

Abstract
The association between conotruncal heart defects (CTHDs) and maternal genetic and environmental exposures is well studied. However, little is known about paternal genetic or environmental exposures and risk of CTHDs. We assessed the effect of paternal genetic variants in the folate, homocysteine, and transsulfuration pathways on risk of CTHDs in offspring. We utilized National Birth Defects Prevention Study data to conduct a family-based case only study using 616 live-born infants with CTHDs, born October 1997 - August 2008. Maternal, paternal and infant DNA was genotyped using an Illumina® Golden Gate custom single nucleotide polymorphism (SNP) panel. Relative risks (RR) and 95% confidence intervals (CI) from log-linear models determined parent of origin effects for 921 SNPs in 60 candidate genes involved in the folate, homocysteine, and transsulfuration pathways on risk of CTHDs. The risk of CTHD among children who inherited a paternally derived copy of the A allele on GLRX (rs17085159) or the T allele of GLRX (rs12109442) was 0.23 (95% CI: 0.12, 0.42; P=1.09×10-6) and 0.27 (95% CI: 0.14, 0.50; P=2.06×10-5) times the risk among children who inherited a maternal copy of the same allele. The paternally inherited copy of the GSR (rs7818511) A allele had a 0.31 (95% CI: 0.18, 0.53; P=9.94×10-6] risk of CTHD compared to children with the maternal copy of the same allele.
The risk of CTHD is less influenced by variants in paternal genes involved in the folate, homocysteine or transsulfuration pathways than variants in maternal genes in those pathways.
Keywords: Conotruncal heart defects, congenital heart defect, genetic variants, parent-of-origin, paternal genetic variants
INTRODUCTION
Congenital heart defects (CHDs) are the most common birth defect, affecting about one percent of live births in the United States (U.S.) annually [Bernier, Stefanescu, Samoukovic, & Tchervenkov, 2010; van der Linde et al., 2011]; they are the most common cause of infant mortality, increase the risk of neurodevelopmental delay in affected children, and increased lifelong morbidity [Bernier, 2010; Marelli, 2007; Reller, 2008]. Unfortunately, most CHDs are diagnosed without a known cause [Fahed, Gelb, Seidman, and Seidman, 2013]. Less than 10% of CHD cases are attributed to chromosomal abnormalities and approximately 85% of cases are attributed to a multi-factorial etiology [Hobbs, MacLeod, Jill James, & Cleves, 2011; Jenkins et al., 2007; Matthews et al., 2003]. Maternal factors implicated with increased risk of CHDs include diabetes mellitus, [Casson et al., 1997; Towner et al., 1995] obesity, [Shaw, Nelson, & Moore, 2002; Waller et al., 1994] prenatal cigarette smoking, [Kallen, 1999; Karatza et al., 2011; Malik et al., 2008; Maurano et al., 2012; Patel et al., 2012] low folate levels, [ L. D. Botto, Mulinare, & Erickson, 2003; Czeizel, 1998; Scanlon et al., 1998;] hyperhomocysteinemia, [Hobbs, Cleves, Melnyk, Zhao, & James, 2005; Verkleij-Hagoort et al., 2006; Wenstrom, Johanning, Johnston, & DuBard, 2001] and genetic polymorphisms in metabolic pathways including the folate, homocysteine and glutathionine/transsulfuration pathways [Chowdhury et al., 2012; Hobbs, Cleves, Karim, Zhao, & MacLeod, 2010].
In contrast, the possible influence of paternal environmental and genetic factors on the risk of CHDs is much less investigated. Some of the few studies conducted suggest associations between young or advanced paternal age and increased risk of atrial septal defects, [Lian, Zack, & Erickson, 1986; Olshan, Schnitzer, & Baird, 1994] ventricular septal defects, [Lian, 1986; Olshan, 1994] right ventricular outflow tract defects, [Green et al., 2010] pulmonary valve atresia, [Green et al., 2010] patent ductus arteriosus, [Su, Yuan, Huang, Olsen, & Li, 2015] situs inversus [Olshan, 1994] and CHDs overall in their children [Hollier, Leveno, Kelly, DD, & Cunningham, 2000; Olshan et al., 1994; Yang et al., 2007; Zhan, Lian, Zheng, & Gao, 1991]. Other studies report no association between paternal age and risk of CHDs in their children [Cedergren, Selbing, & Kallen, 2002; Su et al., 2015; Zhan et al., 1991]. Other paternal exposures associated with increased risk of CHDs in their offspring include cigarette smoking, [Cresci et al., 2011; Deng et al., 2013] alcohol consumption [Ou et al., 2016] and occupational exposure to endocrine disruptors [Cresci et al., 2011; C. Wang et al., 2015]. Although the specific biological mechanisms are unclear, some hypothesize that these exposures may share a similar mechanism: germline mutations and epigenetic alterations to sperm haploid DNA [Kong et al., 2012; Sharma et al., 2015; Wyrobek et al., 2006; Beal, Yauk, & Marchetti, 2017; Linschooten et al., 2013; Marchetti et al., 2011; Yauk et al., 2007].
Given that extant environmental exposure may induce changes in paternal DNA that can result in CHDs, we postulated that inherent paternal genetic factors, i.e., genetic polymorphisms, may also increase CHD risk. Numerous studies confirm that genetic polymorphisms in maternal and infant genes are directly or indirectly associated with risk of CHDs, particularly genes involved in folate, homocysteine and transsulfuration pathways [Chowdhury et al., 2012; Hobbs et al., 2010; Zhu et al., 2012]. Conversely, none of the published literature to date in English in PubMed, assess the influence of paternal genetic variants in folate, homocysteine or transsulfuration pathways and CHD risk in offspring. Thus, we investigated parent-of-origin effect for genetic variants in folate, homocysteine or transsulfuration pathways and the risk of conotruncal heart defects (CTHD) in offspring.
MATERIALS and METHODS
We used data from the National Birth Defects Prevention Study (NBDPS) a population-based, case-control study conducted in the U.S. to investigate the contribution of genetic, environmental, and behavioral factors on the occurrence of major, non-syndromic birth defects [Yoon et al., 2001]. Methods of the NBDPS have been previously described [Yoon et al., 2001] but in brief, NBDPS families of cases and controls were identified from population-based birth defects surveillance systems in 10 states in the US, which included: Arkansas, California, Georgia, Iowa, Massachusetts, New Jersey, New York, North Carolina, Texas and Utah. The NBDPS was conducted from October 1, 1997 until December 2011 and enrolled 44,000 non-Hispanic (NH) white, NH-black and Hispanic women and their families; however, our study only included enrolled families of infants with estimated dates of delivery between October 1997 and August 2008.
