

Abstract
Identification of molecular ligands that recognize peptides or proteins is significant but poses a fundamental challenge in chemical biology and biomedical sciences. Development of cyclic peptidomimetic library is scarce, and thus discovery of cyclic peptidomimetic ligands for protein targets is rare. Herein we report the unprecedented one-bead–two-compound (OBTC) combinatorial library based on a novel class of the macrocyclic peptidomimetics γ-AApeptides. In the library, we utilized the coding peptide tags synthesized with Dde-protected α-amino acids, which were orthogonal to solid phase synthesis of γ-AApeptides. Employing the thioether linkage, the desired macrocyclic γ-AApeptides were found to be effective for ligand identification. Screening the library against the receptor tyrosine kinase EphA2 led to the discovery of one lead compound that tightly bound to EphA2 (Kd = 81 nM) and potently antagonized EphA2-mediated signaling. This new approach of macrocyclic peptidomimetic library may lead to a novel platform for biomacromolecular surface recognition and function modulation.
Graphical Abstract
INTRODUCTION
Chemical biology and biomedical sciences are undergoing a new era of vigorous development due to the rapid discovery of new protein targets and unveiling of their biological importance. Consequently, it is an unmet need to identify ligands that recognize peptide or protein targets with high specificity and affinity,1 since such an effort could help in understanding function of proteins and lead to potential therapeutic agents for diagnosis and treatment. Combinatorial chemistry, which serves as a powerful tool for ligand screening in basic medicinal chemistry, has emerged to meet this challenge.2 The diverse library of compounds provides concurrent but independent chances for target screening. Since it provides the possibility to yield important therapeutic agents from various target proteins, the progress of drug discovery has been largely accelerated.3 As peptides are versatile molecules, which have modular chemical diversity and favorable binding activity, they are natural building blocks for combinatorial libraries for screening against various targets. Among them, macrocyclic peptides that have enhanced conformational constraint and binding affinity are widely recognized for exploring ligand–receptor interactions. Several general methods have been successfully developed to construct the macrocyclic ring systems.4,5,8–12
Recent efforts have contributed to the creation of nonnatural sequence-specific peptidomimetics.13 These peptidomimetics are developed based on the mimicry of peptide primary structure and possess a modified peptide backbone for the introduction of diverse functional side chains. Compared with natural peptides, peptidomimetics possess improved protease resistance, chemodiversity, and bioavailability.14 The past decade has witnessed noteworthy progress in the development of biomimetic oligomers, including β-peptides,15–17,19 peptoids, 18,20–23 α-aminoxypeptides,24 α/β-peptides,25,26 azapeptides, 27 and others. However, to date only a handful of peptidomimetic combinatorial libraries were systematically investigated for protein ligand identification.28 The development of the macrocyclic peptidomimetic combinatorial library is even rarer.29–31 Motivated by these findings, we have recently developed the new class of peptidomimetics, γ-AApeptides (oligomers of γ-substituted-N-acylated-N-aminoethylamino acids), which were inspired by the backbone structure of the chiral PNA.32,33 γ-AApeptides are highly resistant to proteolytic degradation and possess cellular translocation capability and have enhanced chemodiversity, making them ideal candidates as molecular probes or therapeutic agents.34 Indeed, these features have been demonstrated by our previously developed one-bead–one-compound (OBOC) linear γ-AApeptides-based combinatorial libraries capable of inhibiting Aβ aggregation and disrupting STAT3/DNA interaction.35,36 We thus envisioned that the macrocyclic combinatorial library of γ-AApeptides would be endowed with enhanced conformational rigidity and constraints compared with the linear counterparts and could be even more beneficial in the identification of bioactive ligands.
RESULTS AND DISCUSSION
Design of the Cyclic Library
The thioether moiety is widely found in nature. For instance, methionine has a thioether side chain and is virtually present in all proteins. Other natural products such as antibiotic lanthipeptides directly utilize thioether bridges to form multimacrocylic ring structures. Heinis et al. also adopted the thioether linkage to develop phage-display mediated bicyclic peptide libraries.4–7 This thioether-bridge mediated cyclization has proven to be highly efficient in cyclization, leading to cyclic peptides with not only rigidified conformational freedom but also enhanced metabolic stability. These features make them promising affinitive candidates for biological applications. By employment of a thioether-bridge approach, a novel cyclic γ-AApeptide library could be synthesized and tested against target proteins. To test our hypothesis, we embarked on the design of the cyclic γ-AApeptide library by introducing a Dmt (4,4′-dimethoxytrityl) protected mercaptoethyl carbonyl group to the secondary amine in the first γ-AApeptide building block (Figure 1) on the solid phase. After another three γ-AApeptide building blocks were assembled subsequently, the 4-(bromomethyl)benzoyl group was attached to the N-terminal amino group of the sequence. Next, the Dmt group was removed, and the γ-AApeptide could be cyclized under nearly neutral conditions with virtually quantitative yield due to the high efficiency of sulfur-mediated SN2 reaction.
Figure 1.

Synthesis of the thioether bridged one-bead–two-compound macrocyclic γ-AApeptide library. See Supporting Information for details.
Molecular Dynamics (MD)
In order to assess the potential conformational rigidity of this class of cyclic γ-AApeptides, we conducted molecular dynamics (MD) simulations of the cyclic backbone and linear backbone in explicit water (Supporting Information, Figure S5). Consistent with our hypothesis, MD simulation (Supporting Information, Figure S5) suggested the backbone of thioether bridged cyclic γ-AApeptides is much more conformationally rigid than that of the linear one, leading to possibility of identification of novel ligands with high affinity.
Library Diversity and Decoding
To ensure the development of relatively unbiased cyclic library, a diverse set of hydrophobic, cationic, and negatively charged side chains were chosen. Briefly, due to the structural nature of γ-AApeptides, four chiral side chains came from the side chains of five different N-Alloc protected γ-AApeptide building blocks (R, Figure 1). The other three side chains were introduced by acylating the secondary amino group with six different carboxylic acids or acyl chlorides after deprotection of the alloc group (R′, Figure 1). Thus, by using the split and pool method, the theoretical diversity of the library was expected to be 135 000 (Figure 1), and 405 000 beads were used for the preparation of the library.
