

Abstract
Benzidine, a known carcinogen, is closely associated with the development of bladder cancer (BC). Epithelial–mesenchymal transition (EMT) is a critical pathophysiological process in BC progression. The underlying molecular mechanisms of mitogen-activated protein kinase (MAPK) pathway, especially extracellular regulated protein kinases 5 (ERK5), in regulating benzidine-induced EMT remains unclarified. Hence, two human bladder cell lines, T24 and EJ, were utilized in our study. Briefly, cell migration was assessed by wound healing assay, and cell invasion was determined by Transwell assay. Quantitative PCR and western blot were utilized to determine both gene expressions as well as protein levels of EMT and MAPK, respectively. Small interfering RNA (siRNA) was transfected to further determine ERK5 function. As a result, the migration and invasion abilities were enhanced, epithelial marker expression was decreased while mesenchymal marker expression was increased in human BC cell lines. Meanwhile, benzidine administration led to activation of ERK5 and activator protein 1 (AP-1) proteins, without effective stimulation of the Jun N-terminal kinase (JNK) or p38 pathways. Moreover, Benzidine-induced EMT and ERK5 activation were completely suppressed by XMD8-92 and siRNAs specific to ERK5. Of note, ERK1/2 was activated in benzidine-treated T24 cells, while benzidine-induced EMT could not be reversed by U0126, an ERK1/2 inhibitor, as indicated by further study. Collectively, our findings revealed that ERK5-mediated EMT was critically involved in benzidine-correlated BC progression, indicating the therapeutic significance of ERK5 in benzidine-related BC.
Keywords: benzidine, bladder cancer, epithelial-mesenchymal transition, ERK5
INTRODUCTION
Bladder cancer (BC), the most frequently-seen urinary tract malignancy, appears to be one of the costliest diseases to manage from diagnosis to death (Siegel et al., 2016). Benzidine, an aromatic amine, is commercially produced and predominantly used in the production of dyes (Chung, 2016). Benzidine can cause various types of cancers, which, therefore, is confirmed as definite human carcinogens by the International Agency for Cancer Research (IARC) for several decades (IARC, 1987). To date, the positive association of benzidine with BC has been revealed by multiple epidemiological as well as experimental researches (Baan et al., 2008; Ma et al., 2012; Weistenhofer et al., 2008). Nevertheless, a lack of comprehensive understanding of molecular mechanism is one of the biggest obstacles to identify effective diagnostic and therapeutic approaches for benzidine-associated BC.
The process of epithelial–mesenchymal transition (EMT), initially described as an essential developmental program for embryogenesis, implantation and organogenesis, is now defined as a vital contributor to cancer progression and metastasis. It is featured by a lack of epithelial characteristics and the attainment of a motile, invasive, as well as migratory mesenchymal phenotype (Yeung and Yang, 2017). Studies have demonstrated the important involvement of EMT in tumorigenicity, tumor invasion, distant metastasis (Hui et al., 2017). Many carcinogens are involved in BC development, including benzidine, demonstrated to enhance benzidine-induced EMT (Liu et al., 2017; Zhao et al., 2015). Nevertheless, the potential mechanisms of benzidine-induced-EMT remains unclarified.
Conventional MAPKs are mainly divided into four major groups: ERK1/2, JNK, p38 and ERK5. ERK5 signaling is one of the less-studied MAPK cascades, despite its increasingly demonstrated relevance in growth, survival, and differentiation of cancer cells (Hoang et al., 2017). Recently, the dysregulation of ERK5 has been documented in multiple malignancies, including breast cancer, glioma, lung cancer, colorectal cancer, renal cancer as well as prostate cancer (Liu et al., 2017; Salinas-Sánchez et al., 2017; Won et al., 2017; Wu et al., 2016; Zhuang et al., 2016). However, the function of ERK5 in BC is still unknown.
Here, we reported that benzidine enhanced migration and invasion in human BC cells T24 and EJ. In addition, benzidine-induced EMT had a correlation with activation of ERK5 pathway, which could be reversed by XMD8-92 and siRNAs specific to ERK5. In spite of activation of ERK1/2 signaling in T24 cells, inhibition of ERK1/2 by U0126 could not restore benzidine-induced EMT. Collectively, these data suggest the effect of ERK5 in modulation of benzidine-stimulated EMT and benzidine-related BC promoting effects.
MATERIALS AND METHODS
Reagents and antibodies
Benzidine (4, 4′-diaminobiphenyl; ≥98.0%, RT), methanol as well as DMSO were purchased from Merck (USA). Benzidine was dissolved in DMSO, which was subsequently preserved at −20°C. The final concentration of DMSO administered in cells was under 1‰. Polyclonal antibodies against p-ERK1/2, p-p38, p-JNK, p-ERK5, p-c-Jun, p-c-Fos, and Fra-1 were purchased from Cell Signaling Technology (USA). Polyclonal antibodies against ZO-1, E-cadherin, Snail, N-cadherin, Vimentin, XMD8-92, U0126, as well as GAPDH were commercially obtained from Santa Cruz (USA). Primers for Snail, N-cadherin, Vimentin, ZO-1, E-cadherin as well as GAPDH were synthesized by Invitrogen (USA). Sources of unmentioned materials were described in the following.
