

Sun et al. establish the importance of transmission of changes in β-integrin transmembrane domain (TMD) topology in physiological integrin affinity modulation and biological function. Introduction of a flexible kink in the β7 integrin TMD blocks talin-mediated agonist-induced α4β7 integrin activation and function in gut lymphoid tissue development.
Abstract
Integrin activation regulates adhesion, extracellular matrix assembly, and cell migration, thereby playing an indispensable role in development and in many pathological processes. A proline mutation in the central integrin β3 transmembrane domain (TMD) creates a flexible kink that uncouples the topology of the inner half of the TMD from the outer half. In this study, using leukocyte integrin α4β7, which enables development of gut-associated lymphoid tissue (GALT), we examined the biological effect of such a proline mutation and report that it impairs agonist-induced talin-mediated activation of integrin α4β7, thereby inhibiting rolling lymphocyte arrest, a key step in transmigration. Furthermore, the α4β7(L721P) mutation blocks lymphocyte homing to and development of the GALT. These studies show that impairing the ability of an integrin β TMD to transmit talin-induced TMD topology inhibits agonist-induced physiological integrin activation and biological function in development.
Graphical Abstract
Introduction
Regulation of the affinity of integrins for adhesive ligands is central to cell adhesion, migration, and assembly of the extracellular matrix. Blood cells, such as leukocytes and platelets, have played a pivotal role in establishing current paradigms of this process because their integrins are expressed in a low-affinity form and intracellular signals initiated by agonists acting via distinct excitatory receptors, resulting in increased integrin affinity often referred to as “activation.” In these cells, talin binding to the integrin β2 or β3 cytoplasmic domains is a critical final common step in integrin activation both in vitro (Tadokoro et al., 2003; Shattil et al., 2010) and in vivo (Simonson et al., 2006; Nieswandt et al., 2007; Petrich et al., 2007a,b).
Mutational studies suggest that activation of integrin αΙΙbβ3 requires disrupting interactions of the α and β cytoplasmic and transmembrane domains (TMDs; Hughes et al., 1996; Luo et al., 2004). The αIIb and β3 TMD association is maintained by specific helical packing TMD interactions near the extracellular face of the membrane, termed the outer membrane clasp (Kim et al., 2009; Lau et al., 2009). Analyses of talin interactions with the β3 and β1D cytoplasmic domains (Wegener et al., 2007; Anthis et al., 2009) and molecular dynamic simulations (Kalli et al., 2011; Arcario and Tajkhorshid, 2014) suggest that talin disrupts the outer membrane clasp by altering the topology of the β3 TMD. Work with isolated integrin TMD-cytoplasmic domain fragments in phospholipid nanodiscs provided direct experimental evidence that talin binding can change the membrane embedding and therefore the topology of integrin β3 TMD (Kim et al., 2012).
Prolines perturb the structure of α helices by introducing a kink between the segments preceding and following the proline residue because of steric clash between the ring of the proline at position (i) and the backbone carbonyl at position (i-4). In addition, the lack of an amide proton also eliminates a helix backbone H-bond. The disruption of helical pattern and the reduction in H-bond stabilization lead to the flexibility of the kink in a proline-containing transmembrane α helix (Woolfson and Williams, 1990; von Heijne, 1991; Nilsson and von Heijne, 1998; Visiers et al., 2000). The structure of an integrin β3 TMD containing such a proline mutation [β3(A711P)], confirmed the expected flexible kink and showed that it prevents the transmission of talin-induced change in β3 TMD topology across the membrane (Kim et al., 2011, 2012). The physiological importance of transmission of such topology changes through the integrin β TMD has not been tested nor has it been examined in integrins other than β3. The latter issue is particularly compelling in light of recent research stressing that mechanisms of integrin activation may differ markedly among different integrin classes (Lu et al., 2016).