For these analyses we conducted a case-parent trio study using 616 case families (1298 samples (not all were trios)) with a singleton, live-born infant diagnosed with a CTHD within the first year of life. Infants with CTHDs affected by a known single gene disorder, chromosomal abnormality, or syndrome were excluded. In each surveillance program, medical records were abstracted by trained staff who actively ascertained cases from hospitals, birthing and other facilities.
CTHD Ascertainment
For NBDPS, all CHD cases were identified based on having at least one of the following diagnostic procedures: echocardiograms, surgical reports, cardiac catherizations, or autopsies. All diagnostic procedure information was then reviewed by a pediatric cardiologist at each study center to ensure uniform criteria for diagnoses. CHD classification was done by a panel of pediatric cardiologists from each study centers led by a pediatric cardiologist at the Center for Disease Control and Prevention (CDC) using the classification system specifically developed for the NBDPS which incorporates cardiac phenotype, cardiac complexity, and extra-cardiac anomalies [L. D. Botto et al., 2007]. In our study, conotruncal defects included truncus arteriosus, interrupted aortic arch type B, transposition of the great arteries, double outlet right ventricle, conoventricular septal defects, tetralogy of Fallot and pulmonary atresia with ventricular septal defect [L. D. Botto et al., 2007].
Maternal Interview
After informed consent, case mothers completed a one-hour computer-assisted telephone interview administered in English or Spanish [Yoon et al., 2001]. During the interview women were queried about their prenatal exposures and information about the baby’s father, including, the father’s age, and race, occupation and health behaviors.
DNA Sample Collection
After completing the telephone interview, case families were mailed buccal swab kits for collection of maternal, infant and paternal DNA samples [Yoon et al., 2001]. DNA sample storage and processing methods for the NBDPS are described in detail in a prior publication [Rasmussen et al., 2002]. All cases who had maternal, paternal and infant DNA samples available were included in our study.
Genotyping and Quality Assessment
Genotyping was conducted on a total of 635 case families using 200 ng of DNA on the Illumina Golden Gate platform [Fan, Chee, & Gunderson, 2006; Tang et al., 2014]. A total of 297 individuals were removed due to study ineligibility (n=33), high no-call rates (n=63), or high rates of Mendelian inconsistency (n=201). To ensure high-quality genotypes, we applied stringent quality control measures and excluded SNPs with obviously poor clustering behavior (60 SNPs), no-call rates >10% (328 SNPs), Mendelian error rates >5% (11 SNPs), MAF <5% (204 SNPs), or significant deviation from Hardy-Weinberg Equilibrium in at least one racial group (p<10-4, 12 SNPs). More detailed information regarding genotyping and quality assessment is described in Tang et al. [Tang et al., 2014]. For the current study, the final dataset included 1298 individuals from 616 case families, each with 921 SNPs. Thirty-seven percent of the study population were family trio samples.
Statistical Method
Summary statistics were expressed as mean (standard deviation) for continuous variables, and count (percentage) for categorical variables. In investigate the parent-of-origin effect; a log-linear model was fitted for the counts of each SNP as a function of mating types, maternal genetic effect, infant genetic effect, and imprinting parameter [Weinberg, 1999]. Based on the log-linear model for counts and assuming a Poisson distribution, the imprinting effect was estimated as the relative risk (RR) of a CTHD in a child who inherited a paternally derived copy of the minor allele compared to a child who inherits a copy of the minor allele from the mother. Bonferroni correction was used to adjust for multiple testing. Statistical significance level was set at P <5.43×10-5. Data were analyzed using statistical software SAS 9.4 (SAS Institute Inc., Cary, NC) for computing descriptive statistics and PREMIM/EMIM for fitting imprinting models [Howey & Cordell, 2012].
Human Subjects Review
This study was approved by the University of Arkansas for Medical Sciences’ Institutional Review Board and the National Birth Defects Prevention Network with protocol oversight by the CDC, Center for Birth Defects and Developmental Disabilities. All study participants provided written informed consent and legal guardians provided written informed consent for participants who were minors.
RESULTS
The distributions of maternal and paternal characteristics of infants with CTHDs are presented in Table 1. Approximately 70% of the mothers were non-Hispanic white, 27% had a high school education and 28% had some college education; 24% were overweight and 21% were obese. Fifty-one percent of mothers reported periconceptional use of folic acid, 24.5% consumed alcohol during pregnancy and 19% smoked cigarettes during pregnancy.
Table I.
Summary of characteristics from Chi-squared analyses for mothers of infants with (cases) conotruncal heart defects, The National Birth Defects Prevention Study, U.S.A., October 1997 – August 2008 births (n=616).