Cell culture and treatment
Human BC cells T24 and EJ were purchased from American Type Culture Collection (ATCC, USA) and cultured in RPMI-1640 medium containing 10% FBS, penicillin, streptomycin (HyClone, USA) at 37°C in a humidified atmosphere with 5% CO2. Cells were kept in 25-cm2 flasks, with the medium changing every two days. When cells grown to 80–90% confluency, cells were administered with indicated concentrations of benzidine for 4 days (T24) or 5 days (EJ), with or without XMD8-92 (5 μM) or U0126 (5 μM). All procedures were conducted in triplicate.
Cell viability assay
In brief, cells were seeded into 96-well plates (2000 cells/well) for 24 h, followed by incubation with indicated concentrations of benzidine for 4 days (T24) or 5 days (EJ). Then, MTT solution was given to each well for solubilization of formazan crystals, followed by incubation at 37°C for 4 h. After discarding MTT solution and solubilization of crystals in DMSO, the optical density was detected at a wavelength of 490 nm by a microplate reader. Each treatment was conducted in triplicate for statistical analysis.
Wound healing assay
Human BC cells were initially treated with or without benzidine for 4 days (T24) or 5 days (EJ), which were further seeded into 6-well dishes. When cells reached to 90% confluency, they were scratched with a 2-mm-wide tip. Cells were allowed to migrate, followed by photography under an inverted microscope.
Transwell invasion assay
Cell invasion was performed with transwell chamber (Millipore, USA) with Matrigel (BD, USA). Briefly, T24 cells were pretreated for 4 days, then cells resuspended in 100 μl medium with 2% serum (containing 1 × 104 cells) were cultured in upper chambers, with 800 μl of medium with 10% serum in lower chambers. After incubating at 37°C for 24 h, cells in upper chamber were discarded with cotton swabs, and 0.1% crystal violet was employed to stain the invaded cells after fixing in methanol. Finally, cells of each membrane were counted and photographed in five random fields of 100 magnification under microscope.
Transfection with small interfering RNA (siRNA)
For ERK5 silencing, we transiently transfected cells with 70 pmol/L of specific human siRNAs against ERK5 using Lipofectamine 2000 reagent (Invitrogen) according to standard protocols. Targeting sequences of siRNA were described as follows: human ERK5 siRNA, 5′-GGGCCTATATCC AGAGCUU-3′. Non-specific control siRNA was commercially obtained from Santa Cruz Biotechnology (USA) to serve as a negative control. Cells were harvested 96 hours (T24) or 120 h (EJ) after siRNA transfection, followed by Western blot to determine protein expression.
Western blotting analysis
Briefly, cell protein was extracted from T24 and EJ cells by washing with PBS, followed by lysis in 100 μl of RIPA solution (Thermo Scientific, USA), centrifugation as well as mixture with 5× SDS sample buffer. Protein samples were subjected to 10% SDS-PAGE, transferred onto PVDF membranes (Millipore, USA), and blocked in 5% milk. Then, the membranes were reacted with appropriate primary and secondary antibodies, followed by visualization using Super-Signal West Pico Chemiluminescent Substrate.
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
RNA extraction was carried out from T24 and EJ cells using RNAiso Plus (TaKaRa, Japan) in line with standard protocols. cDNA was synthesized from 2 mg of total RNA with AMV reverse transcriptase (Takara) in accordance with the manufacturer’s instructions. qRT-PCR was conducted using Power SYBR Green Master Mix (TaKaRa, Japan) with ABI 7300 real-time PCR detection system (Applied Biosystems, USA) under default conditions: 95°C for 10 s, and 40 cycles of 95°C for 10 s and 72°C for 30 s. Comparative Ct method was employed for quantification, and the mRNA expression of every gene was normalized to its respective GAPDH. The primers used were as follows: GAPDH, forward 5′-GCTGCCCAACGC ACCGAATA-3′ and reverse 5′-GAGTCAACGGATTTGGTCGT-3′; ZO-1, forward 5′-GCAGCCACAACCAATTCATAG-3′ and reverse 5′-GCAGACGATGTTCATAGTTTC-3′; E-cadherin, forward 5′-TCGACACCCGATTCAAAGTGG-3′ and reverse 5′-TTCCAGAAACGGAGGCCTGAT-3′; N-cadherin forward 5′-ATCAAGTGCCATTAGCCAAG-3′ and reverse 5′-CTGAGCAG TGAATGTTGTCA-3′; Vimentin, forward 5′-CCTTGACATTGA GATTGCCA-3′ and reverse 5′-GTATCAACCAGAGGGAGTGA-3′; Snail, forward 5′-TTCCAGCAGCCCTACGACCAG-3′ and reverse 5′-CGGACTCTTGGTGCTTGTGGA-3′.
Statistical analysis
Results presented in figures were representative from triplicate. Data were expressed as mean ± standard deviation after analyzing with SPSS 16.0 software (SPSS, Inc., USA). Unpaired Student’s t-test was employed for comparison between two groups. One-way ANOVA was utilized in multiple groups comparison, followed by LSD significant difference test. P values < 0.05 implicated statistical significance.
