

Abstract
Infectious diseases caused by pathogenic protozoa are among the most significant causes of death in humans. Therapeutic options are scarce and massively challenged by the emergence of resistant parasite strains. Many of the current anti-parasite drugs target soluble enzymes, generate unspecific oxidative stress, or act by an unresolved mechanism within the parasite. In recent years, collections of drug-like compounds derived from large-scale phenotypic screenings, such as the malaria or pathogen box, have been made available to researchers free of charge boosting the identification of novel promising targets. Remarkably, several of the compound hits have been found to inhibit membrane proteins at the periphery of the parasites, i.e., channels and transporters for ions and metabolites. In this review, we will focus on the progress made on targeting channels and transporters at different levels and the potential for use against infections with apicomplexan parasites mainly Plasmodium spp. (malaria) and Toxoplasma gondii (toxoplasmosis), with kinetoplastids Trypanosoma brucei (sleeping sickness), Trypanosoma cruzi (Chagas disease), and Leishmania ssp. (leishmaniasis), and the amoeba Entamoeba histolytica (amoebiasis).
Keywords: drug target, transport, infection, resistance, parasite, malaria, protozoa
Human-pathogenic protozoa, current treatment, resistance
Apicomplexa
With more than 200 million new infections per year, malaria-causing Plasmodium spp. are the most prominent parasites. The death toll of malaria is still >400,000 per year. About 90% of the cases occur in the WHO African Region and are caused almost exclusively by the species Plasmodium falciparum. In the subtropical zones outside of Africa, Plasmodium vivax is responsible for up to 64% of the cases. Ongoing efforts to eradicate malaria are hampered by the lack of effective antisera and the spreading of resistant strains against antimalarial treatment (WHO, 2017b). Hence, current research aims at identifying suitable epitopes for future immunization programs, and discovery of new drug targets to establish novel modes of antimalarial drug action.
The current first-line treatment of uncomplicated P. falciparum malaria is an oral artemisinin-based combination therapy (WHO, 2015). The mechanisms of how artemisinin and its derivatives, such as artesunate and artemether, attain antimalarial activity are thought to reside in generating oxidative stress by liberating reactive oxygen species from an internal peroxo-moiety, and in affecting a calcium ATPase (SERCA or PfATP6) of the sarcoplasmic-endoplasmic reticulum (Moore et al., 2011). The half-life of the fast-acting artemisinins is very short, typically around 1 h. To maintain this highly efficient therapeutic option in view of an increasing number of mutant parasite strains with varying degrees of resistance to the artemisinins (Jambou et al., 2005), these compounds are combined with drugs that exhibit longer half-lives (WHO, 2001; Kavishe et al., 2017). Patients infected with P. vivax, Plasmodium ovale, Plasmodium malariae, or Plasmodium knowlesi are equally treated with an artemisinin combination therapy or with chloroquine depending on the sensitivity of the infecting strain. For a number of decades, chloroquine was used for monotherapy until massive resistance occurred. Chloroquine accumulates in the parasite's digestive vacuole and interferes with heme-detoxification during hydrolysis of hemoglobin from the host (Slater, 1993; Thomé et al., 2013; WHO, 2015). For preventing relapse from dormant liver stages after infections with P. vivax or P. ovale, the use of primaquine is recommended (Fernando et al., 2011; Mikolajczak et al., 2015; Lalève et al., 2016). Complicated infections require rapid administration via intravenous or intramuscular injections of artesunate for at least 24 h followed by artemisinin-based combination therapy (Abiodun et al., 2013; WHO, 2015).
A malaria-related, tick-borne disease, babesiosis, normally occurs in livestock and domestic animals, and only occasionally emerges in humans. Most cases of human babesiosis are caused by Babesia microti. First-line treatment is a combination of the ubiquinone analog atovaquone and the antibiotic macrolide azithromycin (Krause et al., 2000).
Another apicomplexan parasite, Toxoplasma gondii, causes the food-borne disease toxoplasmosis. It is estimated that 30–50% of the world's population are infected with this parasite. It persists, often life-long, in the host in a dormant, cystic bradyzoite form. Although the infection usually occurs asymptomatic, it can evolve to a life-threating illness in immune-compromised patients. Infections of pregnant women can be transmitted to the fetus giving rise to spontaneous abortion or stillbirth (Flegr et al., 2014). Toxoplasmosis is treated with the dihydrofolate reductase inhibitor pyrimethamine or the antibiotic sulfadiazine. Second-line drugs are azithromycin, clarithromycin, atovaquone, dapsone, and cotrimoxazole. Due to side effects and ineffectiveness against the dormant bradyzoite form novel therapeutics are urgently needed (Petersen and Schmidt, 2003).
Kinetoplastids
Parasites of the phylum euglenozoa, i.e., the kinetoplastids, are the causative agents of various infections that are classified as neglected tropical diseases. Overall, it is estimated that one billion people in tropical and subtropical countries are affected. Infections with Leishmania spp. lead to cutaneous (Leishmania major, Leishmania tropica) mucocutaneous (Leishmania braziliensis), or visceral leishmaniasis (Leishmania donovani, Leishmania infantum), which is spread by sandflies. About 250,000 new cases are registered per year in 87 countries (WHO, 2017a). For treatment, sodium stibogluconate, amphotericin B, miltefosine, paromomycin, and pentamidine are used; yet, the therapy needs major improvement as it is characterized by high levels of toxicity for the patient. Further, resistance against the drugs, in particular to the pentavalent antimonial stibogluconate, strongly limits their usability (Loiseau and Bories, 2006; Ponte-Sucre et al., 2017).
Parasites of a related kinetoplastid species, Trypanosoma, cause life-threatening infections, i.e., human African trypanosomiasis or sleeping sickness (Trypanosoma brucei) and Chagas disease (Trypanosoma cruzi). Human African trypanosomiasis is spread by the tsetse fly in tropical Africa. Approximately 3,000 cases were reported in 2016 (WHO, 2016). In the first hemolytic stage, T. brucei replicates extracellularly in the host blood causing fever and joint pain among other symptoms. In the second, severe neurological stage of the disease, the parasite reaches the central nervous system. Patients suffer from disruption of the sleep-wake cycle and irreparable neurological damage. Parasites in the peripheral blood stream can be attacked by the drugs suramin and pentamidine. For the central nervous system form, only the mercurial melarsoprol and eflornithine are available (Brun et al., 2010). As in the case of leishmaniasis, modern, i.e., less toxic and more effective drugs are needed. T. cruzi-derived Chagas disease is prevalent in Latin-America claiming 14.000 deaths per year. An estimated 6 million people are infected by T. cruzi spread by the bug Triatoma infestans (also kissing bug or winchuka). As a treatment, the chemical radical-producing drug benznidazole and nifurtimox are available. With only two compounds at hand, limited success rates and severe side-effects, new drugs are required against this parasite (Castro et al., 2006).
Amoebae
The free-living amoebozoan parasite Entamoeba histolytica causes amoebiasis. With an estimated death toll of 40,000–100,000 per year, it ranges second behind Plasmodium infections (Stanley, 2003). The disease is most prevalent in but not restricted to the tropics when sanitation is poor. Although the majority of infections progress asymptomatically, a life-threatening amoebic colitis can manifest. E. histolytica forms hardy, infectious cysts that are ingested by the host via contaminated food or water. After reaching the colon, the cysts transform into trophozoites that are capable of invading the intestinal mucosa. When breaching the mucosa, trophozoites can disseminate, among others, to the liver and the central nervous system causing serious complications, i.e., amoebic abscesses (Shirley and Moonah, 2016). First line treatment is a combination of the antibiotics metronidazole and paromomycin. Alternative, second line treatments for metronidazole are other nitroimidazoles, e.g., tinidazole or ornidazole, and the broad-spectrum anti-parasitic nitazoxanide. Paromomycin can be substituted by diiodohydroxyquinoline and diloxanide, both drugs act by a so-far unresolved mechanism and may not be effective against all strains (McAuley and Juranek, 1992).
The severity of infections by protozoan parasites, the limited arsenal of drugs, often with hardly tolerable side effects, and the increasing resistance problem call for novel approaches. The scope of this review is, thus, to discuss the potential of channel and transport proteins as novel targets for anti-parasite chemotherapy in terms of druggability, selectivity, and proneness to resistance. There is considerably more data available from the malaria research field compared to the more neglected parasite-caused diseases. A major criterion for inclusion of a channel or transporter into this review was existing proof of principle involving first small-molecule inhibitors that exhibit anti-parasitic potency.
Targeting transport processes of parasites at different levels
Transmembrane transporters and channels are usually classified based on their biophysical and biochemical properties, such as mechanism of transport and substrate selectivity. We decided to provide a pharmacological and pharmaceutical view on the topic and structured this manuscript based on the location of the transporter of interest and, accordingly, the site of action of a respective drug (Figure 1). We will approach the parasite from the outside, first hitting the host cell in the case of intracellular parasites. i. Indirect targeting. If it is possible to address infected host cells selectively by targeting transport proteins of the host cell plasma membrane this would leave the parasite little options for defending itself against the attack. Malaria parasites for instance are known to modify the functionality of red blood cell proteins and to integrate plasmodial membrane proteins into the erythrocyte membrane. ii. Peripheral targeting. Target proteins residing in a parasite's plasma membrane possibly can be inhibited from the outside. In this case and in indirect targeting, resistance mechanisms would be limited to changing the drug binding site of the target protein. iii. Internal targeting. For targeting transporters within a parasite, i.e., at organelles such as the digestive vacuole or mitochondria, respective drugs would need to enter the parasite's cytosol. In this case, the parasite has additional means to generate resistance, either by preventing uptake of the compound, by altering it chemically, or by pumping it out via efflux transporters. Hence, we will also address iv. Targeting drug efflux transporters.
Figure 1.

Channels and transporters of parasites as targets for indirect, peripheral, and internal therapeutic attacks. HPM, host plasma membrane; HVM, host vacuolar membrane; PPM, parasite plasma membrane; POMs, parasite organelle membranes; Mito, mitochondrion; Vac, vacuole. Abbreviations of the channel and transporter proteins are explained in the text.
Indirect targeting—channels and transporters of the infected host cell membrane
It was recognized early on in malaria research that the transport of ions, amino acids, and other nutrients across the plasma membrane of infected red blood cells increases compared to uninfected cells. This way, the parasite actively adapts the ionic environment inside the erythrocyte to its needs and ensures access to nutrients from the host blood. Over the years, it has become evident that such new permeability pathways (NPP) are not only due to infection-dependent alteration of the host membrane proteins but also to export of Plasmodium-derived proteins and integration into the host cell membrane (Overman, 1947; Ginsburg et al., 1985; Desai et al., 2000; Huber et al., 2002). Proteins at the erythrocyte plasma membrane pose attractive targeting sites as the respective inhibitor compounds would not be in direct contact with the parasite. Interference by the parasite with drug action would be restricted to alteration of the protein resulting from gene mutations, whereas other means, such as expedited drug export or metabolism would not be applicable. It remains to be shown, however, whether this indirect approach can reliably kill parasites (Cohn et al., 2003). The efficiency of compounds that indirectly target parasites is summarized in Table 1.
Table 1.
Efficiency of compounds for indirect targeting.
| Target | Parasite species | Compound name | Effect on protein | Effect on parasite | Cell stage in vitro | Effect in vivo | Host species | Reference |
|---|---|---|---|---|---|---|---|---|
| PSAC | P. falciparum |
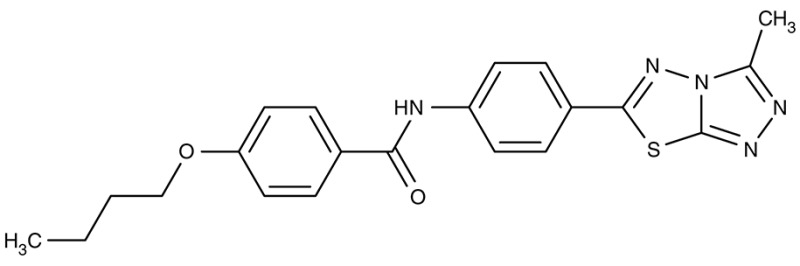 PRT1-20 |
– | IC50 5 μM |
Trophozoites | – | Human | Pain et al., 2016 |
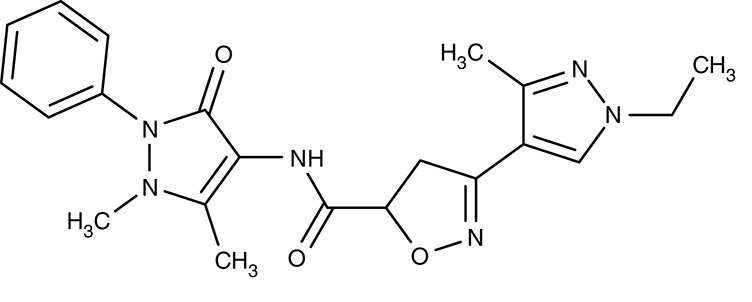 ISPA-28 |
– | IC50 0.06 μM |
Trophozoites | – | Human | Nguitragool et al., 2011 | ||
| VRAC (host) | P. berghei |
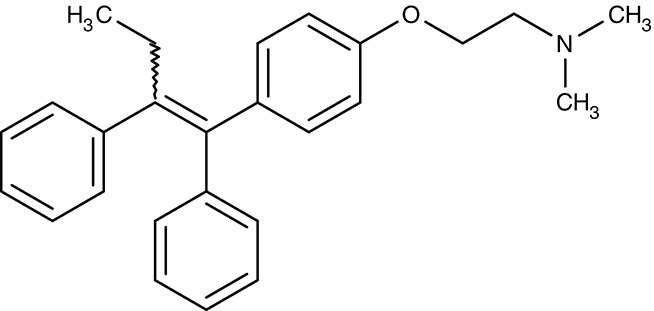 Tamoxifen |
IC50 4 μM |
– | Liver-stage | – | Human | Prudêncio et al., 2009 |
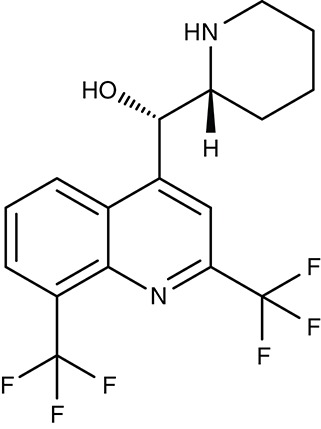 Mefloquine |
IC50 2 μM |
– | Liver-stage | – | Human | Prudêncio et al., 2009 | ||
| ATP6V0C (host) | L. donovani |
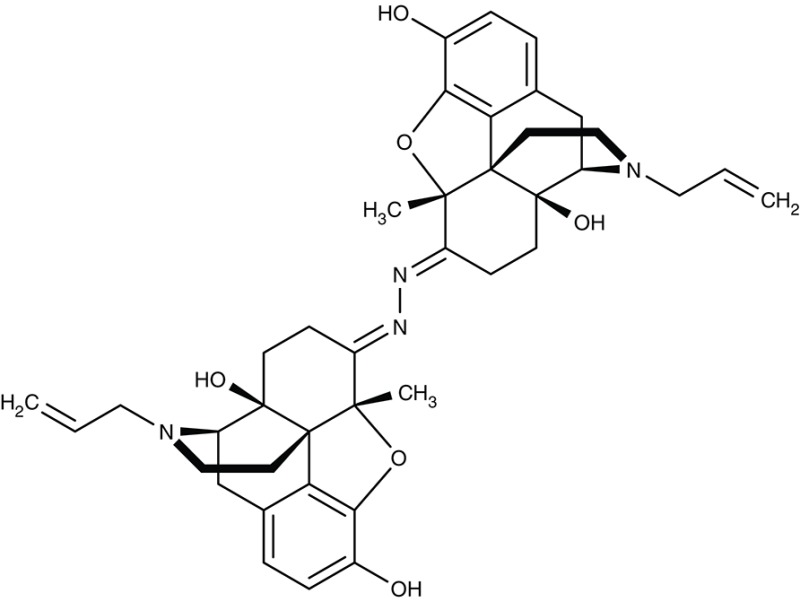 Naloxonazine |
– | IC50 3.5 μM |
Amastigotes (Intracellular) | – | Human | de Muylder et al., 2011 |
Plasmodial surface anion channel
One extensively studied type of conductivity of the red blood cell membrane that is brought about upon infection is derived from the plasmodial surface anion channel (PSAC; Figure 1). Despite a still elusive protein identity, permeability for various substrates, such as sugars, amino acids, nucleosides, and inorganic anions and cations, has been attributed to this voltage-dependent channel (Ginsburg et al., 1985; Kirk and Horner, 1995; Upston and Gero, 1995; Saliba et al., 1998; Hill et al., 2007). PSAC seems to transport the mentioned substrates via two different routes within the protein or protein complex. One path is said to be used primarily for alanine and sorbitol uptake and can be blocked by furosemide, whereas the other one conducts mainly proline and the unnatural substrate phenyltrimethylammonium (PhTMA). The latter path is sensitive to so-called PSAC residual transport inhibitors, abbreviated as PRT (Alkhalil et al., 2004; Pain et al., 2016). Based on this finding, full blockade of PSAC may require a combination of a PRT inhibitor plus a furosemide derivative. For example, PRT1-20 alone (Table 1) yielded an IC50 on P. falciparum growth of 5 μM; in combination with furosemide it was 10 times lower (Pain et al., 2016). Toward identification of the PSAC protein or regulators thereof, the finding could become helpful that certain parasite strains are susceptible to one particular group of PSAC inhibitors. This pointed to two genes, CLAG3.1 and CLAG3.2 (cytoadherence linked asexual protein) of which only one appears to be active at a time. Mutations in the genes led to modified PSAC activity. Further, epigenetic regulation of the CLAG3 genes was suggested to modulate PSAC (Sharma et al., 2013; Nguitragool et al., 2014). To illustrate this, ISPA-28 (Table 1), an isolate-specific PSAC antagonist, exhibited an IC50 of 56 nM against the P. falciparum Dd2 strain and 43 μM against the HB3 strain, i.e., a value higher by three orders of magnitude. When the CLAG3 gene of the Dd2 strain was transferred to the HB3 strain, it showed the same nanomolar susceptibility (Nguitragool et al., 2011, 2014). Clearly, identification of the true nature of the PSAC protein and/or components as well as expression or reconstitution in a heterologous or artificial system would be highly appreciated for in-depth structure-function analyses, inhibitor screening and development.
Host ion channels
Besides the parasite-derived PSAC, host encoded transport proteins of the erythrocyte may be exploited as drug targets if their functionality changes with infection. Oxidative stress was shown to alter potassium and chloride conductance of infected red blood cells (Staines et al., 2001; Huber et al., 2004). However, specific inhibitors are yet to be found to determine their potential as anti-parasite drugs.