We selected lymphocyte α4β7 to test the importance of talin-induced topology change because the role of its cytoplasmic domain in adhesion regulation has long been known (Crowe et al., 1994); however, there is little understanding of the mechanism of the final steps in its activation or of transmission of the signal across the membrane. α4β7 integrin plays a crucial role in lymphocyte trafficking to gastrointestinal mucosa by binding to its ligand, MAdCAM-1, which is displayed on endothelial cells in intestinal postcapillary venules (Berlin et al., 1995; Sun et al., 2014), thereby enabling construction of the gut immune system (Wagner et al., 1996; Gorfu et al., 2009). Integrin α4β7 has therefore emerged as proven therapeutic target in diseases involving the gut immune system such as inflammatory bowel disease (IBD; Feagan et al., 2013; Sandborn et al., 2013; Cherry et al., 2015) and as a potential target in HIV-AIDS (Arthos et al., 2008; Byrareddy et al., 2016). Agonist stimulation, important in lymphocyte trafficking to gut-associated lymphoid tissues (GALTs; Gorfu et al., 2009; Sun et al., 2014), can activate integrin α4β7; however, direct proof of an integrin activation requirement by itself for development is lacking. In this study, we report that talin binding to the β7 cytoplasmic domain activates α4β7 in a fashion analogous to αIIbβ3 and that creation of a flexible kink in the β7 TMD by insertion of proline, but not other mutations, impairs activation. We find that the β7(L721P) mutation prevents the activation of α4β7 by chemokine agonists, thereby inhibiting arrest of rolling lymphocytes in a flow chamber and their homing to GALT. Moreover, we report that GALT development is disrupted in a mouse bearing a β7(L720P) mutation. These studies show, for the first time, that impairing the ability of the integrin β TMD to transmit talin-induced TMD topology inhibits physiological integrin activation and biological functions in development.
Results
Talin binding to the β7 cytoplasmic domain activates integrin α4β7
Although it has long been known that agonists can activate T cell integrin α4β7 (Berlin et al., 1995), the role of talin in that process has not been evaluated. We used CRISPR/Cas9 mutagenesis to delete talin in Jurkat T cells, which lack endogenous β7 expression. Then, β7 was stably expressed in both Jurkat T cells and Jurkat T–talin knockout (KO) cells (Fig. 1 A). Addition of 12-O-tetradecanoylphorbol-13-acetate PMA failed to increase the low constitutive binding of soluble MAdCAM-1 to talin-null cells (Fig. 1 B), whereas the exogenous activator, Mn2+, stimulated binding to talin-null cells. Talin deletion also abolished PMA-stimulated increase in firm adhesion of Jurkat-β7 cells on MAdCAM-1, confirming the loss of α4β7 activation (Fig. 1 C).
Figure 1.

