules (Fig. 4) but to determine the localization of the immobile/slow-moving and the mobile fractions. The expectation was to find many immobile proteins at the division septum for SftA and possibly a mildly increased fraction of SpoIIIE molecules at the cell center upon the addition of MMC. We defined immobile/slow moving as molecules that do not leave an area of 3 by 3 pixels for 5 frames, and we used 30-ms stream acquisition. It must be kept in mind that any freely diffusing molecule alternates between periods of rapid and slow movement and can pause for some time. We therefore set the threshold to 5 frames, because freely diffusing molecules are unlikely to arrest their movement for such an extended period of time. Figure 6 shows slow-moving molecules in red and fast-moving molecules in blue. For SftA, midcell and cell poles represent positions of frequent arrests/slow movement (Fig. 6C), as expected, and also for FtsA, static molecules are localized to the midcell position (Fig. 6B, note that for FtsA-YFP, midcell localization was better seen using 15-ms stream acquisition, rather than 30 ms; see Fig. S2), while PfkA shows very few static tracks throughout the cells (Fig. 6A). Midcell localization for FtsA and for SftA can also be clearly seen in the x axis scans (Fig. 6G and H). As predicted, there is a visible reduction in the number of septum-associated stops in the ftsA deletion strain (Fig. 6D, 1,000 frames were used for comparison of the two conditions) and an increase in stops throughout the cells. For SpoIIIE, no septum enrichment can be seen during exponential growth (Fig. 6E and J). Using epifluorescence, it was reported that 5% of cells contain visible fluorescent spots after the addition of MMC, due to the assembly of SpoIIIE double hexamers at the division site where DNA is entrapped (2); this is reflected by a moderate visual increase in static tracks in MMC-treated cells (Fig. S3, middle panels). This enrichment is most easily seen when slow and fast tracks are sorted along the long axis (x) of the cells (Fig. S4, middle panels). As described above, SpoIIIE-YFP tracks are detected away from the membrane because the focal plane is not always perfectly in the middle plane of cells, and because due to focal depth, molecules are visible that are several hundred microns away from the actual focal plane. This caveat notwithstanding, there is a clear difference in the spatial distribution of PfkA, FtsA, SftA, and SpoIIIE as viewed along the y (short) axis of cells: while SpoIIIE-YFP localization is enriched at the outer edges (i.e., the membrane, Fig. 6J), soluble PfkA-YFP is mostly present at the cell center of the tube-like cell (Fig. 6A). FtsA and SftA are somewhat in between these patterns (Fig. 6B and C), indicative of peripheral membrane association. This makes sense, because the Z-ring that carries many FtsA and SftA molecules is at the membrane for a prolonged time during the cell cycle, before it constricts.
FIG 6.

(A to E) Determination of the subcellular localization of slow- and of fast-moving molecules. Tracks were randomly sampled from PfkA-YFP (A), FtsA-YFP (B), and SftA-YFP (C) in wild-type cells or in cells deleted for ezrA and ftsA (D), and from cells expressing SpoIIIE-YFP (E). Tracks were projected into a standardized cell of 3 by 1 μm and were sorted into slow-moving (not leaving an area of 3 by 3 pixels) molecules, indicated by red lines, and fast-moving molecules, indicated by blue lines. Note that stream rate was 30 ms, except for 15 ms for FtsA-YFP. (F to J) Superimposition of slow-moving (red line) and fast-moving (blue line) molecules sorted in x- and y-orientation (long and short axis of the cell). Shown are the mean and the standard deviation of a bootstrap analysis sampling 50 times tracks with replacement from PfkA-YFP (F), FtsA-YFP (G), or SftA-YFP (H) in wild-type cells or in cells deleted for ezrA and ftsA (I), as well as from cells expressing SpoIIIE-YFP (J). Except for SftA-YFP and SpoIIIE-YFP, for which only 750 tracks were sampled, bootstrapping was performed with 1,000 tracks.