Unlike linear γ-AApeptide library,35,36 the direct sequencing of cyclic γ-AApeptides was no longer feasible using MS/MS. Instead, the strategy of a one-bead–two-compound (OBTC) library containing both ligands and analyzable coding sequences on the same bead was desired.37,38 Peptides consisting of α- amino acids could serve well as coding sequences since the MS/MS pattern of amino acid fragments is unambiguous. To this end, we developed a highly effective approach to synthesize coding peptides by using Dde ((1-(4,4-dimethyl-2,6-dioxacyclohexylidene) ethyl) protected α-amino acids. The deprotection of Dde was very mild using NH2OH·HCl and imidazole39 and fully orthogonal to chemistry engaged in the synthesis of thioether-bridged cyclic γ-AApeptides.
Synthesis of the Library
To prepare the desired OBTC cyclic γ-AApeptide combinatorial library, TentaGel beads (200–250 μm; 1.5 nmol/bead)) were soaked overnight in water and then exposed to 1:1 (v/v) DCM/Et2O containing 0.5 equiv of di-tert-butyl decarbonate (Boc2O) (Figure 1).40,41 This was expected to possess Boc protection of amino groups on the outer surface of the beads since the interior of the beads still remains in water. After washing with DMF, the interior of the beads was allowed to react with the Met, the amino acid facilitating coding peptide cleavage upon cyanogen bromide (CNBr) treatment. After removal of the Fmoc protecting group, the beads were split into five equal aliquots and reacted with five different Dde protected amino acids, respectively, so as to establish a coding tag representing the first γ-AApeptide building block on the outer layer. Subsequently, the Boc group of the outer layer was removed by TFA, followed by the attachment of five Alloc γ-AApeptide protected building blocks. Next, the Alloc group was removed by Pd(PPh3)4 and Me2NH·BH3, and the Dmt protected 3-mercaptopropanoic acid was added to react with the secondary amino group. The beads were pooled and split into five aliquots again. After the Dde group was removed, the second set of the Dde protected amino acids was added to introduce the coding tag for the second γ-AApeptide building block on the outer layer of the beads. These steps were repeated 3 more times. Compared with the cycle of the first building block, the only difference in the subsequent synthetic process on the outer layer was that after the Alloc group was removed, the N group of the γ-AApeptide building block was reacted with a different carboxylic acid or acyl chloride to introduce diverse side chains. Since each γ-AApeptide building block bears two side chains, two Dde protected amino acids were used to code each building block (Figure 1). Finally, the 4-(bromomethyl)benzoyl chloride was used to cap the N-terminus of γ-AApeptides on the outer layer, followed by selective removal of the Dmt group on the thiol linker with 2% TFA in DCM. The cyclization of γ-AApeptides was achieved in the presence of the ammonium carbonate ((NH4)2CO3), which occurred on the surface of the beads only due to the lack of cyclization linker in the interior of the beads. The deprotection of side chain protecting groups was finally conducted in 94% TFA, 2% triisopropylsilane, 2% water, and 2% thioanisole (v:v:v:v). Quality of the beads was excellent, as evidenced by the fact that 8 out 10 randomly selected beads showed unambiguous MS/MS fragmentation patterns by MALDI and were able to provide information on the coding peptide sequence almost instantly.
Library Screening and Binding Affinity
With the success of obtaining the cyclic γ-AApeptide combinatorial library, we moved forward to examine its potential for identification of valuable biological ligands. EphA2 (ephrin type-A receptor 2) was chosen due to its prominent role in the pathogenesis of various tumors.42 EphA2 (Figure 2) belongs to the family of Eph receptor tyrosine kinases that regulate tissue development and patterning of the visual system and play a critical role in mediating the cell–cell communication and angiogenesis.43,44 Recent findings suggest that EphA2 overexpression is a key factor contributing to multiple cancers such as melanoma, ovarian, lung, and breast.45–47 Therefore, EphA2 is a promising target for cancer therapeutic development. Considerable efforts have started to be made to identify inhibitors that block the capability of EphA2 for the phosphorylation of its downstream protein substrates, thereby dampening EphA2 mediated cell signaling. However, to date, only limited success was achieved.48 Thus, it is compelling to identify ligands from our macrocyclic γ-AApeptide library that bind to EphA2 with high affinity.
Figure 2.

EphA2 signaling and inhibition pathway.
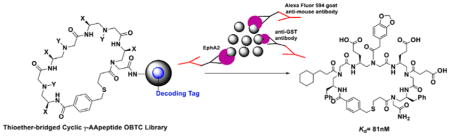
To conduct the library screening, we first incubated beads with GST tagged intracellular domain of EphA2 protein (Figure 3A and Supporting Information). After the thorough wash and the following incubation with the mouse anti-GST antibody, the beads were washed again and treated with the Alexa Fluor 594 labeled goat-anti mouse secondary antibody. Ten beads emitting red fluorescence were identified under the fluorescence microscope and picked up as the putative positive hits. Subsequently, these beads were incubated with guanidium chloride (GdmCl) to denature any potential proteins stuck on the surfaces of the beads. The coding peptides in the inner layers of the beads were then cleaved off the beads by treatment with CNBr and subsequently sequenced by tandem MS/MS of MALDI. Among the 10 hits, seven different peptide structures could be determined unambiguously. (The rest to the three hits had poor fragmentation which were very difficult for us to work out the structures. This may be caused by the cleavage step.) Next, the corresponding sequences of the seven cyclic γ-AApeptides conjugated with fluorescein isothiocyanate (FITC) labels were resynthesized and measured for their binding affinity toward EphA2 by fluorescence polarization (FP) assay. Fortunately, the most potent hit, AApeptide 2 (FITC labeled AApeptide 1) (Figure 3B), exhibited excellent binding affinity to EphA2 with a Kd value of 81 nM (Figure 3C). The potential binding to the GST tag was excluded as 2 only showed negligible binding affinity to GST protein with a Kd of 14.2 μM, which is ~170-fold weaker than that of EphA2 binding (Figure 3D). Besides, the GST protein, another two kinases AKT1-PKBα and NFKBIA were chosen to test the selectivity of the 1, which showed Kd of 3.62 μM and 0.741 μM (Supporting Information, Figure S8), respectively, demonstrating the binding of 1 toward EphA2 is selective. Moreover, we treated C13 cells with 1 μM and 5 μM 2, and the diffuse fluorescence was clearly visible in 1 h (Supporting Information, Figure S4), suggesting the compound is cell permeable.
Figure 3.

(A) Screening of the γ-AApeptide library. (B) Structure of 2 (FITC labeled lead compound 1). (C) Kd of the compounds to the EphA2. (D) Kd of the compounds to the GST.