Talin binding to the β7 cytoplasmic domain activates integrin α4β7. (A) Expression of talin and β7 in parental or talin KO β7-expressing Jurkat T cells. Top: Total expression by Western blot. Bottom: Surface expression by flow cytometry. The filled histograms represent untransfected Jurkat cells, whereas open histograms represent β7-expressing Jurkat cells or talin KO cells. (B) Binding of soluble MAdCAM-1 to β7 expressing Jurkat T cells or talin KO cells. PMA (100 nM) markedly increased binding to parental but not talin KO cells. Mn2+ (0.5 mM) stimulated binding to both cell types. Stimulated cells were compared with resting (None) for each cell line using one-way ANOVA. (C) Adhesion of β7-expressing Jurkat T cells or Jurkat-talin KO cells to MAdCAM-1 substrate in the presence or absence of PMA (100 nM) under a wall shear stress of 2 dyn/cm2. Nontransfected Jurkat T cells (Jurkat) provided a negative control. Jurkat-β7-Talin KO or Jurkat were compared with the Jurkat-β7 for each condition using one-way ANOVA. (D) Structural model of the talin F3 domain in complex with integrin β7 tail (Arg728 to Thr766). Talin F3 domain is shown by a surface representation and colored by charge. A ribbon diagram of the docked β7 tail is highlighted in red. β7-Leu758 and -Tyr759 in the NPLY motif are shown as light blue–colored stick figures. (E) Soluble MAdCAM-1 binding to 293T cells transfected with WT or mutant α4β7 with or without THD cotransfection. Mn2+ (0.5 mM) was used as a positive control for integrin activation. Nontransfected 293T cells (MOCK) provided a negative control. Mutant integrins were compared with the WT for each condition using one-way ANOVA. (F) Adhesion of 293T cells transfected with WT or mutant α4β7 with or without THD cotransfection on MAdCAM-1 under a wall shear stress of 2 dyn/cm2. Nontransfected 293T cells (MOCK) provided a negative control. Mutant integrins were compared with the WT for each condition using one-way ANOVA. (G) Binding of soluble MAdCAM-1 to 293T-α4β7 cells transfected with EGFP vector, EGFP-THD, EGFP-THD(L325R), or EGFP-THD(W359A). THD-stimulated cells were compared with vector control (α4β7 + EGFP vector) for each cell line using two-way ANOVA. MFI, mean fluorescent intensity. (H) Adhesion of 293T-α4β7 cells transfected with EGFP vector, EGFP-THD, EGFP-THD(L325R), or EGFP-THD(W359A) on MAdCAM-1 under a wall shear stress of 2 dyn/cm2. 293T cells transfected with THD only and nontransfected 293T cells (MOCK) were used as negative controls. THD-stimulated cells were compared with vector control (α4β7 + EGFP vector) for each cell line using one-way ANOVA. Error bars show means ± SD. n = 5. NS, P > 0.05; *, 0.01 < P < 0.05; **, 0.001 < P < 0.01; ***, P < 0.001.
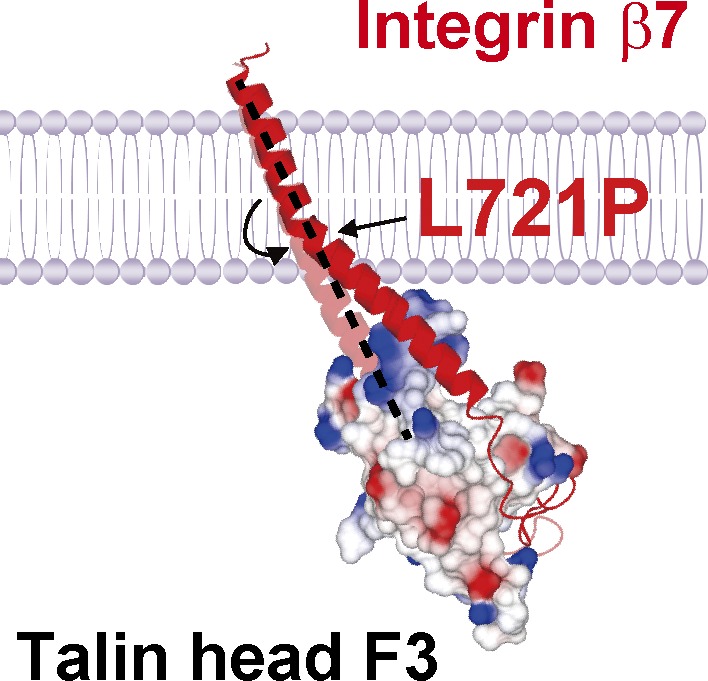
Talin binding to the integrin β3 cytoplasmic domain depends on engagement of the F3 subdomain of the talin FERM domain with a “membrane distal” site in the β3 tail (García-Alvarez et al., 2003; Wegener et al., 2007). The β3(L746A) mutation selectively blocks talin binding, whereas β3(Y747A) blocks the binding of talin and other proteins (García-Alvarez et al., 2003; Petrich et al., 2007a; Haling et al., 2011). We aligned the β3 and β7 cytoplasmic tails and used that alignment to construct a homology model of β7 bound to the talin F3 domain (Fig. 1 D). The model predicted that β7 Leu758 and Tyr759 form critical hydrophobic contacts with talin F3, thereby fulfilling the role of β3 Leu746 and β3 Tyr747. The finding that β7(L758A) and β7(Y759A) mutations greatly reduced the binding of recombinant talin head domain (THD; Fig. S1, A and B) or full-length talin (Fig. S1 C) validated this model.