These experiments verify the idea that FtsA contributes to the recruitment of SftA to the division machinery and that only few of the total number of SpoIIIE molecules are recruited to the division site upon induction of DNA damage, while a majority continue to move throughout the cell membrane.
DISCUSSION
Our data strongly support the idea of SftA being a soluble component of the division machinery, which is recruited to the cell center in B. subtilis via protein-protein interactions (37), while SpoIIIE is not. We show that SftA is a cytosolic protein when it is expressed in a eukaryotic cell line and shows no sign of membrane recruitment, in spite of the fact that it contains a predicted transmembrane helix within its N terminus (31). We also visualized SftA molecules using single-molecule microscopy. A major fraction of the molecules (more than two-thirds) was statically positioned at midcell, while a minor fraction of molecules moved freely within the cytoplasm, consistent with the idea that non-septum-bound SftA molecules are soluble. In contrast, a majority of FtsA molecules are freely diffusing through the cell, and only a minority are statically located at midcell. Compared with the cytosolic enzyme PfkA, mobile SftA molecules moved more slowly, suggesting that SftA migrates as a hexamer within the cell, in agreement with the finding that purified SftA forms stable hexamers in vitro (2).
SftA fails to localize to midcell in the absence of FtsZ (2). Here, we show that SftA is still recruited to midcell in the absence of early division proteins EzrA, ZapA, and SepF but showed reduced recruitment when FtsA was absent (in an otherwise ezrA mutant background). In single-molecule experiments, the dynamic fraction of SftA was increased in the ftsA mutant strain. This indicates that FtsA acts as an important element that helps recruit SftA to the septum. In S2 Schneider cells, SftA interacted with FtsA, but no interaction was detected between SftA and FtsZ, while FtsA was able to recruit FtsZ to the cell membrane, as well as SftA. These findings reinforce the idea that SftA is part of the divisome and suggest that it may be recruited via interactions with several proteins, including FtsA. The function of FtsA as one of the recruitment factors shows that FtsA plays an additional role in B. subtilis besides tethering FtsZ to the membrane (38), by recruiting the DNA translocase SftA and other proteins.
In contrast to SftA, SpoIIIE molecules showed movement within the membrane. Under the conditions used, SpoIIIE moved largely as a slow-moving population, most likely hexamers, and a more rapid-moving population, and the fraction of mobile molecules did not change markedly upon the induction of DNA double-strand breaks. We did not observe a specific localization pattern of SpoIIIE at the division septum or any other site within the membrane; however, we observed a noticeable increase in the number of static SpoIIIE molecules at midcell after the induction of DNA damage, reflecting the 5% of cells that show visible assembly of SpoIIIE double hexamers under this condition (2). SpoIIIE tracks were distributed throughout the cell membrane and did not show displacement over a large distance. SpoIIIE moved very slowly, suggesting that it also moves as a hexamer. It will be interesting to investigate how a divisome-associated or membrane-diffusing hexamer opens up to bind to DNA, or if the faster-diffusing SpoIIIE monomers observed in this study play an important role in DNA binding.
SMT can reveal the binding kinetics of proteins at a single-molecule level and often reveals striking differences between in vitro and in vivo behaviors of proteins (39). Our analysis shows that SMT is also a powerful method to determine if low-abundance proteins are soluble, membrane attached, or membrane integral, and to reveal areas of preferred localization, even for very fast-moving molecules. From the integration of 1,000 or more tracks into a standard-sized cell, scans along the x axis can show quantitative differences in recruitment to the Z-ring at midcell and along the y axis membrane localization (SpoIIIE), membrane enrichment (FtsA and SftA), or free cytosolic movement (PfkA). Further, SMT is able to quantify the number of static (target-bound) and mobile molecules and to pick up even small changes in the dynamic behavior of proteins. Our analysis is also able to visualize subcellular areas of slow-moving and of fast-moving proteins and should therefore be highly relevant for a multitude of other cell biological experiments.