Inhibition Kinase Activity of EPhA2
Given the strong binding affinity of 1 (Figure 4A) toward EphA2, we next performed assays to evaluate its biological activity. The initial in vitro kinase assay (Figure 4B) showed that 2 μM 1 could completely inhibit EphA2 kinase activity by preventing phosphorylation of its substrate poly(Glu-Tyr). Another compound 9 (Figure S1, Supporting Information), with the same molecular scaffold, failed to inhibit activity of EphA2, suggesting that recognition of 1 toward EphA2 was specific. At the same concentration, 1 also greatly suppressed the autocatalytic activity of EphA2 (Figure 4C). Both above-mentioned in vitro kinase assays suggested 1 is a potent inhibitor of EphA2 kinase activity. Inspired by the results, we further investigated the ability of 1 for the regulation of catalytic activity in an ovarian cancer cell line C13 cells which displays high expression of EphA2.49 As shown in Figure 4D, the phosphorylation level of EphA2 was inhibited in a dose-dependent fashion with the increased concentrations of 1. The IC50 of 1 for the inhibition of EphA2 inhibition was approximately 5 μM.
Figure 4.

(A) Structure of lead compound 1. (B) In vitro kinase assay was performed using 50 ng of EphA2 and 50 μg of poly(Glu-Tyr) as a substrate containing 2 μM 1. Quantification of substrate phosphorylation was shown. (C) Autophosphorylation of EphA2 was detected by incubating 50 ng of EphA2 with 2 μM 1 in the kinase assay buffer. Immunoblot analysis with pTyr antibody was performed. (D) C13 cells were treated with the indicated concentrations of 1 for 24 h. Immunoblot analysis of EphA2-Ser897 was performed. Total EphA2 was used as a control. The extent of phosphorylation was quantified using ImageJ software. Densitometric analysis was performed by SPSS.
Cell Migration and Invasion
Since EphA2 was shown to play a critical role in cell migration,50 we next tested the effect of 1 on cell migration and invasion. 1 was applied to C13 cells and analyzed using a scratch-wound motility assay. The data in Figure 5A showed a time- and dose-dependent downregulation of cell migration in response 1 treatment, as shown by as much as 70% delay in wound closure at 24 h post-treatment with the highest dose of 1 (Figure 5A). Similar results were seen in Matrigel-coated Transwell assays, treatment of 10 μM 1 led to around 70% and 80% decrease in cell migration (Figure 5B) and invasion (Figure 5C), respectively, suggesting EphA2 mediated cell signaling was significantly suppressed. The inactive compound 9 was also tested for comparison (Supporting Information, Figure S2), which as expected did not show capability to prevent migration. A linear compound AApeptide 10 was also tested (Figure S3), and the result was consistent with the MD simulation because 10 was much less potent. Taken together, the ability of 1 for the strong inhibition of EphA2 activity in cells suggests that cyclic γ-AApeptides possess excellent cell permeability, augmenting their future development in biomedical sciences.
Figure 5.

(A) Wound-healing assay. Images were taken 0, 12, and 24 h after wound formation. Data are presented as the mean ± SD of triplicate experiments and using image J software. C13 cells treated with 10 μM 1 or vehicle and were subjected to migration (B) and invasion (C) assays. Transwell data were determined by the number of the migrated cells, and the value from parental cells was arbitrarily set at 100%.
CONCLUSION
In summary, a new class of macrocyclic peptidomimetic combinatorial library has been developed. With the unique γ-AApeptide backbone, which ensures the chemodiversity in the library, this new method has great potential to be a rich source in protein/peptide ligands identification. In addition, compared with previous linear peptide library developed by our group, the thioether bridged macrocyclic γ-AApeptide library has the dual advantages of significantly enhanced conformal rigidity of the backbones and cell permeability. This led to higher promise in the identification of more potent and useful ligands/molecular probes. Furthermore, the new encoding approach of Dde peptide tags also greatly increases the possibility and ease of the structural elucidation of putative hits. The promise of this macrocyclic library was manifested by the identification of potent ligands that specifically target the EphA2 receptor tyrosine kinase. With a Kd value of 81 nM, compound 1 was found to be a potent inhibitor of the EphA2 signaling in both in vitro and cellular assays. Currently, the library is being used as a new platform for screening against various targets in our group, and the in vivo study of compound 1 is underway.
EXPERIMENTAL SECTION
General Information
Fmoc-protected amino acids were purchased from Chem-impex (Wood Dale, IL). TentaGel resin (0.23 mmol/g) was purchased from RAPP Polymere (Tuebingen, Bermany). Rink amide-MBHA resin (0.55 mmol/g) was purchased from GL Biochem (Shanghai, China). 1-Hydroxybenzotriazole wetted with no less than 20% wt water (HOBt), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), 5,5-dimethyl-1,3-cyclohexanedione, and 4,4′-dimethoxytrityl chloride were purchased from Oakwood Chemical (Estill, SC). 4-(Bromomethyl)benzoic acid was purchased from AK-Scientific (Union City, CA). 3-Mercaptopropionic acid was purchased from TCI (Tokyo, Japan). Fluorescein isothiocyanate (FITC) was purchased from Chemodex (Gallen, Switzerland). Solid phase synthesis was conducted in peptide synthesis vessels on a Burrell Wrist-Action shaker. γ-AApeptides were analyzed and purified on a Waters Breeze 2 HPLC system and then lyophilized on a Labcono lyophilizer. The purity of the compounds was determined to be >95% by analytical HPLC. Masses of γ-AApeptides and the MS/MS analysis were obtained on an Applied Biosystems 4700 Proteomics analyzer. 1H NMR spectra were recorded at 500 MHz using TMS as the internal standard. 13C NMR spectra were recorded at 125 MHz using TMS as the internal standard. The multiplicities are reported as follows: singlet (s), doublet (d), doublet of doublets (dd), triplet (t), quartet (q), multiplet (m). Coupling constants are reported in hertz (Hz). High resolution mass spectra were obtained on an Agilent 6220 using electrospray ionization time-of-flight (ESI-TOF). Cell culture medium was purchased from Gibco (Rockford, IL), fetal bovine serum (FBS) was purchased from Peak Serum (Fort Collins, CO), and penicillin–streptomycin was purchased from Invitrogen (Carlsbad, CA). GST-EphA2 recombinant protein, poly(Glu-Tyr), and Alexa Fluor 594 goat anti-mouse antibody were purchased from Life Technologies (Carlsbad, CA) and Sigma-Aldrich (St. Louis, MO), respectively. Phosphotyrosine HRP conjugated antibody (pTyr) was purchased from R&D Biosystems (Minneapolis, MN). pEphA2-Ser897 and EphA2 antibodies were purchased from Cell Signaling Technologies (Danvers, MA). Anti-GST antibody was purchased from Santa Cruz Biotechnology (Dallas, TX). All solvents and other chemical reagents were obtained from Sigma-Aldrich (St. Louis, MO) and were used without further purification.