There is evidence that in the liver-stage, Plasmodium berghei infection leads to a sevenfold increase in chloride conductance of the host's volume-regulated anion channel, VRAC, as found using a human hepatoma cell line (Figure 1; Prudêncio et al., 2009). Conductivity was inhibited by tamoxifen, and mefloquine at single-digit micromolar concentrations (Table 1). The underlying mechanism of channel activation by the parasite, the effect of estrogen receptor modulators on VRAC conductivity, and, ultimately, whether indirect targeting of VRAC would be suitable for malarial therapy of the liver stage is not clear at this time. With the system and compounds at hand further investigations will be possible that may shed some light on the phenomenon.
Host V-type proton ATPase
In the search of new drug targets against L. donovani, Muylder et al. chose a strategy of host-directed therapy. The amastigote form of the parasite develops primarily inside phagocytic cells and is inert to digesting enzymes. A screening assay using a human macrophage cell line infected with L. donovani yielded one hit compound, a μ-opioid receptor antagonist naloxonazine (de Muylder et al., 2011; Table 1). It turned out that naloxonazine upregulates expression of the V-type proton ATPase subunit C, ATP6V0C. This upregulation was linked to an increase in the volume of intracellular acidic vacuoles suggesting an indirect effect on Leishmania amastigotes through host cell vacuolar remodeling (de Muylder et al., 2016). How such a therapy would be tolerated by the host and whether the parasites will find ways to adapt to the remodeled vacuoles is not known.
Host nutrient channels and transporters
We describe two examples illustrating that nutrient transport of the host cell affects growth of P. berghei parasites, i.e., in the blood-stage depending on the glycerol permeability of an aquaporin (Liu et al., 2007), and in the liver-stage via arginine transport (Meireles et al., 2017). Aquaporin-9 knockout mice lack a functional glycerol channel in their erythrocytes. In this environment, P. berghei, grew considerably slower compared to wildtype erythrocytes, and infected AQP9-null mice survived longer. The authors attribute the effect to reduced glycerol levels in the parasite impeding glyceroplipid biosynthesis for the build-up of membranes during growth. Similarly, knockdown of the arginine-transporting SLC7A2 of the solute carrier family decreased intra-hepatic growth and multiplication of P. berghei parasites in vivo and ex vivo (Meireles et al., 2017). A sufficient supply of arginine is required for the vital polyamine synthesis of the parasites. Today, glycerol or arginine transport-modulating small molecules have not been found and/or tested.
Together, although an indirect approach holds strong potential against parasite infections, several gaps in basic knowledge need to be filled with regard to the identity of the involved transport proteins, selectivity of inhibitors, and susceptibility/adaptability of the parasites.
Peripheral targeting—channels and transporters of the parasite plasma membrane
The substrate spectrum of the parasite-induced new permeation pathways at the host cell membrane is broad. Transport proteins at the parasite's plasma membrane (Figure 1), in turn, appear much more specific. These membrane proteins facilitate the uptake of the main energy source, glucose, and precursors for biosynthesis, such as nucleosides for DNA/RNA, or glycerol for glycerolipids. Equally vital is the efficient release of waste molecules derived from energy metabolism, e.g., lactic acid, or from protein degradation, i.e., ammonia and urea. Nutrient and metabolite transport often depends on transmembrane ion gradients, e.g., of protons or sodium, generated by ATPases and is further modulated by ion channels. Efficiency data on compounds for peripheral parastite targeting are displayed in Table 2.
Table 2.
Efficiency of compounds for peripheral targeting.
| Target | Parasite species | Compound name | Effect on protein | Effect on parasite | Cell stage in vitro | Effect in vivo | Host species | Reference |
|---|---|---|---|---|---|---|---|---|
| HT | Plasmodium spp. |
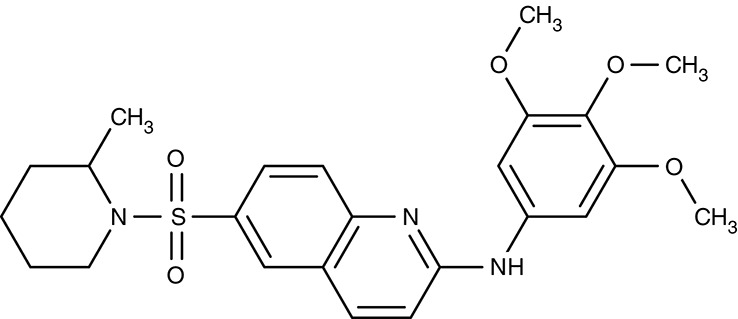 TCMDC-125163 |
IC50 39 nM |
IC50 1.24 μM |
Trophozoites | – | – | Ortiz et al., 2015 |
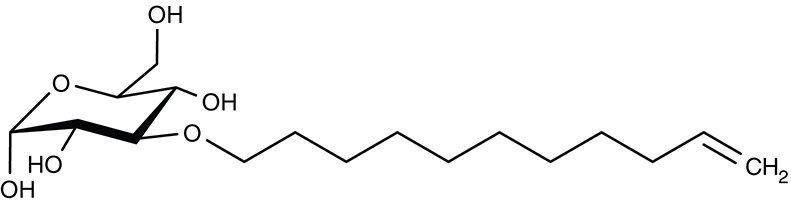 C3361 |
Ki 8.6–53 μM |
IC50 15–16 μM |
Trophozoites | – | – | Joet et al., 2003; Blume et al., 2011 | ||
| – | IC50 11 μM |
Liver-stage | – | Human | Slavic et al., 2011 | |||
| HT1 | B. bovis | Ki 4.1 μM |
No inhibition at 100 μM | Trophozoites | – | – | de Muylder et al., 2011 | |
| GT1 | T. gondii | Ki 82 μM |
No inhibition at 200 μM | Tachyzoites | – | – | Blume et al., 2011 | |
| FNT | P. falciparum |
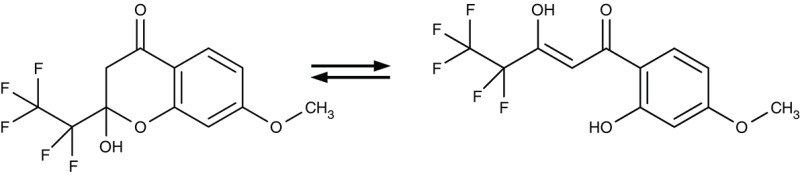 MMV007839 |
IC50 0.02–0.17 μM |
IC50 0.14 μM |
Trophozoites | – | – | Golldack et al., 2017; Hapuarachchi et al., 2017 |
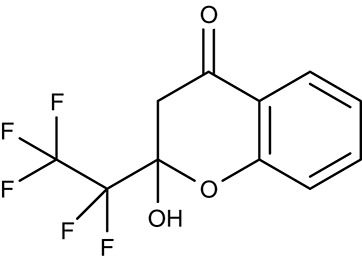 MMV000972 |
IC50 0.05–0.17 μM |
IC50 1.70 μM |
Trophozoites | – | – | |||
| PT0 | T. brucei |
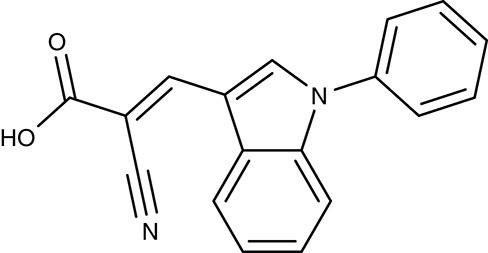 UK5099 |
Inhibition at 250 μM | – | – | – | – | Sanchez, 2013 |
| PAT12 | T.cruzi |
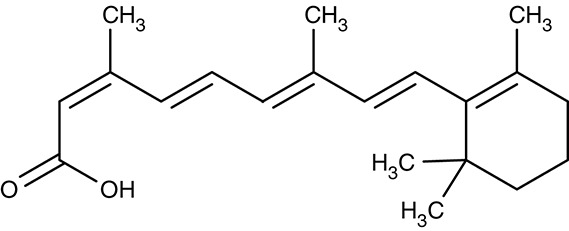 Isotretinoin |
– | IC50 0.13 μM |
Epimastigotes | – | – | Reigada et al., 2017 |
| IC50 30.6 μM |
Trypomastigotes | – | – | |||||
| ENT1 | Plasmodium spp. |
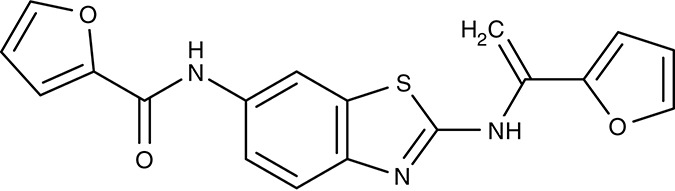 ChemBridge 9001893 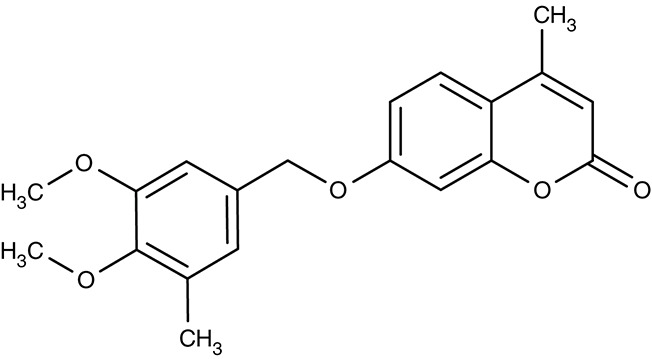 ChemBridge 6946484 |
IC50 2.5–30 nM |
IC50 3–55 μM |
Trophozoites | – | – | Frame et al., 2015b |
| ATP4 | P. falciparum |
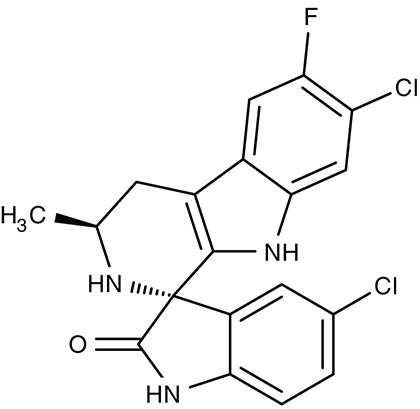 Cipargamin |
– | IC50 0.5–1.4 nM |
Trophozoites | Clearance with 3-day dosing, 30 mg per day | Human | Rottmann et al., 2010; Spillman and Kirk, 2015 |
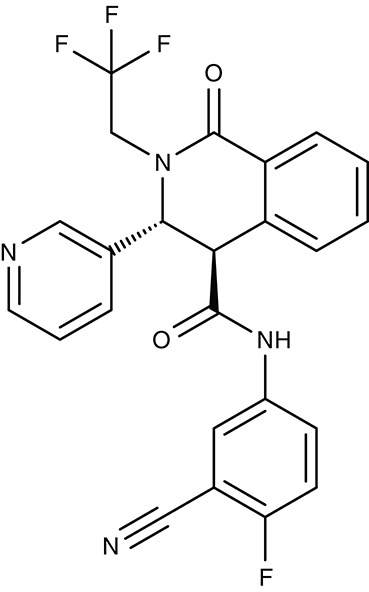 SJ733 |
– | IC50 30 μM |
Trophozoites | – | – | Spillman and Kirk, 2015 | ||
| Calcium channels | Leishmania spp. Trypanosoma spp. |
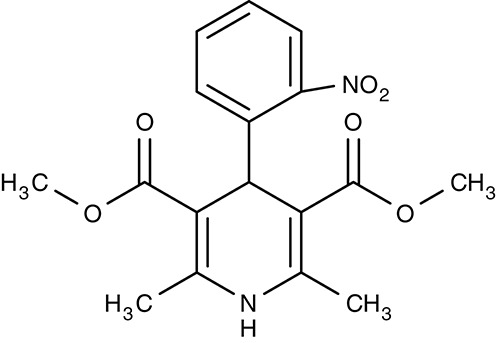 1,4-dihydropyridines (e.g., Nifedipine) |
−− | IC50 2.6–181 μM |
Promastigotes/Amastigotes Trypomastigotes | −− | −− | Tempone et al., 2009 Reimão et al., 2010, 2011 |
| A. castellanii |
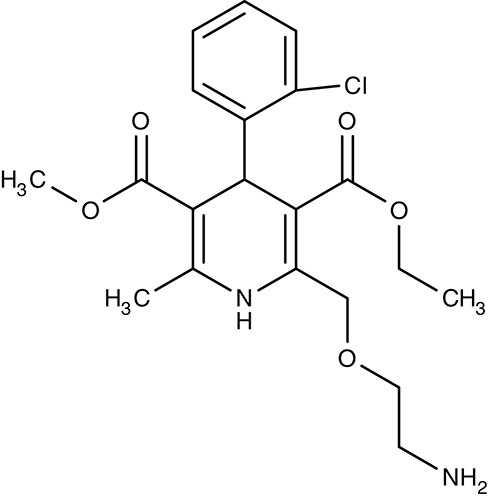 Amlodipine |
– | Large inhibition at 1.2 μM | Trophozoites | – | – | Baig et al., 2013 | |
| L. infantum |
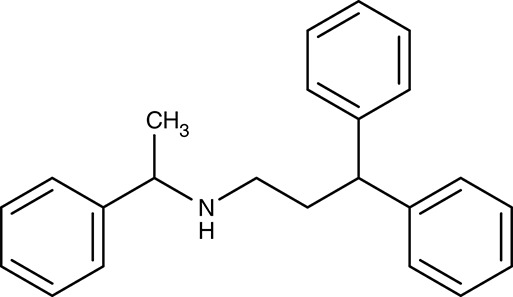 Non-dihydropyridines (e.g., fendiline) |
– | IC50 2–16 μM |
Promastigotes | – | – | Reimão et al., 2016 | |
| T. cruzi | – | Epimastigotes | – | – | Reimão et al., 2016 | |||
| K1/K2 | T. brucei |
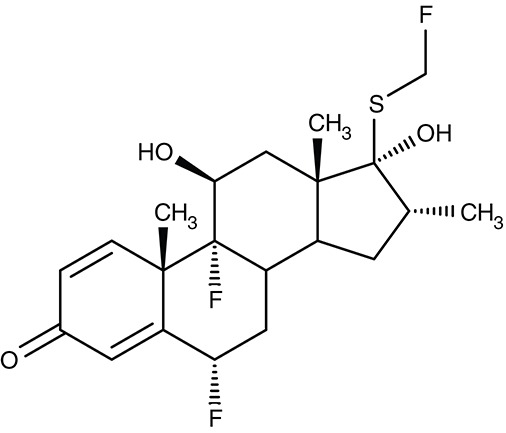 Fluticasone |
IC50 0.7 μM |
– | – | – | – | Schmidt et al., 2017 |
Targeting peripheral nutrient and metabolite transporters
Considering their significance for survival, it seems quite surprising that plasmodia rely on a single hexose transporter, HT, and a single lactic acid transporter, the latter being a member of the microbial formate-nitrite-transporter family, FNT. Both transporters are present at the plasma membrane and both have been validated as novel antimalarial drug targets using cultured parasites.
Glucose transporters
Soon after the identification of HT, first weak glucose-analog inhibitors were described (Krishna and Woodrow, 1999; Woodrow et al., 1999; Joet et al., 2003). One of these compounds, C3361 (Table 2), yielded Ki-values in the μM range on glucose transport of Plasmodium berghei, Plasmodium falciparum, Plasmodium yoelii, Plasmodium vivax, Plasmodium knowlesi, Babesia bovis, and T. gondii (Joet et al., 2003; Blume et al., 2011). C3361 was not only active in the blood-stage but also inhibited parasite development in the liver-stage of P. berghei with an IC50 of 11 μM (Slavic et al., 2011). The vector stages, however, were much less susceptible, and a transmission block required 1 mM. Interestingly, C3361 failed to inhibit growth of the related apicomplexan Babesia parasites suggesting an alternative glucose transport pathway. In fact, the B. bovis genome contains two putative hexose transporter genes, of which only one has been characterized so-far (Derbyshire et al., 2008). Knockout of the homologous Toxoplasma glucose transporter, GT, led to moderate growth inhibition. Apparently, it is dispensable for the survival of the parasite. A search for alternative transporters in Toxoplasma produced three more putative sugar transporters of which one was found to be located at the plasma membrane. Yet, a knockout failed to effect parasite growth. Contrary to plasmodia, Toxoplasma seems not to rely exclusively on glucose as an energy source. It is discussed that glutamine can be used as a potent alternative energy source (Blume et al., 2009).
More recent screenings for inhibitors of the plasmodial HT using the Tres Cantos antimalarial compound set (TCAMS) and the malaria box led to the discovery of nanomolar inhibitors, e.g., TCMDC-125163 (Table 2) has an IC50 of 39 nM for heterologously expressed HT, 3.2 μM for the human red blood cell glucose transporter GLUT1, and an EC50 of 1.24 μM for growth of cultured P. falciparum parasites. Binding of the identified compounds occurred mostly non-competitive with glucose and, hence, likely to a site different from the glucose binding pocket (Ortiz et al., 2015).
Lactate and pyruvate transporters
The end products of glucose-based energy metabolism are lactic acid in plasmodia, and pyruvic acid in trypanosomes. In order to prevent detrimental acidification of the cytosol and inhibition of the metabolic pathway by accumulating product, such molecules need to be swiftly released from the cells. Lactate transport in living P. falciparum parasites was experimentally shown in the early 1990s (Kanaani and Ginsburg, 1991). It took until 2015 that the responsible transporter was identified by our and Kiaran Kirk's group (Marchetti et al., 2015; Wu et al., 2015). The protein is structurally and in terms of transport mechanism unrelated to human lactate transporters from the monocarboxylate transporter family (MCT). Instead, the plasmodial lactic acid transporter is a member of the microbial formate-nitrite transporter family, FNT. Besides l-lactate, it transports d-lactate, as well as formate, acetate and pyruvate by a proton cotransport mechanism (Wiechert and Beitz, 2017; Wiechert et al., 2017).