MATERIALS AND METHODS
Growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 3, and the nucleotides are listed in Table S1. Escherichia coli strain XL1-Blue (Stratagene) was used for the construction and propagation of plasmids and E. coli strain BL21 Star DE3 (Invitrogen) for the heterologous overexpression of proteins. All B. subtilis strains were derived from the prototrophic wild-type strain PY79. Cells were grown in lysogeny broth (LB)-rich medium at 37°C or 30°C or in minimal medium containing S750 salts (40) at 25°C. Media were supplemented with antibiotics where appropriate (100 mg · ml−1 ampicillin, 5 mg · ml−1 chloramphenicol, 100 mg · ml−1 spectinomycin, 10 mg · ml−1 kanamycin, and/or 10 mg · ml−1 tetracycline).
TABLE 3.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotype | Resistancea | Reference or source |
|---|---|---|---|
| Strains | |||
| E. coli | |||
| XL1-Blue | endA1 gyrA96(nalr) thi-1 recA1 relA1 lac glnV44 F′[::Tn10 proAB+ lacIq Δ(lacZ)M15] hsdR17(rK− mK+) | Tet | Stratagene |
| BL21 Star (DE3) | BF− ompT gal dcm lon hsdSB(rB− mB−) λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) [malB+]K-12(λs) | None | Invitrogen |
| B. subtilis | |||
| PY79 | Wild type | None | 46 |
| CK70 | sftA-yfp | Cm | 2 |
| CK83 | sftA-yfp | Tet | This study |
| WW01 | ftsH::mls | Mls | 41 |
| NEJ13 | ftsH::mls sftA-yfp | Mls, Cm | This study |
| pFG1 | divIVA::spc | Spec | 47 |
| CK100 | sftA-yfp divIVA::spc | Cm, Spec | This study |
| FG375 | ezrA::spc | Spec | 47 |
| CK185 | sftA-yfp ezrA::spc | Cm, Spec | This study |
| BFA 2863 | sepF::mls | Mls | 27 |
| CK187 | sftA-yfp sepF::mls | Mls, Cm | This study |
| FG343 | zapA-yshB::tet | Tet | 47 |
| CK186 | sftA-yfp zapA-yshB::tet | Cm, Tet | This study |
| FG718 | ΔftsA amyE::Pxyl ftsA | Cm | 48 |
| PG2664 | pfkA-yfp | Cm | This study |
| CK55 | spoIIIE-yfp | Cm | 2 |
| MD137 | ΔezrA::spc ΔftsA::erm (Pspac-ftsZ) | Spec, Ery | 26 |
| NEJ14 | sftA-yfp ΔezrA::spc ΔftsA::erm (Pspac-ftsZ) | Spec, Ery, Cm | This study |
| Plasmids for S2 cells | |||
| pFD1 | yfp, ampr (E. coli) | 42 | |
| pFD1-SftA1-439-YFP | sftA1-439-yfp, ampr (E. coli) | This study | |
| pFD1-SftA-YFP | sftA-yfp, ampr (E. coli) | This study | |
| pFD1-FtsA-YFP | ftsA-yfp, ampr (E. coli) | This study | |
| pFD1-FtsA | ftsA, ampr (E. coli) | This study | |
| pFD1-FtsZ-YFP | ftsZ-yfp, ampr (E. coli) | This study | |
| pFD1-FtsZ | ftsZ, ampr (E. coli) | This study |
Tet, tetracycline; Cm, chloramphenicol; Spec, spectinomycin; Ery, erythromycin.
Strain construction.
A strain with YFP-tagged SftA in an ftsH deletion background (ftsH::mls sftA-yfp) was generated by transformation of CK70 (2) competent cells with chromosomal DNA from strain WW01 (41). For the S2 Schneider cell experiment, the pFD1 plasmid was used for all clonings (42). The 5′ region of sftA encoding the N terminus (residues 1 to 439) of the protein was cloned between ApaI and ClaI. Full-length sftA was cloned between ApaI and EcoRV. Full-length ftsA was cloned between ApaI and XhoI, cleaving out the yfp gene. To create a vector with ftsA-yfp, ftsA was cloned upstream of the yfp gene between ApaI and PstI. Full-length ftsZ was cloned between ApaI and XhoI; in this construct, the yfp gene was cleaved out. For ftsZ-yfp, ftsZ was cloned between ApaI and PstI.
