

 This work is licensed under a
This work is licensed under a Summary
Co-existence of craniopharyngioma and acromegaly has been very rarely reported. A 65-year-old man presented with visual deterioration, fatigue and frontal headaches. Magnetic resonance imaging revealed a suprasellar heterogeneous, mainly cystic, 1.9 × 2 × 1.9 cm mass compressing the optic chiasm and expanding to the third ventricle; the findings were consistent with a craniopharyngioma. Pituitary hormone profile showed hypogonadotropic hypogonadism, mildly elevated prolactin, increased insulin-like growth factor 1 (IGF-1) and normal thyroid function and cortisol reserve. The patient had transsphenoidal surgery and pathology of the specimen was diagnostic of adamantinomatous craniopharyngioma. Post-operatively, he had diabetes insipidus, hypogonadotropic hypogonadism and adrenocorticotropic hormone and thyroid-stimulating hormone deficiency. Despite the hypopituitarism, his IGF-1 levels remained elevated and subsequent oral glucose tolerance test did not show complete growth hormone (GH) suppression. Further review of the pre-operative imaging revealed a 12 × 4 mm pituitary adenoma close to the right carotid artery and no signs of pituitary hyperplasia. At that time, he was also diagnosed with squamous cell carcinoma of the left upper lung lobe finally managed with radical radiotherapy. Treatment with long-acting somatostatin analogue was initiated leading to biochemical control of the acromegaly. Latest imaging has shown no evidence of craniopharyngioma regrowth and stable adenoma. This is a unique case report of co-existence of craniopharyngioma, acromegaly and squamous lung cell carcinoma that highlights diagnostic and management challenges. Potential effects of the GH hypersecretion on the co-existent tumours of this patient are also briefly discussed.
Learning points:
Although an extremely rare clinical scenario, craniopharyngioma and acromegaly can co-exist; aetiopathogenic link between these two conditions is unlikely.
Meticulous review of unexpected biochemical findings is vital for correct diagnosis of dual pituitary pathology.
The potential adverse impact of GH excess due to acromegaly in a patient with craniopharyngioma (and other neoplasm) mandates adequate biochemical control of the GH hypersecretion.
Patient Demographics: Adult, Male, White, United Kingdom
Clinical Overview: Pituitary, Pituitary, Gonadotropins, IGF1, Prolactin, TSH, ACTH, GH, Testosterone, Craniopharyngioma, Acromegaly, Hypergonadotropic hypogonadism, Pituitary adenoma
Diagnosis and Treatment: Visual impairment, Headache, Fatigue, Hypogonadism, Vision - acuity reduction, Diabetes insipidus, Hands - enlargement, Face - coarse features, Breathing difficulties, MRI, Prolactin, IGF1, Histopathology, Gonadotrophins, ACTH, TSH, Glucose tolerance (oral), GH, GH suppression, Visual field assessment, X-ray, CT scan, Fine needle aspiration biopsy, Immunohistochemistry, Synaptophysin, PET scan, Transsphenoidal surgery, Radiotherapy, Resection of tumour, Somatostatin analogues, Desmopressin, Hydrocortisone, Glucocorticoids, Levothyroxine, Testosterone, Lanreotide
Related Disciplines: Oncology
Publication Details: Unique/unexpected symptoms or presentations of a disease, March, 2018
Background
Craniopharyngiomas are rare benign epithelial sellar/parasellar tumours with annual incidence of 0.13–0.20 cases/100 000 (1, 2). They can be detected at any age (from the prenatal and neonatal period until late adulthood) and are associated with significant morbidity and mortality making them one of the most challenging tumours to manage (2). Acromegaly is also a rare condition with prevalence of 2.8–13.7 cases/100 000 people and annual incidence of 0.2–1.1 cases/100 000 (3).
Co-existence of a craniopharyngioma with pituitary adenomas or other (para)sellar tumours has been rarely described.
Herein, we present a challenging case of a patient with a craniopharyngioma co-existent with acromegaly who during the pre-operative assessment for the removal of the craniopharyngioma was also diagnosed with squamous cell lung carcinoma.
Case presentation
A 65-year-old man presented with a three-month history of visual deterioration, fatigue and frontal headaches. His medical history included hypertension. He was a smoker (55 pack-years). Ophthalmological assessment revealed right optic neuropathy, decreased visual acuity (right and left eye 6/60 and 6/9, respectively) and defects in both temporal fields, as well as in the right nasal field. Subsequent head magnetic resonance imaging (MRI) disclosed a suprasellar heterogeneous, mainly cystic, 1.9 × 2 × 1.9 cm mass compressing and displacing the optic chiasm and expanding to the third ventricle without causing hydrocephalus; these findings were considered to be consistent with a craniopharyngioma (Fig. 1A), and he was referred for surgery. On initial clinical review, there were no typical manifestations consistent with acromegaly.