RESULTS
Benzidine induced EMT of human BC cells
In exploration of the relationship of benzidine and BC metastasis, human BC cells were culture with indicated concentrations of benzidine for 4 days (T24) or 5 days (EJ). As a result, benzidine at a concentration up to 0.1 μM did not exert an obvious effect on cell viability (Fig. 1A). Hence, benzidine concentrations of 0.001, 0.005, 0.01, 0.05 μM were selected for the following procedures in our study. EMT is critically involved in tumor invasion as well as metastasis. In our study, benzidine treatment led to an obvious morphological alteration, from round-shaped to spindle-like mesenchymal form (Figs. 1B and 1C). Wound healing assays revealed that benzidine increased migration ability of T24 and EJ cells (Figs. 1D and 1E), and Transwell assays showed that benzidine enhanced invasion ability in T24 cell (Fig. 1F). Therefore, we analyzed the role of benzidine on EMT by detecting the expression levels of EMT markers. Western blot showed that benzidine exposure reduced the protein expression of epithelial indicators, such as E-cadherin and ZO-1, but enhanced the levels of mesenchymal indicators, including Vimentin, N-cadherin as well as Snail (Fig. 2A), which were consistent with outcomes of qRT-PCR assay (Fig. 2B). Together, these data showed that benzidine administration triggered EMT in human BC cells.
Fig. 1. Benzidine induced EMT morphological alterations and increased migratory and invasive abilities of human BC cells.
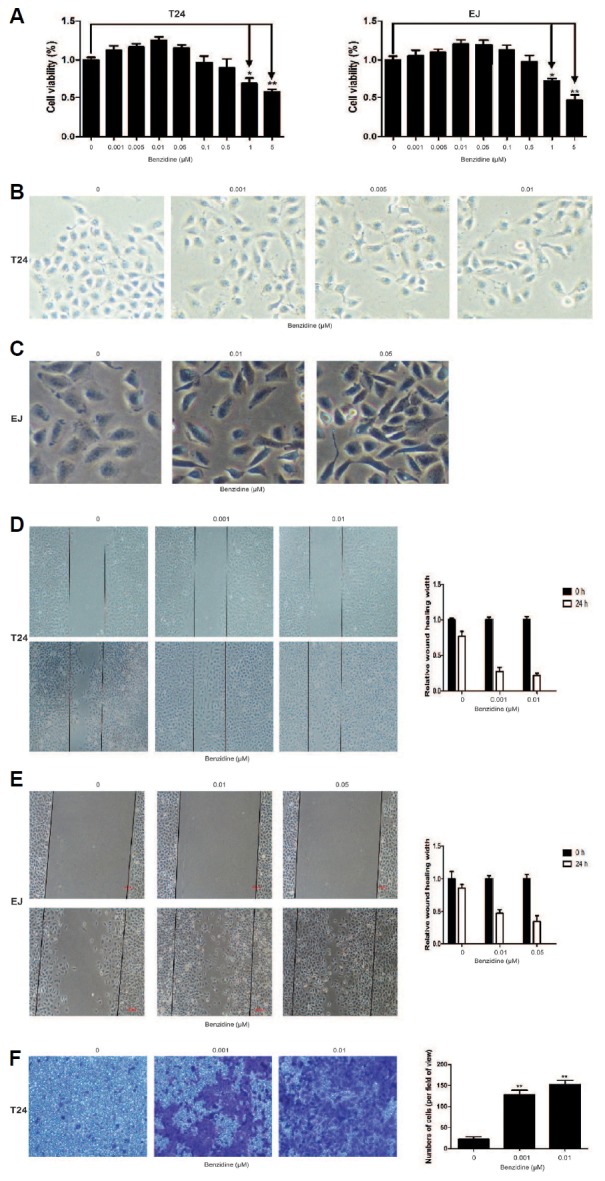
(A) Human BC cells were exposed to 0–5 μM of benzidine for 4 days (T24) or 5 days (EJ), followed by cell viability assessment by MTT. (B, C) Morphological examination of human BC cells showed that benzidine led to morphological alterations from epithelial to spindle-like mesenchymal form. (D, E) Wound healing assay indicated that benzidine increased migratory ability of human BC cells. (F) Transwell invasion assay showed that invasive ability of T24 cells were elevated by benzidine. *P < 0.05 and **P < 0.01, compared to control group.
Fig. 2. Benzidine modulated the expressions of EMT markers in human BC cells.
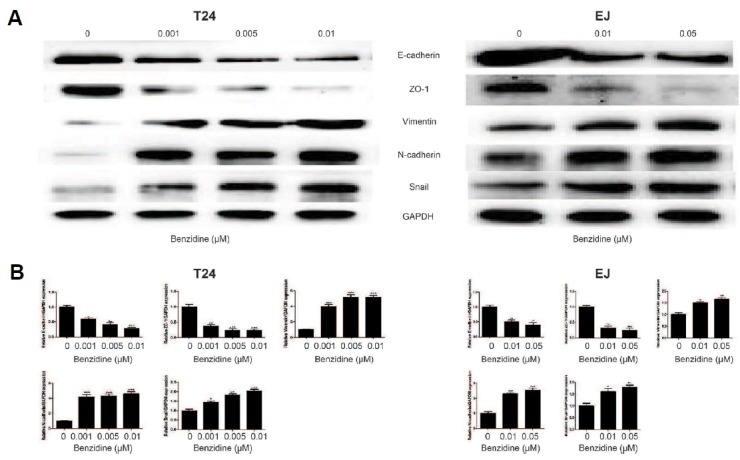
(A) Protein levels of ZO-1, E-cadherin, Snail, N-cadherin as well as Vimentin by Western blot. GAPDH was employed as an internal control. (B) mRNA levels of ZO-1, E-cadherin, Snail, N-cadherin as well as Vimentin by qRT-PCR, after normalization to GAPDH. *P < 0.05, **P < 0.01, ***P < 0.001, compared to control group.