Screening of the malaria box yielded two compounds, MMV007839 and MMV000972 (Table 2), that efficiently block PfFNT at nanomolar concentrations (Spangenberg et al., 2013; Golldack et al., 2017). In vitro selection of a resistant P. falciparum strain helped to locate the binding site at the intracellular face of the transporter. The compounds, thus, need to enter the parasite where they assume a lactate substrate-like form carrying a negative charge for efficient binding. Transport across consecutive membranes that shield the parasite is achieved by a cyclic hemiketal form that is neutral and lipophilic, see Table 2 (Golldack et al., 2017). FNTs are absent in humans, however, some other protozoan parasites carry single or multiple copies of FNT genes, e.g., Babesia spp., T. gondii, and E. histolytica, representing putative targets. Kinetoplastids, in turn, do not encode FNTs in their genomes, raising the question of how monocarboxylate transport is achieved in these organisms. In T. brucei, a high-affinity pyruvate transporter, TbPT0, was recently discovered that is more related to polytopic proteins from plants than to mammalian MCTs (Sanchez, 2013). This transporter at the plasma membrane plus two additional mitochondrial pyruvate transporters of T. brucei were found to be inhibitable by the pyruvate-reminiscent compound UK5099 (Štáfková et al., 2016; Table 2).
Nucleobase and nucleoside transporters
Apart from nutrients and metabolites of energy metabolism, precursors, and components of biosynthetic pathways are typical substrates of parasite transporters. In this sense, a group of transporters found at the plasma membrane of plasmodia imports nucleobases and nucleosides. Four P. falciparum genes encode equilibrative nucleoside transporters, ENT1–4 of which ENT1 seems to provide the major uptake route (Downie et al., 2006, 2008, 2010; Frame et al., 2012, 2015a). Small molecule inhibitors were found by high throughput screening, e.g., ChemBridge no. 9001893 and 8946464 (Table 2), that inhibited the P. falciparum PfENT1 with IC50 values in the low nanomolar range. Efficiency was similar with the P. vivax and P. berghei ENT1 proteins (Arora et al., 2016; Deniskin et al., 2016). The compounds were less potent, however, in parasite cultures with EC50-values from 0.8 to 6.5 μM (Frame et al., 2015b).
Aquaporin solute channels
Plasmodium and Toxoplasma parasites express a single aquaglyceroporin channel, AQP, at the plasma membrane (Hansen et al., 2002; Pavlovic-Djuranovic et al., 2006). These AQPs conduct water and small, uncharged solutes that are relevant in glycerolipid biosynthesis (glycerol), protein degradation (urea, ammonia), and oxidative stress (hydrogen peroxide) (Hansen et al., 2002; Beitz et al., 2004; Zeuthen et al., 2006; Wu et al., 2010; Wree et al., 2011; Almasalmeh et al., 2014). Small, drug-like inhibitors for apicomplexan AQPs are missing (Song et al., 2012), but their potential as drug targets is underscored by a P. berghei PbAQP knockout strain that exhibited strongly reduced growth, virulence, and progression through the liver stage (Promeneur et al., 2007, 2018). T. brucei expresses three AQPs of which TbAQP2 is a key factor for the uptake of the anti-trypanosomal drug pentamidine (Uzcategui et al., 2004; Song et al., 2016). The L. major AQP facilitates uptake of antimonite into the parasite released from the anti-leishmanial drug stibogluconate (Mukhopadhyay and Beitz, 2010; Mukhopadhyay et al., 2011).
Drug repurposing/polyamine transporters
An attempt to repurpose already used drugs revealed that retinoids, an established class for the pharmacotherapy of severe acne, target parasitic nutrient transporters. Initially, retinoic acid and retinol acetate were shown to inhibit the growth of L. donovani (Mukhopadhyay and Madhubala, 1994). More specifically, isotretinoin (Table 2) was found to block a polyamine transporter, PAT12, when adding to cultures of T. cruzi epimastigotes. In vitro growth of emerging trypomastigotes and epimastigotes was inhibited with IC50 values of 0.13 and 30.6 μM, respectively. PAT12 is a member of the polyamine and amino acid transporter family, AAAP, for which isotretinoin displayed activity as a multi-target inhibitor (Reigada et al., 2017). This shows that repurposing is a valid tool and can promote research in the field of neglected diseases.
Targeting peripheral ion transporters and channels
The establishment and maintenance of ion gradients across the parasite plasma membrane is vital for the membrane potential, osmotic balance, as well as for driving transport processes. P-type ATPases are single protein units that convert energy from ATP hydrolysis into cation transport (Weiner and Kooij, 2016). ATP4 of P. falciparum was recently shown to act as a sodium pump at the plasma membrane (Dyer et al., 1996; Spillman et al., 2013). There is evidence that sodium export by ATP4 is coupled to proton import. Whether cell death upon blockade of ATP4 occurs due to cytosol acidification, osmotic swelling, collapse of the electrochemical potential, or a combination thereof is unknown (Spillman et al., 2013; Spillman and Kirk, 2015). For whatever reason, blocking of ATP4 is lethal for malaria parasites rendering ATP4 a most attractive novel drug target. Analysis of the 400 malaria box compounds yielded the surprisingly high number of 28 hits, which further underscores the central role of ATP4 for parasite viability.
ATP4/P-type sodium ATPase
Two previously identified ATP4 inhibitors already entered the clinical trial stage. The clinical candidate cipargamin with a spiroindolone scaffold (Table 2) is thought to bind to the transport path of ATP4 from the intracellular entry site as deduced from in vitro selection of resistance mutations (Spillman and Kirk, 2015). Growth of sensitive P. falciparum strains was inhibited with IC50-values in the range of 0.5–1.4 nM. Application of a single 100 mg kg−1 dose in an in vivo mouse model study killed all P. berghei parasites. In human trials, a 3-day dosing regime with 30 mg per day led to parasite clearance (Rottmann et al., 2010; White et al., 2014). Cipargamin exhibited low toxicity in humans, high oral bioavailability and suitable half-life. Other related ATP4-inhibiting spiroindolones have been found to be similarly potent (Spillman et al., 2013). The second promising candidate undergoing a clinical trial is the dihydroisoquinolone SJ733 (Jiménez-Díaz et al., 2014; Table 2). It shows more distant structure similarities to cipargamin with the 5-membered heterocycle of the indolone moiety replaced by a 6-membered ring (Jiménez-Díaz et al., 2014; Spillman and Kirk, 2015; Crawford et al., 2017). Although resistance mutations were selectable in vitro by sub-lethal concentrations, ATP4 inhibitors, when dosed properly, might prove advantageous against the rise of resistant strains in the clinic due to their fast acting property.
Drug repurposing/calcium channels
Drug repurposing approaches aim at ion channels at the plasma membrane of kinetoplastids. Several established 1,4-dihydropyridine calcium channel blockers used for the treatment of hypertension in humans were tested on various Leishmania and Trypanosoma species. Nifedipine, amlodipine, bepridil, nimodipine, and others showed weak effects in vitro with IC50-values in the micromolar range (Maya et al., 2000; Tempone et al., 2009; Reimão et al., 2010, 2011). Amlodipine and lacidipine administered in four weekly single doses of 10 mg kg−1 reduced the parasite burden of L. donovani-infected BALB/c mice by 75–85% (Palit and Ali, 2008). Amlodipine was also tested for activity against cultured amoebae of Acanthamoeba castellanii and largely inhibited growth at 1.2 μM (Baig et al., 2013). The non-dihydropyridine calcium channel blockers fendiline, mibrefadil, and lidoflazine inhibited in vitro growth of L. infantum promastigotes and T. cruzi epimastigotes with IC50-values from 2–16 μM (Reimão et al., 2016). Verapamil, however, failed to inhibit growth of L. donovani promastigotes, but seemed to reverse the resistance against stibogluconate by an unknown mechanism (Neal et al., 1989; Valiathan et al., 2006). Although being calcium channel blockers in humans, the target in the tested parasites remains to be established.
Drug repurposing/potassium channels
Recently, a screening by the National Center for Advancing Translational Sciences Small Molecule Resource identified fluticasone, an established corticosteroid for the treatment of asthma, to inhibit the T. brucei potassium channels TbK1 and TbK2. The proteins were localized to the parasite's plasma membrane and electrophysiologically characterized in Xenopus oocytes (Steinmann et al., 2015). Fluticasone was found to inhibit the TbK1/TbK2-mediated currents at an IC50 of 0.7 μM (Schmidt et al., 2017).
Internal targeting—channels and transporters of parasite organelle membranes
Drugs that need to enter the parasite's cytosol encounter several more challenges than compounds acting from the outside. Diffusional uptake across the plasma membrane requires high lipophilicity, small molecule size, and absence of charged moieties. Alternatively, compounds can be shuttled into the cell by a more active type of transport via endogenous channels and transporters. This route is taken for instance by antimonite released from stibogluconate in Leishmania therapy or by pentamidine against trypanosomes. In both cases, resistance mutations of a transporting aquaglyceroporin efficiently prevent the drugs from entering the parasites. Another factor is the metabolic stability of the drug within the parasite cell. This area is not well studied, yet it is conceivable that inactivation by chemical modification may well occur in a similar fashion as in the host cells, which detoxify xenobiotics e.g., by oxidation and conjugation reactions. Finally, drugs that actually made it to the site of action in a functional form may be pumped out of the parasite cell by drug resistance transporters. The prominent example is the chloroquine resistance transporter, CRT. Table 3 gives an overview on the new developments of compound targeting channels and transporters of internal organelle membranes.
Table 3.
Efficiency of compounds for internal targeting.
| Target | Parasite species | Compound name | Effect on protein | Effect on parasite | Cell stage in vitro | Effect in vivo | Host species | Reference |
|---|---|---|---|---|---|---|---|---|
| ATP6/SERCA | P. falciparum |
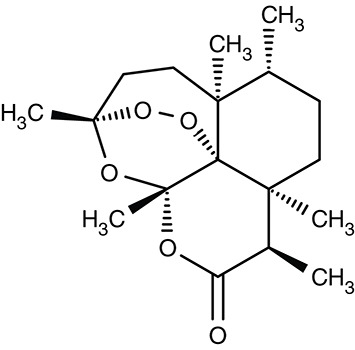 Artemisinin |
– | IC50 11 – 13 nM |
Trophozoites | – | – | del Pilar Crespo et al., 2008 |
| T. gondii | Inhibition at 10 μM | IC50 0.36–8 μM |
Tachyzoites | – | – | Berens et al., 1998; Jones-Brando et al., 2006; Nagamune et al., 2007; Hencken et al., 2010 | ||
| Trypanosoma spp. | – | IC50 13–20 μM |
Trypomastigotes/Epimastigotes | – | – | Yang and Liew, 1993; Mishina et al., 2007; Sen et al., 2010 | ||
| Leishmania spp. | – | IC50 0.75–120 μM |
Promastigotes/Amastigotes | Reduction of parasite burden with oral dose of 10 mg kg−1 | Mouse/Hamster | |||
| B. gibsoni | – | IC50 2.2 μM |
Trophozoites | – | – | Iguchi et al., 2015 | ||
| P. falciparum |
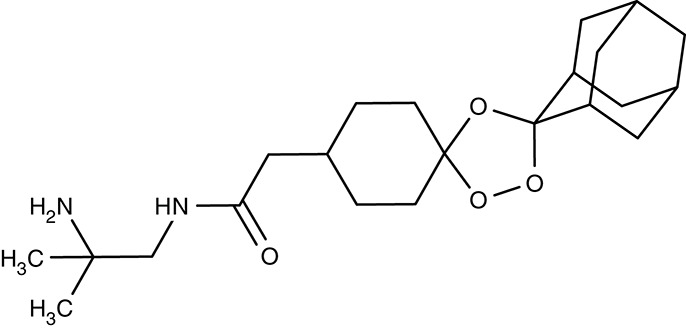 Arterolane |
Ki 7.7 μM |
IC50 1.5 nM |
Trophozoites | – | – | Abiodun et al., 2013 | |
| P. falciparum |
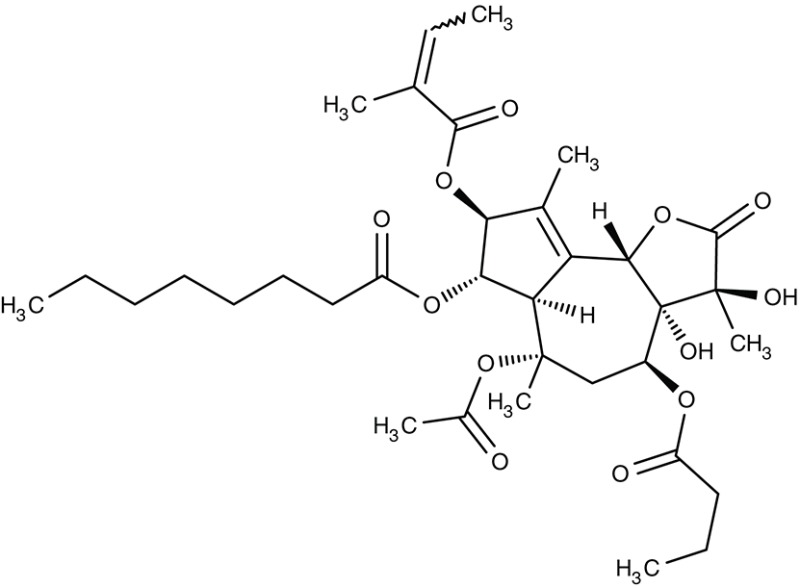 Thapsigargin |
– | IC50 0.25– 0.30 μM |
Trophozoites | – | – | del Pilar Crespo et al., 2008; Abiodun et al., 2013 | |
| T. gondii | Inhibition at 1 μM | – | – | – | – | Nagamune et al., 2007 | ||
| Trypanosoma spp. | – | IC50 30–34 μM |
Trypomastigotes/Epimastigotes | – | – | Mishina et al., 2007 | ||
| L. donovani | – | 28.1 μM | Promastigotes | – | – | Mishina et al., 2007 | ||
| E. invadens | – | Inhibition of encystation at 0.5 μM | Trophozoites | – | – | Martínez-Higuera et al., 2015 | ||
| N. canium | – | Growth Inhibition at 0.1 μg ml−1 | Tachyzoites | – | – | Kim et al., 2002 | ||
| Cytochrome bc1 complex | P. falciparum |
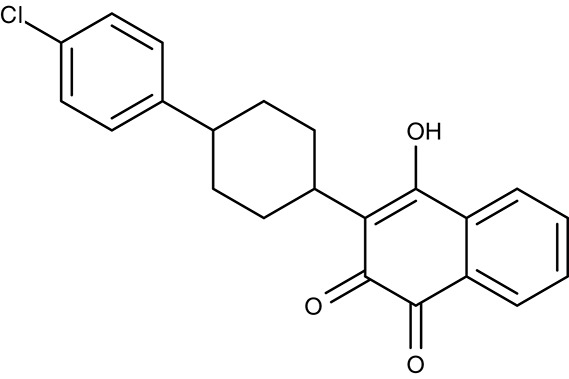 Atovaquone |