| Characteristics | Maternal | Paternal |
|---|---|---|
| N (%) | ||
| Age at Delivery (mean ± standard deviation) | 28.3 (6.1) | 31.4 (7.3) |
| <35 years | 504 (82.4%) | 372 (71.0%) |
| ≥35 years | 108 (17.7%) | 152 (29.0%) |
| Missing information | 4 | 92 |
| Race/Ethnicity | ||
| Non-Hispanic white | 401 (65.5%) | 386 (63.7%) |
| Non-Hispanic black | 49 (8.0%) | 58 (9.6%) |
| Hispanic | 123 (20.1%) | 125 (20.6%) |
| Other | 39 (6.4%) | 37 (6.1%) |
| Missing | 4 | 10 |
| Education | ||
| < 12 years | 83 (13.5%) | 94 (15.6%) |
| High school diploma or equivalent | 167 (27.2%) | 190 (31.5%) |
| < 4 years of college education | 173 (28.2%) | 135 (22.4%) |
| At least 4 years of college or Bachelor’s degree | 190 (31.0%) | 185 (30.6%) |
| Missing | 3 | 12 |
| Mean Household Income | ||
| < $10,000 | 94 (16.2%) | |
| $10,000 - $29,999 | 150 (25.9%) | |
| $30,000 – $49,999 | 118 (20.4%) | |
| $50,000 + | 217 (37.5%) | |
| Missing | 37 | |
| Body Mass Index | ||
| Underweight (< 18.5 kg/m2) | 31 (5.3%) | |
| Normal weight (18.5 to < 25.0 kg/m2) | 298 (50.4%) | |
| Overweight (25.0 to <30.0 kg/m2) | 141 (23.9%) | |
| Obese (≥ 30.0 kg/m2) | 121 (20.5%) | |
| Missing | 25 | |
| Periconceptional Folic acid Supplementation | ||
| Yes | 314 (51.2%) | N/A |
| No | 299 (48.8%) | N/A |
| Missing | 3 | N/A |
| Alcohol Consumption During Pregnancy | ||
| Yes | 149 (24.5%) | N/A |
| No | 460 (75.5%) | N/A |
| Missing | 7 | N/A |
| Cigarette Smoking During Pregnancy | ||
| Yes | 114 (18.6%) | N/A |
| No | 498 (81.4%) | N/A |
| Missing | 4 | |
| Cigarette smoking in home during first trimester | 521 (85.3%) | |
| No | N/A | 90 (14.7%) |
| Yes | N/A | 5 |
| Missing information | N/A | |
| Currently employed? | 31 (5.2%) | |
| No | N/A | 570 (94.8%) |
| Yes | N/A | 15 |
| Missing information | N/A | |
| Health problem at birth or birth defect? | 512 (85.2%) | |
| No | N/A | 89 (14.8%) |
| Yes | N/A | 15 |
| Missing information | N/A | |
| Mother blood relative of baby’s father? | 607 (99.5%) | |
| No | N/A | 3 (0.5%) |
| Yes | N/A | 6 |
| Missing information | N/A |
A total of 921 SNPs within 60 genes were included in the final analyses. We observed a statistically significant, decreased risk of CTHDs for paternally derived effects for three SNPs in two genes—glutaredoxin (GLRX) and glutathione-disulfide reductase (GSR) (Table 2). These two genes are involved in numerous cellular metabolic and homeostatic processes including the transsulfuration pathway, oxidative-reduction processes, cellular redox homeostasis, and nucleobase-containing small molecular interconversion reactions. For a child who inherited a paternally derived copy of the A allele for rs17085159 in the GLRX gene, the risk of a CTHD was 0.23 (95% CI: 0.12, 0.44; P=2.66 x 10-4) compared to a child who inherits the maternal A allele. For the SNP rs12109442, also in GLRX, the risk of developing a CTHD for a child who inherited the paternal copy of the T allele was 0.27 (95% CI: 0.14, 0.50; P=2.06 x 10-5, when compared to a child who inherited the maternally derived T allele. A similar magnitude of risk was also observed in children who inherited the paternal copy of the rs7818511 A allele, a SNP in the GSR gene, with a decrease in CTHD risk compared to children who inherited the maternal copy of the A allele (RR=0.31; 95% CI: 0.18, 0.53; P=9.94 × 10-4).
Table II.
Risk Ratios (RR) and 95% Confidence Intervals (CI) with P-values for Paternally-derived Effects for the Top 20 SNPs Identified from Hybrid Analyses compared to Maternally-derived Effects for Common Variants in Genes Involved in Folate, Homocysteine and Transsulfuration Pathways and Risk of Conotruncal Heart Defects among XXX, The National Birth Defects Prevention Study, USA, October 1997 – August 2008 Births (n=616 case families) (highlighted SNPs are significant SNPs with p-value ≤ 5.43×10-5).
| Pathway | Gene Symbol | dbSNP ID | Chromosome | Genotype | Maternally-derived effect | Paternally-derived Relative Risk (95% CI) | P-value for paternal vs. maternal effect |
|---|---|---|---|---|---|---|---|
| Transsulfuration | GLRX | rs17085159 | 5 | A/G | referent | 0.23 (0.12, 0.44) | 2.66×10-6 |
| Transsulfuration | GSR | rs7818511 | 8 | A/G | referent | 0.31 (0.18, 0.53) | 9.94×10-6 |
| Transsulfuration | GLRX | rs12109442 | 5 | T/A | referent | 0.27 (0.14, 0.50) | 2.06×10-5 |
| Homocysteine | MTRR | rs1801394 | 5 | G/A | referent | 0.48 (0.33, 0.71) | 8.72×10-5 |
| Transsulfuration | GSTA3 | rs614765 | 6 | G/C | referent | 0.41 (0.25, 0.67) | 2.70×10-4 |
| Folate | MTHFD2 | rs6745054 | 2 | G/A | referent | 0.47 (0.31, 0.72) | 3.05×10-4 |
| Transsulfuration | GCLC | rs13437395 | 6 | G/A | referent | 0.40 (0.24, 0.67) | 3.68×10-4 |
| Transsulfuration | OGG1 | rs2072668 | 3 | G/C | referent | 0.47 (0.31, 0.73) | 3.95×10-4 |
| Transsulfuration | MGMT | rs483959 | 10 | A/G | referent | 0.53 (0.36, 0.77) | 5.05×10-4 |
| Homocysteine | DNMT3B | rs7360212 | 20 | C/A | referent | 0.22 (0.09, 0.53) | 5.23×10-4 |
| Transsulfuration | GLRX | rs11953653 | 5 | A/G | referent | 1.78 (1.29, 2.45) | 5.98×10-4 |
| Transsulfuration | MGMT | rs4751106 | 10 | A/C | referent | 0.55 (0.39, 0.79) | 6.93×10-4 |
| Transsulfuration | GCLC | rs2277108 | 6 | A/G | referent | 0.43 (0.26, 0.71) | 6.96×10-4 |
| Transsulfuration | SOD1 | rs16988427 | 21 | G/A | referent | 0.32 (0.16, 0.63) | 7.29×10-4 |
| Transsulfuration | GSTP1 | rs7927381 | 11 | A/G | referent | 0.46 (0.28, 0.73) | 8.35×10-4 |
| Transsulfuration | GCLC | rs606548 | 6 | A/G | referent | 0.42 (0.25, 0.72) | 1.14×10-3 |
| Transsulfuration | GCLC | rs12524494 | 6 | G/A | referent | 0.41 (0.23, 0.71) | 1.29×10-3 |
| Folate | RFC1 | rs2062228 | 4 | A/G | referent | 0.23 (0.09, 0.59) | 1.46×10-3 |
| Transsulfuration | MGMT | rs544217 | 10 | A/G | referent | 0.58 (0.41, 0.82) | 1.50×10-3 |
| Transsulfuration | GCLC | rs13437220 | 6 | C/G | referent | 0.45 (0.27, 0.74) | 1.55×10-3 |
Overall we observed non-statistically significant decreased risks of CTHDs in children who inherited the paternally derived copy of the several folate, homocysteine and transulfuration pathway alleles compared to children who inherited the maternally derived copy of the allele (Table 2 and Figure 1). There was one exception in this observation concerning the SNP rs11953653 in the GLRX gene which showed an increased risk of CTHDs (RR=1.78; 95% CI: 1.29, 2.45; P=5.98 x 10-4) for children who inherited the paternal copy of the A allele versus those children who inherited the maternal copy of the allele (Table 2); however, the confidence interval contained the null value after the Bonferroni adjustment for multiple comparisons.