Strains CK100, CK185, CK186, and CK187 were constructed by transformation of CK70 (sftA-yfp) competent cells with chromosomal DNA of strains carrying the corresponding mutation of cell division proteins (listed in Table 3). Strain NEJ14 was created by transforming the strain CK185 with the genomic DNA of the strain MD137. The subsequent strain expressing SftA-YFP from the original locus and harboring an ezrA ftsA double deletion was selected for growth on the antibiotics chloramphenicol, erythromycin, and spectinomycin. The growth of the strain was supplemented with 0.01 mM IPTG. The pfkA-yfp fusion was generated through PCR amplification of 500 bp of the 3′ end of the pfkA gene (lacking the stop codon) into pSG1164yfp (43) using the ApaI and EcoRI enzymes.
Schneider cell culture and transient transfection.
Drosophila melanogaster S2 Schneider cells (30) were grown in Schneider's Drosophila medium (Lonza Group Ltd.) supplemented with 5 to 10% (vol/vol) fetal calf serum (FCS) at 25°C. Cells were transferred every 48 h and transfected with the above-described pFD1 plasmids using the X-tremeGENE HP DNA transfection reagent (Roche), as described in detail by El Andari et al. (30). Protein expression was induced with a final concentration of 1 mM CuSO4, and cells were analyzed after 19 h.
Fluorescence microscopy.
For fluorescence microscopy analysis of strains CK100, CK185, CK186, CK187, CK188, and NEJ14, B. subtilis cells were grown in S750 minimal medium at 25°C under shaking conditions until exponential growth. Three microliters of cells was transferred on a glass slide (microscope slides standard; Roth) coated with an agarose layer (S750 minimal medium, 10 mg/ml agarose) and covered with a coverslip (Roth). Wide-field fluorescence microscopy was performed using a Zeiss Observer A1 microscope (Carl Zeiss) with an oil immersion objective (×100 magnification, 1.45 numerical aperture, alpha Plan-FLUAR; Carl Zeiss) a charge-coupled-device (CCD) camera (CoolSNAP EZ; Photometrics) and an HXP 120 metal halide fluorescence illumination with intensity control. Cells were treated with red fluorescent membrane stain FM 4-64 (excitation, 560 nm; emission, 640 nm; final concentration, 1 nM) and the DNA-intercalating blue fluorescent dye DAPI (4′,6-diamidino-2-phenylindole) (excitation, 358 nm; emission, 461 nm; final concentration, 0.72 nM) and incubated for 2 min at room temperature. Microscopy was done with an exposure time of 1 s under the excitation wavelength for YFP (excitation, 514 nm; emission, 527 nm) and 200 ms for excitation of each of the dyes. Data were processed using the MetaMorph 7.5.5.0 software (Molecular Devices, Sunnyvale, CA, USA), which also allows the calibration of fluorescence intensity and of pixel size to determine cell length.
Single-molecule microscopy and tracking.
In contrast to the wide-field illumination used in conventional epifluorescence microscopy, the excitation laser beam used in our setup is directed to underfill the back aperture of the objective lens. The effect of this light path is to generate a concentrated parallel illumination profile at the level of the sample, leading to a strong excitation followed by rapid bleaching of the fluorophores. When only a few unbleached molecules are present, their movement can be tracked. In addition, freshly synthesized and folded fluorophores become visible when the sample is excited again. When an observed molecule is bleached in a single step during the imaging, it is assumed to be a single molecule (44, 45). Image acquisition was done continuously during laser excitation with the electron-multiplying CCD (EMCCD) camera iXon Ultra (Andor Technology, Belfast, UK). A total of 1,500 images were taken per field, with an exposure time of 0.03 s, or 0.015 s for FtsA-YFP.