We then tested the effect of talin and integrin mutations in 293T cells because of technical ease of transfection. The β7 mutants (Y759A or L758A) markedly inhibited the capacity of recombinant α4β7 to bind soluble MAdCAM-1 (Fig. 1 E). Expression of THD in these α4β7-expressing cells increased MAdCAM-1 binding to α4β7; however, this increase was severely blunted by both β7 mutations (Fig. 1 E). The effects of these mutations on binding of soluble MAdCAM-1 were reflected in a marked reduction in cell adhesion to a MAdCAM-1 substrate (Fig. 1 F) and increase in rolling velocity (Fig. S1 D) at a wall shear stress of 2 dyn/cm2 using a parallel wall flow chamber as described previously (Sun et al., 2014).
The homology model also indicated, similar to β3, that talin Leu325 and Trp359 would be important in the binding of talin to the membrane proximal helical segment and the membrane distal segment of the β7 tail, respectively. The β7–talin interaction was again similar to the β3–talin interaction (Wegener et al., 2007) in that THD(W359A) showed negligible binding to the β7 tail, whereas THD(L325R) exhibited a more modest reduction in affinity (Fig. S1, E and F). Both of these talin mutants abrogated the capacity of THD to increase the binding of soluble MAdCAM-1 to recombinant α4β7 (Figs. 1 G and S1, G and H). The effects of the mutations on soluble MAdCAM-1 binding were reflected by the reduction in adhesion (Fig. 1 H) and increased velocity of rolling (Fig. S1 I) on a MAdCAM-1 substrate. Notably, THD(L325R) expression reduced soluble MAdCAM-1 binding and increased rolling velocity relative to vector-transfected cells (Figs. 1 G and S1 I). This is ascribable to the dominant-negative effect of THD(L325R) competition with endogenous talin for binding to the β7 cytoplasmic domain. In sum, these data show that talin interacts with the β7 tail in a fashion analogous to β3 and that, in doing so, it activates α4β7 to promote the arrest of rolling cells in flow.
Blocking talin-induced change in β7 TMD topology disrupts THD-induced integrin α4β7 activation
An α-β TMD association stabilizes the low-affinity state of αIIbβ3 integrin (Li et al., 2002; Luo et al., 2004; Partridge et al., 2005), and talin binding disrupts this association, in part, by altering the topology of the β3 TMD, resulting in destabilization of the α-β TMD complex and integrin activation (Kim et al., 2012). To decouple talin-induced changes in inner β7 TMD topology from the outer half, we first aligned the TMD sequence of β3 and β7, and predicted that β7 Leu723 could correspond with β3 Ala711 (Fig. 2 A). Because the crossing angle of the β7 TMD is uncertain, we constructed a homology model of the structure of α4β7 TMD complex based on the αIIbβ3 TMD (Lau et al., 2008). Based on this model, we also selected Leu721 and Gly722 and mutated each of these three residues individually to proline and transfected 293T cells with the three mutants resulting in similar levels of expression to WT (Fig. S2 A). Cells expressing either α4β7(L723P) or α4β7(L721P) exhibited markedly reduced adhesion to a MAdCAM-1 substrate at 2 dyn/cm2 shear stress, whereas those expressing α4β7(G722P) exhibited a more mild reduction in adhesion (Fig. 2 B). The effect of the β7 Pro mutations was largely mirrored in the marked reduction of binding of soluble MAdCAM-1 to cells expressing α4 in combination with either β7(L721P) or β7(L723P) and a lesser reduction for α4β7(G722P) (Figs. 2 C and S2 B). Moreover, both of the leucine replacement mutants abolished the capacity of THD to increase soluble MAdCAM-1 binding (Fig. 2 C), and these results were mirrored in dramatically reduced effect of THD on adhesion (Fig. S2 C). However, cells expressing α4β7(G722P) showed only a mild reduction in THD-induced soluble MAdCAM-1 binding (Figs. 2 C and S2 C). Notably, we found that the association of the THD with each of the mutant α4β7 integrins was similar to that observed with the WT integrin (Fig. 2 D). In addition, we also substituted Ala (nonpolar) or Gly (polar) for Leu721 and Leu723 and found they had no effects on α4β7 integrin-mediated adhesion to MAdCAM-1 with or without THD coexpression (Fig. S3). However, substitution Arg for Leu723 increased α4β7−mediated adhesion to MAdCAM-1. In sum, introduction of a proline but not other mutations at specific sites in the midpoint of the β7 TMD disrupts the capacity of talin to activate this integrin, without perturbing talin binding to the integrin, suggesting that a flexible kink in the β7 TMD blocks transmission of talin’s activation signal across the plasma membrane (Fig. 2 E).