Figure I.
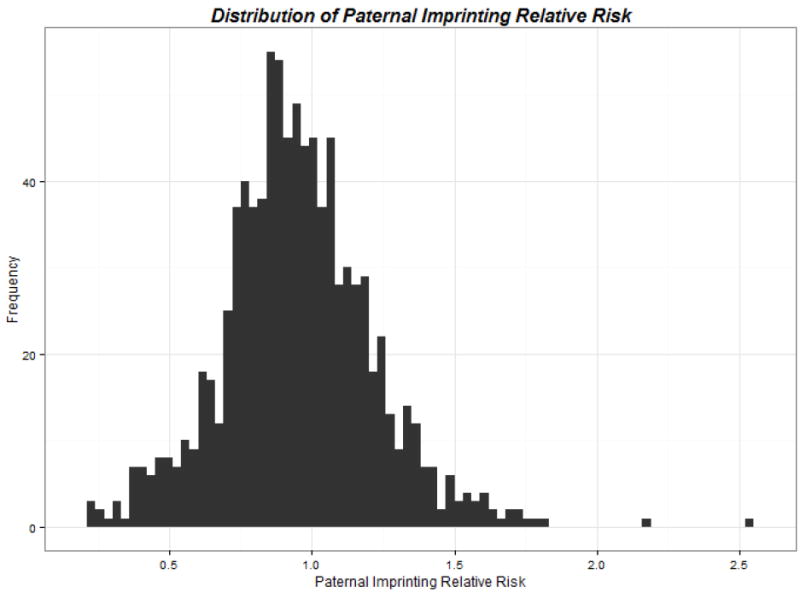
Distribution of Paternally-derived Effects for the SNPs Identified from Hybrid Analyses compared to Maternally-derived Effects for Common Variants in Genes Involved in Folate, Homocysteine and Transsulfuration Pathways and Risk of Conotruncal Heart Defects, The National Birth Defects Prevention Study, USA, October 1997 – August 2008 Births
DISCUSSION
Our intent was to determine parent-of-origin effects for genetic variants in folate, homocysteine and transsulfuration pathways and risk of CTHDs in offspring. Little is known about the role of paternally derived genetic variants on pregnancy outcomes including their role in the etiology of CHDs. In our study, we found no statistically significant increased risk of CTHDs for a child who inherited a paternally derived copy of an allele from a SNP in genes involved in the folate, homocysteine or transsulfuration pathways compared to a child who inherited a maternal copy of the allele. However, we did identify three SNPs for which there was decreased risk of CTHDs for a child who inherited a paternally derived copy of an allele from rs17085159, rs12109442 and rs11953653 compared to a child who inherited a maternal copy of the allele (Figure 2). One possible explanation for these findings is a parent-of-origin genetic effect in the etiology of CTHDs.
Figure II.
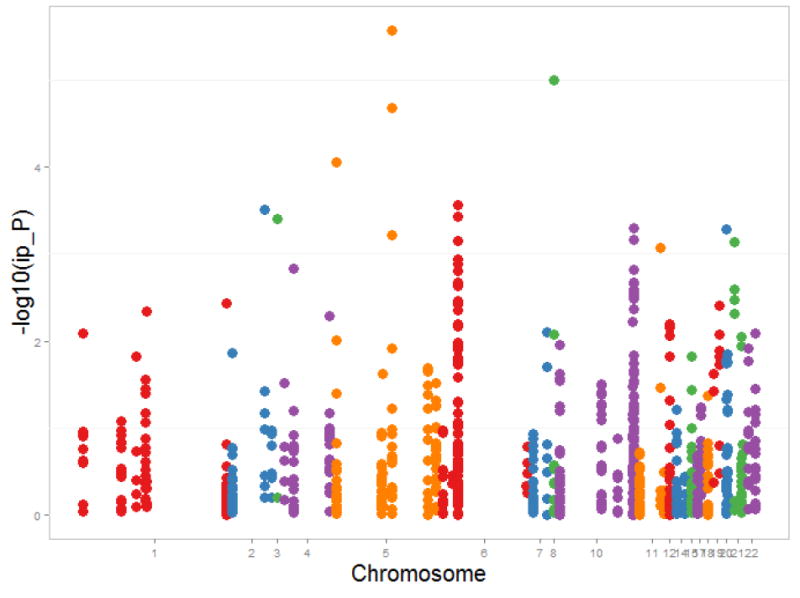
Manhattan plot for Paternally-derived Effects for the SNPs Identified from Hybrid Analyses compared to Maternally-derived Effects for Common Variants in Genes Involved in Folate, Homocysteine and Transsulfuration Pathways and Risk of Conotruncal Heart Defects, The National Birth Defects Prevention Study, USA, October 1997 – August 2008 Births
Parent-of-origin genetic effects comprise a growing field of developmental genetics and occur, primarily, through two mechanisms: genomic imprinting and trans-generational effects [Connolly & Heron, 2015; Elhamamsy, 2017; Lawson, Cheverud, & Wolf, 2013]. Genomic imprinting occurs when gene expression of a particular locus depends exclusively on the parent-of-origin [Plasschaert & Bartolomei, 2014]. Imprinting defects occur when this parental origin specific expression has been interfered with through mechanisms like uniparental disomy, translocations, inversions, or a deletion of one parental allele that allows for expression of only one copy of the imprinted gene [Elhamamsy, 2017; Lawson et al., 2013]. Trans-generational genomic effects occur through complex interactions in utero or during spermatogenesis and can involve gene-environment or transgenerational gene-gene interactions [Connolly & Heron, 2015; Lawson et al., 2013]. GWAS studies have successfully identified both types of parent-of-origin effects in several phenotypes including attention deficit hyperactivity disorder, [K. S. Wang, Liu, Zhang, Aragam, & Pan, 2012] schizophrenia, [Palmer et al., 2006] testicular germ cell tumors, [Karlsson et al., 2013] cleft lip with/without palate, [Sull et al., 2009] body mass, [Hoggart et al., 2014] and autism spectrum disorder [Connolly, Anney, Gallagher, & Heron, 2017].