IC50 0.2 nM |
IC50 2 nM |
Trophozoites | – | – | da Cruz et al., 2012 |
| T. gondii | – | IC50 0.1–0.5 μM |
Tachyzoites | IC50 0.14–0.85 μM | Mouse | Doggett et al., 2012 | ||
| Babesia spp. | – | IC50 94 nM |
Trophozoites | Effective at dose of 100 mg kg−1 d−1 | Hamster | Hughes and Oz, 1995; Wittner et al., 1996; Matsuu et al., 2008 | ||
| – | – | – | Effective at dose of 1500 mg d−1 plus azithromycin 500 mg on day 1 and 250 mg per day thereafter | Human | Krause et al., 2000 | |||
| L. donovani | – | – | – | 30 % reduction of parasite burden with 100 mg kg−1 for 5 days | Mouse | Croft et al., 1992 | ||
| Theileria spp. |
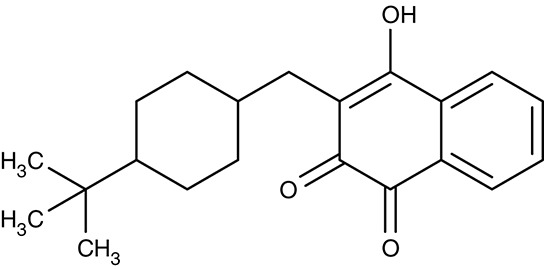 Buparvaquone |
– | – | – | Effective at 2.5–6 mg kg−1 | Cattle, Horse | McHardy et al., 1985; Zaugg and Lane, 1989; Muraguri et al., 1999; Mhadhbi et al., 2010 | |
| Leishmania spp. | – | IC50 0.001–5.495 μM |
Promastigotes/Amastigotes | 60% reduction of parasite burden with 100 mg kg−1 for 5 days | Mouse | Croft et al., 1992; Mäntylä et al., 2004a; Reimão et al., 2012; Jamal et al., 2015 | ||
| P. falciparum |
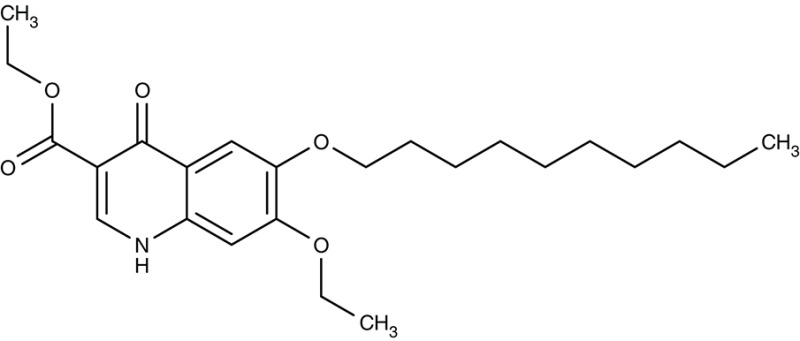 Decoquinate |
IC50 2 nM |
IC50 2.6–36 nM |
Trophozoites | – | – | da Cruz et al., 2012 | |
| P. falciparum |
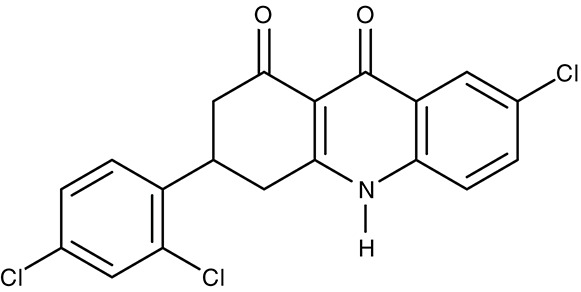 WR249685 |
– | IC50 3 nM |
Trophozoites | – | – | Biagini et al., 2008 | |
| P. falciparum |
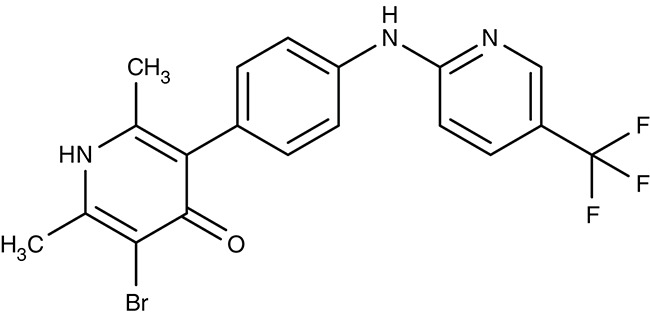 TCMDC-135546 |
– | IC50 22 nM |
Trophozoites | – | – | Raphemot et al., 2015 | |
| Mitochondrial F1F0 ATPase | T. brucei |
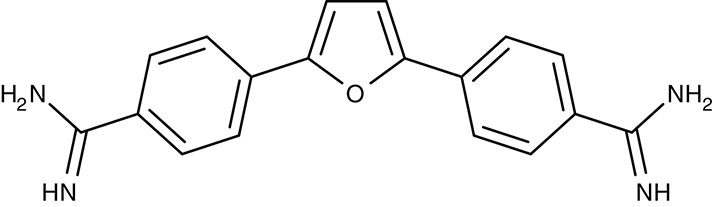 Furamidine (DB75) |
Inhibition at 10 μM | IC50 4.5 nM |
Trypomastigotes | Effective as pentamidine at 3 mg kg−1 d−1, for 7 days | Gerbil | Steck et al., 1982; Ismail et al., 2003; Lanteri et al., 2008 |
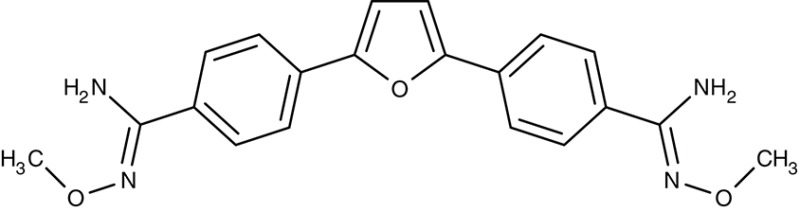 Pafuramidine (DB289) |
– | IC50 14.6 μM |
Trypomastigotes | Effective at a dose of 400 mg kg−1 p.o. | Mouse | Ansede et al., 2004 | ||
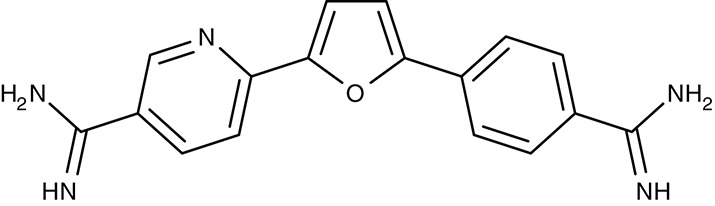 DB820 |
– | IC50 7.9–141 nM |
Trypomastigotes | Effective at 10 mg kg−1 i.p. | Mouse | Wenzler et al., 2009 | ||
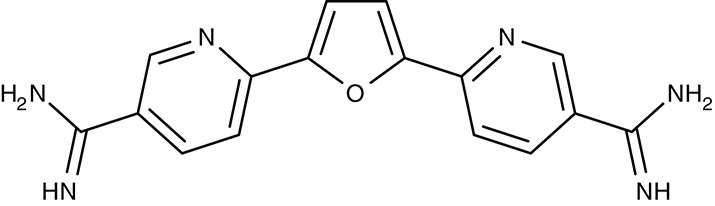 DB829 |
– | IC50 20–346 nM |
Trypomastigotes | Effective at 10 mg kg−1 i.p. | Mouse | Wenzler et al., 2009 | ||
| Mitochondrial choline transporter | P. falciparum |
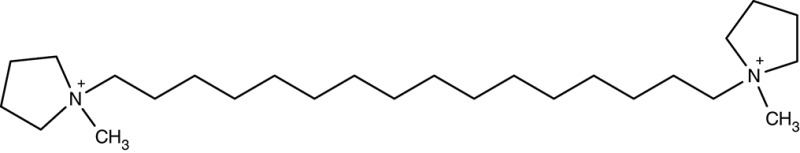 G25 |
– | – | – | – | – | Wengelnik et al., 2002; Biagini et al., 2004 |
| T. brucei | – | EC98 0.16 μM |
Trypomastigotes | – | – | de Macêdo et al., 2015 | ||
| VPPase VP1 | T. gondii |
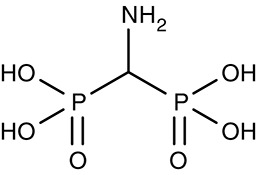 AMDP |
IC50 0.9 μM |
Inhibition at 5–10 μM | – | – | – | Drozdowicz et al., 2003 |
| CRT | P. falciparum |
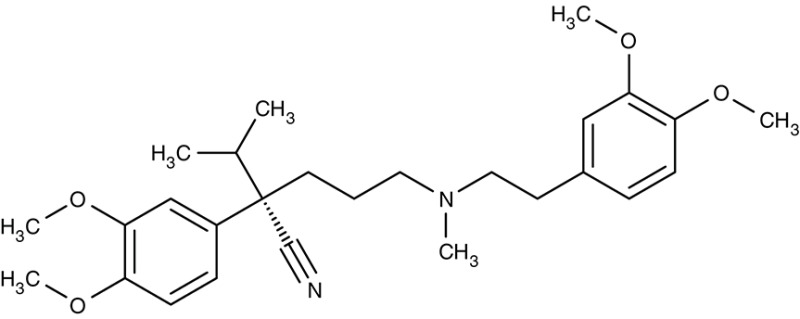 Verapamil |
IC50 30 μM |
– | – | – | – | Ye and van Dyke, 1994 |
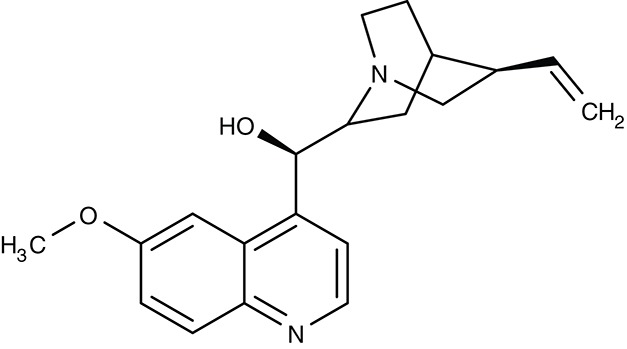 Quinine |
IC50 48 μM |
– | – | – | – | Martin et al., 2009 | ||
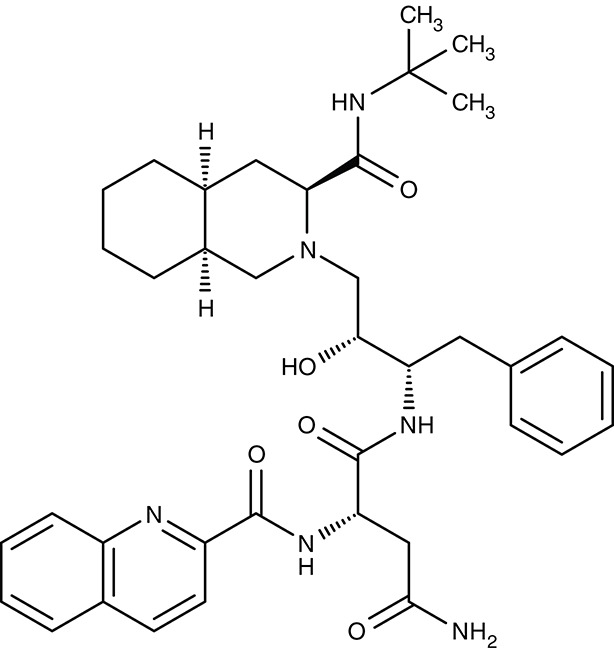 Saquinavir |
IC50 13 μM |
– | – | – | – | Martin et al., 2012 | ||
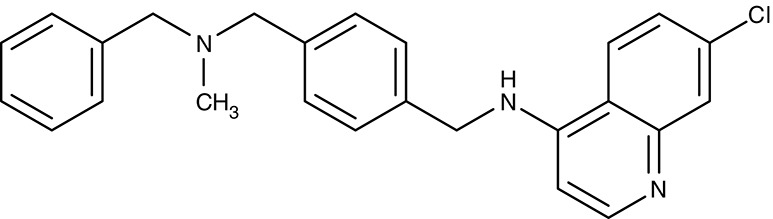 Dibemethin 6a |
IC50 69 μM |
IC50 26 nM |
Trophozoites | – | – | Zishiri et al., 2011 | ||
| MDR1 | P. falciparum |
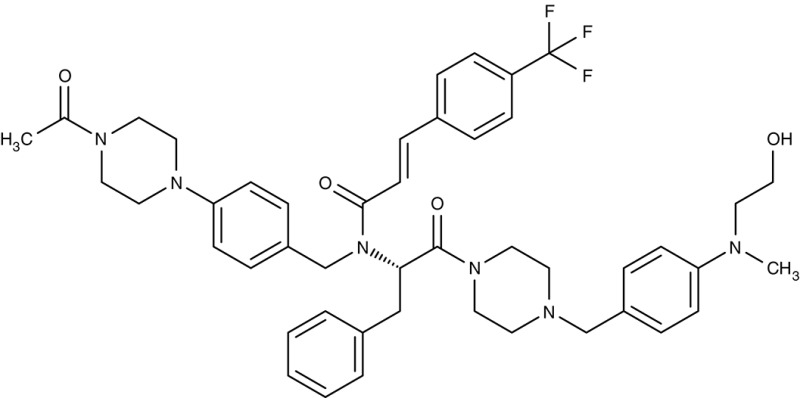 ACT-213615 |
– | IC50 4 nM |
Trophozoites | ED90 8.4 mg kg−1 as effective as chloroquine | Mouse | Brunner et al., 2012, 2013 |
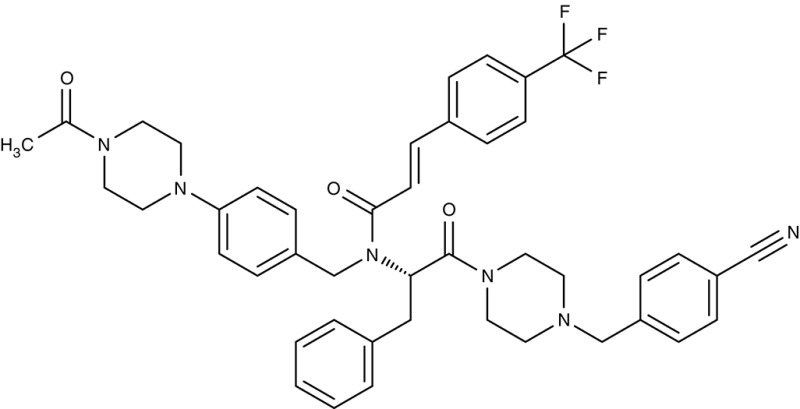 ACT-451840 |
– | IC50 4 nM |
Gametocytes | IC50 2.7 ng ml−1 (3.6 nM) | Human | Krause et al., 2016; Le Bihan et al., 2016; Ng et al., 2016 |
Despite these challenges, all currently used drugs in anti-parasite therapy act at internal sites. It remains to be seen how a shift to peripheral or even indirect attacks will affect resistance formation.
Artemisinins/sarcoplasmic P-type calcium ATPase SERCA/ATP6
Artemisinin (Table 3) and derivatives are the most important antimalarials today. The molecules contain a peroxo moiety. It is discussed that ironII from heme when released from hemoglobin during degradation in the digestive vacuole chemically activates the artemisinins producing reactive oxygen species that damage proteins more or less specifically inside the mitochondria (Asawamahasakda et al., 1994; Moore et al., 2011) or at other sites in the cell including DNA as a target. Evidence further points to a P-type calcium ATPase, SERCA, or ATP6, at the sarcoplasmic endoplasmic reticulum as one of the affected proteins (Eckstein-Ludwig et al., 2003; Naik et al., 2011; Abiodun et al., 2013; Pulcini et al., 2013; Krishna et al., 2014; Nunes et al., 2016). However, there is a mismatch of artemisinin efficiency on the parasites and on heterologously expressed ATP6. In P. falciparum cultures, IC50-values were 11 and 13 nM tested on a chloroquine resistant (K1) and a sensitive (NF54) strain, respectively (del Pilar Crespo et al., 2008). The artemisinins also showed some potency on several other protozoan parasites, i.e., T. gondii (Berens et al., 1998; Jones-Brando et al., 2006; Hencken et al., 2010), T. brucei, T. cruzi, L. donovani, L. major (Yang and Liew, 1993; Mishina et al., 2007), and Babesia gibsoni (Iguchi et al., 2015) yielding EC50-values from 0.36 to 120 μM. For Leishmania spp., artemisinin exhibited in vivo activity in infected hamster and BALB/c mice models (Ma et al., 2004; Sen et al., 2010; Ghaffarifar et al., 2015). Naegleria fowleri, a problematic, cyst-forming amoeba, has been shown to be sensitive to artemisinin in vitro (Cooke et al., 1987), whereas treatment failed in a mouse model (Gupta et al., 1995). Purified recombinant P. falciparum ATP6, however, could not be directly inhibited by artemisinin or derivatives (Cardi et al., 2010; Arnou et al., 2011), and full inhibition of yeast expressed SERCA of T. gondii required high concentrations of 10 μM (Nagamune et al., 2007). The small molecule arterolane (Table 3) has a different scaffold than the artemisinins but equally contains a peroxo group and, thus, should be capable of releasing reactive oxygen species. When tested on P. falciparum ATP6 expressed in Xenopus oocytes it was found to be clearly less potent than artemisinin with a Ki-value of 7.7 μM; yet, parasite growth was inhibited a very low IC50 of 1.5 nM similar to artemisinin. These mixed results show that ATP6/SERCA is probably not the main target of the artemisinins.
Still, parasite SERCA/ATP6 seems to hold potential as a therapeutic target, because thapsigargin (Table 3), a plant sesquiterpene lactone and general SERCA inhibitor, killed cultured chloroquine resistant and sensitive P. falciparum parasites with IC50 of 246 and 298 nM (del Pilar Crespo et al., 2008; Abiodun et al., 2013). Thapsigargin further inhibited growth of T. gondii, Trypanosoma spp., L. donovani, E. invadens, and Neospora canium with EC50-values in the range of 0.5–39 μM (Kim et al., 2002; Mishina et al., 2007; Martínez-Higuera et al., 2015). Yeast expressed T. gondii SERCA was fully inhibited by thapsigargin at 1 μM (Nagamune et al., 2007).
Atovaquone/mitochondrial cytochrome bc1 complex
A key function of mitochondria in general is the build-up of a steep proton gradient across the inner mitochondrial membrane, which is used to drive ATP synthesis by the F-type ATPase, or ATP synthase. To this end, the inner membrane harbors a cytochrome bc1 complex. The bc1 proteins use ubiquinone, also called coenzyme Q, as a redox cofactor in electron transfer reactions (Q-cycle), which free four protons that are transported to the intermembrane space in the process (Crofts et al., 1999a,b; Crofts, 2004).
The drug atovaquone (Table 3), a ubiquinone analog and cytochrome bc1 complex inhibitor, is in use against malaria, toxoplasmosis and babesiosis for many years. It interferes with ubiquinone cofactor binding to the Q0 site as shown by manifesting resistance mutations in this region of the protein (Fry and Pudney, 1992; Srivastava et al., 1999; McFadden et al., 2000; Kessl et al., 2007; Vallières et al., 2012; Siregar et al., 2015). Combination of atovaquone with proguanil lowers the IC50 from 2 nM to 400 pM probably by a synergistic mechanism as proguanil destroys the mitochondrial membrane potential in the presence of an electron transport inhibitor (Srivastava and Vaidya, 1999). Several resistant strains have formed during the use of atovaquone (Hutchinson et al., 1996; Painter et al., 2007; da Cruz et al., 2012). Since the cytochrome bc1 complex is common to mitochondria of all species is was possible to inhibit the growth of other parasites as well. Potency against T. gondii was sub-micromolar in vitro, i.e., tachyzoites in human foreskin fibroplasts, and in vivo using a mouse model with IC50-values of 0.14 and 0.85 μM, respectively (Doggett et al., 2012). Similarly, atovaquone acted on Babesia spp. in vitro and in vivo in hamsters (Hughes and Oz, 1995; Wittner et al., 1996; Matsuu et al., 2008). In dog and human studies, combination with the antibiotic azithromycin turned out to be positive (Krause et al., 2000; Birkenheuer et al., 2004; Di Cicco et al., 2012; Checa et al., 2017). Treatment of L. donovani infections in a mouse model were less successful resulting only in a 30% lower parasite burden (Croft et al., 1992).
A related hydroxynaphthoquinone compound, buparvaquone (Table 3), is used for the treatment of theileriosis in cattle (McHardy et al., 1985; Minami et al., 1985; Muraguri et al., 1999; Mhadhbi et al., 2010), and horses (Zaugg and Lane, 1989). Anti-leishmanial activity was evaluated in vitro for L. donovani, Leishmania aethiopica, L. major, Leishmania amazonensis, Leishmania mexicana, Leishmania panamensis, L. infantum, Leishmania chagasi, L. braziliensis, and L. tropica promastigotes and amastigotes resulting in IC50-values of 0.001–5.495 μM (Mäntylä et al., 2004a,b; Reimão et al., 2012; Jamal et al., 2015). Animal models showed that the in vivo efficiency was higher when prodrugs of buparvaquone were applied (Croft et al., 1992; Garnier et al., 2007) or when a nanoliposomal drug preparation was used (da Costa-Silva et al., 2017).
In the search for alternative chemical scaffolds, decoquinate (Table 3), a 4-oxo-quinoline, was tested. However, the overall structure is still similar to ubiquinone. The compound is in use in veterinary medicine against coccidia (Miner and Jensen, 1976; Ricketts and Pfefferkorn, 1993). When tested against blood-stage as well as liver-stage P. falciparum, in either case nanomolar IC50 values were found at low host cell toxicity (da Cruz et al., 2012). Another potent inhibitor of the cytochrome bc1 complex with good selectivity for P. falciparum is the dihydroacridinedione WR249685 (Table 3), which has an IC50 of 3 nM on the in vitro growth of the parasites (Biagini et al., 2008). Screening of the TCAMS library for cytochrome bc1 complex inhibition yielded one efficient compound, TCMDC-135546 (Table 3), with an IC50 of 22 nM on parasite growth (Raphemot et al., 2015). Naphthoquinone esters were derived from the anticancer drug rhinacanthin and showed low nanomolar IC50-values on the growth of P. falciparum. Notably, the molecules seem to bind to the Qi ubiquinone site of cytochrome bc1 rather than Q0 as the above-mentioned compounds (Kongkathip et al., 2010). In a diversity oriented synthesis approach, macrolactame derivatives were generated that yielded nanomolar EC50-values and are thought to target the Qi site as well (Comer et al., 2014; Lukens et al., 2015). Finally, more compounds were derived from endochin, an experimental antimalarial of the 1940s, with the aim to improve solubility and metabolic stability in the host. Such compounds were found not to inhibit the human cytochrome bc1 complex but to very efficiently target the Qi site of the cytochrome bc1 complex of falciparum and vivax plasmodia from various clinical field isolates as well as T. gondii and Babesia microti (Doggett et al., 2012; Nilsen et al., 2013; Lawres et al., 2016). Leishmania spp., however, were much less susceptible to this type of inhibitors with IC50-values in the micromolar range (Ortiz et al., 2016).