The exposed chip size corresponded to 256 by 256 pixels down to 128 by 128 pixels. The settings of recording conditions were made with the camera's program AndorSolis 4.2. The microscope used in the process was an Olympus IX71, with a ×100 objective (UAPON 100×OTIRF; numerical aperture [NA], 1.49; oil immersion). A 514-nm laser diode was used as excitation source, and the band corresponding to the fluorophore was filtered out. All imaged proteins are tagged with monomeric YFP, expressed from original gene locus, and the exposure time and laser intensity were the same in all experiments, at 30 ms (33 frames per second [fps]) or 15 ms (66 fps) and 10 mW, respectively.
Single-molecule data analyses.
The tracking analysis was done with U-track-2.3.1, which was specifically written for Matlab (MathWorks, Natick, MA, USA). The generated data were analyzed with an MSD analyzer in Matlab. The acquired movies were first cropped, and their format was converted from RAW to TIFF with ImageJ. Only trajectories consisting of a minimum of 5 frames were considered tracks and included for further analysis. Generation of heat maps, analyses of molecule dwell times, and visualization of slow and fast tracks in a standardized cell are based on a custom-written Matlab script that is available on request.
Calculation of diffusion constants.
A widely accepted method to analyze the diffusive behavior of molecules is by using the mean squared displacement (MSD)-versus-time-lag curve. This provides an estimate of the diffusion coefficient as well as of the kind of motion, e.g., diffusive, subdiffusive, or directed. However, the method requires that within a complete trajectory there be only one type of homogeneous motion and that the trajectory is preferably of infinite length. Currently used fluorophores show rather short lifetimes, limiting the time of observation to short time intervals, which in turn limit the statistical significance of the results. Additionally, this method provides information on whether different kinds of diffusive molecules are present within one population. To distinguish immobile and mobile molecules from each other, we compare the frame-to-frame displacement of all molecules in x and the y directions. Using a Gaussian mixture model to fit the probability density distribution function of all frame-to-frame displacements, determine the standard deviations σ1 and σ2, as well as the percentages F1 and F2 of the slow and the fast subfractions of molecules, respectively. Finally, the diffusion constants were calculated according to Di = σi2/4Δt (i = 1,2), where Δt is the time interval between subsequent imaging frames.
Supplementary Material
ACKNOWLEDGMENTS
This work has been supported by the Deutsche Forschungsgemeinschaft (DFG), by the BMBF-funded graduate training group NANOKAT, and by the LOEWE-funded Centre for Synthetic Microbiology (SYNMIKRO) at the Philipps Universität Marburg.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02610-17.
REFERENCES
- 1.Biller SJ, Burkholder WF. 2009. The Bacillus subtilis SftA (YtpS) and SpoIIIE DNA translocases play distinct roles in growing cells to ensure faithful chromosome partitioning. Mol Microbiol 74:790–809. doi: 10.1111/j.1365-2958.2009.06893.x. [DOI] [PubMed] [Google Scholar]
- 2.Kaimer C, Gonzaler-Pastor E, Graumann PL. 2009. SpoIIIE and a novel type of DNA translocase, SftA, couple chromosome segregation with cell division in Bacillus subtilis. Mol Microbiol 74:810–825. doi: 10.1111/j.1365-2958.2009.06894.x. [DOI] [PubMed] [Google Scholar]
- 3.Nigg EA. 2001. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol 2:21–32. doi: 10.1038/35048096. [DOI] [PubMed] [Google Scholar]
- 4.Errington J, Bath J, Wu LJ. 2001. DNA transport in bacteria. Nat Rev Mol Cell Biol 2:538. doi: 10.1038/35080005. [DOI] [PubMed] [Google Scholar]