Benzidine enhanced MAPK/AP-1 activation in human BC cells
In investigation of the potential association between MAPKs activation and benzidine-induced EMT in human BC cells, the expressions of total as well as phosphorylated ERK5, JNK, ERK1/2, p38 were determined by Western blot. Consequently, benzidine significantly increased p-ERK1/2 and p-ERK5 expression, while had little effect on p-JNK and p-p38 in T24 cells (Fig. 3A), while benzidine exposure only stimulated p-ERK5 in EJ cells (Fig. 3A). Moreover, Western blot also revealed elevated expressions of downstream target AP-1 proteins, including Fra-1, p-c-Fos, as well as p-c-Jun (Fig. 3B).
Fig. 3. Benzidine-induced EMT was related to MAPK/AP-1 activation in human BC cells.
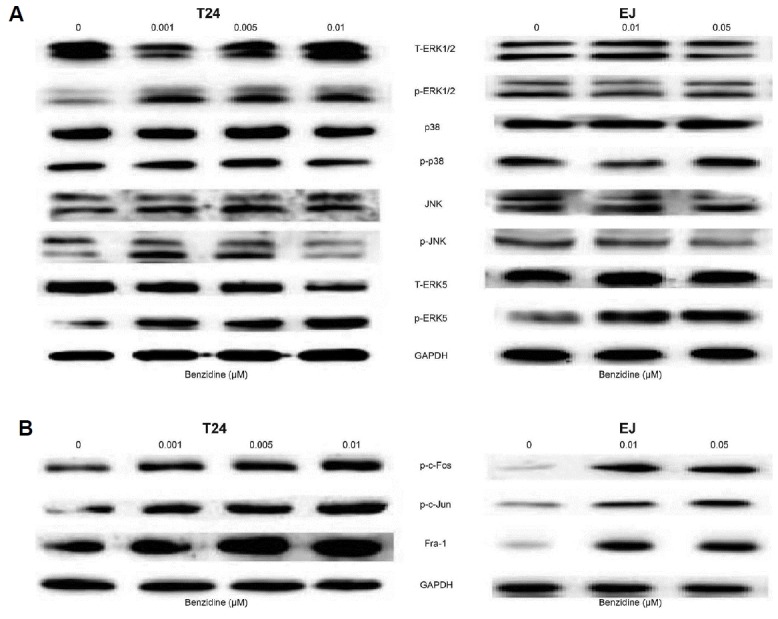
(A) Protein expressions of T-ERK1/2, p-ERK1/2, p38, p-p38, JNK, p-JNK, T-ERK5 as well as p-ERK5 by Western blot. GAPDH was utilized as an internal control. (B) AP-1 protein expressions (p-c-Fos, p-c-Jun as well as Fra-1) by Western blot. GAPDH was employed as an internal control.
Blockade of ERK5 by XMD8-92 prevented benzidine-induced EMT in human BC cells
Next, XMD8-92 (a specific inhibitor of ERK5) was used to further determine whether benzidine-triggered EMT could be suppressed by blocking ERK5 signaling in vitro. As a result, XMD8-92 decreased the protein expressions of p-ERK5, p-c-Fos as well as p-c-Jun in T24 cells, as indicated by Western blot (Figs. 4A and 4B). Morphological evaluations showed that XMD8-92 rescued the mensenchymal-like morphological alterations (Fig. 4C). XMD8-92 suppressed the invasion capacities of T24, as the representative micrographs demonstrated in Fig. 4D. Besides, XMD8-92 restored the migratory ability of EJ cells, as revealed by wound healing assays (Fig. 4E). Moreover, XMD8-92 reversed benzidine-stimulated reduction of E-cadherin, ZO-1 expressions, and enhancement of Vimentin, N-cadherin, Snail in T24 cells using Western blot (Fig. 4F).
Fig. 4. Suppression of ERK5 by XMD8-92 reversed benzidine-triggered EMT in human BC cells.

(A, B) Cells were administered various concentrations of benzidine with or without 5 μM XMD8-92 for 4 days (T24) or 5 days (EJ), followed by Western blot, which implicated that XMD8-92 diminished benzidine-induced ERK5 and AP-1 protein activation. GAPDH was utilized as an internal control. (C) Morphological alterations of human BC cells induced by benzidine were attenuated by XMD8-92. (D) Transwell invasion assay revealed that XMD8-92 decreased benzidine-triggered invasive capacity of T24 cells. (E) Wound healing assay revealed that XMD8-92 restored benzidine-triggered migratory capacity of the EJ cells. (F) Western blotting analyses showed that XMD8-92 reversed benzidine-triggered down-regulation of protein levels of ZO-1, E-cadherin, and up-regulation of protein expressions of N-cadherin, Snail and Vimentin. GAPDH was employed as an internal control. **P < 0.01, compared to control group; ##P < 0.01, compared to 0.01 μM benzidine-treated group.