Synthesis of the Dmt Protected Mercaptopropionic Acid (11)
4,4′-Dimethoxytrityl chloride (6.38 g, 18.82 mmol) was dissolved in 40 mL of CH2Cl2 containing 3-mercaptopropionic Acid (1.64 mL, 18.82 mmol) at room temperature. The triethylamine (3.93, 22.58 mmol) was slowly added to the above solution. The solution was stirred at room temperature for 4–6 h. After that the mixture was evaporated under reduced pressure, and the residue was washed with saturated citric acid and extracted with ethyl acetate (30 mL ×3). The organic layer was dried over anhydrous Na2SO4, and the solvent was evaporated. The residue was purified by flash column chromatography (hexane/ethyl acetate 1:1) to afford the desired product as a light yellow solid (80% yield). 1H NMR (500 MHz, CDCl3): δ 7.42–7.44 (d, J = 8.00 Hz, 2H), 7.34–7.35 (d, J = 9.00 Hz, 2H), 7.29(t, J = 7.50 Hz, 2H), 7.21 (t, J = 7.50 Hz, 1H), 6.84–6.82 (d, J = 9.00 Hz, 2H), 3.79 (s, 6H), 2.49 (t, J = 7.50 Hz, 2H), 2.30 (t, J = 7.50 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ 178.2, 158.1, 145.2, 137.0, 130.7, 129.4, 127.9, 126.6, 113.2, 66.0, 55.2, 33.5, 26.6.
Synthesis of the 4-(Bromomethyl)benzoyl Chloride (12)
The 4-(bromomethyl)benzoic acid (5 g, 23.25 mmol) was dissolved in 10 mL of thionyl chloride and reflux for 5 h. The excess thionyl chloride was removed under reduced pressure to afford the desired product as a white solid and directly use without purification (85% yield).51 1H NMR (500 MHz, CDCl3): δ 8.06 (d, J = 5.00 Hz, 2H), 7.52 (d, J = 10.00 Hz, 2H), 4.50 (s, 2H). 13C NMR (125 MHz, CDCl3): δ 167.7, 145.3, 132.9, 131.8, 129.6, 31.4.
Synthesis of 2-Acetyl-5,5-dimethylcyclohexane-1,3-dione (13)
To a 100 mL round-bottom flask was added 5,5-dimethylcyclohexane-1,3-dione (10 g, 71.34 mmol), N,N-diisopropylethylamine (14.91 mL, 85.6 mmol), 4-dimethylaminopyridine (435.76 mg, 3.57 mmol), and 50 mL of DCM. The mixture was stirred in an ice bath to which acetyl chloride (6.08 mL, 85.6 mmol) was added. The reaction was warmed up to room temperature and allowed to stir for 8 h. The solvent was evaporated, and the residue was washed with 1 M HCl and then extracted with ethyl acetate (30 mL × 3). The organic layer was dried over anhydrous Na2SO4 and then removed in vacuo. The residue was purified by flash column chromatography (hexane/ethyl acetate 1:1) to afford the 2-acetyl-5,5-dimethylcyclohexane-1,3-dione as a yellowish solid (11 g, yield 85%).52 1H NMR (500 MHz, CDCl3): δ 2.54 (s, 3H), 2.48 (s, 3H), 2.3 (s, 2H), 1.01 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 202.3, 197.8, 195.1, 112.3, 52.4, 46.8, 30.6.
Synthesis of Dde Protected Amino Acids
The L-amino acid (1 equiv) was suspended in a solution of the 2-acetyl-5,5-dimethylcyclohexane-1,3-dione (1.3 equiv) in absolute ethanol (~50 mL). Triethylamine (1.5 equiv) was added, and the reaction mixture was refluxed for 18 h. The resulting yellow solution was cooled and concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (50 mL) and washed with 1 M HCl (50 mL × 2). The organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. Addition of Et2O (~40 mL) to the residue resulted in immediate white precipitate, which was filtered and washed with cold Et2O to afford the title compound as an off-white crystalline solid (~70%).53
Dde-Ala-OH (14)
White solid. 1H NMR (500 MHz, DMSO-d6): δ 13.51 (d, J = 5.00 Hz, 1H), 4.61 (t, J = 5.00 Hz, 1H), 2.48 (s, 3H), 2.27 (s, 4H), 1.41 (d, J = 5.00 Hz, 3H), 0.92 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 197.4, 172.9, 172.5, 107.6, 52.8, 51.6, 30.3, 28.3,19.1, 18.1. HRMS (ESI) ([M + H]+) calcd for C13H20NO4, 254.1392; found, 254.1396.
Dde-Val-OH (15)
White solid. 1H NMR (500 MHz, CDCl3): δ 13.6 (d, J = 5.00 Hz, 1H), 10.97 (s, 1H), 4.61 (t, J = 5.00 Hz, 1H), 2.5 (s, 3H),2.39 (s, 4H), 2.36 (m, 1H), 1.08 (d, J = 5.00 Hz, 3H), 1.04 (d, J = 5.00 Hz, 3H), 1.0 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 174.3, 171.6, 107.9, 62.3, 51.9, 31.1, 30.1, 28.1, 19.1, 18.7, 17.0. HRMS (ESI) ([M + H]+) calcd for C15H24NO4, 282.1705; found, 282.1717.
Dde-Phe-OH (16)
White solid. 1H NMR (500 MHz, CDCl3): δ 13.71 (d, J = 5.00 Hz, 1H), 7.18–7.27 (m, 5H), 4.57–4.61(m, 1H), 3.05–3.09 (m, 2H), 2.36 (s, 4H), 2.20 (s, 3H), 1.00 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 198.1, 173.6, 171.0,135.5, 129.4, 128.6, 127.4, 107.9, 58.3, 52.4, 45.5, 39.3, 30.1, 28.0, 18.1, 8.5. HRMS (ESI) ([M + H]+) calcd for C19H24NO4, 330.1705; found, 330.1714.