- 5.Sciochetti SA, Piggot PJ, Blakely GW. 2001. Identification and characterization of the dif site from Bacillus subtilis. J Bacteriol 183:1058–1068. doi: 10.1128/JB.183.3.1058-1068.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu LJ, Errington J. 1997. Septal localization of the SpoIIIE chromosome partitioning protein in Bacillus subtilis. EMBO J 16:2161–2169. doi: 10.1093/emboj/16.8.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bath J, Wu LJ, Errington J, Wang JC. 2000. Role of Bacillus subtilis SpoIIIE in DNA transport across the mother cell-prespore division septum. Science 290:995–997. doi: 10.1126/science.290.5493.995. [DOI] [PubMed] [Google Scholar]
- 8.Yen Shin J, Lopez-Garrido J, Lee S-H, Diaz-Celis C, Fleming T, Bustamante C, Pogliano K. 2015. Visualization and functional dissection of coaxial paired SpoIIIE channels across the sporulation septum. Elife 4:e06474. doi: 10.7554/eLife.06474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Veiga H, G Pinho M. 2017. Staphylococcus aureus requires at least one FtsK/SpoIIIE protein for correct chromosome segregation. Mol Microbiol 103:504–517. doi: 10.1111/mmi.13572. [DOI] [PubMed] [Google Scholar]
- 10.Massey TH, Mercogliano CP, Yates J, Sherratt DJ, Löwe J. 2006. Double-stranded DNA translocation: structure and mechanism of hexameric FtsK. Mol Cell 23:457–469. doi: 10.1016/j.molcel.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 11.Cattoni DI, Thakur S, Godefroy C, Le Gall A, Lai-Kee-Him J, Milhiet P-E, Bron P, Nöllmann M. 2013. Structure and DNA-binding properties of the Bacillus subtilis SpoIIIE DNA translocase revealed by single-molecule and electron microscopies. Nucleic Acids Res 42:2624–2636. doi: 10.1093/nar/gkt1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fiche J-B, Cattoni DI, Diekmann N, Langerak JM, Clerte C, Royer CA, Margeat E, Doan T, Nöllmann M. 2013. Recruitment, assembly, and molecular architecture of the SpoIIIE DNA pump revealed by superresolution microscopy. PLoS Biol 11:e1001557. doi: 10.1371/journal.pbio.1001557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bigot S, Saleh OA, Cornet F, Allemand J-F, Barre F-X. 2006. Oriented loading of FtsK on KOPS. Nat Struct Mol Biol 13:1026. doi: 10.1038/nsmb1159. [DOI] [PubMed] [Google Scholar]
- 14.Ptacin JL, Nöllmann M, Bustamante C, Cozzarelli NR. 2006. Identification of the FtsK sequence-recognition domain. Nat Struct Mol Biol 13:1023. doi: 10.1038/nsmb1157. [DOI] [PubMed] [Google Scholar]
- 15.Bose B, Reed SE, Besprozvannaya M, Burton BM. 2016. Missense mutations allow a sequence-blind mutant of SpoEIII to successfully translocate chromosomes during sporulation. PLoS One 11:e0148365. doi: 10.1371/journal.pone.0148365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Löwe J, Ellonen A, Allen MD, Atkinson C, Sherratt DJ, Grainge I. 2008. Molecular mechanism of sequence-directed DNA loading and translocation by FtsK. Mol Cell 31:498–509. doi: 10.1016/j.molcel.2008.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bigot S, Saleh OA, Lesterlin C, Pages C, El Karoui M, Dennis C, Grigoriev M, Allemand JF, Barre FX, Cornet F. 2005. KOPS: DNA motifs that control E. coli chromosome segregation by orienting the FtsK translocase. EMBO J 24:3770–3780. doi: 10.1038/sj.emboj.7600835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blakely G, May G, McCulloch R, Arciszewska LK, Burke M, Lovett ST, Sherratt DJ. 1993. Two related recombinases are required for site-specific recombination at dif and cer in E. coli K12. Cell 75:351–361. doi: 10.1016/0092-8674(93)80076-Q. [DOI] [PubMed] [Google Scholar]