Suppression of ERK5 by a specific small interfering RNA rescued benzidine-induced EMT in human BC cells
To further characterize regulation of EMT by ERK5, loss of function was performed using siERK5 and non-silencing siRNA. Initially, Western blot was utilized to confirm the efficacy of siERK5, which efficiently reduced ERK5 protein expression, but did not affect ERK1/2 protein expression (Fig. 5A). siERK5 decreased levels of p-ERK5, p-c-Fos as well as pc-Jun in T24 cells (Fig. 5B). Moreover, suppression of ERK5 by siERK5 led to elevated protein levels of epithelial indicators, including E-cadherin, ZO-1, and decreased levels of mesenchymal indicators, such as Vimentin, N-cadherin and Snail (Fig. 5C). Taken together, these observations demonstrated a critical role of ERK5 activity in benzidine-triggered EMT in human BC cells.
Fig. 5. Inhibition of ERK5 by siRNA repressed benzidine-induced EMT of human BC cells.

(A) Human BC cells were transfected with 70 pmol/L siERK5, 70 pmol/L non-silencing siRNA or left untransfected. Cells were lysed 4 days (T24) or 5 days (EJ) after transfection, followed by Western blot to illustrate the role of siERK5 on ERK5 and ERK1/2 expressions. (B) siERK5 inhibited benzidine-induced ERK5 as well as AP-1 protein activation in human BC cells. After exposure to various concentrations of benzidine with or without 70 pmol/L siERK5 for 4 days (T24) or 5 days (EJ), the expression levels of p-c-Jun, p-c-Fos as well as p-ERK5 were determined by western blot. GAPDH was employed as an internal control. (C) Treatment of siERK5 in human BC cells attenuated benzidine-stimulated down-regulation of protein expressions of ZO-1, E-cadherin, and up-regulation of protein levels of Snail, N-cadherin and Vimentin, indicated by Western blot. GAPDH was employed as an internal control.
Inhibition of ERK1/2 by U0126 could not reverse benzidine-triggered EMT in T24
Next, we tendentiously investigated whether ERK1/2 contributed to benzidine-induced EMT in T24. To this end, U0126 (a selective inhibitor of MEK1 and MEK2) was ultilized to pretreat the cells. Consequently, U0126 restored benzidine-induced increase of p-ERK1/2, p-c-Fos, as well as p-c-Jun in T24 cells, as indicated by Western blot (Fig. 5A and 5B). However, U0126 treatment did not reverse the mensenchymal-like morphological alterations (Fig. 5C), neither did it have effect on invasive capacity of T24 cells (Fig. 5D). Further study demonstrated that U0126 had no significant effect on benzidine-triggered reduction of E-cadherin, ZO-1 expressions, or enhancement of N-cadherin, Snail and Vimentin in T24 cells (Fig. 5E). Therefore, ERK1/2 was not likely to play an essential role in benzidine-induced EMT in T24.
DISCUSSION
Recently, the participation of MAPK pathway in EMT has been a hot topic, and dysregulation of MAPK exhibits a crucial function in cancer progression, which provides novel insights into therapeutics against cancers. Nevertheless, the roles of MAPKs, especially ERK5, in the development of benzidine-related BC are far from fully understood. Numerous studies have proved that EMT contributes to the malignancy of BC. Thus, exploring the mechanism underlying the relationship between MAPKs and EMT in benzidine-associated BC is particularly important for developing novel therapeutic drugs and improving patient survival rate.
EMT is an integral part of metastatic progression whereby cells gain motile and invasive capabilities through loss of epithelial markers, namely E-cadherin and ZO-1, and obtainment of mesenchymal markers, including Vimentin as well as N-cadherin. Accumulating evidence has shown that EMT is of paramount importance in BC development and progression (Kiselyov et al., 2016), and the effects of benzidine on carcinogenesis as well as EMT induction (Liu et al., 2017; Zhao et al., 2015). In agreement with previous researches, we found that benzidine exposure triggered EMT in human BC cells T24 and EJ, as evidenced by morphological alteration from epithelial to mesenchymal form, enhanced migratory as well as invasive abilities, and changes in expressions of EMT markers, including reduced epithelial markers, ZO-1 and E-cadherin, as well as elevated mesenchymal markers N-cadherin, Vimentin and Snail.
The ERK5 pathway harbors many features that are structurally and functionally distinct from other MAPKs, all of which make it possible to be an ideal therapeutic target (Hoang et al., 2017). Recently, there is increasing evidence regarding the involvement of ERK5 signaling in cancer progression. ERK5 overexpression in cancer cells, such as human prostate cancer PC3 cells, exhibited a migratory and invasive phenotype, therefore contributing to increased capacities of metastasis (Ramsay et al., 2011). Moreover, pharmacological suppression of ERK5 inhibited cell motility and invasion in several kinds of tumor cells in vitro (Rovida et al., 2015; Sawhney et al., 2009), and reduced metastasis of prostate and liver cancer xenografts in vivo (Ramsay et al., 2011; Rovida et al., 2015). Loveridge et al. (2017) indicated that ERK5 deletion could increase T-cell infiltration in a PTEN-deficient mouse model of prostate carcinogenesis, suggesting that ERK5 may affect immune therapy in prostate cancer. ERK5 has also been demonstrated to regulate TS-mediated urocystic, pulmonary and hepatic EMT in vivo and in vitro (Geng et al., 2015; Liang et al., 2015a; 2015b; 2017; Min et al., 2017). Our team previously reported that benzidine induced EMT, activated ERK5, and transactivated AP-1 proteins in SV-40, an immortalized human urothelial cell (Liu et al., 2017). Likewise, in our research, we verified that benzidine-elicited EMT of human BC cells was linked with activation of ERK5. We observed that utilizing XMD8-92 or siRNA targeting ERK5 contributed to the inhibition of the ERK5/AP-1 signaling pathway, consequently reversing benzidine-stimulated EMT. These researches revealed the underlying mechanisms of benzidine-facilitated or -induced migration, invasion, and EMT process in human BC cells.