Dde-Leu-OH (17)
White solid. 1H NMR (500 MHz, CDCl3): δ 13.60 (d, J = 10.00 Hz, 1H), 10.01 (s, 1H), 4.57–4.61 (m, 1H), 2.50 (s, 3H), 2.37 (s, 4H), 1.82 (m, 2H), 1.77 (m, 1H), 0.99 (s, 6H), 0.95 (d, J = 5.00 Hz, 3H), 0.89 (d, J = 5.00 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 198.9, 173.9, 107.9, 54.9, 52.3, 45.6, 41.3, 30.1, 28.2, 24.8, 22.7, 21.7, 18.7, 8.4. HRMS (ESI) ([M + H]+) calcd for C16H26NO4, 296.1862; found, 296.1873.
Dde-Glu(OBn)-OH (18)
Pale yellow solid. 1H NMR (500 MHz, DMSO-d6): δ 13.77 (d, J = 5.00 Hz, 1H), 10.50 (s, 1H), 7.33 (s, 5H), 5.10 (s, 2H), 4.55 (m, 1H), 2.53–2.59 (m, 2H), 2.51 (s, 3H), 2.39 (s, 4H), 2.21–2.25 (m, 2H), 1.01 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 174.3, 171.9, 171.2, 135.4, 128.6, 128.4, 128.3, 128.2, 66.7, 55.4, 52.3, 30.2, 29.6, 28.2, 27.7, 18.7. HRMS (ESI) ([M + H]+) calcd for C22H28NO6, 402.1917; found, 402.1925.
Dde-Asp-OH (19)
White solid. 1H NMR (500 MHz, DMSO-d6): δ 13.56 (d, J = 10.00 Hz, 1H), 4.84 (m, 1H), 2.90 (dd, J = 15.00, 5.00 Hz, 1H), 2.78 (dd, J = 15.00, 5.00 Hz, 1H), 2.46 (s, 3H), 2.27 (s, 4H), 1.36 (s, 9H), 0.92 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 172.3, 170.7, 168.7, 107.7, 81.5, 52.5, 38.2, 30.1, 38.3, 28.0, 17.9. HRMS (ESI) ([M + H]+) calcd for C18H28NO6, 354.1917; found, 354.1929.
Dde-Glu-OH (20)
White solid. 1H NMR (500 MHz, DMSO-d6): δ 13.54 (d, J = 10.00 Hz, 1H), 4.26 (q, J = 5.00 Hz, 1H), 2.43 (s, 3H), 2.28 (s, 4H), 2.24–2.26 (m, 2H), 1.90–2.10 (m, 2H), 1.36 (s, 9H), 0.93 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 172.7, 171.8, 107.4, 80.3, 54.2, 52.1, 45.2, 30.8, 30.1, 28.3, 28.1, 27.8. HRMS (ESI) ([M + H]+) calcd for C19H30NO6, 368.2073; found, 368.2075.
Dde-Lys-OH (21)
White solid. 1H NMR (500 MHz, CDCl3): δ 13.69 (m, 1H), 10.24 (s, 1H), 4.80 (m, 1H), 4.40 (m, 2H), 3.09 (m, 2H), 2.50 (s, 3H), 2.34 (s, 4H), 1.95 (m, 2H), 1.50 (m, 2H), 1.40 (s, 9H), 1.01 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 173.3, 172.9, 172.0, 171.6, 155.9, 107.6, 80.9, 78.9, 60.1, 56.1, 40.9, 39.8, 32.1, 30.3, 29.1, 27.9, 21.6, 18.4, 13.9. HRMS (ESI) ([M + H]+) calcd for C21H35N2O6, 411.2495; found, 411.2497.
Fmoc-Lys(Dde)-OH (22).54
Pale yellow solid. 1H NMR (500 MHz, CDCl3): δ 13.29 (s, 1H), 7.73–7.74 (d, J = 5.00 Hz, 2H), 7.58 (t, J = 5.00 Hz, 2H), 7.37 (t, J = 5.00 Hz, 2H), 7.27 (t, J = 5.00 Hz, 2H), 5.79 (d, J = 10.00 Hz, 1H), 4.42–4.46 (m, 1H), 4.35–4.37 (d, J = 10.00 Hz, 2H), 4.18 (t, J = 5.00 Hz, 1H), 3.38–3.39 (m, 2H), 2.53 (s, 3H), 2.35 (s, 4H), 1.78–1.80 (m, 2H), 1.70–1.72 (m, 2H), 1.47–1.57 (m, 2H), 1.00 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 198.5, 174.7, 174.3, 156.2, 143.8, 143.7, 141.3, 127.7, 127.0, 125.1, 119.9, 107.8, 67.1, 53.4, 52.2, 47.1, 43.4, 31.9, 30.2, 28.3, 28.2, 22.4, 21.1, 18.3, 14.2. HRMS (ESI) ([M + H]+) calcd for C31H37N2O6, 533.2652; found, 533.2661.
Synthesis of Cyclic γ-AApeptides Hits
After structures of putative hits were determined by MALDI MS/MS, the hits and its FITC labeled analogues were resynthesized on the Rink amide resin and confirmed by Applied Biosystems 4700 Proteomics analyzer (Scheme S2). For the synthesis of the fluorescent cyclic peptide, the Fmoc-Lys(Dde)-OH was first attached to the Rink amide resin. The Fmoc protection group was then removed, followed by the desired building blocks needed for the sequence synthesis. After the γ- AApeptides were cyclized, the Dde group was removed. Then FITC (2 equiv) and DIPEA (6 equiv) in DMF were added to the resin and shaken for 12 h at room temperature. The FITC labeled cyclic γ-peptide was cleaved by 1:1 (v/v) DCM/TFA containing 2% triisopropylsilane. The crude was purified by the Waters HPLC system with flow rate of 0.8 mL/min with a linear gradient from 5% to 100% (CH3CN in water) in 40 min (Figure S5).
Fluorescence Polarization (FP)
The binding affinity (Kd) of the hits was obtained by fluorescence polarization (FP). The FP experiment was performed by incubating 50 nM FITC labeled AApeptide with EphA2 (0.0625–2 μM) in 1× PBS. The binding affinity of the lead compound to the GST protein (Kd) was obtained by incubating 50 nM FITC labeled AApeptide in GST ranging from 0.3125 to 55 μM. Dissociation constants (Kd) were determined by plotting the fluorescence anisotropy values as a function of protein concentration, and the plots were fitted to the following equation (Figures S7 and S8).37
The Lst is the concentration of the AApeptide and the x stands for the concentration of the protein. The experiments were conducted in triplicates and repeated three times.