Figure 1.
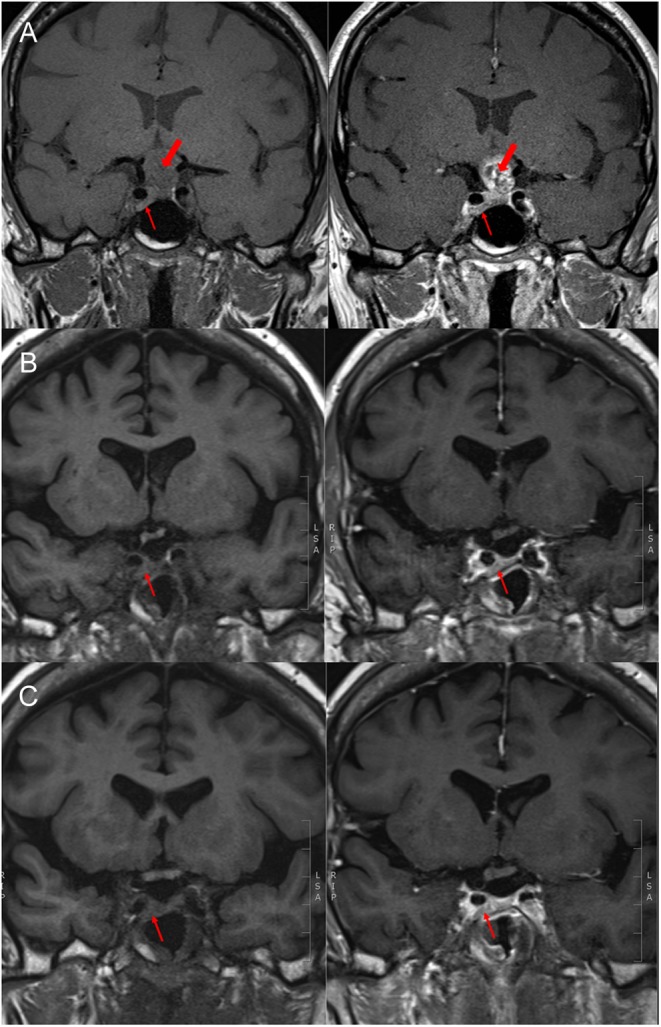
(A) Pre-operative T1-weighted images showing an isointense lesion at the suprasellar cistern (craniopharyngioma – thick arrow) and an isointense to marginally hyperintense lesion related to the right carotid artery (pituitary adenoma – thin arrow), with mixed intensity after contrast enhancement, (B) post-operative T1-weighted images, before and after gadolinium administration, showing the adenoma (thin arrow) and no residual craniopharyngioma tissue, (C) pre- and post-contrast-enhanced T1-weighted images one year after the initiation of long-acting somatostatin analogue treatment showing no craniopharyngioma regrowth and stable appearances of the adenoma (thin arrow).
Investigation
Hormonal investigations revealed hypogonadotropic hypogonadism, mildly elevated prolactin levels (334 mU/L, reference range 85–325), increased insulin-like growth hormone 1 (IGF-1) (365.6 ng/mL, reference range: 68.1–244.8) and normal thyroid function and cortisol reserve. There was no evidence of diabetes insipidus.
Treatment
An endoscopic transsphenoidal resection of the suprasellar mass was performed and histopathological examination of the removed specimen was diagnostic of an adamantinomatous craniopharyngioma WHO grade 1. After surgery, the patient’s vision was markedly improved and he had diabetes insipidus, adrenocorticotropic hormone (ACTH) deficiency, thyroid-stimulating hormone (TSH) deficiency and hypogonadotropic hypogonadism, requiring desmopressin, hydrocortisone, levothyroxine and testosterone replacement therapy. Post-operative MRI was reported as showing no residual craniopharyngioma tissue. Despite the hypopituitarism, the patient’s IGF-1 levels remained elevated (369.5 ng/mL, reference range: 68.1–244.8) and GH was not supressed on a 75 g oral glucose tolerance test (nadir: 1.18 µg/L). Notably, when manifestations of acromegaly were discussed at this stage, enlargement of the hands and development of more coarse facial features over the years were noticed. On further review of the pre-operative MRI, a 12 × 4 mm pituitary adenoma close to the right carotid artery was noted (Fig. 1); this was also evident in the post-operative scan (Fig. 1B). No signs of pituitary hyperplasia were demonstrated. During this period, the patient was also further investigated for a left lung upper lobe consolidation found incidentally in his chest X-ray performed in his pre-operative evaluation. Chest computed tomography demonstrated a 3 × 2.6 cm endobronchial tumour in the left upper lobe and bronchoscopy/fine-needle aspiration biopsy confirmed the presence of a necrotic tumour characterised as squamous cell lung carcinoma, T1bN1M0, with no neuroendocrine features; immunohistochemistry showed that tumour cells were negative for synaptophysin and strongly expressed p63. Staging positron emission tomography was not consistent with metastatic disease. Cardiac assessment had also revealed a non-specific cardiomyopathy (NYHA II) associated with mid-ventricular obstruction and apical akinesis. Due to the high peri-operative risk, surgical management was not considered a safe option and radical radiotherapy treatment was offered (55 Gy in 20 fractions). For the management of acromegaly, the pituitary multidisciplinary team also took into account the patient’s co-morbidities and suggested medical treatment with long-acting somatostatin analogue, which the patient commenced (lanreotide autogel: 90 mg/4 weeks).