- 19.Sherratt DJ, Søballe B, Barre FX, Filipe S, Lau I, Massey T, Yates J. 2004. Recombination and chromosome segregation. Philos Trans R Soc Lond B Biol Sci 359:61–69. doi: 10.1098/rstb.2003.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blakely GW, Davidson AO, Sherratt DJ. 2000. Sequential strand exchange by XerC and XerD during site-specific recombination at dif. J Biol Chem 275:9930–9936. doi: 10.1074/jbc.275.14.9930. [DOI] [PubMed] [Google Scholar]
- 21.Barre F-X, Aroyo M, Colloms SD, Helfrich A, Cornet F, Sherratt DJ. 2000. FtsK functions in the processing of a Holliday junction intermediate during bacterial chromosome segregation. Genes Dev 14:2976–2988. doi: 10.1101/gad.188700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Recchia GD, Sherratt DJ. 1999. Conservation of xer site-specific recombination genes in bacteria. Mol Microbiol 34:1146–1148. doi: 10.1046/j.1365-2958.1999.01668.x. [DOI] [PubMed] [Google Scholar]
- 23.Sciochetti SA, Piggot PJ, Sherratt DJ, Blakely G. 1999. The ripX locus of Bacillus subtilis encodes a site-specific recombinase involved in proper chromosome partitioning. J Bacteriol 181:6053–6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Britton RA, Grossman AD. 1999. Synthetic lethal phenotypes caused by mutations affecting chromosome partitioning in Bacillus subtilis. J Bacteriol 181:5860–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaimer C, Schenk K, Graumann PL. 2011. Two DNA translocases synergistically affect chromosome dimer resolution in Bacillus subtilis. J Bacteriol 193:1334–1340. doi: 10.1128/JB.00918-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duman R, Ishikawa S, Celik I, Strahl H, Ogasawara N, Troc P, Löwe J, Hamoen LW. 2013. Structural and genetic analyses reveal the protein SepF as a new membrane anchor for the Z ring. Proc Natl Acad Sci U S A 110:E4601–E4610. doi: 10.1073/pnas.1313978110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamoen LW, Meile JC, De Jong W, Noirot P, Errington J. 2006. SepF, a novel FtsZ-interacting protein required for a late step in cell division. Mol Microbiol 59:989–999. doi: 10.1111/j.1365-2958.2005.04987.x. [DOI] [PubMed] [Google Scholar]
- 28.Edwards DH, Errington J. 1997. The Bacillus subtilis DivIVA protein targets to the division septum and controls the site specificity of cell division. Mol Microbiol 24:905–915. doi: 10.1046/j.1365-2958.1997.3811764.x. [DOI] [PubMed] [Google Scholar]
- 29.Pichoff S, Lutkenhaus J. 2005. Tethering the Z ring to the membrane through a conserved membrane targeting sequence in FtsA. Mol Microbiol 55:1722–1734. doi: 10.1111/j.1365-2958.2005.04522.x. [DOI] [PubMed] [Google Scholar]
- 30.El Andari J, Altegoer F, Bange G, Graumann PL. 2015. Bacillus subtilis bactofilins are essential for flagellar hook- and filament assembly and dynamically localize into structures of less than 100 nm diameter underneath the cell membrane. PLoS One 10:e0141546. doi: 10.1371/journal.pone.0141546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crozat E, Rousseau P, Fournes F, Cornet F. 2014. The FtsK family of DNA translocases finds the ends of circles. J Mol Microbiol Biotechnol 24:396–408. doi: 10.1159/000369213. [DOI] [PubMed] [Google Scholar]
- 32.Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, Danuser G. 2008. Robust single-particle tracking in live-cell time-lapse sequences. Nat Methods 5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garner EC. 2011. MicrobeTracker: quantitative image analysis designed for the smallest organisms. Mol Microbiol 80:577–579. doi: 10.1111/j.1365-2958.2011.07580.x. [DOI] [PubMed] [Google Scholar]