In Vitro Kinase Assays and Immunoblotting
In vitro kinase assay to evaluate EphA2 kinase activity was performed as described before.46 Briefly, 50 μL of kinase reaction containing 50 ng of recombinant EphA2, 50 μg of poly(Glu-Tyr) and 200 μM ATP, and 2 μM 1 and 9 (negative control) were incubated at 30 °C for 40 min. The reaction was stopped by addition of gel loading buffer. The samples were boiled for 5 min and SDS–PAGE was performed. Immunoblotting with pTyr-HRP antibody (1:2000 dilution) was performed to detect substrate phosphorylation. The extent of phosphorylation was quantified using ImageJ software. For studying the activity of the compounds in cells, C13 cells were treated at indicated doses for 24 h. The cells were lysed in lysis buffer, and immunoblot analysis was performed. Densitometric analysis was performed by SPSS. The experiments were conducted in triplicates and repeated three times.
Scratch-Wound Motility Assay
C13 cells were trypsinized, and 105 cells were reseeded on a 12-well tissue culture plate. After 12 h, the attached cells were scratched with a 200 mL pipet tip and cell migration was observed for up to 24 h. 0 h images were captured using a Nikon ECLIPSE microscope. The plates were placed back at 37 °C and 5% CO2 for 24 h, and another set of images were captured of the same wounds. The wound widths were measured by ImageJ (version 1.50). The experiments were conducted in triplicates and repeated three times.
Matrigel-Coated Transwell Assays
The assays were performed by transfected the C13 cell line with control EphA2 for 48 h. After that the cells were plated into the top chambers and treated with either vehicle or 10 μM 1. Twenty-four hours later cells in the top chamber were scraped and migrated cells were fixed with crystal violet staining. Transwell data were determined by the number of the migrated cells, and the value from parental cells was arbitrarily set at 100%. The experiments were conducted in triplicates and repeated three times.
Supplementary Material
Acknowledgments
This work was generously supported by NSF CAREER Grant 1351265 (J.C.), NIH Grant 1R01GM112652-01A1 (J.C.), National Natural Science Foundation of China (Grants 81520108031, 81473478), Shanghai Outstanding Academic Leaders Plan (Grant 16XD1403600), The Science Foundation for Shanghai Committee of Science Project (Grant 14430722900) Shanghai Outstanding Medical Academic Leader, Shanghai Three-Year Action Plan of Traditional Chinese Medicine (Grants ZY3-CCCX-3-3012, ZY3-CCCX-2-1003). We thank Ning Ma for technical assistance with the parametrization. Computer time was provided by USF Research Computing, sponsored in part by NSF MRI CHE-1531590.
ABBREVIATIONS USED
- EphA2
ephrin type-A receptor 2
- DMF
dimethylformamide
- DCM
dichloromethane
- HOBt
hydroxybenzotriazole
- DIPEA
N,N-diisopropylethylamine
- DIC
N,N′-diisopropylcarbodiimide
- TFA
trifluoroacetic acid
- FITC
fluorescein isothiocyanate
- Kd
dissociation constant
- Tris
tris(hydroxymethyl)-aminomethane
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem. 7b01280.
Procedures for library preparation, library screening, the bioactivity assay, cell permeability test, molecular dynamics simulations, list of hits, the determination of decoding sequences, the Kd data of other hits and kinases, 1H and 13C NMR spectra, and HPLC traces (PDF)
Molecular formula strings and some data (CSV)
References
- 1.Kodadek T. The rise, fall and reinvention of combinatorial chemistry. Chem Commun. 2011;47:9757–9763. doi: 10.1039/c1cc12102b. [DOI] [PubMed] [Google Scholar]
- 2.Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. A new type of synthetic peptide library for identifying ligand-binding activity. Nature. 1991;354:82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 3.Aquino C, Sarkar M, Chalmers MJ, Mendes K, Kodadek T, Micalizio GC. A biomimetic polyketide-inspired approach to small-molecule ligand discovery. Nat Chem. 2012;4:99–104. doi: 10.1038/nchem.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen S, Rentero Rebollo I, Buth SA, Morales-Sanfrutos J, Touati J, Leiman PG, Heinis C. Bicyclic peptide ligands pulled out of cysteine-rich peptide libraries. J Am Chem Soc. 2013;135:6562–6569. doi: 10.1021/ja400461h. [DOI] [PubMed] [Google Scholar]
- 5.Heinis C, Rutherford T, Freund S, Winter G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat Chem Biol. 2009;5:502–507. doi: 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]
- 6.Lee JH, Meyer AM, Lim HS. A simple strategy for the construction of combinatorial cyclic peptoid libraries. Chem Commun. 2010;46:8615–8617. doi: 10.1039/c0cc03272g. [DOI] [PubMed] [Google Scholar]
- 7.Hipolito CJ, Suga H. Ribosomal production and in vitro selection of natural product-like peptidomimetics: The FIT and RaPID systems. Curr Opin Chem Biol. 2012;16:196–203. doi: 10.1016/j.cbpa.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 8.Qian Z, Xu X, Amacher JF, Madden DR, Cormet-Boyaka E, Pei D. Intracellular delivery of peptidyl ligands by reversible cyclization: discovery of a PDZ domain inhibitor that rescues CFTR activity. Angew Chem, Int Ed. 2015;54:5874–5878. doi: 10.1002/anie.201411594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Upadhyaya P, Qian Z, Selner NG, Clippinger SR, Wu Z, Briesewitz R, Pei D. Inhibition of ras signaling by blocking Ras–effector interactions with cyclic peptides. Angew Chem, Int Ed. 2015;54:7602–7606. doi: 10.1002/anie.201502763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawamura A, Munzel M, Kojima T, Yapp C, Bhushan B, Goto Y, Tumber A, Katoh T, King ON, Passioura T, Walport LJ, Hatch SB, Madden S, Muller S, Brennan PE, Chowdhury R, Hopkinson RJ, Suga H, Schofield CJ. Highly selective inhibition of histone demethylases by de novo macrocyclic peptides. Nat Commun. 2017;8:14773–14782. doi: 10.1038/ncomms14773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaniraj PJ, Maayan G. A facile strategy for the construction of cyclic peptoids under microwave irradiation through a simple substitution reaction. Org Lett. 2015;17:2110–2113. doi: 10.1021/acs.orglett.5b00696. [DOI] [PubMed] [Google Scholar]