One type of parent-of-origin effect is genomic imprinting which is a mechanism by which epigenetic modifications lead to silencing of one of the two inherited alleles leading to parental-origin determined expression. There are instances where an expressed gene’s parent-of-origin can lead to disease. The most common example of this is in Prader-Willi syndrome [Shemer et al., 2000] and Angelman syndrome [Buiting, Williams, & Horsthemke, 2016; Shemer et al., 2000] in which either the paternal or maternal locus in region 15q11-13 is silenced or deleted, respectively [Elhamamsy, 2017]. To date, only a handful of imprinted loci have been discovered encompassing a small portion of the human genome. Using the Catalogue of Imprinted Genes [Morison, Ramsay, & Spencer, 2005] we discovered that these three SNPs with significant paternal versus maternal effect (rs17085159, rs12109442 and rs11953653) are close (<7kb) to a known imprinted gene, RHOBTB3 that is paternally expressed in the human placenta [Metsalu et al., 2014] and involved in embryonal development [Salas-Vidal, Meijer, Cheng, & Spaink, 2005]. Because imprinted genes tend to cluster together, it has been efficacious to examine regions within 500kb of imprinted genes to identify parent of origin effects, therefore, these GLRX SNPs might well be within the imprinted cluster [Kong et al., 2009]. These findings suggest that the risk of CTHD associated with maternally inherited GLRX SNPs may be linked to the parent-of-origin imprinting of the nearby gene RHOBTB3. While our results are suggestive of this, further study of this region using gene expression profiling of the parental-trio will be necessary to confirm the role of imprinting in CTHD risk. There is also evidence to suggest a genetic basis for CTHDs [Andersen, Troelsen Kde, & Larsen, 2014; Fahed et al., 2013; Gelb & Chung, 2014; Pierpont et al., 2007] but the causal genetic mutation has been identified in only a fraction of cases [Gelb & Chung, 2014]. These results highlight the necessity of further investigating the role of imprinting in human development and as an etiological explanation for CHDs.
The SNP rs7818511 was significant (P=9.94E-6) when comparing paternal versus maternal inheritance. Evaluation of this SNP using the mutation prediction tool Mutation Taster [Schwarz, Cooper, Schuelke, & Seelow, 2014] revealed that this polymorphism may cause splice site changes in the assembly of the Glutathione-Disulfide Reductase (GSR) protein leading to potential haploinsufficiency of the protein product. GSR is an essential component of the oxidative machinery used in multiple processes in the body. GSR production of glutathione, a significant antioxidant, is crucial to effective host defense against bacterial infections [Yan et al., 2013]. Febrile illness and infections are known risk factors for the development of CHDs and other developmental pathologies [Lorenzo D. Botto, Lynberg, & Erickson, 2001]. Maternal haploinsufficiency of GSR, a critical enzyme of the innate immune system, might predispose the mother to infectious disease, prolonged infection, or allow for disease associated sequela to occur.
Our search of the published English language literature in PubMed to date produced no other studies which conducted parent-of-origin analyses for risk of CHDs as the primary aim. However, one study did conduct parent-of-origin analyses in relation to risk of CHDs as ad hoc analyses. Long et al. [Long, Lupo, Goldmuntz, & Mitchell, 2011] assessed the associations between maternal SNPs in folate-related genes and the risk of CTHD and left-sided heart defects. In their analyses they found that the maternal genotype, MTR A2756G, was associated with the studied cardiac defect phenotypes. They then tested for the presence of parental imprinting effects in this genotype using the parent-of-origin likelihood-ratio-test and the log-linear likelihood-ratio-test. The imprinting parameters from the parent-of-origin likelihood-ratio-test were elevated for both CTHDs and left-sided defects but were not statistically significant [Samanek et al., 1989]. But the parameter from the log-linear likelihood-ratio-test, for CTHD triads was statistically significant (Im=2.27, 95% CI: 1.06-4.87; p = 0.03). These findings further support the possibility of parent-of-origin effects in the etiology of CTHDs.
Our study results should be interpreted in consideration of potential limitations. DNA samples were obtained from self-collected buccal cheek cell samples and there was disparate quality of the DNA samples. Additionally, gene expression data was not available in the case-parental trios to confirm the role of imprinting on CTHD risk. In spite of these limitations our study has several strengths. Our study population is a large population-based, multi-ethnic sample, the CTHDs cases were verified by pediatric cardiologists and there was standardization of the CTHD classification across study centers. We also used a very conservative method (Bonferroni correction) to determine statistical significance.
Although our study did not identify any paternal genetic variants in the folate, homocysteine or glutathione/transsulfuration pathways that increased risk of CTHDs, we did find suggestive evidence of parent-of-origin effects for maternal genetic variants in the GSR and GLRX genes. Further research with larger sample sizes and multi-omic data is need to validate our findings.
Supplementary Material
Acknowledgments
The authors wish to thank the families who participated in the National Birth Defects Prevention Study (NBDPS) who made this research study possible. We also thank the faculty and staff of the Arkansas Center for Birth Defects Research and Prevention for working so hard to recruit, enroll and collection data from families for the NBDPS. We also thank the Centers for Birth Defects Research and Prevention in California, Georgia, Iowa, Massachusetts, New York, North Carolina, Texas and Utah for generously contributing their DNA samples for these analyses.
The authors also wish to thank Dr. Claire Weinberg for her assistance and suggestions in the analysis of this manuscript. The authors thank Lindsey Overman for assistance with the submission of this manuscript.
The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the Centers for Disease Control and Prevention.
Funding Source: This study was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development (#5R01HD039054-12), the CDC National Center for Birth Defects and Developmental Disabilities (#5U01DD000491-05) and the Arkansas Biosciences Institute.