Several reports have demonstrated the importance of ERK1/2 pathway in BC progression (Sun et al., 2016; 2017; Zhao et al., 2015). Studies by Zhao revealed that benzidine triggered ERK1/2 activation and EMT in human uroepithelial cells, which could be suppressed by U0126, an ERK1/2 inhibitor (Zhao et al., 2015). Our team previously confirmed that benzidine promoted proliferation of human BC T24 cells via ERK1/2 signaling (Sun et al., 2016). In addition, ERK1/2 is critically involved in CSE-induced EMT of T24 (Sun et al., 2017). Taken together, we speculated that ERK1/2 might regulate benzidine-stimulated EMT in human BC cells. Interestingly, our data showed an activation of ERK1/2 activated in benzidine-triggered EMT in T24 cells, but not in EJ cells. Moreover, using ERK1/2 inhibitor, U0126, could significantly down-regulate p-ERK1/2 expression. Further study demonstrated that U0126 had no obvious effect on benzidine-induced EMT phenotype, cell invasion ability, and EMT markers expressions. These observations seem inconsistent with our previous report. In CSE-treated T24 cells, inhibition of ERK1/2 can reverse EMT process (Sun et al., 2017). However, the results from these two studies are not contradictory, since T24 cells were treated with different stimuli (i.e., cigarette smoke or benzidine). In comparison to benzidine, cigarette smoke is a complex chemical mixture that contains over 4,500 compounds, including more than 60 carcinogens (Boffetta, 2008). Because of the different oncogenic mechanisms of each carcinogen in cigarette smoke, the role of distinct signaling pathways varies widely. In our studies, we have showed that ERK1/2 pathway responds differently to CSE or benzidine in T24 cells. Taking all of these facts into account, in spite of the enhanced ERK1/2 experssion in benzidine-treated T24 cells, ERK1/2 is not likely to be critically involved in EMT process.
AP-1 has been demonstrated to participate in progression of a number of malignancies, including BC. Increased expressions of AP-1 proteins were also found in multiple human cancer samples, such as colorectal cancer, endometrial carcinoma, and so on (Shivtia and Yehudit, 2017). As a downstream target of MAPKs, studies showed that AP-1 exerted a directly and indirectly transcriptional regulation of EMT markers during EMT induction (Bakiri et al., 2015; Lu et al., 2016; Yoshinaga et al., 2016). Here, we found that benzidine-mediated MAPK activation increased the activation of AP-1 proteins. Meanwhile, benzidine-stimulated AP-1 activation was decreased after administration of XMD8-92, siRNA, and U0126 treatment. These results suggested the importance of AP-1 in benzidine-elicited EMT of human BC cells. However, there are still many uncertainties about the molecular mechanism of AP-1 on EMT, which requires further investigation.
In conclusion, to the best our knowledge, our work was the first to reported that benzidine induced EMT in human BC cells, primarily via activation of ERK5, but not ERK1/2 signaling. Our findings implicate that the targeted blockade of ERK5 pathway might be if effect as therapeutic approaches against benzidine-related BC progression, which is worth of further investigation.
Fig. 6. ERK1/2 inhibition had little influence on benzidine-induced EMT in T24 cells.

(A, B) After exposure of T24 cells to various concentrations of benzidine with/or 5 μM U0126, an ERK1/2 inhibitor for 4 days, Western blot implicated that U0126 suppressed benzidine-triggered ERK1/2 and AP-1 protein activation. GAPDH was employed as an internal control. (C) Morphological alterations of T24 cells induced by benzidine could not be restored by U0126. (D) Benzidine-triggered invasion ability of T24 cells could not be decreased by U0126, detected by Transwell invasion assay. (E) Western blot revealed that U0126 failed to ameliorate benzidine-stimulated down-regulation of protein levels of ZO-1, E-cadherin, and up-regulation of protein expressions of Snail, N-cadherin and Vimentin. GAPDH was employed as an internal control. **P < 0.01, compared to control group; ##P < 0.01, compared to 0.01 μM benzidine-treated group.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (No.81373005, No. 81072330, No. 81202194).