Diamidines/mitochondrial ATP synthase
Similar to the artemisinins in plasmodia, the mode of diamidine action, e.g., pentamidine, in trypanosomes is not fully resolved. Screening of an diamidine library yielded furamidine (Table 3) and related compounds that act comparably to pentamidine against T. brucei parasites in mice and rhesus monkeys (Rane et al., 1976; Steck et al., 1982). Further, a prodrug of furamidine, DB289 (Table 3), with better oral availability was developed (Ismail et al., 2003; Ansede et al., 2004). Regarding transporter targeting, it was found that the F1F0-ATPase, i.e., the mitochondrial proton gradient-driven ATP synthase, was inhibited at concentrations around 10 μM and caused a collapse of the mitochondrial membrane potential. The efficiency of diamidines on the growth of trypanosomes, however, is clearly lower, i.e., in the submicromolar range, indicating that ATP synthase is not the main target. It was also suggested that the compounds inhibit other ATPases (Lanteri et al., 2008). DB289 entered phase III clinical trials as the first orally available drug against blood stage human African trypanosomiasis. However, due to manifestation of delayed renal insufficiency in a number of recipients, further development was terminated (Harrill et al., 2012). Two related aza analogs of furamidine (DB820, CPD0801) are still followed up on as they have shown efficiency against T. brucei in a mouse model of second stage trypanosomiasis (Wenzler et al., 2009; Ward et al., 2011).
G25/mitochondrial choline transport
The notion that a choline-related compound, named G25 (Table 3) carrying two quaternary ammonium moieties spaced by a 16-carbon linker, efficiently kills malaria parasites led to the idea that choline transport might be a valid antimalarial target (Wengelnik et al., 2002; Biagini et al., 2004). Besides P. falciparum, also T. brucei and L. mexicana turned out to be sensitive to G25 (Ibrahim et al., 2011). The mode of action remains unclear. Besides the notion that these compounds inhibit choline transport, it was reported that the mitochondrial structure and function of trypanosomes was affected (Ibrahim et al., 2011; Macêdo et al., 2013). An RNA interference approach in the blood stream form of T. brucei hinted at the involvement of a member of a mitochondrial carrier protein family, TbMCP14, which is unrelated to mammalian carriers (Schumann Burkard et al., 2011; de Macêdo et al., 2015).
Aminomethylenediphosphonate/vacuolar-type inorganic pyrophosphatase
Vacuolar-type pyrophosphatases, V-PPases, seem to be absent in vertebrates, yet have vital functions in protozoan energy conservation and membrane transport. Accordingly, they may represent suitable drug targets (Rodrigues et al., 2000; Drozdowicz et al., 2003). Yeast-expressed V-PPase from T. gondii, TgVP1, was inhibitable by aminomethylenediphosphonate, AMDP (Table 3), with an IC50 of 0.9 μM. The presence of V-PPases has also been shown in P. falciparum and T. cruzi (Urbina et al., 1999; McIntosh et al., 2001). Anti-parasitic, drug-like molecules targeting V-PPases are yet to be found.
Targeting drug efflux transporters
Drug resistance due to expedited export of the compounds is a key factor in pharmacotherapy not only of anti-infectives but in general. The loss of drug action may be reversed by inhibition of the responsible efflux transporter.
Verapamil, (dimeric) quinine, saquinavir/digestive vacuole chloroquine resistance transporter
The discovery of chloroquine was a breakthrough in malaria therapy. It acts by inhibiting heme detoxification in the form of polymerized hemozoin in the digestive vacuole (Foley and Tilley, 1997). Promoted by monotherapeutic use and widespread underdosing in eradication programs, however, resistant P. falciparum strains were selected over the years rendering chloroquine largely useless today (Wellems et al., 1991; Waller et al., 2003). Resistance mutations were found in one particular gene of unknown function at that time. It turned out that the mutations resulted in a gain-of-function transporter shuttling chloroquine out of the digestive vacuole at a strongly increased rate (Fidock et al., 2000; Lakshmanan et al., 2005). The physiological role of the chloroquine-resistance-transporter, CRT, is still elusive. However, a variety of amino acids, polyamines and peptides have been found to be CRT substrates (Juge et al., 2015). Several attempts have been undertaken to block CRT with the aim to reverse chloroquine resistance. The calcium channel blocker verapamil (Table 3) was shown to inhibit CRT expressed in Xenopus oocytes with an IC50 of 30 μM (Ye and van Dyke, 1988, 1994; Tanabe et al., 1989). In the same system, quinine, a natural product and chloroquine analog, yielded an IC50 of 48 μM (Martin et al., 2009), and the antiretroviral drug saquinavir was effective at 13 μM (Martin et al., 2012). Chemical synthesis of dimeric quinines lowered the IC50 to 1 μM on CRT-expressing oocytes and efficiently inhibited parasite growth in the nanomolar range (Hrycyna et al., 2014).
The resistance reversing effect of verapamil cannot be exploited in humans, because the required dose would be too high to be tolerable (Ye and van Dyke, 1988). One approach was to develop a drug-like compound with dual functionality, i.e., blockade of hemozoin formation plus inhibition of CRT (Burgess et al., 2006; Kelly et al., 2009). The obtained dibemethin derivates (Table 3) showed IC50 values of 26 nM on parasite growth, yet potency on the isolated CRT protein was only 69 μM. Bioavailability appeared sufficient after oral administration in mice (Zishiri et al., 2011).
ACT-213615, ACT-451840/digestive vacuole multidrug resistance transporter 1
Based on similarity to human multidrug resistance transporters in terms of sequence and function another transporter of the digestive vacuole of malaria parasites was termed multidrug resistance transporter 1, MDR1 (Foote et al., 1989; Cowman et al., 1991). MDR1 transport is directed into the digestive vacuole. This way, mefloquine, artemisinin, and artesunate are thought to be trapped by compartmentalization preventing the drugs from hitting their target in the sarcoplasmic endoplasmic reticulum (Reed et al., 2000; Pickard et al., 2003; Price et al., 2004; Rohrbach et al., 2006). Resistance was increased in strains with multiple copies of the pfmdr1 gene (Cowman et al., 1994; Pickard et al., 2003). For quinine in turn, it is required that MDR1 is functional to deliver the compound to its site of action inside the digestive vacuole. Therefore, a transport decreasing N1042D mutation of MDR1 was made responsible for quinine resistance (Rohrbach et al., 2006). Apart from transport of antimalarial drugs, MDR1 appears to be vital for the malaria parasite, because in cell viability screening and subsequent analyses, two small-molecule compounds were found to target MDR1. ACT-213615 (Table 3) is proposed to inhibit either PfMDR1 directly or a regulating protein upstream of it yielding an EC50 value of 4 nM on parasite growth (Brunner et al., 2012, 2013). The second, highly related compound, ACT-451840 (Table 3), was even more potent and a single nucleotide polymorphism in the MDR1 gene was identified that turned out to be responsible for resistance against the compound. The compound was well tolerated in first clinical trials, however, none of the eight tested subjects could be completely freed from parasites (Krause et al., 2016; Le Bihan et al., 2016; Ng et al., 2016). The physiological substrates and function of MDR1 remain to be established.
Conclusion
The identification of anti-parasitic targets at the parasite's plasma membrane or even at host membranes opens up options for peripheral or indirect therapeutic attacks (Figure 1). This approach holds the potential to limit resistance formation because the only remaining line of defense is mutational modification of the drug binding site at the target protein. It is thinkable that massive upregulation of target protein expression may also reduce efficiency, yet at high energetic cost for the parasite.
Still, most currently addressed targets are located inside the parasite. Here, specificity must be taken into account. Drugs affecting vital functions by hitting multiple targets, e.g., the artemisinins by eliciting oxidative stress, are hard to defend by the parasite at the various target sites. However, several ways exist to interfere with drug action by acting directly on the molecular entity. Drug uptake can be prevented if transporters are required to deliver the compound to the cytosol, e.g., pentamidine via TbAQP2. The drug can be chemically modified or compartmentalized, e.g., artesunate transport into the digestive vacuole by MDR1. Finally, the compound can be shuttled out to keep the concentration below a harmful level, e.g., chloroquine via CRT. Drug metabolism and transport issues cannot be fully appreciated beforehand. Hence, in the search for potent anti-infectives against parasites and bacteria alike, phenotypic screenings have proven more successful than specific approaches in designing inhibitors for a selected target protein.
The ideal anti-parasitic drug compound should i. reach the site of action independent of parasite transport proteins, ii. be specific for the parasite but not necessarily for a single target protein, iii. act fast, iv. be metabolically stable, and v. be safe for the patient. Meeting such demands and circumventing the influence by the parasite may be eased by addressing parasite-specific proteins at the periphery.
Author contributions
AM, HE, and EB jointly wrote the manuscript and prepared the figures.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was funded by the Deutsche Forschungsgemeinschaft (Be2253/7-1 and Be2253/8-1).
References
- Abiodun O. O., Brun R., Wittlin S. (2013). In vitro interaction of artemisinin derivatives or the fully synthetic peroxidic anti-malarial OZ277 with thapsigargin in Plasmodium falciparum strains. Malar. J. 12:43. 10.1186/1475-2875-12-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhalil A., Cohn J. V., Wagner M. A., Cabrera J. S., Rajapandi T., Desai S. A. (2004). Plasmodium falciparum likely encodes the principal anion channel on infected human erythrocytes. Blood 104, 4279–4286. 10.1182/blood-2004-05-2047 [DOI] [PubMed] [Google Scholar]
- Almasalmeh A., Krenc D., Wu B., Beitz E. (2014). Structural determinants of the hydrogen peroxide permeability of aquaporins. FEBS J. 281, 647–656. 10.1111/febs.12653 [DOI] [PubMed] [Google Scholar]
- Ansede J. H., Anbazhagan M., Brun R., Easterbrook J. D., Hall J. E., Boykin D. W. (2004). O-alkoxyamidine prodrugs of furamidine: in vitro transport and microsomal metabolism as indicators of in vivo efficacy in a mouse model of Trypanosoma brucei rhodesiense infection. J. Med. Chem. 47, 4335–4338. 10.1021/jm030604o [DOI] [PubMed] [Google Scholar]
- Arnou B., Montigny C., Morth J. P., Nissen P., Jaxel C., Møller J. V., et al. (2011). The Plasmodium falciparum Ca2+-ATPase PfATP6: insensitive to artemisinin, but a potential drug target. Biochem. Soc. Trans. 39, 823–831. 10.1042/BST0390823 [DOI] [PubMed] [Google Scholar]
- Arora A., Deniskin R., Sosa Y., Nishtala S. N., Henrich P. P., Kumar T. R. S., et al. (2016). Substrate and inhibitor specificity of the Plasmodium berghei equilibrative nucleoside transporter type 1. Mol. Pharmacol. 89, 678–685. 10.1124/mol.115.101386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asawamahasakda W., Ittarat I., Pu Y. M., Ziffer H., Meshnick S. R. (1994). Reaction of antimalarial endoperoxides with specific parasite proteins. Antimicrob. Agents Chemother. 38, 1854–1858. 10.1128/AAC.38.8.1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig A. M., Iqbal J., Khan N. A. (2013). In vitro efficacies of clinically available drugs against growth and viability of an Acanthamoeba castellanii keratitis isolate belonging to the T4 genotype. Antimicrob. Agents Chemother. 57, 3561–3567. 10.1128/AAC.00299-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beitz E., Pavlovic-Djuranovic S., Yasui M., Agre P., Schultz J. E. (2004). Molecular dissection of water and glycerol permeability of the aquaglyceroporin from Plasmodium falciparum by mutational analysis. Proc Natl Acad Sci U.S.A. 101, 1153–1158. 10.1073/pnas.0307295101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berens R. L., Krug E. C., Nash P. B., Curiel T. J. (1998). Selection and characterization of Toxoplasma gondii mutants resistant to artemisinin. J. Infect. Dis. 177, 1128–1131. 10.1086/517411 [DOI] [PubMed] [Google Scholar]
- Biagini G. A., Pasini E. M., Hughes R., Koning H. P., de Vial H. J., O'Neill P. M., et al. (2004). Characterization of the choline carrier of Plasmodium falciparum: a route for the selective delivery of novel antimalarial drugs. Blood 104, 3372–3377. 10.1182/blood-2004-03-1084 [DOI] [PubMed] [Google Scholar]
- Biagini G. A., Fisher N., Berry N., Stocks P. A., Meunier B., Williams D. P., et al. (2008). Acridinediones: selective and potent inhibitors of the malaria parasite mitochondrial bc1 complex. Mol. Pharmacol. 73, 1347–1355. 10.1124/mol.108.045120 [DOI] [PubMed] [Google Scholar]
- Birkenheuer A. J., Levy M. G., Breitschwerdt E. B. (2004). Efficacy of combined atovaquone and azithromycin for therapy of chronic Babesia gibsoni (Asian genotype) infections in dogs. J. Vet. Intern. Med. 18, 494–498. 10.1111/j.1939-1676.2004.tb02573.x [DOI] [PubMed] [Google Scholar]
- Blume M., Rodriguez-Contreras D., Landfear S., Fleige T., Soldati-Favre D., Lucius R., et al. (2009). Host-derived glucose and its transporter in the obligate intracellular pathogen Toxoplasma gondii are dispensable by glutaminolysis. Proc. Natl. Acad. Sci. U.S.A. 106, 12998–13003. 10.1073/pnas.0903831106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume M., Hliscs M., Rodriguez-Contreras D., Sanchez M., Landfear S., Lucius R., et al. (2011). A constitutive pan-hexose permease for the Plasmodium life cycle and transgenic models for screening of antimalarial sugar analogs. FASEB J. 25, 1218–1229. 10.1096/fj.10-173278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun R., Blum J., Chappuis F., Burri C. (2010). Human African trypanosomiasis. Lancet 375, 148–159. 10.1016/S0140-6736(09)60829-1 [DOI] [PubMed] [Google Scholar]
- Brunner R., Aissaoui H., Boss C., Bozdech Z., Brun R., Corminboeuf O., et al. (2012). Identification of a new chemical class of antimalarials. J. Infect. Dis. 206, 735–743. 10.1093/infdis/jis418 [DOI] [PubMed] [Google Scholar]
- Brunner R., Ng C. L., Aissaoui H., Akabas M. H., Boss C., Brun R., et al. (2013). UV-triggered affinity capture identifies interactions between the Plasmodium falciparum multidrug resistance protein 1 (PfMDR1) and antimalarial agents in live parasitized cells. J. Biol. Chem. 288, 22576–22583. 10.1074/jbc.M113.453159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess S. J., Selzer A., Kelly J. X., Smilkstein M. J., Riscoe M. K., Peyton D. H. (2006). A chloroquine-like molecule designed to reverse resistance in Plasmodium falciparum. J. Med. Chem. 49, 5623–5625. 10.1021/jm060399n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardi D., Pozza A., Arnou B., Marchal E., Clausen J. D., Andersen J. P., et al. (2010). Purified E255L mutant SERCA1a and purified PfATP6 are sensitive to SERCA-type inhibitors but insensitive to artemisinins. J. Biol. Chem. 285, 26406–26416. 10.1074/jbc.M109.090340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro J. A., de Mecca M. M., Bartel L. C. (2006). Toxic side effects of drugs used to treat Chagas' disease (American trypanosomiasis). Hum. Exp. Toxicol. 25, 471–479. 10.1191/0960327106het653oa [DOI] [PubMed] [Google Scholar]
- Checa R., Montoya A., Ortega N., González-Fraga J. L., Bartolomé A., Gálvez R., et al. (2017). Efficacy, safety and tolerance of imidocarb dipropionate versus atovaquone or buparvaquone plus azithromycin used to treat sick dogs naturally infected with the Babesia microti-like piroplasm. Parasit. Vectors 10, 145. 10.1186/s13071-017-2049-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn J. V., Alkhalil A., Wagner M. A., Rajapandi T., Desai S. A. (2003). Extracellular lysines on the plasmodial surface anion channel involved in Na+ exclusion. Mol. Biochem. Parasitol. 132, 27–34. 10.1016/j.molbiopara.2003.08.001 [DOI] [PubMed] [Google Scholar]
- Comer E., Beaudoin J. A., Kato N., Fitzgerald M. E., Heidebrecht R. W., Lee M. D., et al. (2014). Diversity-oriented synthesis-facilitated medicinal chemistry: Toward the development of novel antimalarial agents. J. Med. Chem. 57, 8496–8502. 10.1021/jm500994n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke D. W., Lallinger G. J., Durack D. T. (1987). In vitro sensitivity of Naegleria fowleri to qinghaosu and dihydroqinghaosu. J. Parasitol. 73, 411. 10.2307/3282098 [DOI] [PubMed] [Google Scholar]