References
- Andersen TA, Troelsen Kde L, Larsen LA. Of mice and men: molecular genetics of congenital heart disease. Cell Mol Life Sci. 2014;71(8):1327–1352. doi: 10.1007/s00018-013-1430-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MA, Yauk CL, Marchetti F. From Sperm to Offspring: Assessing the Heritable Genetic Consequences of Paternal Smoking and Potential Public Health Impacts. Mutation Research/Reviews in Mutation Research. 2017 doi: 10.1016/j.mrrev.2017.04.001. [DOI] [PubMed] [Google Scholar]
- Bernier PL, Stefanescu A, Samoukovic G, Tchervenkov CI. The Challenge of Congenital Heart Disease Worldwide: Epidemiologic and Demographic Facts. Seminars in Thoracic and Cardiovascular Surgery: Pediatric Cardiac Surgery Annual. 2010;13(1):26–34. doi: 10.1053/j.pcsu.2010.02.005. https://doi.org/10.1053/j.pcsu.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Botto LD, Lin AE, Riehle-Colarusso T, Malik S, Correa A National Birth Defects Prevention S. Seeking causes: Classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Res A Clin Mol Teratol. 2007;79(10):714–727. doi: 10.1002/bdra.20403. [DOI] [PubMed] [Google Scholar]
- Botto LD, Lynberg MC, Erickson JD. Congenital Heart Defects, Maternal Febrile Illness, and Multivitamine Use: A Population-Based Study. Epidemiology. 2001;12(5):485–490. doi: 10.1097/00001648-200109000-00004. [DOI] [PubMed] [Google Scholar]
- Botto LD, Mulinare J, Erickson JD. Do multivitamin or folic acid supplements reduce the risk for congenital heart defects? Evidence and gaps. Am J Med Genet A. 2003;121A(2):95–101. doi: 10.1002/ajmg.a.20132. [DOI] [PubMed] [Google Scholar]
- Buiting K, Williams C, Horsthemke B. Angelman syndrome - insights into a rare neurogenetic disorder. Nat Rev Neurol. 2016;12(10):584–593. doi: 10.1038/nrneurol.2016.133. [DOI] [PubMed] [Google Scholar]
- Casson IF, Clarke CA, Howard CV, McKendrick O, Pennycook S, Pharoah PO, Walkinshaw S, et al. Outcomes of pregnancy in insulin dependent diabetic women: results of a five year population cohort study. BMJ. 1997;315(7103):275–278. doi: 10.1136/bmj.315.7103.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedergren MI, Selbing AJ, Kallen BA. Risk factors for cardiovascular malformation--a study based on prospectively collected data. Scand J Work Environ Health. 2002;28(1):12–17. doi: 10.5271/sjweh.641. [DOI] [PubMed] [Google Scholar]
- Chowdhury S, Hobbs CA, MacLeod SL, Cleves MA, Melnyk S, James SJ, Erickson SW, et al. Associations between maternal genotypes and metabolites implicated in congenital heart defects. Mol Genet Metab. 2012;107(3):596–604. doi: 10.1016/j.ymgme.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly S, Anney R, Gallagher L, Heron EA. A genome-wide investigation into parent-of-origin effects in autism spectrum disorder identifies previously associated genes including SHANK3. Eur J Hum Genet. 2017;25(2):234–239. doi: 10.1038/ejhg.2016.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly S, Heron EA. Review of statistical methodologies for the detection of parent-of-origin effects in family trio genome-wide association data with binary disease traits. Brief Bioinform. 2015;16(3):429–448. doi: 10.1093/bib/bbu017. [DOI] [PubMed] [Google Scholar]
- Cresci M, Foffa I, Ait-Ali L, Pulignani S, Gianicolo EA, Botto N, Andreassi MG, et al. Maternal and paternal environmental risk factors, metabolizing GSTM1 and GSTT1 polymorphisms, and congenital heart disease. Am J Cardiol. 2011;108(11):1625–1631. doi: 10.1016/j.amjcard.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Czeizel AE. Periconceptional folic acid containing multivitamin supplementation. Eur J Obstet Gynecol Reprod Biol. 1998;78(2):151–161. doi: 10.1016/s0301-2115(98)00061-x. [DOI] [PubMed] [Google Scholar]
- Deng K, Liu Z, Lin Y, Mu D, Chen X, Li J, Zhu J, et al. Periconceptional paternal smoking and the risk of congenital heart defects: a case-control study. Birth Defects Res A Clin Mol Teratol. 2013;97(4):210–216. doi: 10.1002/bdra.23128. [DOI] [PubMed] [Google Scholar]
- Elhamamsy AR. Role of DNA methylation in imprinting disorders: an updated review. J Assist Reprod Genet. 2017 doi: 10.1007/s10815-017-0895-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahed AC, Gelb BD, Seidman JG, Seidman CE. Genetics of congenital heart disease: the glass half empty. Circ Res. 2013;112(4):707–720. doi: 10.1161/CIRCRESAHA.112.300853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan JB, Chee MS, Gunderson KL. Highly parallel genomic assays. Nat Rev Genet. 2006;7(8):632–644. doi: 10.1038/nrg1901. [DOI] [PubMed] [Google Scholar]
- Gelb BD, Chung WK. Complex genetics and the etiology of human congenital heart disease. Cold Spring Harb Perspect Med. 2014;4(7):a013953. doi: 10.1101/cshperspect.a013953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RF, Devine O, Crider KS, Olney RS, Archer N, Olshan AF, Shapira SK. Association of paternal age and risk for major congenital anomalies from the National Birth Defects Prevention Study, 1997 to 2004. Ann Epidemiol. 2010;20(3):241–249. doi: 10.1016/j.annepidem.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs CA, Cleves MA, Karim MA, Zhao W, MacLeod SL. Maternal folate-related gene environment interactions and congenital heart defects. Obstet Gynecol. 2010;116(2 Pt 1):316–322. doi: 10.1097/AOG.0b013e3181e80979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs CA, Cleves MA, Melnyk S, Zhao W, James SJ. Congenital heart defects and abnormal maternal biomarkers of methionine and homocysteine metabolism. Am J Clin Nutr. 2005;81(1):147–153. doi: 10.1093/ajcn/81.1.147. [DOI] [PubMed] [Google Scholar]
- Hobbs CA, MacLeod SL, Jill James S, Cleves MA. Congenital heart defects and maternal genetic, metabolic, and lifestyle factors. Birth Defects Res A Clin Mol Teratol. 2011;91(4):195–203. doi: 10.1002/bdra.20784. [DOI] [PubMed] [Google Scholar]
- Hoggart CJ, Venturini G, Mangino M, Gomez F, Ascari G, Zhao JH, Kutalik Z, et al. Novel approach identifies SNPs in SLC2A10 and KCNK9 with evidence for parent-of-origin effect on body mass index. PLoS Genet. 2014;10(7):e1004508. doi: 10.1371/journal.pgen.1004508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollier LM, Leveno KJ, Kelly MA, DD MC, Cunningham FG. Maternal age and malformations in singleton births. Obstet Gynecol. 2000;96(5 Pt 1):701–706. doi: 10.1016/s0029-7844(00)01019-x. [DOI] [PubMed] [Google Scholar]