- 34.Kumar M, Mommer MS, Sourjik V. 2010. Mobility of cytoplasmic, membrane, and DNA-binding proteins in Escherichia coli. Biophys J 98:552–559. doi: 10.1016/j.bpj.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haeusser DP, Garza AC, Buscher AZ, Levin PA. 2007. The division inhibitor EzrA contains a seven-residue patch required for maintaining the dynamic nature of the medial FtsZ ring. J Bacteriol 189:9001–9010. doi: 10.1128/JB.01172-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ben-Yehuda S, Rudner DZ, Losick R. 2003. Assembly of the SpoIIIE DNA translocase depends on chromosome trapping in Bacillus subtilis. Curr Biol 13:2196–2200. doi: 10.1016/j.cub.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 37.El Najjar N, Kaimer C, Rosch T, Graumann PL. 2017. Requirements for septal localization and chromosome segregation activity of the DNA translocase SftA from Bacillus subtilis. J Mol Microbiol Biotechnol 27:29–42. doi: 10.1159/000450725. [DOI] [PubMed] [Google Scholar]
- 38.Jensen SO, Thompson LS, Harry EJ. 2005. Cell division in Bacillus subtilis: FtsZ and FtsA association is Z-ring independent, and FtsA is required for efficient midcell Z-ring assembly. J Bacteriol 187:6536–6544. doi: 10.1128/JB.187.18.6536-6544.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schenk K, Hervas AB, Rosch TC, Eisemann M, Schmitt BA, Dahlke S, Kleine-Borgmann L, Murray SM, Graumann PL. 2017. Rapid turnover of DnaA at replication origin regions contributes to initiation control of DNA replication. PLoS Genet 13:e1006561. doi: 10.1371/journal.pgen.1006561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaacks K, Healy J, Losick R, Grossman A. 1989. Identification and characterization of genes controlled by the sporulation-regulatory gene spo0H in Bacillus subtilis. J Bacteriol 171:4121–4129. doi: 10.1128/jb.171.8.4121-4129.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zellmeier S, Zuber U, Schumann W, Wiegert T. 2003. The absence of FtsH metalloprotease activity causes overexpression of the σW-controlled pbpE gene, resulting in filamentous growth of Bacillus subtilis. J Bacteriol 185:973–982. doi: 10.1128/JB.185.3.973-982.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dempwolff F, Reimold C, Reth M, Graumann PL. 2011. Bacillus subtilis MreB orthologs self-organize into filamentous structures underneath the cell membrane in a heterologous cell system. PLoS One 6:e27035. doi: 10.1371/journal.pone.0027035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kidane D, Sanchez H, Alonso JC, Graumann PL. 2004. Visualization of DNA double-strand break repair in live bacteria reveals dynamic recruitment of Bacillus subtilis RecF, RecO and RecN proteins to distinct sites on the nucleoids. Mol Microbiol 52:1627–1639. doi: 10.1111/j.1365-2958.2004.04102.x. [DOI] [PubMed] [Google Scholar]
- 44.Plank M, Wadhams GH, Leake MC. 2009. Millisecond timescale slimfield imaging and automated quantification of single fluorescent protein molecules for use in probing complex biological processes. Integr Biol (Camb) 1:602–612. doi: 10.1039/b907837a. [DOI] [PubMed] [Google Scholar]
- 45.Reyes-Lamothe R, Sherratt DJ, Leake MC. 2010. Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science 328:498–501. doi: 10.1126/science.1185757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Youngman PJ, Perkins JB, Losick R. 1983. Genetic transposition and insertional mutagenesis in Bacillus subtilis with Streptococcus faecalis transposon Tn917. Proc Natl Acad Sci U S A 80:2305–2309. doi: 10.1073/pnas.80.8.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gueiros-Filho FJ, Losick R. 2002. A widely conserved bacterial cell division protein that promotes assembly of the tubulin-like protein FtsZ. Genes Dev 16:2544–2556. doi: 10.1101/gad.1014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tavares JR, de Souza RF, Meira GLS, Gueiros-Filho FJ. 2008. Cytological characterization of YpsB, a novel component of the Bacillus subtilis divisome. J Bacteriol 190:7096–7107. doi: 10.1128/JB.00064-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.