Figure 2.

Blocking talin-induced change in β7 TMD topology abolished α4β7 activation. (A) Sequence alignment of partial TMDs of integrin β3 and β7 subunits. β3 Pro mutant site Ala711 is highlighted in red. The sites in β7 that were mutated to Pro (Leu721 and Leu723) are highlighted in green and Gly722 is highlighted in pink and are projected onto a homology model of the α4β7 TMD (α4 in blue and β7 in red). (B) Adhesion of 293T cells transfected with β7 WT or mutations (L721P, G722P, or L723P) plus α4 on MAdCAM-1 under a wall shear stress of 2 dyn/cm2. Nontransfected 293T cells (MOCK) were used as a negative control (Ctrl). Mutant integrins were compared with the WT using one-way ANOVA. (C) Binding of soluble MAdCAM-1 to 293T cells transfected with WT α4β7 or α4 in combination with mutants (L721P, G722P, and L723P) in the presence or absence of THD. Nontransfected 293T cells (MOCK) were used as a negative control. Mutant integrins were compared with the WT for each condition using one-way ANOVA. Error bars show means ± SD. n = 5 (A and B) or 4 (C). *, 0.01 < P < 0.05; **, 0.001 < P < 0.01; ***, P < 0.001. MFI, mean fluorescence intensity. (D) Coimmunoprecipitation of THD with α4β7 WT or mutants. Lysates of 293T cells were transfected as in C, in combination with THD-GFP, and α4β7 was isolated by immunoprecipitation (IP). Precipitated proteins were analyzed by Western blotting with the indicated antibodies and confirmed similar THD association with the mutant and WT integrins. Data are representative of at least three independent experiments. Note that a shorter exposure time was used for the THD input. Molecular masses are given in kilodaltons. (E) A model of how a Pro mutation can prevent transmission of altered topology of the β7 TMD by talin. The complex formed between the β7 cytoplasmic tail/TMD (red) and cytoplasmic talin F3 domain (surface representation; colored by charge) alters the topology of the inner portion of the transmembrane helix, which is transmitted to the outer moiety, where it can disrupt the outer membrane clasp (Lau et al., 2009), resulting in destabilization of the α-β TMD complex and integrin activation. β7(L721P) breaks the TMD helix into two helices connected by a flexible kink; the flexible kink prevents transmission of the talin-induced change in intracellular TMD topology to stabilize the α-β TMD interaction and block talin-induced activation of integrin α4β7.