- 12.Lee KJ, Lim HS. Facile method to sequence cyclic peptides/peptoids via one-pot ring-opening/cleavage reaction. Org Lett. 2014;16:5710–5713. doi: 10.1021/ol502788e. [DOI] [PubMed] [Google Scholar]
- 13.Wu YD, Gellman S. Peptidomimetics. Acc Chem Res. 2008;41:1231–1232. doi: 10.1021/ar800216e. [DOI] [PubMed] [Google Scholar]
- 14.Patch JA, Barron AE. Mimicry of bioactive peptides via nonnatural, sequence-specific peptidomimetic oligomers. Curr Opin Chem Biol. 2002;6:872–877. doi: 10.1016/s1367-5931(02)00385-x. [DOI] [PubMed] [Google Scholar]
- 15.Cheng RP, Gellman SH, DeGrado WF. β-Peptides: From structure to function. Chem Rev. 2001;101:3219–3232. doi: 10.1021/cr000045i. [DOI] [PubMed] [Google Scholar]
- 16.Karlsson AJ, Pomerantz WC, Weisblum B, Gellman SH, Palecek SP. Antifungal activity from 14-helical β-peptides. J Am Chem Soc. 2006;128:12630–12631. doi: 10.1021/ja064630y. [DOI] [PubMed] [Google Scholar]
- 17.Kritzer JA, Lear JD, Hodsdon ME, Schepartz A. Helical β-peptide inhibitors of the p53-hDM2 interaction. J Am Chem Soc. 2004;126:9468–9469. doi: 10.1021/ja031625a. [DOI] [PubMed] [Google Scholar]
- 18.Laursen JS, Engel-Andreasen J, Olsen CA. β-Peptoid foldamers at last. Acc Chem Res. 2015;48:2696–2704. doi: 10.1021/acs.accounts.5b00257. [DOI] [PubMed] [Google Scholar]
- 19.Seebach D, Gardiner J. β-peptidic peptidomimetics. Acc Chem Res. 2008;41:1366–1375. doi: 10.1021/ar700263g. [DOI] [PubMed] [Google Scholar]
- 20.Armand P, Kirshenbaum K, Goldsmith RA, Farr-Jones S, Barron AE, Truong KTV, Dill KA, Mierke DF, Cohen FE, Zuckermann RN, Bradley EK. NMR determination of the major solution conformation of a peptoid pentamer with chiral side chains. Proc Natl Acad Sci U S A. 1998;95:4309–4314. doi: 10.1073/pnas.95.8.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu CW, Kirshenbaum K, Sanborn TJ, Patch JA, Huang K, Dill KA, Zuckermann RN, Barron AE. Structural and spectroscopic studies of peptoid oligomers with α-chiral aliphatic side chains. J Am Chem Soc. 2003;125:13525–13530. doi: 10.1021/ja037540r. [DOI] [PubMed] [Google Scholar]
- 22.Stringer JR, Crapster JA, Guzei IA, Blackwell HE. Extraordinarily robust polyproline type I peptoid helices generated via the incorporation of α-chiral aromatic N-1-naphthylethyl side chains. J Am Chem Soc. 2011;133:15559–15567. doi: 10.1021/ja204755p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang ML, Benson MA, Shin SBY, Torres VJ, Kirshenbaum K. Amphiphilic cyclic peptoids that exhibit antimicrobial activity by disrupting staphylococcus aureus membranes. Eur J Org Chem. 2013;2013:3560–3566. [Google Scholar]
- 24.Li X, Wu YD, Yang D. α-Aminoxy Acids: New possibilities from foldamers to anion receptors and channels. Acc Chem Res. 2008;41:1428–1438. doi: 10.1021/ar8001393. [DOI] [PubMed] [Google Scholar]
- 25.Berlicki Ł, Pilsl L, Wéber E, Mándity IM, Cabrele C, Martinek TA, Fülöp F, Reiser O. Unique α, β- and αα, β, β-peptide foldamers based on cis-β-aminocyclopentanecarboxylic acid. Angew Chem Int Ed. 2012;51:2208–2212. doi: 10.1002/anie.201107702. [DOI] [PubMed] [Google Scholar]
- 26.Horne WS, Johnson LM, Ketas TJ, Klasse PJ, Lu M, Moore JP, Gellman SH. Structural and biological mimicry of protein surface recognition by α/β-peptide foldamers. Proc Natl Acad Sci U S A. 2009;106:14751–14756. doi: 10.1073/pnas.0902663106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee HJ, Song JW, Choi YS, Park HM, Lee KB. A theoretical study of conformational properties of N-methyl azapeptide derivatives. J Am Chem Soc. 2002;124:11881–11893. doi: 10.1021/ja026496x. [DOI] [PubMed] [Google Scholar]
- 28.Trader DJ, Simanski S, Kodadek T. A reversible and highly selective inhibitor of the proteasomal ubiquitin receptor Rpn13 Is toxic to multiple myeloma cells. J Am Chem Soc. 2015;137:6312–6319. doi: 10.1021/jacs.5b02069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simpson LS, Kodadek T. A cleavable scaffold strategy for the synthesis of one-bead one-compound cyclic peptoid libraries that can be sequenced by tandem mass spectrometry. Tetrahedron Lett. 2012;53:2341–2344. doi: 10.1016/j.tetlet.2012.02.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JH, Kim HS, Lim HS. Design and facile solid-phase synthesis of conformationally constrained bicyclic peptoids. Org Lett. 2011;13:5012–5015. doi: 10.1021/ol201773f. [DOI] [PubMed] [Google Scholar]
- 31.Oh M, Lee JH, Moon H, Hyun YJ, Lim HS. A chemical inhibitor of the Skp2/p300 interaction that promotes p53-mediated apoptosis. Angew Chem, Int Ed. 2016;55:602–606. doi: 10.1002/anie.201508716. [DOI] [PubMed] [Google Scholar]