- Howey R, Cordell HJ. PREMIM and EMIM: tools for estimation of maternal, imprinting and interaction effects using multinomial modelling. BMC Bioinformatics. 2012;13:149. doi: 10.1186/1471-2105-13-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins KJ, Correa A, Feinstein JA, Botto L, Britt AE, Daniels SR, et al. American Heart Association Council on Cardiovascular Disease in the, Y. Noninherited risk factors and congenital cardiovascular defects: current knowledge: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115(23):2995–3014. doi: 10.1161/CIRCULATIONAHA.106.183216. [DOI] [PubMed] [Google Scholar]
- Kallen K. Maternal smoking and congenital heart defects. Eur J Epidemiol. 1999;15(8):731–737. doi: 10.1023/a:1007671631188. [DOI] [PubMed] [Google Scholar]
- Karatza AA, Giannakopoulos I, Dassios TG, Belavgenis G, Mantagos SP, Varvarigou AA. Periconceptional tobacco smoking and isolated congenital heart defects in the neonatal period. Int J Cardiol. 2011;148(3):295–299. doi: 10.1016/j.ijcard.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Karlsson R, Andreassen KE, Kristiansen W, Aschim EL, Bremnes RM, Dahl O, Wiklund F, et al. Investigation of six testicular germ cell tumor susceptibility genes suggests a parent-of-origin effect in SPRY4. Hum Mol Genet. 2013;22(16):3373–3380. doi: 10.1093/hmg/ddt188. [DOI] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Stefansson K, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488(7412):471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Steinthorsdottir V, Masson G, Thorleifsson G, Sulem P, Besenbacher S, Stefansson K, et al. Parental origin of sequence variants associated with complex diseases. Nature. 2009;462(7275):868–874. doi: 10.1038/nature08625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson HA, Cheverud JM, Wolf JB. Genomic imprinting and parent-of-origin effects on complex traits. Nat Rev Genet. 2013;14(9):609–617. doi: 10.1038/nrg3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian ZH, Zack MM, Erickson JD. Paternal age and the occurrence of birth defects. Am J Hum Genet. 1986;39(5):648–660. [PMC free article] [PubMed] [Google Scholar]
- Linschooten JO, Verhofstad N, Gutzkow K, Olsen AK, Yauk C, Oligschlager Y, Godschalk RW, et al. Paternal lifestyle as a potential source of germline mutations transmitted to offspring. FASEB J. 2013;27(7):2873–2879. doi: 10.1096/fj.13-227694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J, Lupo PJ, Goldmuntz E, Mitchell LE. Evaluation of heterogeneity in the association between congenital heart defects and variants of folate metabolism genes: conotruncal and left-sided cardiac defects. Birth Defects Res A Clin Mol Teratol. 2011;91(10):879–884. doi: 10.1002/bdra.22849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, Cleves MA, Honein MA, Romitti PA, Botto LD, Yang S, et al. National Birth Defects Prevention, S. Maternal smoking and congenital heart defects. Pediatrics. 2008;121(4):e810–816. doi: 10.1542/peds.2007-1519. [DOI] [PubMed] [Google Scholar]
- Marchetti F, Rowan-Carroll A, Williams A, Polyzos A, Berndt-Weis ML, Yauk CL. Sidestream tobacco smoke is a male germ cell mutagen. Proc Natl Acad Sci U S A. 2011;108(31):12811–12814. doi: 10.1073/pnas.1106896108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews V, Schuster B, Schutze S, Bussmeyer I, Ludwig A, Hundhausen C, Rose-John S, et al. Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 [TACE] J Biol Chem. 2003;278(40):38829–38839. doi: 10.1074/jbc.M210584200. [DOI] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Stamatoyannopoulos JA, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metsalu T, Viltrop T, Tiirats A, Rajashekar B, Reimann E, Koks S, Salumets A, et al. Using RNA sequencing for identifying gene imprinting and random monoallelic expression in human placenta. Epigenetics. 2014;9(10):1397–1409. doi: 10.4161/15592294.2014.970052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morison IM, Ramsay JP, Spencer HG. A census of mammalian imprinting. Trends Genet. 2005;21(8):457–465. doi: 10.1016/j.tig.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Olshan AF, Schnitzer PG, Baird PA. Paternal age and the risk of congenital heart defects. Teratology. 1994;50(1):80–84. doi: 10.1002/tera.1420500111. [DOI] [PubMed] [Google Scholar]
- Ou Y, Mai J, Zhuang J, Liu X, Wu Y, Gao X, Lin S, et al. Risk factors of different congenital heart defects in Guangdong, China. Pediatr Res. 2016;79(4):549–558. doi: 10.1038/pr.2015.264. [DOI] [PubMed] [Google Scholar]
- Palmer CG, Hsieh HJ, Reed EF, Lonnqvist J, Peltonen L, Woodward JA, Sinsheimer JS. HLA-B maternal-fetal genotype matching increases risk of schizophrenia. Am J Hum Genet. 2006;79(4):710–715. doi: 10.1086/507829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SS, Burns TL, Botto LD, Riehle-Colarusso TJ, Lin AE, Shaw GM, et al. National Birth Defects Prevention, S. Analysis of selected maternal exposures and non-syndromic atrioventricular septal defects in the National Birth Defects Prevention Study, 1997-2005. Am J Med Genet A. 2012;158A(10):2447–2455. doi: 10.1002/ajmg.a.35555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierpont ME, Basson CT, Benson DW, Jr, Gelb BD, Giglia TM, Goldmuntz E, et al. American Heart Association Congenital Cardiac Defects Committee, C. o. C. D. i. t. Y. . Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115(23):3015–3038. doi: 10.1161/CIRCULATIONAHA.106.183056. [DOI] [PubMed] [Google Scholar]
- Plasschaert RN, Bartolomei MS. Genomic imprinting in development, growth, behavior and stem cells. Development. 2014;141(9):1805–1813. doi: 10.1242/dev.101428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SA, Lammer EJ, Shaw GM, Finnell RH, McGehee RE, Jr, Gallagher M, Murray JC, et al. Integration of DNA sample collection into a multi-site birth defects case-control study. Teratology. 2002;66(4):177–184. doi: 10.1002/tera.10086. [DOI] [PubMed] [Google Scholar]