Outcome and follow-up
Pituitary MRI one year after the long-acting somatostatin analogue initiation showed no craniopharyngioma regrowth and stable adenoma appearances (Fig. 1C). Somatostatin analogue treatment was well tolerated and led to biochemical control of the acromegaly (IGF-1 levels within the reference range and random GH <1 µg/L). On cardiology review, improvement of the breathlessness was documented with exercise tolerance of 300 yards before stopping. The patient has regular oncology follow-up and no evidence of lung cancer progression.
Discussion
We have presented a very rare case of two co-existent (para)sellar tumours, an adamantinomatous craniopharyngioma and one causing acromegaly. The latter, although not histologically proven, was an incidental and rather surprising diagnosis pointing out the importance of meticulous review of unexpected biochemical findings. Retrospective review of the pituitary MRI suggested the presence of an adenoma, which is most likely the source of GH hypersecretion.
Co-existence of craniopharyngioma and pituitary adenoma, either as collision tumours or as two clearly distinct lesions, has been very rarely reported. The published cases are mostly adamantinomatous craniopharyngiomas, whilst the pituitary adenomas are of various types, including prolactinomas, non-functioning gonadotroph adenomas, a TSH-secreting adenoma and a silent ACTH adenoma (4, 5, 6). There are only two reports of craniopharyngioma associated with acromegaly due to a GH-secreting adenoma, one as collision tumour (7) and the second with the diagnosis of craniopharyngioma established 18 years after the initial diagnosis of the adenoma (with the patient, however, being lost to follow-up during this period) (8).
Mutations in exon 3 of the CTNNB1 gene that prevent phosphorylation and degradation of β-catenin, a component of the adherens junction and a mediator of Wnt signalling pathway have been identified in adamantinomatous craniopharyngiomas and are possibly implicated in their pathogenesis (9). On the other hand, pituitary adenomas are monoclonal in origin and their pathogenesis is complex and still unclear: genetic mutations and epigenetic modifications resulting in abnormal cell cycle regulation, defects in signalling pathways or loss of tumour suppressor factors have been proposed as possible underlying mechanisms (10). An aetiological link between these two entities seems unlikely and their co-existence in our patient may be entirely co-incidental.
The impact of GH excess on the evolution of craniopharyngioma cannot be excluded. Strong IGF-1 receptor expression has been demonstrated in cell lines and paraffin-embedded material in a subset of craniopharyngiomas; in this group, treatment of cells with an IGF-1 receptor inhibitor was associated with decreased Akt phosphorylation and growth arrest suggesting that the IGF-1 receptor may promote the proliferation of craniopharyngioma cells (11).
Furthermore, the GH–IGF axis regulates cell proliferation, differentiation, angiogenesis, motility, and apoptosis and may contribute to cancer development and progression (12). A variety of malignancies have been described in association with acromegaly and two recent meta-analyses have demonstrated increased risk of colon and thyroid neoplasms in these patients (13, 14). Interestingly, over-expression of IGF-1 receptors has been suggested in squamous cell lung carcinoma, and this has been related with poor prognosis (15).
In our patient, medical treatment of acromegaly was considered to be the most pragmatic approach and proved to be beneficial for the control of the disease. Successful management of the GH hypersecretion was also deemed vital given the presence of other tumours (craniopharyngioma and lung cancer).
Our case is characterised by a very rare presentation of unexpected dual pituitary pathology and highlights challenges in its diagnosis and management, particularly, in a patient with co-existent lung malignancy.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
Written informed consent has been obtained from the patient for publication of the submitted article and accompanying images.
Author contribution statement
A Fountas, S T Chai contributed to the writing of the manuscript. N Gittoes and J Ayuk were involved in the care of the patient. S Chavda conducted the imaging review and prepared the figures of the manuscript. N Karavitaki was involved in the care of the patient, contributed to the writing of the manuscript and supervised the preparation of this work. All listed authors contributed to the editing process.