- Cowman A. F., Karcz S., Galatis D., Culvenor J. G. (1991). A P-glycoprotein homologue of Plasmodium falciparum is localized on the digestive vacuole. J. Cell Biol. 113, 1033–1042. 10.1083/jcb.113.5.1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowman A. F., Galatis D., Thompson J. K. (1994). Selection for mefloquine resistance in Plasmodium falciparum is linked to amplification of the pfmdr1 gene and cross-resistance to halofantrine and quinine. Proc. Natl. Acad. Sci. U.S.A. 91, 1143–1147. 10.1073/pnas.91.3.1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford E. D., Quan J., Horst J. A., Ebert D., Wu W., DeRisi J. L. (2017). Plasmid-free CRISPR/Cas9 genome editing in Plasmodium falciparum confirms mutations conferring resistance to the dihydroisoquinolone clinical candidate SJ733. PLoS ONE 12:e0178163. 10.1371/journal.pone.0178163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft S. L., Hogg J., Gutteridge W. E., Hudson A. T., Randall A. W. (1992). The activity of hydroxynaphthoquinones against Leishmania donovani. J. Antimicrob. Chemother. 30, 827–832. 10.1093/jac/30.6.827 [DOI] [PubMed] [Google Scholar]
- Crofts A. R., Barquera B., Gennis R. B., Kuras R., Guergova-Kuras M., Berry E. A. (1999a). Mechanism of ubiquinol oxidation by the bc1 complex: different domains of the quinol binding pocket and their role in the mechanism and binding of inhibitors. Biochemistry 38, 15807–15826. 10.1021/bi990962m [DOI] [PubMed] [Google Scholar]
- Crofts A. R., Guergova-Kuras M., Huang L., Kuras R., Zhang Z., Berry E. A. (1999b). Mechanism of ubiquinol oxidation by the bc1 complex: role of the iron sulfur protein and its mobility. Biochemistry 38, 15791–15806. 10.1021/bi990961u [DOI] [PubMed] [Google Scholar]
- Crofts A. R. (2004). The cytochrome bc1 complex: function in the context of structure. Annu. Rev. Physiol. 66, 689–733. 10.1146/annurev.physiol.66.032102.150251 [DOI] [PubMed] [Google Scholar]
- da Costa-Silva T. A., Galisteo A. J., Lindoso J. A. L., Barbosa L. R. S., Tempone A. G. (2017). Nanoliposomal buparvaquone immunomodulates Leishmania infantum-infected macrophages and is highly effective in a murine model. Antimicrob. Agents Chemother. 61:e02297–16. 10.1128/AAC.02297-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cruz F. P., Martin C., Buchholz K., Lafuente-Monasterio M. J., Rodrigues T., Sönnichsen B., et al. (2012). Drug screen targeted at Plasmodium liver stages identifies a potent multistage antimalarial drug. J. Infect. Dis. 205, 1278–1286. 10.1093/infdis/jis184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Macêdo J. P., Schumann Burkard G., Niemann M., Barrett M. P., Vial H., Mäser P., et al. (2015). An atypical mitochondrial carrier that mediates drug action in Trypanosoma brucei. PLoS Pathog. 11:e1004875. 10.1371/journal.ppat.1004875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Muylder G., Ang K. K. H., Chen S., Arkin M. R., Engel J. C., McKerrow J. H. (2011). A screen against Leishmania intracellular amastigotes: comparison to a promastigote screen and identification of a host cell-specific hit. PLoS Negl. Trop. Dis. 5:e1253. 10.1371/journal.pntd.0001253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Muylder G., Vanhollebeke B., Caljon G., Wolfe A. R., McKerrow J., Dujardin J.-C. (2016). Naloxonazine, an amastigote-specific compound, affects Leishmania parasites through modulation of host-encoded functions. PLoS Negl. Trop. Dis. 10:e0005234. 10.1371/journal.pntd.0005234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Pilar Crespo M., Avery T. D., Hanssen E., Fox E., Robinson T. V., Valente P., et al. (2008). Artemisinin and a series of novel endoperoxide antimalarials exert early effects on digestive vacuole morphology. Antimicrob. Agents Chemother. 52, 98–109. 10.1128/AAC.00609-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniskin R., Frame I. J., Sosa Y., Akabas M. H. (2016). Targeting the Plasmodium vivax equilibrative nucleoside transporter 1 (PvENT1) for antimalarial drug development. Int. J. Parasitol. Drugs Drug Resist. 6, 1–11. 10.1016/j.ijpddr.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbyshire E. T., Franssen F. J., de Vries E., Morin C., Woodrow C. J., Krishna S., et al. (2008). Identification, expression and characterisation of a Babesia bovis hexose transporter. Mol. Biochem. Parasitol. 161, 124–129. 10.1016/j.molbiopara.2008.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai S. A., Bezrukov S. M., Zimmerberg J. (2000). A voltage-dependent channel involved in nutrient uptake by red blood cells infected with the malaria parasite. Nature 406, 1001–1005. 10.1038/35023000 [DOI] [PubMed] [Google Scholar]
- Di Cicco M. F., Downey M. E., Beeler E., Marr H., Cyrog P., Kidd L., et al. (2012). Re-emergence of Babesia conradae and effective treatment of infected dogs with atovaquone and azithromycin. Vet. Parasitol. 187, 23–27. 10.1016/j.vetpar.2012.01.006 [DOI] [PubMed] [Google Scholar]
- Doggett J. S., Nilsen A., Forquer I., Wegmann K. W., Jones-Brando L., Yolken R. H., et al. (2012). Endochin-like quinolones are highly efficacious against acute and latent experimental toxoplasmosis. Proc. Natl. Acad. Sci. U.S.A. 109, 15936–15941. 10.1073/pnas.1208069109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downie M. J., Saliba K. J., Howitt S. M., Bröer S., Kirk K. (2006). Transport of nucleosides across the Plasmodium falciparum parasite plasma membrane has characteristics of PfENT1. Mol. Microbiol. 60, 738–748. 10.1111/j.1365-2958.2006.05125.x [DOI] [PubMed] [Google Scholar]
- Downie M. J., Saliba K. J., Bröer S., Howitt S. M., Kirk K. (2008). Purine nucleobase transport in the intraerythrocytic malaria parasite. Int. J. Parasitol. 38, 203–209. 10.1016/j.ijpara.2007.07.005 [DOI] [PubMed] [Google Scholar]
- Downie M. J., El Bissati K., Bobenchik A. M., Nic Lochlainn L., Amerik A., Zufferey R., et al. (2010). PfNT2, a permease of the equilibrative nucleoside transporter family in the endoplasmic reticulum of Plasmodium falciparum. J. Biol. Chem. 285, 20827–20833. 10.1074/jbc.M110.118489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozdowicz Y. M., Shaw M., Nishi M., Striepen B., Liwinski H. A., Roos D. S., et al. (2003). Isolation and characterization of TgVP1, a type I vacuolar H+-translocating pyrophosphatase from Toxoplasma gondii. the dynamics of its subcellular localization and the cellular effects of a diphosphonate inhibitor. J. Biol. Chem. 278, 1075–1085. 10.1074/jbc.M209436200 [DOI] [PubMed] [Google Scholar]
- Dyer M., Jackson M., McWhinney C., Zhao G., Mikkelsen R. (1996). Analysis of a cation-transporting ATPase of Plasmodium falciparum. Mol. Biochem. Parasitol. 78, 1–12. 10.1016/S0166-6851(96)02593-5 [DOI] [PubMed] [Google Scholar]
- Eckstein-Ludwig U., Webb R. J., van Goethem I. D. A., East J. M., Lee A. G., Kimura M., et al. (2003). Artemisinins target the SERCA of Plasmodium falciparum. Nature 424, 957–961. 10.1038/nature01813 [DOI] [PubMed] [Google Scholar]
- Fernando D., Rodrigo C., Rajapakse S. (2011). Primaquine in vivax malaria: an update and review on management issues. Malar. J. 10, 351. 10.1186/1475-2875-10-351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidock D. A., Nomura T., Talley A. K., Cooper R. A., Dzekunov S. M., Ferdig M. T., et al. (2000). Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell 6, 861–871. 10.1016/S1097-2765(05)00077-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flegr J., Prandota J., Sovičková M., Israili Z. H. (2014). Toxoplasmosis—a global threat. correlation of latent toxoplasmosis with specific disease burden in a set of 88 countries. PLoS ONE 9:e90203. 10.1371/journal.pone.0090203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley M., Tilley L. (1997). Quinoline antimalarials: mechanisms of action and resistance. Int. J. Parasitol. 27, 231–240. 10.1016/S0020-7519(96)00152-X [DOI] [PubMed] [Google Scholar]
- Foote S. J., Thompson J. K., Cowman A. F., Kemp D. J. (1989). Amplification of the multidrug resistance gene in some chloroquine-resistant isolates of P. falciparum. Cell 57, 921–930. 10.1016/0092-8674(89)90330-9 [DOI] [PubMed] [Google Scholar]
- Frame I. J., Deniskin R., Arora A., Akabas M. H. (2015a). Purine import into malaria parasites as a target for antimalarial drug development. Ann. N.Y. Acad. Sci. 1342, 19–28. 10.1111/nyas.12568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame I. J., Deniskin R., Rinderspacher A., Katz F., Deng S.-X., Moir R. D., et al. (2015b). Yeast-based high-throughput screen identifies Plasmodium falciparum equilibrative nucleoside transporter 1 inhibitors that kill malaria parasites. ACS Chem. Biol. 10, 775–783. 10.1021/cb500981y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame I. J., Merino E. F., Schramm V. L., Cassera M. B., Akabas M. H. (2012). Malaria parasite type 4 equilibrative nucleoside transporters (ENT4) are purine transporters with distinct substrate specificity. Biochem. J. 446, 179–190. 10.1042/BJ20112220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry M., Pudney M. (1992). Site of action of the antimalarial hydroxynaphthoquinone, 2-trans-4-(4′-chlorophenyl) cyclohexyl-3-hydroxy-1,4-naphthoquinone (566C80). Biochem. Pharmacol. 43, 1545–1553. [DOI] [PubMed] [Google Scholar]
- Garnier T., Mäntylä A., Järvinen T., Lawrence J., Brown M., Croft S. L. (2007). In vivo studies on the antileishmanial activity of buparvaquone and its prodrugs. J. Antimicrob. Chemother. 60, 802–810. 10.1093/jac/dkm303 [DOI] [PubMed] [Google Scholar]
- Ghaffarifar F., Esavand Heydari F., Dalimi A., Hassan Z. M., Delavari M., Mikaeiloo H. (2015). Evaluation of apoptotic and antileishmanial activities of artemisinin on promastigotes and BALB/C mice infected with Leishmania major. Iran. J. Parasitol. 10, 258–267. [PMC free article] [PubMed] [Google Scholar]
- Ginsburg H., Kutner S., Krugliak M., Ioav Cabantchik Z. (1985). Characterization of permeation pathways appearing in the host membrane of Plasmodium falciparum infected red blood cells. Mol. Biochem. Parasitol. 14, 313–322. 10.1016/0166-6851(85)90059-3 [DOI] [PubMed] [Google Scholar]
- Golldack A., Henke B., Bergmann B., Wiechert M. I., Erler H., Blancke Soares A., et al. (2017). Substrate-analogous inhibitors exert antimalarial action by targeting the Plasmodium lactate transporter PfFNT at nanomolar scale. PLoS Pathog. 13, e1006172. 10.1371/journal.ppat.1006172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Ghosh P. K., Dutta G. P., Vishwakarma R. A. (1995). In vivo study of artemisinin and its derivatives against primary amebic meningoencephalitis caused by Naegleria fowleri. J. Parasitol. 81, 1012–1013. 10.2307/3284060 [DOI] [PubMed] [Google Scholar]
- Hansen M., Kun J. F. J., Schultz J. E., Beitz E. (2002). A single, bi-functional aquaglyceroporin in blood-stage Plasmodium falciparum malaria parasites. J. Biol. Chem. 277, 4874–4882. 10.1074/jbc.M110683200 [DOI] [PubMed] [Google Scholar]
- Hapuarachchi S. V., Cobbold S. A., Shafik S. H., Dennis A. S. M., McConville M. J., Martin R. E., et al. (2017). The malaria parasite's lactate transporter PfFNT is the target of antiplasmodial compounds identified in whole cell phenotypic screens. PLoS Pathog. 13:e1006180. 10.1371/journal.ppat.1006180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrill A. H., Desmet K. D., Wolf K. K., Bridges A. S., Eaddy J. S., Kurtz C. L., et al. (2012). A mouse diversity panel approach reveals the potential for clinical kidney injury due to DB289 not predicted by classical rodent models. Toxicol. Sci. 130, 416–426. 10.1093/toxsci/kfs238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hencken C. P., Jones-Brando L., Bordón C., Stohler R., Mott B. T., Yolken R., et al. (2010). Thiazole, oxadiazole, and carboxamide derivatives of artemisinin are highly selective and potent inhibitors of Toxoplasma gondii. J. Med. Chem. 53, 3594–3601. 10.1021/jm901857d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill D. A., Pillai A. D., Nawaz F., Hayton K., Doan L., Lisk G., et al. (2007). A blasticidin S-resistant Plasmodium falciparum mutant with a defective plasmodial surface anion channel. Proc. Natl. Acad. Sci. U.S.A. 104, 1063–1068. 10.1073/pnas.0610353104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrycyna C. A., Summers R. L., Lehane A. M., Pires M. M., Namanja H., Bohn K., et al. (2014). Quinine dimers are potent inhibitors of the Plasmodium falciparum chloroquine resistance transporter and are active against quinoline-resistant P. falciparum. ACS Chem. Biol. 9, 722–730. 10.1021/cb4008953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber S. M., Uhlemann A.-C., Gamper N. L., Duranton C., Kremsner P. G., Lang F. (2002). Plasmodium falciparum activates endogenous Cl− channels of human erythrocytes by membrane oxidation. EMBO J. 21, 22–30. 10.1093/emboj/21.1.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber S. M., Duranton C., Henke G., van de Sand C., Heussler V., Shumilina E., et al. (2004). Plasmodium induces swelling-activated ClC-2 anion channels in the host erythrocyte. J. Biol. Chem. 279, 41444–41452. 10.1074/jbc.M407618200 [DOI] [PubMed] [Google Scholar]
- Hughes W. T., Oz H. S. (1995). Successful prevention and treatment of babesiosis with atovaquone. J. Infect. Dis. 172, 1042–1046. 10.1093/infdis/172.4.1042 [DOI] [PubMed] [Google Scholar]
- Hutchinson D. B., Viravan C., Kyle D. E., Looareesuwan S., Canfield C. J., Webster H. K. (1996). Clinical studies of atovaquone, alone or in combination with other antimalarial drugs, for treatment of acute uncomplicated malaria in Thailand. Am. J. Trop. Med. Hyg. 54, 62–66. 10.4269/ajtmh.1996.54.62 [DOI] [PubMed] [Google Scholar]
- Ibrahim H. M. S., Al-Salabi M. I., El Sabbagh N., Quashie N. B., Alkhaldi A. A. M., Escale R., et al. (2011). Symmetrical choline-derived dications display strong anti-kinetoplastid activity. J. Antimicrob. Chemother. 66, 111–125. 10.1093/jac/dkq401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi A., Matsuu A., Matsuyama K., Hikasa Y. (2015). The efficacy of artemisinin, artemether, and lumefantrine against Babesia gibsoni in vitro. Parasitol. Int. 64, 190–193. 10.1016/j.parint.2014.12.006 [DOI] [PubMed] [Google Scholar]
- Ismail M. A., Brun R., Easterbrook J. D., Tanious F. A., Wilson W. D., Boykin D. W. (2003). Synthesis and antiprotozoal activity of aza-analogues of furamidine. J. Med. Chem. 46, 4761–4769. 10.1021/jm0302602 [DOI] [PubMed] [Google Scholar]
- Jamal Q., Khan N. H., Wahid S., Awan M. M., Sutherland C., Shah A. (2015). In-vitro sensitivity of Pakistani Leishmania tropica field isolate against buparvaquone in comparison to standard anti-leishmanial drugs. Exp. Parasitol. 154, 93–97. 10.1016/j.exppara.2015.04.017 [DOI] [PubMed] [Google Scholar]
- Jambou R., Legrand E., Niang M., Khim N., Lim P., Volney B., et al. (2005). Resistance of Plasmodium falciparum field isolates to in-vitro artemether and point mutations of the SERCA-type PfATPase6. Lancet 366, 1960–1963. 10.1016/S0140-6736(05)67787-2 [DOI] [PubMed] [Google Scholar]
- Jiménez-Díaz M. B., Ebert D., Salinas Y., Pradhan A., Lehane A. M., Myrand-Lapierre M.-E., et al. (2014). (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proc. Natl. Acad. Sci. U.S.A. 111, E5455–E5462. 10.1073/pnas.1414221111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joet T., Eckstein-Ludwig U., Morin C., Krishna S. (2003). Validation of the hexose transporter of Plasmodium falciparum as a novel drug target. Proc. Natl. Acad. Sci. U.S.A. 100, 7476–7479. 10.1073/pnas.1330865100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones-Brando L., D'Angelo J., Posner G. H., Yolken R. (2006). In vitro inhibition of Toxoplasma gondii by four new derivatives of artemisinin. Antimicrob. Agents Chemother. 50, 4206–4208. 10.1128/AAC.00793-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juge N., Moriyama S., Miyaji T., Kawakami M., Iwai H., Fukui T., et al. (2015). Plasmodium falciparum chloroquine resistance transporter is a H+-coupled polyspecific nutrient and drug exporter. Proc. Natl. Acad. Sci. U.S.A. 112, 3356–3361. 10.1073/pnas.1417102112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaani J., Ginsburg H. (1991). Transport of lactate in Plasmodium falciparum-infected human erythrocytes. J. Cell. Physiol. 149, 469–476. 10.1002/jcp.1041490316 [DOI] [PubMed] [Google Scholar]
- Kavishe R. A., Koenderink J. B., Alifrangis M. (2017). Oxidative stress in malaria and artemisinin combination therapy: pros and cons. FEBS J. 284, 2579–2591. 10.1111/febs.14097 [DOI] [PubMed] [Google Scholar]
- Kelly J. X., Smilkstein M. J., Brun R., Wittlin S., Cooper R. A., Lane K. D., et al. (2009). Discovery of dual function acridones as a new antimalarial chemotype. Nature 459, 270–273. 10.1038/nature07937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessl J. J., Meshnick S. R., Trumpower B. L. (2007). Modeling the molecular basis of atovaquone resistance in parasites and pathogenic fungi. Trends Parasitol. 23, 494–501. 10.1016/j.pt.2007.08.004 [DOI] [PubMed] [Google Scholar]