- Salas-Vidal E, Meijer AH, Cheng X, Spaink HP. Genomic annotation and expression analysis of the zebrafish Rho small GTPase family during development and bacterial infection. Genomics. 2005;86(1):25–37. doi: 10.1016/j.ygeno.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Scanlon KS, Ferencz C, Loffredo CA, Wilson PD, Correa-Villasenor A, Khoury MJ, Willett WC. Preconceptional folate intake and malformations of the cardiac outflow tract. Baltimore-Washington Infant Study Group. Epidemiology. 1998;9(1):95–98. [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- Sharma R, Agarwal A, Rohra VK, Assidi M, Abu-Elmagd M, Turki RF. Effects of increased paternal age on sperm quality, reproductive outcome and associated epigenetic risks to offspring. Reprod Biol Endocrinol. 2015;13:35. doi: 10.1186/s12958-015-0028-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw GM, Nelson V, Moore CA. Prepregnancy body mass index and risk of multiple congenital anomalies. Am J Med Genet. 2002;107(3):253–255. doi: 10.1002/ajmg.10164. [DOI] [PubMed] [Google Scholar]
- Shemer R, Hershko AY, Perk J, Mostoslavsky R, Tsuberi B, Cedar H, Razin A, et al. The imprinting box of the Prader-Willi/Angelman syndrome domain. Nat Genet. 2000;26(4):440–443. doi: 10.1038/82571. [DOI] [PubMed] [Google Scholar]
- Su XJ, Yuan W, Huang GY, Olsen J, Li J. Paternal age and offspring congenital heart defects: a national cohort study. PLoS One. 2015;10(3):e0121030. doi: 10.1371/journal.pone.0121030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sull JW, Liang KY, Hetmanski JB, Wu T, Fallin MD, Ingersoll RG, Beaty TH, et al. Evidence that TGFA influences risk to cleft lip with/without cleft palate through unconventional genetic mechanisms. Hum Genet. 2009;126(3):385–394. doi: 10.1007/s00439-009-0680-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Nick TG, Cleves MA, Erickson SW, Li M, Li J, Hobbs CA, et al. Maternal obesity and tobacco use modify the impact of genetic variants on the occurrence of conotruncal heart defects. PLoS One. 2014;9(9):e108903. doi: 10.1371/journal.pone.0108903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towner D, Kjos SL, Leung B, Montoro MM, Xiang A, Mestman JH, Buchanan TA. Congenital malformations in pregnancies complicated by NIDDM. Diabetes Care. 1995;18(11):1446–1451. doi: 10.2337/diacare.18.11.1446. [DOI] [PubMed] [Google Scholar]
- van der Linde D, Konings EEM, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJM, Roos-Hesselink JW. Birth Prevalence of Congenital Heart Disease Worldwide: A Systematic Review and Meta-Analysis. Journal of the American College of Cardiology. 2011;58(21):2241–2247. doi: 10.1016/j.jacc.2011.08.025. https://doi.org/10.1016/j.jacc.2011.08.025. [DOI] [PubMed] [Google Scholar]
- Verkleij-Hagoort AC, Verlinde M, Ursem NT, Lindemans J, Helbing WA, Ottenkamp J, Steegers-Theunissen RP, et al. Maternal hyperhomocysteinaemia is a risk factor for congenital heart disease. BJOG. 2006;113(12):1412–1418. doi: 10.1111/j.1471-0528.2006.01109.x. [DOI] [PubMed] [Google Scholar]
- Waller DK, Mills JL, Simpson JL, Cunningham GC, Conley MR, Lassman MR, Rhoads GG. Are obese women at higher risk for producing malformed offspring? Am J Obstet Gynecol. 1994;170(2):541–548. doi: 10.1016/s0002-9378(94)70224-1. [DOI] [PubMed] [Google Scholar]
- Wang C, Zhan Y, Wang F, Li H, Xie L, Liu B, Hua Y, et al. Parental occupational exposures to endocrine disruptors and the risk of simple isolated congenital heart defects. Pediatr Cardiol. 2015;36(5):1024–1037. doi: 10.1007/s00246-015-1116-6. [DOI] [PubMed] [Google Scholar]
- Wang KS, Liu X, Zhang Q, Aragam N, Pan Y. Parent-of-origin effects of FAS and PDLIM1 in attention-deficit/hyperactivity disorder. J Psychiatry Neurosci. 2012;37(1):46–52. doi: 10.1503/jpn.100173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg CR. Methods for detection of parent-of-origin effects in genetic studies of case-parents triads. Am J Hum Genet. 1999;65(1):229–235. doi: 10.1086/302466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenstrom KD, Johanning GL, Johnston KE, DuBard M. Association of the C677T methylenetetrahydrofolate reductase mutation and elevated homocysteine levels with congenital cardiac malformations. Am J Obstet Gynecol. 2001;184(5):806–812. doi: 10.1067/mob.2001.113845. discussion 812-807. [DOI] [PubMed] [Google Scholar]
- Wyrobek AJ, Eskenazi B, Young S, Arnheim N, Tiemann-Boege I, Jabs EW, Evenson D, et al. Advancing age has differential effects on DNA damage, chromatin integrity, gene mutations, and aneuploidies in sperm. Proc Natl Acad Sci U S A. 2006;103(25):9601–9606. doi: 10.1073/pnas.0506468103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Ralston MM, Meng X, Bongiovanni KD, Jones AL, Benndorf R, Liu Y, et al. Glutathione reductase is essential for host defense against bacterial infection. Free Radic Biol Med. 2013;61:320–332. doi: 10.1016/j.freeradbiomed.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Wen SW, Leader A, Chen XK, Lipson J, Walker M. Paternal age and birth defects: how strong is the association? Hum Reprod. 2007;22(3):696–701. doi: 10.1093/humrep/del453. [DOI] [PubMed] [Google Scholar]
- Yauk CL, Berndt ML, Williams A, Rowan-Carroll A, Douglas GR, Stampfli MR. Mainstream tobacco smoke causes paternal germ-line DNA mutation. Cancer Res. 2007;67(11):5103–5106. doi: 10.1158/0008-5472.CAN-07-0279. [DOI] [PubMed] [Google Scholar]
- Yoon PW, Rasmussen SA, Lynberg MC, Moore CA, Anderka M, Carmichael SL, Edmonds LD, et al. The National Birth Defects Prevention Study. Public Health Rep. 2001;116(Suppl 1):32–40. doi: 10.1093/phr/116.S1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan SY, Lian ZH, Zheng DZ, Gao L. Effect of fathers’ age and birth order on occurrence of congenital heart disease. J Epidemiol Community Health. 1991;45(4):299–301. doi: 10.1136/jech.45.4.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Yang W, Lu W, Etheredge AJ, Lammer EJ, Finnell RH, Shaw GM, et al. Gene variants in the folate-mediated one-carbon metabolism [FOCM] pathway as risk factors for conotruncal heart defects. Am J Med Genet A. 2012;158A(5):1124–1134. doi: 10.1002/ajmg.a.35313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.