References
- 1.Al-Dahmani K, Mohammad S, Imran F, Theriault C, Doucette S, Zwicker D, Yip CE, Clarke DB, Imran SA. Sellar masses: an Epidemiological Study. Canadian Journal of Neurological Sciences 2016. 43 291–297. ( 10.1017/cjn.2015.301) [DOI] [PubMed] [Google Scholar]
- 2.Karavitaki N, Cudlip S, Adams CB, Wass JA. Craniopharyngiomas. Endocrine Reviews 2006. 27 371–397. ( 10.1210/er.2006-0002) [DOI] [PubMed] [Google Scholar]
- 3.Lavrentaki A, Paluzzi A, Wass JA, Karavitaki N. Epidemiology of acromegaly: review of population studies. Pituitary 2017. 20 4–9. ( 10.1007/s11102-016-0754-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finzi G, Cerati M, Marando A, Zoia C, Ferreli F, Tomei G, Castelnuovo P, La Rosa S, Capella C. Mixed pituitary adenoma/craniopharyngioma: clinical, morphological, immunohistochemical and ultrastructural study of a case, review of the literature, and pathogenetic and nosological considerations. Pituitary 2014. 17 53–59. ( 10.1007/s11102-013-0465-5) [DOI] [PubMed] [Google Scholar]
- 5.Guaraldi F, Prencipe N, di Giacomo V, Scanarini M, Gasco V, Gardiman MP, Berton AM, Ghigo E, Grottoli S. Association of craniopharyngioma and pituitary adenoma. Endocrine 2013. 44 59–65. ( 10.1007/s12020-013-9892-3) [DOI] [PubMed] [Google Scholar]
- 6.Karavitaki N, Scheithauer BW, Watt J, Ansorge O, Moschopoulos M, Llaquno AV, Wass JA. Collision lesions of the sella: co-existence of craniopharyngioma with gonadotroph adenoma and of Rathke’s cleft cyst with corticotroph adenoma. Pituitary 2008. 11 317–323. ( 10.1007/s11102-007-0070-6) [DOI] [PubMed] [Google Scholar]
- 7.Prabhakar V, Rao BD, Subramanyam MV. Pituitary adenoma associated with craniopharyngioma. Journal of Pathology 1971. 103 185–187. ( 10.1002/path.1711030307) [DOI] [PubMed] [Google Scholar]
- 8.El-Bilbeisi H, Ghannam M, Nimri CF, Ahmad AT. Craniopharyngioma in a patient with acromegaly due to a pituitary macroadenoma. Annals of Saudi Medicine 2010. 30 485–488. ( 10.4103/0256-4947.70581) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larkin S, Karavitaki N. Recent advances in molecular pathology of craniopharyngioma. F1000Research 2017. 6 1202 ( 10.12688/f1000research.11549.1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melmed S. Pathogenesis of pituitary tumors. Nature Reviews Endocrinology 2011. 7 257–266. ( 10.1038/nrendo.2011.40) [DOI] [PubMed] [Google Scholar]
- 11.Ulfarsson E, Karström A, Yin S, Girnita A, Vasilcanu D, Thoren M, Kratz G, Hillman J, Axelson M, Larsson O, et al Expression and growth dependency of the insulin-like growth factor I receptor in craniopharyngioma cells: a novel therapeutic approach. Clinical Cancer Research 2005. 11 4674–4680. ( 10.1158/1078-0432.CCR-05-0129) [DOI] [PubMed] [Google Scholar]
- 12.Clayton PE, Banerjee I, Murray PG, Renehan AG. Growth hormone, the insulin-like growth factor axis, insulin and cancer risk. Nature Reviews Endocrinology 2011. 7 11–24. ( 10.1038/nrendo.2010.171) [DOI] [PubMed] [Google Scholar]
- 13.Wolinski K, Czarnywojtek A, Ruchala M. Risk of thyroid nodular disease and thyroid cancer in patients with acromegaly – meta-analysis and systematic review. PLoS ONE 2014. 9 e88787 ( 10.1371/journal.pone.0088787) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rokkas T, Pistiolas D, Sechopoulos P, Margantinis G, Koukoulis G. Risk of colorectal neoplasm in patients with acromegaly: a meta-analysis. World Journal of Gastroenterology 2008. 14 3484–3489. ( 10.3748/wjg.14.3484) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao J, Shi X, Wang T, Ying C, He S, Chen Y. The prognostic and clinicopathological significance of IGF-1R in NSCLC: a meta-analysis. Cellular Physiology and Biochemistry 2017. 43 697–704. ( 10.1159/000480655) [DOI] [PubMed] [Google Scholar]