- Kim J.-T., Park J.-Y., Seo H.-S., Oh H.-G., Noh J.-W., Kim J.-H., et al. (2002). In vitro antiprotozoal effects of artemisinin on Neospora caninum. Vet. Parasitol. 103, 53–63. 10.1016/S0304-4017(01)00580-5 [DOI] [PubMed] [Google Scholar]
- Kirk K., Horner H. A. (1995). In search of a selective inhibitor of the induced transport of small solutes in Plasmodium falciparum-infected erythrocytes: effects of arylaminobenzoates. Biochem. J. 311, 761–768. 10.1042/bj3110761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kongkathip N., Pradidphol N., Hasitapan K., Grigg R., Kao W.-C., Hunte C., et al. (2010). Transforming rhinacanthin analogues from potent anticancer agents into potent antimalarial agents. J. Med. Chem. 53, 1211–1221. 10.1021/jm901545z [DOI] [PubMed] [Google Scholar]
- Krause P. J., Lepore T., Sikand V. K., Gadbaw J., Burke G., Telford S. R., et al. (2000). Atovaquone and azithromycin for the treatment of babesiosis. New Engl. J. Med. 343, 1454–1458. 10.1056/NEJM200011163432004 [DOI] [PubMed] [Google Scholar]
- Krause A., Dingemanse J., Mathis A., Marquart L., Möhrle J. J., McCarthy J. S. (2016). Pharmacokinetic/pharmacodynamic modelling of the antimalarial effect of Actelion-451840 in an induced blood stage malaria study in healthy subjects. Br. J. Clin. Pharmacol. 82, 412–421. 10.1111/bcp.12962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna S., Woodrow C. J. (1999). Expression of parasite transporters in Xenopus oocytes. Novartis Found. Symp. 226, 126–139. [DOI] [PubMed] [Google Scholar]
- Krishna S., Pulcini S., Moore C. M., Teo B. H.-Y., Staines H. M. (2014). Pumped up: reflections on PfATP6 as the target for artemisinins. Trends Pharmacol. Sci. 35, 4–11. 10.1016/j.tips.2013.10.007 [DOI] [PubMed] [Google Scholar]
- Lakshmanan V., Bray P. G., Verdier-Pinard D., Johnson D. J., Horrocks P., Muhle R. A., et al. (2005). A critical role for PfCRT K76T in Plasmodium falciparum verapamil-reversible chloroquine resistance. EMBO J. 24, 2294–2305. 10.1038/sj.emboj.7600681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalève A., Vallières C., Golinelli-Cohen M.-P., Bouton C., Song Z., Pawlik G., et al. (2016). The antimalarial drug primaquine targets Fe-S cluster proteins and yeast respiratory growth. Redox Biol. 7, 21–29. 10.1016/j.redox.2015.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanteri C. A., Tidwell R. R., Meshnick S. R. (2008). The mitochondrion is a site of trypanocidal action of the aromatic diamidine DB75 in bloodstream forms of Trypanosoma brucei. Antimicrob. Agents Chemother. 52, 875–882. 10.1128/AAC.00642-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawres L. A., Garg A., Kumar V., Bruzual I., Forquer I. P., Renard I., et al. (2016). Radical cure of experimental babesiosis in immunodeficient mice using a combination of an endochin-like quinolone and atovaquone. J. Exp. Med. 213, 1307–1318. 10.1084/jem.20151519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bihan A., de Kanter R., Angulo-Barturen I., Binkert C., Boss C., Brun R., et al. (2016). Characterization of novel antimalarial compound ACT-451840: preclinical assessment of activity and dose-efficacy modeling. PLoS Med. 13, e1002138. 10.1371/journal.pmed.1002138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Promeneur D., Rojek A., Kumar N., Frøkiaer J., Nielsen S., et al. (2007). Aquaporin 9 is the major pathway for glycerol uptake by mouse erythrocytes, with implications for malarial virulence. Proc. Natl. Acad. Sci. U.S.A. 104, 12560–12564. 10.1073/pnas.0705313104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loiseau P. M., Bories C. (2006). Mechanisms of drug action and drug resistance in Leishmania as basis for therapeutic target identification and design of antileishmanial modulators. Curr. Top. Med. Chem. 6, 539–550. 10.2174/156802606776743165 [DOI] [PubMed] [Google Scholar]
- Lukens A. K., Heidebrecht R. W., Mulrooney C., Beaudoin J. A., Comer E., Duvall J. R., et al. (2015). Diversity-oriented synthesis probe targets Plasmodium falciparum cytochrome b ubiquinone reduction site and synergizes with oxidation site inhibitors. J. Infect. Dis. 211, 1097–1103. 10.1093/infdis/jiu565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y., Lu D.-M., Lu X.-J., Liao L., Hu X.-S. (2004). Activity of dihydroartemisinin against Leishmania donovani both in vitro and vivo. Chin. Med. J. 117, 1271–1273. [PubMed] [Google Scholar]
- Macêdo J. P., Schmidt R. S., Mäser P., Rentsch D., Vial H. J., Sigel E., et al. (2013). Characterization of choline uptake in Trypanosoma brucei procyclic and bloodstream forms. Mol. Biochem. Parasitol. 190, 16–22. 10.1016/j.molbiopara.2013.05.007 [DOI] [PubMed] [Google Scholar]
- Mäntylä A., Garnier T., Rautio J., Nevalainen T., Vepsälainen J., Koskinen A., et al. (2004a). Synthesis, in vitro evaluation, and antileishmanial activity of water-soluble prodrugs of buparvaquone. J. Med. Chem. 47, 188–195. 10.1021/jm030868a [DOI] [PubMed] [Google Scholar]
- Mäntylä A., Rautio J., Nevalainen T., Vepsälainen J., Juvonen R., Kendrick H., et al. (2004b). Synthesis and antileishmanial activity of novel buparvaquone oxime derivatives. Bioorg. Med. Chem. 12, 3497–3502. 10.1016/j.bmc.2004.04.032 [DOI] [PubMed] [Google Scholar]
- Marchetti R. V., Lehane A. M., Shafik S. H., Winterberg M., Martin R. E., Kirk K. (2015). A lactate and formate transporter in the intraerythrocytic malaria parasite, Plasmodium falciparum. Nat. Commun. 6:6721. 10.1038/ncomms7721 [DOI] [PubMed] [Google Scholar]
- Martin R. E., Marchetti R. V., Cowan A. I., Howitt S. M., Bröer S., Kirk K. (2009). Chloroquine transport via the malaria parasite's chloroquine resistance transporter. Science 325, 1680–1682. 10.1126/science.1175667 [DOI] [PubMed] [Google Scholar]
- Martin R. E., Butterworth A. S., Gardiner D. L., Kirk K., McCarthy J. S., Skinner-Adams T. S. (2012). Saquinavir inhibits the malaria parasite's chloroquine resistance transporter. Antimicrob. Agents Chemother. 56, 2283–2289. 10.1128/AAC.00166-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Higuera A., Herrera-Martínez M., Chávez-Munguía B., Valle-Solís M., Muñiz-Lino M. A., Cázares-Apátiga J., et al. (2015). Entamoeba invadens: Identification of a SERCA protein and effect of SERCA inhibitors on encystation. Microb. Pathog. 89, 18–26. 10.1016/j.micpath.2015.08.016 [DOI] [PubMed] [Google Scholar]
- Matsuu A., Yamasaki M., Xuan X., Ikadai H., Hikasa Y. (2008). In vitro evaluation of the growth inhibitory activities of 15 drugs against Babesia gibsoni (Aomori strain). Vet. Parasitol. 157, 1–8. 10.1016/j.vetpar.2008.07.023 [DOI] [PubMed] [Google Scholar]
- Maya J. D., Morello A., Repetto Y., Tellez R., Rodriguez A., Zelada U., et al. (2000). Effects of 3-chloro-phenyl-1,4-dihydropyridine derivatives on Trypanosome cruzi epimastigotes. Comp. Biochem. Physiol. C 125, 103–109. 10.1016/S0742-8413(99)00096-1 [DOI] [PubMed] [Google Scholar]
- McAuley J. B., Juranek D. D. (1992). Luminal agents in the treatment of amebiasis. Clin. Infect. Dis. 14, 1161–1162. 10.1093/clinids/14.5.1161 [DOI] [PubMed] [Google Scholar]
- McFadden D. C., Tomavo S., Berry E. A., Boothroyd J. C. (2000). Characterization of cytochrome b from Toxoplasma gondii and Q(o) domain mutations as a mechanism of atovaquone-resistance. Mol. Biochem. Parasitol. 108, 1–12. 10.1016/S0166-6851(00)00184-5 [DOI] [PubMed] [Google Scholar]
- McHardy N., Wekesa L. S., Hudson A. T., Randall A. W. (1985). Antitheilerial activity of BW720C (buparvaquone): a comparison with parvaquone. Res. Vet. Sci. 39, 29–33. [PubMed] [Google Scholar]
- McIntosh M. T., Drozdowicz Y. M., Laroiya K., Rea P. A., Vaidya A. B. (2001). Two classes of plant-like vacuolar-type H+-pyrophosphatases in malaria parasites. Mol. Biochem. Parasitol. 114, 183–195. 10.1016/S0166-6851(01)00251-1 [DOI] [PubMed] [Google Scholar]
- Meireles P., Mendes A. M., Aroeira R. I., Mounce B. C., Vignuzzi M., Staines H. M., et al. (2017). Uptake and metabolism of arginine impact Plasmodium development in the liver. Sci. Rep. 7:4072. 10.1038/s41598-017-04424-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhadhbi M., Naouach A., Boumiza A., Chaabani M. F., BenAbderazzak S., Darghouth M. A. (2010). In vivo evidence for the resistance of Theileria annulata to buparvaquone. Vet. Parasitol. 169, 241–247. 10.1016/j.vetpar.2010.01.013 [DOI] [PubMed] [Google Scholar]
- Mikolajczak S. A., Vaughan A. M., Kangwanrangsan N., Roobsoong W., Fishbaugher M., Yimamnuaychok N., et al. (2015). Plasmodium vivax liver stage development and hypnozoite persistence in human liver-chimeric mice. Cell Host Microbe 17, 526–535. 10.1016/j.chom.2015.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami T., Nakano T., Shimizu S., Shimura K., Fujinaga T., Ito S. (1985). Efficacy of naphthoquinones and imidocarb dipropionate on Theileria sergenti infections in splenectomized calves. Jpn. J. Vet. Sci. 47, 297–300. 10.1292/jvms1939.47.297 [DOI] [PubMed] [Google Scholar]
- Miner M. L., Jensen J. B. (1976). Decoquinate in the control of experimentally induced coccidiosis of calves. Am. J. Vet. Res. 37, 1043–1045. [PubMed] [Google Scholar]
- Mishina Y. V., Krishna S., Haynes R. K., Meade J. C. (2007). Artemisinins inhibit Trypanosoma cruzi and Trypanosoma brucei rhodesiense in vitro growth. Antimicrob. Agents Chemother. 51, 1852–1854. 10.1128/AAC.01544-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore C. M., Hoey E. M., Trudgett A., Timson D. J. (2011). Artemisinins act through at least two targets in a yeast model. FEMS Yeast Res. 11, 233–237. 10.1111/j.1567-1364.2010.00706.x [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay R., Beitz E. (2010). Metalloid transport by aquaglyceroporins: consequences in the treatment of human diseases. Adv. Exp. Med. Biol. 679, 57–69. 10.1007/978-1-4419-6315-4_5 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay R., Madhubala R. (1994). Effect of antioxidants on the growth and polyamine levels of Leishmania donovani. Biochem. Pharmacol. 47, 611–615. 10.1016/0006-2952(94)90122-8 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay R., Mandal G., Atluri V. S. R., Figarella K., Uzcategui N. L., Zhou Y., et al. (2011). The role of alanine 163 in solute permeability of Leishmania major aquaglyceroporin LmAQP1. Mol. Biochem. Parasitol. 175, 83–90. 10.1016/j.molbiopara.2010.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraguri G. R., Kiara H. K., McHardy N. (1999). Treatment of East Coast fever: a comparison of parvaquone and buparvaquone. Vet. Parasitol. 87, 25–37. 10.1016/S0304-4017(99)00154-5 [DOI] [PubMed] [Google Scholar]
- Nagamune K., Beatty W. L., Sibley L. D. (2007). Artemisinin induces calcium-dependent protein secretion in the protozoan parasite Toxoplasma gondii. Eukaryotic Cell 6, 2147–2156. 10.1128/EC.00262-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik P. K., Srivastava M., Bajaj P., Jain S., Dubey A., Ranjan P., et al. (2011). The binding modes and binding affinities of artemisinin derivatives with Plasmodium falciparum Ca2+-ATPase (PfATP6). J. Mol. Model. 17, 333–357. 10.1007/s00894-010-0726-4 [DOI] [PubMed] [Google Scholar]
- Neal R. A., van Bueren J., McCoy N. G., Iwobi M. (1989). Reversal of drug resistance in Trypanosoma cruzi and Leishmania donovani by verapamil. Trans. R. Soc. Trop. Med. Hyg. 83, 197–198. 10.1016/0035-9203(89)90642-1 [DOI] [PubMed] [Google Scholar]
- Ng C. L., Siciliano G., Lee M. C. S., Almeida M. J., de Corey V. C., Bopp S. E., et al. (2016). CRISPR-Cas9-modified pfmdr1 protects Plasmodium falciparum asexual blood stages and gametocytes against a class of piperazine-containing compounds but potentiates artemisinin-based combination therapy partner drugs. Mol. Microbiol. 101, 381–393. 10.1111/mmi.13397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguitragool W., Bokhari A. A. B., Pillai A. D., Rayavara K., Sharma P., Turpin B., et al. (2011). Malaria parasite clag3 genes determine channel-mediated nutrient uptake by infected red blood cells. Cell 145, 665–677. 10.1016/j.cell.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguitragool W., Rayavara K., Desai S. A. (2014). Proteolysis at a specific extracellular residue implicates integral membrane CLAG3 in malaria parasite nutrient channels. PLoS ONE 9:e93759. 10.1371/journal.pone.0093759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen A., LaCrue A. N., White K. L., Forquer I. P., Cross R. M., Marfurt J., et al. (2013). Quinolone-3-diarylethers: a new class of antimalarial drug. Sci. Transl. Med. 5, 177ra37. 10.1126/scitranslmed.3005029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes R. R., Costa M. D. S., Santos B. D. R., Fonseca A. L. D., Ferreira L. S., Chagas R. C. R., et al. (2016). Successful application of virtual screening and molecular dynamics simulations against antimalarial molecular targets. Mem. Inst. Oswaldo Cruz 111, 721–730. 10.1590/0074-02760160207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz D., Guiguemde W. A., Johnson A., Elya C., Anderson J., Clark J., et al. (2015). Identification of selective inhibitors of the Plasmodium falciparum hexose transporter PfHT by screening focused libraries of anti-malarial compounds. PLoS ONE 10:e0123598. 10.1371/journal.pone.0123598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz D., Forquer I., Boitz J., Soysa R., Elya C., Fulwiler A., et al. (2016). Targeting the cytochrome bc1 complex of Leishmania parasites for discovery of novel drugs. Antimicrob. Agents Chemother. 60, 4972–4982. 10.1128/AAC.00850-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overman R. R. (1947). Reversible cellular permeability alterations in disease. in vivo studies on sodium, potassium and chloride concentrations in erythrocytes of the malarious monkey. Am. J. Physiol. 152, 113–121. 10.1152/ajplegacy.1947.152.1.113 [DOI] [PubMed] [Google Scholar]
- Pain M., Fuller A. W., Basore K., Pillai A. D., Solomon T., Bokhari A. A. B., et al. (2016). Synergistic malaria parasite killing by two types of plasmodial surface anion channel Inhibitors. PLoS ONE 11:e0149214. 10.1371/journal.pone.0149214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter H. J., Morrisey J. M., Mather M. W., Vaidya A. B. (2007). Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 446, 88–91. 10.1038/nature05572 [DOI] [PubMed] [Google Scholar]
- Palit P., Ali N. (2008). Oral therapy with amlodipine and lacidipine, 1,4-dihydropyridine derivatives showing activity against experimental visceral leishmaniasis. Antimicrob. Agents Chemother. 52, 374–377. 10.1128/AAC.00522-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovic-Djuranovic S., Kun J. F. J., Schultz J. E., Beitz E. (2006). Dihydroxyacetone and methylglyoxal as permeants of the Plasmodium aquaglyceroporin inhibit parasite proliferation. Biochim. Biophys. Acta 1758, 1012–1017. 10.1016/j.bbamem.2005.12.002 [DOI] [PubMed] [Google Scholar]
- Petersen E., Schmidt D. R. (2003). Sulfadiazine and pyrimethamine in the postnatal treatment of congenital toxoplasmosis: what are the options? Expert Rev. Anti Infect. Ther. 1, 175–182. 10.1586/14787210.1.1.175 [DOI] [PubMed] [Google Scholar]
- Pickard A. L., Wongsrichanalai C., Purfield A., Kamwendo D., Emery K., Zalewski C., et al. (2003). Resistance to antimalarials in Southeast Asia and genetic polymorphisms in pfmdr1. Antimicrob. Agents Chemother. 47, 2418–2423. 10.1128/AAC.47.8.2418-2423.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponte-Sucre A., Gamarro F., Dujardin J.-C., Barrett M. P., López-Vélez R., García-Hernández R., et al. (2017). Drug resistance and treatment failure in leishmaniasis: a 21st century challenge. PLoS Negl. Trop. Dis. 11:e0006052. 10.1371/journal.pntd.0006052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price R. N., Uhlemann A.-C., Brockman A., McGready R., Ashley E., Phaipun L., et al. (2004). Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet 364, 438–447. 10.1016/S0140-6736(04)16767-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Promeneur D., Liu Y., Maciel J., Agre P., King L. S., Kumar N. (2007). Aquaglyceroporin PbAQP during intraerythrocytic development of the malaria parasite Plasmodium berghei. Proc. Natl. Acad. Sci. U.S.A. 104, 2211–2216. 10.1073/pnas.0610843104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Promeneur D., Mlambo G., Agre P., Coppens I. (2018). Aquaglyceroporin PbAQP is required for efficient progression through the liver stage of Plasmodium infection. Sci. Rep. 8:655. 10.1038/s41598-017-18987-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudêncio M., Derbyshire E. T., Marques C. A., Krishna S., Mota M. M., Staines H. M. (2009). Plasmodium berghei-infection induces volume-regulated anion channel-like activity in human hepatoma cells. Cell. Microbiol. 11, 1492–1501. 10.1111/j.1462-5822.2009.01342.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulcini S., Staines H. M., Pittman J. K., Slavic K., Doerig C., Halbert J., et al. (2013). Expression in yeast links field polymorphisms in PfATP6 to in vitro artemisinin resistance and identifies new inhibitor classes. J. Infect. Dis. 208, 468–478. 10.1093/infdis/jit171 [DOI] [PubMed] [Google Scholar]