REFERENCES
- Bakiri L, Macho-Maschler S, Custic I, Niemiec J, GuíoCarrión A, Hasenfuss SC, Eger A, Müller M, Beug H, Wagner EF. Fra-1/AP-1 induces EMT in mammary epithelial cells by modulating Zeb1/2 and TGFβ expression. Cell Death Differ. 2015;22:336–350. doi: 10.1038/cdd.2014.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baan R, Straif K, Grosse Y, Secretan B, ElGhissassi F, Bouvard V, Benbrahim-Tallaa L, Cogliano V WHO International Agency for Research on Cancer Monograph Working Group. Carcinogenicity of some aromatic amines, organic dyes, and related exposures. Lancet Oncol. 2008;9:322–323. doi: 10.1016/S1470-2045(08)70089-5. [DOI] [PubMed] [Google Scholar]
- Boffetta P. Tobacco smoking and risk of bladder cancer. Scand J Urol Nephrol Suppl. 2008;218:45–54. doi: 10.1080/03008880802283664. [DOI] [PubMed] [Google Scholar]
- Chung KT. Azo dyes and human health: A review. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2016;34:233–261. doi: 10.1080/10590501.2016.1236602. [DOI] [PubMed] [Google Scholar]
- Geng H, Zhao L, Liang Z, Zhang Z, Xie D, Bi L, Wang Y, Zhang T, Cheng L, Yu D, et al. ERK5 positively regulates cigarette smoke-induced urocystic epithelial-mesenchymal transition in SV-40 immortalized human urothelial cells. Oncol Rep. 2015;34:1581–1588. doi: 10.3892/or.2015.4130. [DOI] [PubMed] [Google Scholar]
- Hoang VT, Yan TJ, Cavanaugh JE, Flaherty PT, Beckman BS, Burow ME. Oncogenic signaling of MEK5-ERK5. Cancer Lett. 2017;392:51–59. doi: 10.1016/j.canlet.2017.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui K, Gao Y, Huang J, Xu S, Wang B, Zeng J, Fan J, Wang X, Yue Y, Wu S, et al. RASAL2, a RAS GTPase-activating protein, inhibits stemness and epithelial-mesenchymal transition via MAPK/SOX2 pathway in bladder cancer. Cell Death Dis. 2017;8:e2600. doi: 10.1038/cddis.2017.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC . Overall evaluations of carcinogenicity. IARC monographs on the evaluation of carcinogenic risk of chemicals to humans, supplement 7. Lyon, France: International Agency for Research on Cancer; 1987. pp. 440–447. [Google Scholar]
- Kiselyov A, Bunimovich-Mendrazitsky S, Startsev V. Key signaling pathways in the muscle-invasive bladder carcinoma: Clinical markers for disease modeling and optimized treatment. Int J Cancer. 2016;138:2562–2569. doi: 10.1002/ijc.29918. [DOI] [PubMed] [Google Scholar]
- Liang Z, Xie W, Wu R, Geng H, Zhao L, Xie C, Li X, Huang C, Zhu J, Zhu M, et al. ERK5 negatively regulates tobacco smoke-induced pulmonary epithelial-mesenchymal transition. Oncotarget. 2015a;6:19605–19618. doi: 10.18632/oncotarget.3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Xie W, Wu R, Geng H, Zhao L, Xie C, Li X, Zhu M, Zhu W, Zhu J, et al. Inhibition of tobacco smoke-induced bladder MAPK activation and epithelial-mesenchymal transition in mice by curcumin. Int J Clin Exp Pathol. 2015b;8:4503–4513. [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Wu R, Xie W, Xie C, Wu J, Geng S, Li X, Zhu M, Zhu W, Zhu J, et al. Effects of Curcumin on Tobacco Smoke-induced Hepatic MAPK Pathway Activation and Epithelial-Mesenchymal Transition In Vivo. Phytother Res. 2017 doi: 10.1002/ptr.5844. 10.1002. [DOI] [PubMed] [Google Scholar]
- Liu F, Zhang H, Song H. Upregulation of MEK5 by Stat3 promotes breast cancer cell invasion and metastasis. Oncol Rep. 2017;37:83–90. doi: 10.3892/or.2016.5256. [DOI] [PubMed] [Google Scholar]
- Liu Z, Liu J, Zhao L, Geng H, Ma J, Zhang Z, Yu D, Zhong C. Curcumin reverses benzidine-induced epithelial-mesenchymal transition via suppression of ERK5/AP-1 in SV-40 immortalized human urothelial cells. Int J Oncol. 2017;50:1321–1329. doi: 10.3892/ijo.2017.3887. [DOI] [PubMed] [Google Scholar]
- Loveridge CJ, Mui EJ, Patel R, Tan EH, Ahmad I, Welsh M, Galbraith J, Hedley A, Nixon C, Blyth K, et al. Increased T-cell Infiltration Elicited by Erk5 Deletion in a Pten-Deficient Mouse Model of Prostate Carcinogenesis. Cancer Res. 2017;77:3158–3168. doi: 10.1158/0008-5472.CAN-16-2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Zhang ZL, Huang D, Tang N, Li Y, Peng Z, Lu C, Dong Z, Tang F. Cdk3-promoted epithelial-mesenchymal transition through activating AP-1 is involved in colorectal cancer metastasis. Oncotarget. 2016;7:7012–7028. doi: 10.18632/oncotarget.6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma QW, Lin GF, Chen JG, Guo WC, Qin YQ, Golka K, Shen JH. N-Acetyltransferase 2 genotype, exfoliated urothelial cells and benzidine exposure. Front Biosci (Elite Ed) 2012;4:1966–1974. doi: 10.2741/517. [DOI] [PubMed] [Google Scholar]