- 32.Shi Y, Teng P, Sang P, She F, Wei L, Cai J. γ-AApeptides: Design, structure, and applications. Acc Chem Res. 2016;49:428–441. doi: 10.1021/acs.accounts.5b00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teng P, Shi Y, Sang P, Cai J. γ-AApeptides as a new class of peptidomimetics. Chem - Eur J. 2016;22:5458–5466. doi: 10.1002/chem.201504936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niu Y, Hu Y, Li X, Chen J, Cai J. [gamma]-AApeptides: design, synthesis and evaluation. New J Chem. 2011;35:542–545. [Google Scholar]
- 35.Teng P, Zhang X, Wu H, Qiao Q, Sebti SM, Cai J. Identification of novel inhibitors that disrupt STAT3-DNA interaction from a [gamma]-AApeptide OBOC combinatorial library. Chem Commun. 2014;50:8739–8742. doi: 10.1039/c4cc03909b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu H, Li Y, Bai G, Niu Y, Qiao Q, Tipton JD, Cao C, Cai J. gamma-AApeptide-based small-molecule ligands that inhibit Abeta aggregation. Chem Commun (Cambridge, U K) 2014;50:5206–5208. doi: 10.1039/c3cc46685j. [DOI] [PubMed] [Google Scholar]
- 37.Lian W, Jiang B, Qian Z, Pei D. Cell-permeable bicyclic peptide inhibitors against intracellular proteins. J Am Chem Soc. 2014;136:9830–9833. doi: 10.1021/ja503710n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu R, Marik J, Lam KS. A Novel Peptide-Based Encoding System for “One-Bead One-Compound” peptidomimetic and small molecule combinatorial libraries. J Am Chem Soc. 2002;124:7678–7680. doi: 10.1021/ja026421t. [DOI] [PubMed] [Google Scholar]
- 39.Díaz-Mochón JJ, Bialy L, Bradley M. Full orthogonality between Dde and Fmoc: the direct synthesis of PNA–peptide conjugates. Org Lett. 2004;6:1127–1129. doi: 10.1021/ol049905y. [DOI] [PubMed] [Google Scholar]
- 40.Lian W, Upadhyaya P, Rhodes CA, Liu Y, Pei D. Screening bicyclic peptide libraries for protein–protein interaction inhibitors: discovery of a tumor necrosis factor-α antagonist. J Am Chem Soc. 2013;135:11990–11995. doi: 10.1021/ja405106u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song A, Zhang J, Lebrilla CB, Lam KSA. Novel and rapid encoding method based on mass spectrometry for “One-Bead- One-Compound” small molecule combinatorial libraries. J Am Chem Soc. 2003;125:6180–6188. doi: 10.1021/ja034539j. [DOI] [PubMed] [Google Scholar]
- 42.Klein R. Eph/ephrin signalling during development. Development. 2012;139:4105–4109. doi: 10.1242/dev.074997. [DOI] [PubMed] [Google Scholar]
- 43.Nievergall E, Lackmann M, Janes PW. Eph-dependent cell-cell adhesion and segregation in development and cancer. Cell Mol Life Sci. 2012;69:1813–1842. doi: 10.1007/s00018-011-0900-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133:38–52. doi: 10.1016/j.cell.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 45.Brantley-Sieders DM, Fang WB, Hwang Y, Hicks D, Chen J. Ephrin-A1 facilitates mammary tumor metastasis through an angiogenesis-dependent mechanism mediated by EphA receptor and vascular endothelial growth factor in mice. Cancer Res. 2006;66:10315–10324. doi: 10.1158/0008-5472.CAN-06-1560. [DOI] [PubMed] [Google Scholar]
- 46.Fang WB, Brantley-Sieders DM, Parker MA, Reith AD, Chen J. A kinase-dependent role for EphA2 receptor in promoting tumor growth and metastasis. Oncogene. 2005;24:7859–7868. doi: 10.1038/sj.onc.1208937. [DOI] [PubMed] [Google Scholar]
- 47.Paraiso KHT, Thakur MD, Fang B, Koomen JM, Fedorenko IV, John JK, Tsao H, Flaherty KT, Sondak VK, Messina JL, Pasquale EB, Villagra A, Rao UN, Kirkwood JM, Meier F, Sloot S, Gibney GT, Stuart D, Tawbi H, Smalley KSM. Ligand independent EphA2 signaling drives the adoption of a targeted therapy-mediated metastatic melanoma phenotype. Cancer Discovery. 2015;5:264–273. doi: 10.1158/2159-8290.CD-14-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miao B, Ji Z, Tan L, Taylor M, Zhang J, Choi HG, Frederick DT, Kumar R, Wargo JA, Flaherty KT, Gray NS, Tsao H. EPHA2 is a mediator of vemurafenib resistance and a novel therapeutic target in melanoma. Cancer Discovery. 2015;5:274–287. doi: 10.1158/2159-8290.CD-14-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu C, Shahzad MMK, Wang H, Landen CN, Kim SW, Allen J, Nick AM, Jennings N, Kinch MS, Bar-Eli M, Sood AK. EphA2 overexpression promotes ovarian cancer growth. Cancer Biol Ther. 2008;7:1098–1103. doi: 10.4161/cbt.7.7.6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brantley-Sieders DM, Caughron J, Hicks D, Pozzi A, Ruiz JC, Chen J. EphA2 receptor tyrosine kinase regulates endothelial cell migration and vascular assembly through phosphoinositide 3-kinase-mediated Rac1 GTPase activation. J Cell Sci. 2004;117:2037–2049. doi: 10.1242/jcs.01061. [DOI] [PubMed] [Google Scholar]
- 51.Liu Z, Wang Q. Radical coupling polymerization (RCP) for synthesis of various polymers. RSC Adv. 2016;6:39568–39572. [Google Scholar]
- 52.Ju HQ, Xiang YF, Xin BJ, Pei Y, Lu JX, Wang QL, Xia M, Qian CW, Ren Z, Wang SY, Wang YF, Xing GW. Synthesis and in vitro anti-HSV-1 activity of a novel Hsp90 inhibitor BJ-B11. Bioorg Med Chem Lett. 2011;21:1675–1677. doi: 10.1016/j.bmcl.2011.01.098. [DOI] [PubMed] [Google Scholar]
- 53.Malkinson JP, Anim MK, Zloh M, Searcey M, Hampshire AJ, Fox KR. Efficient solid-phase-based total synthesis of the bisintercalator tandem. J Org Chem. 2005;70:7654–7661. doi: 10.1021/jo050959a. [DOI] [PubMed] [Google Scholar]
- 54.Bycroft BW, Chan WC, Chhabra SR, Hone ND. A novel lysine-protecting procedure for continuous flow solid phase synthesis of branched peptides. J Chem Soc, Chem Commun. 1993:778–779. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.