- Rane L., Rane D. S., Kinnamon K. E. (1976). Screening large numbers of compounds in a model based on mortality of Trypanosoma rhodesiense infected mice. Am. J. Trop. Med. Hyg. 25, 395–400. 10.4269/ajtmh.1976.25.395 [DOI] [PubMed] [Google Scholar]
- Raphemot R., Lafuente-Monasterio M. J., Gamo-Benito F. J., Clardy J., Derbyshire E. R. (2015). Discovery of dual-stage malaria inhibitors with new targets. Antimicrob. Agents Chemother. 60, 1430–1437. 10.1128/AAC.02110-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed M. B., Saliba K. J., Caruana S. R., Kirk K., Cowman A. F. (2000). Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature 403, 906–909. 10.1038/35002615 [DOI] [PubMed] [Google Scholar]
- Reigada C., Valera-Vera E. A., Sayé M., Errasti A. E., Avila C. C., Miranda M. R., et al. (2017). Trypanocidal effect of isotretinoin through the inhibition of polyamine and amino acid transporters in Trypanosoma cruzi. PLoS Negl. Trop. Dis. 11:e0005472. 10.1371/journal.pntd.0005472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimão J. Q., Scotti M. T., Tempone A. G. (2010). Anti-leishmanial and anti-trypanosomal activities of 1,4-dihydropyridines: In vitro evaluation and structure-activity relationship study. Bioorg. Med. Chem. 18, 8044–8053. 10.1016/j.bmc.2010.09.015 [DOI] [PubMed] [Google Scholar]
- Reimão J. Q., Colombo F. A., Pereira-Chioccola V. L., Tempone A. G. (2011). In vitro and experimental therapeutic studies of the calcium channel blocker bepridil: detection of viable Leishmania (L.) chagasi by real-time PCR. Exp. Parasitol. 128, 111–115. 10.1016/j.exppara.2011.02.021 [DOI] [PubMed] [Google Scholar]
- Reimão J. Q., Colombo F. A., Pereira-Chioccola V. L., Tempone A. G. (2012). Effectiveness of liposomal buparvaquone in an experimental hamster model of Leishmania (L.) infantum chagasi. Exp. Parasitol. 130, 195–199. 10.1016/j.exppara.2012.01.010 [DOI] [PubMed] [Google Scholar]
- Reimão J. Q., Mesquita J. T., Ferreira D. D., Tempone A. G. (2016). Investigation of calcium channel blockers as antiprotozoal agents and their interference in the metabolism of Leishmania (L.) infantum. Evid. Based Complement. Alternat. Med. 2016:1523691. 10.1155/2016/1523691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricketts A. P., Pfefferkorn E. R. (1993). Toxoplasma gondii: Susceptibility and development of resistance to anticoccidial drugs in vitro. Antimicrob. Agents Chemother. 37, 2358–2363. 10.1128/AAC.37.11.2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues C. O., Scott D. A., Bailey B. N., Souza W., de Benchimol M., Moreno B., et al. (2000). Vacuolar proton pyrophosphatase activity and pyrophosphate (PPi) in Toxoplasma gondii as possible chemotherapeutic targets. Biochem. J. 349, 737–745. 10.1042/bj3490737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbach P., Sanchez C. P., Hayton K., Friedrich O., Patel J., Sidhu A. B. S., et al. (2006). Genetic linkage of pfmdr1 with food vacuolar solute import in Plasmodium falciparum. EMBO J. 25, 3000–3011. 10.1038/sj.emboj.7601203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottmann M., McNamara C., Yeung B. K. S., Lee M. C. S., Zou B., Russell B., et al. (2010). Spiroindolones, a potent compound class for the treatment of malaria. Science 329, 1175–1180. 10.1126/science.1193225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba K. J., Horner H. A., Kirk K. (1998). Transport and metabolism of the essential vitamin pantothenic acid in human erythrocytes infected with the malaria parasite Plasmodium falciparum. J. Biol. Chem. 273, 10190–10195. 10.1074/jbc.273.17.10190 [DOI] [PubMed] [Google Scholar]
- Sanchez M. A. (2013). Molecular identification and characterization of an essential pyruvate transporter from Trypanosoma brucei. J. Biol. Chem. 288, 14428–14437. 10.1074/jbc.M113.473157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt R. S., Macêdo J. P., Steinmann M. E., Salgado A. G., Bütikofer P., Sigel E., et al. (2017). Transporters of Trypanosoma brucei: phylogeny, physiology, pharmacology. FEBS J. [Epub ahead of print]. 10.1111/febs.14302 [DOI] [PubMed] [Google Scholar]
- Schumann Burkard G., Jutzi P., Roditi I. (2011). Genome-wide RNAi screens in bloodstream form trypanosomes identify drug transporters. Mol. Biochem. Parasitol. 175, 91–94. 10.1016/j.molbiopara.2010.09.002 [DOI] [PubMed] [Google Scholar]
- Sen R., Ganguly S., Saha P., Chatterjee M. (2010). Efficacy of artemisinin in experimental visceral leishmaniasis. Int. J. Antimicrob. Agents 36, 43–49. 10.1016/j.ijantimicag.2010.03.008 [DOI] [PubMed] [Google Scholar]
- Sharma P., Wollenberg K., Sellers M., Zainabadi K., Galinsky K., Moss E., et al. (2013). An epigenetic antimalarial resistance mechanism involving parasite genes linked to nutrient uptake. J. Biol. Chem. 288, 19429–19440. 10.1074/jbc.M113.468371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirley D.-A., Moonah S. (2016). Fulminant amebic colitis after corticosteroid therapy: a systematic review. PLoS Negl. Trop. Dis. 10:e0004879. 10.1371/journal.pntd.0004879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siregar J. E., Kurisu G., Kobayashi T., Matsuzaki M., Sakamoto K., Mi-Ichi F., et al. (2015). Direct evidence for the atovaquone action on the Plasmodium cytochrome bc1 complex. Parasitol. Int. 64, 295–300. 10.1016/j.parint.2014.09.011 [DOI] [PubMed] [Google Scholar]
- Slater A. F.G. (1993). Chloroquine: mechanism of drug action and resistance in Plasmodium falciparum. Pharmacol. Ther. 57, 203–235. 10.1016/0163-7258(93)90056-J [DOI] [PubMed] [Google Scholar]
- Slavic K., Delves M. J., Prudêncio M., Talman A. M., Straschil U., Derbyshire E. T., et al. (2011). Use of a selective inhibitor to define the chemotherapeutic potential of the plasmodial hexose transporter in different stages of the parasite's life cycle. Antimicrob. Agents Chemother. 55, 2824–2830. 10.1128/AAC.01739-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J., Almasalmeh A., Krenc D., Beitz E. (2012). Molar concentrations of sorbitol and polyethylene glycol inhibit the Plasmodium aquaglyceroporin but not that of E. coli: involvement of the channel vestibules. Biochim. Biophys. Acta 1818, 1218–1224. 10.1016/j.bbamem.2012.01.025 [DOI] [PubMed] [Google Scholar]
- Song J., Baker N., Rothert M., Henke B., Jeacock L., Horn D., et al. (2016). Pentamidine is not a permeant but a nanomolar inhibitor of the Trypanosoma brucei aquaglyceroporin-2. PLoS Pathog. 12:e1005436 10.1371/journal.ppat.1005436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg T., Burrows J. N., Kowalczyk P., McDonald S., Wells T. N. C., Willis P. (2013). The open access malaria box: a drug discovery catalyst for neglected diseases. PLoS ONE 8:e62906. 10.1371/journal.pone.0062906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillman N. J., Allen R. J. W., McNamara C. W., Yeung B. K. S., Winzeler E. A., Diagana T. T., et al. (2013). Na+ regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe 13, 227–237. 10.1016/j.chom.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillman N. J., Kirk K. (2015). The malaria parasite cation ATPase PfATP4 and its role in the mechanism of action of a new arsenal of antimalarial drugs. Int. J. Parasitol. Drugs Drug Resist. 5, 149–162. 10.1016/j.ijpddr.2015.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava I. K., Vaidya A. B. (1999). A mechanism for the synergistic antimalarial action of atovaquone and proguanil. Antimicrob. Agents Chemother. 43, 1334–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava I. K., Morrisey J. M., Darrouzet E., Daldal F., Vaidya A. B. (1999). Resistance mutations reveal the atovaquone-binding domain of cytochrome b in malaria parasites. Mol. Microbiol. 33, 704–711. 10.1046/j.1365-2958.1999.01515.x [DOI] [PubMed] [Google Scholar]
- Štáfková J., Mach J., Biran M., Verner Z., Bringaud F., Tachezy J. (2016). Mitochondrial pyruvate carrier in Trypanosoma brucei. Mol. Microbiol. 100, 442–456. 10.1111/mmi.13325 [DOI] [PubMed] [Google Scholar]
- Staines H. M., Ellory J. C., Kirk K. (2001). Perturbation of the pump-leak balance for Na+ and K+ in malaria-infected erythrocytes. Am. J. Physiol. 280, C1576–C1587. 10.1152/ajpcell.2001.280.6.C1576 [DOI] [PubMed] [Google Scholar]
- Stanley S. L. (2003). Amoebiasis. Lancet 361, 1025–1034. 10.1016/S0140-6736(03)12830-9 [DOI] [PubMed] [Google Scholar]
- Steck E. A., Kinnamon K. E., Davidson D. E., Duxbury R. E., Johnson A. J., Masters R. E. (1982). Trypanosoma rhodesiense: Evaluation of the antitrypanosomal action of 2,5-bis(4-guanylphenyl)furan dihydrochloride. Exp. Parasitol. 53, 133–144. 10.1016/0014-4894(82)90099-6 [DOI] [PubMed] [Google Scholar]
- Steinmann M. E., González-Salgado A., Bütikofer P., Mäser P., Sigel E. (2015). A heteromeric potassium channel involved in the modulation of the plasma membrane potential is essential for the survival of African trypanosomes. FASEB J. 29, 3228–3237. 10.1096/fj.15-271353 [DOI] [PubMed] [Google Scholar]
- Tanabe K., Izumo A., Kato M., Miki A., Doi S. (1989). Stage dependent inhibition of Plasmodiun falciparum falciparum by potent Ca2+ and calmodulin modulators. J. Protozool. 36, 139–143. 10.1111/j.1550-7408.1989.tb01060.x [DOI] [PubMed] [Google Scholar]
- Tempone A. G., Taniwaki N. N., Reimão J. Q. (2009). Antileishmanial activity and ultrastructural alterations of Leishmania (L.) chagasi treated with the calcium channel blocker nimodipine. Parasitol. Res. 105, 499–505. 10.1007/s00436-009-1427-8 [DOI] [PubMed] [Google Scholar]
- Thomé R., Lopes S. C. P., Costa F. T. M., Verinaud L. (2013). Chloroquine: modes of action of an undervalued drug. Immunol. Lett. 153, 50–57. 10.1016/j.imlet.2013.07.004 [DOI] [PubMed] [Google Scholar]
- Upston J. M., Gero A. M. (1995). Parasite-induced permeation of nucleosides in Plasmodium falciparum malaria. Biochim. Biophys. Acta 1236, 249–258. 10.1016/0005-2736(95)00055-8 [DOI] [PubMed] [Google Scholar]
- Urbina J. A., Moreno B., Vierkotter S., Oldfield E., Payares G., Sanoja C., et al. (1999). Trypanosoma cruzi contains major pyrophosphate stores, and its growth in vitro and in vivo is blocked by pyrophosphate analogs. J. Biol. Chem. 274, 33609–33615. 10.1074/jbc.274.47.33609 [DOI] [PubMed] [Google Scholar]
- Uzcategui N. L., Szallies A., Pavlovic-Djuranovic S., Palmada M., Figarella K., Boehmer C., et al. (2004). Cloning, heterologous expression and characterization of three aquaglyceroporins from Trypanosoma brucei. J. Biol. Chem. 279, 42669–42676. 10.1074/jbc.M404518200 [DOI] [PubMed] [Google Scholar]
- Valiathan R., Dubey M. L., Mahajan R. C., Malla N. (2006). Leishmania donovani: effect of verapamil on in vitro susceptibility of promastigote and amastigote stages of Indian clinical isolates to sodium stibogluconate. Exp. Parasitol. 114, 103–108. 10.1016/j.exppara.2006.02.015 [DOI] [PubMed] [Google Scholar]
- Vallières C., Fisher N., Antoine T., Al-Helal M., Stocks P., Berry N. G., et al. (2012). HDQ, a potent inhibitor of Plasmodium falciparum proliferation, binds to the quinone reduction site of the cytochrome bc1 complex. Antimicrob. Agents Chemother. 56, 3739–3747. 10.1128/AAC.00486-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller K. L., Muhle R. A., Ursos L. M., Horrocks P., Verdier-Pinard D., Sidhu A. B. S., et al. (2003). Chloroquine resistance modulated in vitro by expression levels of the Plasmodium falciparum chloroquine resistance transporter. J. Biol. Chem. 278, 33593–33601. 10.1074/jbc.M302215200 [DOI] [PubMed] [Google Scholar]
- Ward C. P., Wong P. E., Burchmore R. J., de Koning H. P., Barrett M. P. (2011). Trypanocidal furamidine analogues: influence of pyridine nitrogens on trypanocidal activity, transport kinetics, and resistance patterns. Antimicrob. Agents Chemother. 55, 2352–2361. 10.1128/AAC.01551-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner J., Kooij T. W. A. (2016). Phylogenetic profiles of all membrane transport proteins of the malaria parasite highlight new drug targets. Microb Cell 3, 511–521. 10.15698/mic2016.10.534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellems T. E., Walker-Jonah A., Panton L. J. (1991). Genetic mapping of the chloroquine-resistance locus on Plasmodium falciparum chromosome 7. Proc. Natl. Acad. Sci. U.S.A. 88, 3382–3386. 10.1073/pnas.88.8.3382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengelnik K., Vidal V., Ancelin M. L., Cathiard A.-M., Morgat J. L., Kocken C. H., et al. (2002). A class of potent antimalarials and their specific accumulation in infected erythrocytes. Science 295, 1311–1314. 10.1126/science.1067236 [DOI] [PubMed] [Google Scholar]
- Wenzler T., Boykin D. W., Ismail M. A., Hall J. E., Tidwell R. R., Brun R. (2009). New treatment option for second-stage African sleeping sickness: In vitro and in vivo efficacy of aza analogs of DB289. Antimicrob. Agents Chemother. 53, 4185–4192. 10.1128/AAC.00225-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White N. J., Pukrittayakamee S., Phyo A. P., Rueangweerayut R., Nosten F., Jittamala P., et al. (2014). Spiroindolone KAE609 for falciparum and vivax malaria. New Engl. J. Med. 371, 403–410. 10.1056/NEJMoa1315860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2001). Antimalarial Drug Combination Therapy: Report of a WHO Technical Consultation.
- WHO (2015). Guidelines for the Treatment of Malaria. Geneva: World Health Organization. [PubMed] [Google Scholar]
- WHO (2016). Report of the Second WHO Stakeholders Meeting on Gambiense Human African Trypanosomiasis Elimination, Geneva, 21–23. [Google Scholar]
- WHO (2017a). Global leishmaniasis update, 2006–2015: a turning point in leishmaniasis surveillance, weekly epidemiological record. Relevé Épidémiologique Hebdomadaire 92, 557–572. [PubMed] [Google Scholar]
- WHO (2017b). World Malaria Report 2017. Geneva: World Health Organization. [Google Scholar]
- Wiechert M. I., Beitz E. (2017). Mechanism of formate-nitrite transporters by dielectric shift of substrate acidity. EMBO J. 36, 949–958. 10.15252/embj.201695776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiechert M., Erler H., Golldack A., Beitz E. (2017). A widened substrate selectivity filter of eukaryotic formate-nitrite transporters enables high-level lactate conductance. FEBS J. 284, 2663–2673. 10.1111/febs.14117 [DOI] [PubMed] [Google Scholar]
- Wittner M., Lederman J., Tanowitz H. B., Rosenbaum G. S., Weiss L. M. (1996). Atovaquone in the treatment of Babesia microti infections in hamsters. Am. J. Trop. Med. Hyg. 55, 219–222. 10.4269/ajtmh.1996.55.219 [DOI] [PubMed] [Google Scholar]
- Woodrow C. J., Penny J. I., Krishna S. (1999). Intraerythrocytic Plasmodium falciparum expresses a high affinity facilitative hexose transporter. J. Biol. Chem. 274, 7272–7277. 10.1074/jbc.274.11.7272 [DOI] [PubMed] [Google Scholar]
- Wree D., Wu B., Zeuthen T., Beitz E. (2011). Requirement for asparagine in the aquaporin NPA signature motifs for cation exclusion. FEBS J. 278, 740–748. 10.1111/j.1742-4658.2010.07993.x [DOI] [PubMed] [Google Scholar]
- Wu B., Song J., Beitz E. (2010). Novel channel-enzyme fusion proteins confer arsenate resistance. J. Biol. Chem. 285, 40081–40087. 10.1074/jbc.M110.184457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B., Rambow J., Bock S., Holm-Bertelsen J., Wiechert M. I., Soares A. B., et al. (2015). Identity of a Plasmodium lactate/H+ symporter structurally unrelated to human transporters. Nat. Commun. 6, 6284. 10.1038/ncomms7284 [DOI] [PubMed] [Google Scholar]
- Yang D. M., Liew F. Y. (1993). Effects of qinghaosu (artemisinin) and its derivatives on experimental cutaneous leishmaniasis. Parasitology 106(Pt 1), 7–11. 10.1017/S0031182000074758 [DOI] [PubMed] [Google Scholar]
- Ye Z., van Dyke K. (1988). Reversal of chloroquine resistance in falciparum malaria independent of calcium channels. Biochem. Biophys. Res. Commun. 155, 476–481. 10.1016/S0006-291X(88)81111-2 [DOI] [PubMed] [Google Scholar]
- Ye Z., van Dyke K. (1994). Reversal of chloroquine resistance in falciparum malaria by some calcium channel inhibitors and optical isomers is independent of calcium channel blockade. Drug Chem. Toxicol. 17, 149–162. 10.3109/01480549409014308 [DOI] [PubMed] [Google Scholar]
- Zaugg J. L., Lane V. M. (1989). Evaluations of buparvaquone as a treatment for equine babesiosis (Babesia equi). Am. J. Vet. Res. 50, 782–785. [PubMed] [Google Scholar]
- Zeuthen T., Wu B., Pavlovic-Djuranovic S., Holm L. M., Uzcategui N. L., Duszenko M., et al. (2006). Ammonia permeability of the aquaglyceroporins from Plasmodium falciparum, Toxoplasma gondii and Trypansoma brucei. Mol. Microbiol. 61, 1598–1608. 10.1111/j.1365-2958.2006.05325.x [DOI] [PubMed] [Google Scholar]
- Zishiri V. K., Joshi M. C., Hunter R., Chibale K., Smith P. J., Summers R. L., et al. (2011). Quinoline antimalarials containing a dibemethin group are active against chloroquinone-resistant Plasmodium falciparum and inhibit chloroquine transport via the P. falciparum chloroquine-resistance transporter (PfCRT). J. Med. Chem. 54, 6956–6968. 10.1021/jm2009698 [DOI] [PubMed] [Google Scholar]