- Min J, Geng H, Liu Z, Liang Z, Zhang Z, Xie D, Wang Y, Zhang T, Yu D, Zhong C. ERK5 regulates tobacco smoke-induced urocystic epithelial-mesenchymal transition in BALB/c mice. Mol Med Rep. 2017;15:3893–3897. doi: 10.3892/mmr.2017.6457. [DOI] [PubMed] [Google Scholar]
- Ramsay AK, McCracken SR, Soofi M, Fleming J, Yu AX, Ahmad I, Morland R, Machesky L, Nixon C, Edwards DR. ERK5 signalling in prostate cancer promotes an invasive phenotype. Br J Cancer. 2011;104:664–672. doi: 10.1038/sj.bjc.6606062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovida E, DiMaira G, Tusa I, Cannito S, Paternostro C, Navari N, Vivoli E, Deng X, Gray NS, Esparís-Ogando A, et al. The mitogen-activated protein kinase ERK5 regulates the development and growth of hepatocellular carcinoma. Gut. 2015;64:1454–1465. doi: 10.1136/gutjnl-2014-306761. [DOI] [PubMed] [Google Scholar]
- Salinas-Sánchez AS, Serrano-Oviedo L, Nam-Cha SY, Roche-Losada O, Sánchez-Prieto R, Giménez-Bachs JM. Prognostic value of the VHL, HIF-1α, and VEGF signaling pathway and associated MAPK (ERK1/2 and ERK5) pathways in clear-cell renal cell carcinoma. a long-term study. Clin Genitourin Cancer. 2017 doi: 10.1016/j.clgc.2017.05.016. pii: S1558-7673(17)30153–2. [DOI] [PubMed] [Google Scholar]
- Sawhney RS, Liu W, Brattain MG. A novel role of ERK5 in integrin-mediated cell adhesion and motility in cancer cells via Fak signaling, J. Cell Physiol. 2009;219:152–161. doi: 10.1002/jcp.21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- Sun X, Deng Q, Liang Z, Zhang Z, Zhao L, Geng H, Xie D, Wang Y, Yu D, Zhong C. Curcumin reverses benzidine-induced cell proliferation by suppressing ERK1/2 pathway in human bladder cancer T24 cells. Exp Toxicol Pathol. 2016;68:215–222. doi: 10.1016/j.etp.2015.12.003. [DOI] [PubMed] [Google Scholar]
- Sun X, Deng Q, Liang Z, Liu Z, Geng H, Zhao Li, Zhou Q, Liu J, Ma J, Wang D, et al. Cigarette smoke extract induces epithelial-mesenchymal transition of human bladder cancer T24 cells through activation of ERK1/2 pathway. Biomed Pharmacother. 2017;86:457–465. doi: 10.1016/j.biopha.2016.12.022. [DOI] [PubMed] [Google Scholar]
- Trop-Steinberg S, Azar Y. AP-1 Expression and its Clinical Relevance in Immune Disorders and Cancer. Am J Med Sci. 2017;353:474–483. doi: 10.1016/j.amjms.2017.01.019. [DOI] [PubMed] [Google Scholar]
- Weistenhofer W, Blaszkewicz M, Bolt HM, Golka K. N-acetyltransferase-2 and medical history in bladder cancer cases with a suspected occupational disease (BK 1301) in Germany. J Toxicol Environ Health A. 2008;71:906–910. doi: 10.1080/15287390801988681. [DOI] [PubMed] [Google Scholar]
- Won KJ, Im JY, Kim BK, Ban HS, Jung YJ, Jung KE, Won M. Stability of the cancer target DDIAS is regulated by the CHIP/HSP70 pathway in lung cancer cells. Cell Death Dis. 2017;8:e2554. doi: 10.1038/cddis.2016.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Cui H, Zhu Z, Wang L. MicroRNA-200b-3p suppresses epithelial-mesenchymal transition and inhibits tumor growth of glioma through down-regulation of ERK5. Biochem Biophys Res Commun. 2016;478:1158–1164. doi: 10.1016/j.bbrc.2016.08.085. [DOI] [PubMed] [Google Scholar]
- Yeung KT, Yang J. Epithelial-mesenchymal transition in tumor metastasis. Mol Oncol. 2017;11:28–39. doi: 10.1002/1878-0261.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshinaga T, Uwabe K, Naito S, Higashino K, Nakano T, Numata Y, Kihara A. AM251 suppresses epithelial-mesenchymal transition of renal tubular epithelial Cells. PLoS One. 2016;11:e0167848. doi: 10.1371/journal.pone.0167848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Geng H, Liang Z, Zhang Z, Zhang T, Yu D, Zhong C. Benzidine induces epithelial-mesenchymal transition in human uroepithelial cells through ERK1/2 pathway. Biochem Biophys Res Commun. 2015;459:643–649. doi: 10.1016/j.bbrc.2015.02.163. [DOI] [PubMed] [Google Scholar]
- Zhuang K, Zhang J, Xiong M, Wang X, Luo X, Han L, Meng Y, Zhang Y, Liao W, Liu S. CDK5 functions as a tumor promoter in human colorectal cancer via modulating the ERK5-AP-1 axis. Cell Death Dis. 2016;7:e2415. doi: 10.1038/cddis.2016.333. [DOI] [PMC free article] [PubMed] [Google Scholar]