Disrupting talin-induced change in β7 TMD topology blocks agonist-induced α4β7 activation
α4β7 is expressed primarily in lymphocytes and can be physiologically activated by chemokine stimulation (Wagner et al., 1996; Mora and Von Andrian, 2006; Agace, 2008; Gorfu et al., 2009). To assess whether a topological change in the β7 TMD is involved in agonist-induced activation of α4β7, we expressed β7(L721P) or β7(L723P) in Jurkat T cells that lack endogenous β7 expression. Jurkat cells lack chemokine receptors used in gut homing, CCR9 and CCR7; hence, we used CXCL12 (SDF1α) to stimulate them via endogenous CXCR4. The proline mutants blocked the capacity of CXCL12 to increase the binding of soluble MAdCAM-1 (Fig. 3 A). Moreover, the Pro mutants also prevented activation by the receptor-independent agonist PMA. The direct integrin activator Mn2+ enhanced the binding of MAdCAM-1 to cells bearing either Pro mutant (Fig. 3 A), consistent with the fact that the mutant and WT integrins were expressed at similar abundance on the cell surface (Fig. 3 B). In addition, similar quantities of endogenous talin and kindlin 3 were associated with the mutant and WT α4β7 in both resting and chemokine-stimulated Jurkat T cells (Fig. 3 C). In the flow chamber, which mimics the interaction of T cells with endothelial MAdCAM-1 in flowing blood, the proline mutants blocked the capacity of both CXCL12 and PMA to induce the arrest of rolling T cells (Fig. 4, A and B). The suppression of α4β7 activation was also mirrored by the marked increase in rolling velocity of stimulated T cells bearing either proline mutant (Fig. 4, C and D; and Fig. S4 A). Similar results were obtained in the static cell adhesion experiments. CXCL12 treatment markedly enhanced the adhesion of WT β7 cells on MAdCAM-1. In contrast, the cells bearing β7(L721P) or β7(L723P) did not exhibit CXCL12 stimulation of α4β7-mediated adhesion to MAdCAM-1 (Fig. S4 B). Thus, blocking TMD transmission of talin-mediated topology change impaired the capacity of physiological agonists to promote α4β7-mediated arrest of rolling T cells.
Figure 3.

Inhibiting talin-induced change in β7 TMD topology impairs agonist-induced α4β7 activation. (A) Binding of soluble MAdCAM-1 to Jurkat T cells stably expressing WT or mutant β7(L721P or L723P) with or without CXCL12 or PMA stimulation. Mutant integrins were compared with the WT for each condition using one-way ANOVA. Error bars show means ± SD. n = 5. NS, P > 0.05; **, 0.001 < P < 0.01; ***, P < 0.001. MFI, mean fluorescence intensity. (B) β7 cell surface expression on Jurkat T cells stably expressing WT or mutant β7(L721P or L723P). Mock-transfected cells are depicted in the filled histograms. (C) α4β7 was immunoprecipitated (IP) from lysates of CXCL12-stimulated cells or unstimulated cells depicted in A, and bound proteins were analyzed by Western blotting with the indicated antibodies. Data are representative of at least three independent experiments. Molecular masses are given in kilodaltons.
Figure 4.

Blocking TMD topology transmission in β7 impaired agonist-promoted arrest of rolling T cells. (A and B) Adhesion of Jurkat T cells that stably expressed WT or mutant β7(L721P or L723P) on MAdCAM-1 with or without CXCL12 (A) or PMA (B) stimulation under a wall shear stress of 2 dyn/cm2. Mutant integrins were compared with the WT for each condition using one-way ANOVA. (C and D) Rolling velocity of Jurkat T cells that stably expressed WT or mutant β7(L721P or L723P) on MAdCAM-1 with or without CXCL12 (C) or PMA (D) stimulation under a wall shear stress of 2 dyn/cm2. Stimulated cells were compared with resting (None) for each cell line using one-way ANOVA. The numbers of cells remaining bound (A) and cell velocity (B) were analyzed. Error bars show means ± SD. n = 5. NS, P > 0.05; ***, P < 0.001.
β7(L721P) mutation inhibits lymphocyte homing to GALT
The foregoing studies established the importance of the changing topology of the β7 subunit TMD in talin-induced α4β7 activation in vitro. To examine its importance in the physiological function of α4β7 integrin, we introduced this mutation into TK1 mouse T cells that express α4β7 (Rüegg et al., 1992; Berlin et al., 1993). We first used CRISPR/Cas9 mutagenesis to generate β7-null TK1 clones. We then used lentivirus-mediated transduction to express β7 and β7(L721P) at similar levels in these TK1-β7-KO cells (Fig. 5 A). We examined the capacity of these TK1 cell lines to bind to soluble MAdCAM-1 in the presence or absence of CXCL12 stimulation (Fig. 5 B). The cells expressing β7(L721P) bound negligible soluble MAdCAM-1 in the presence or absence of CXCL12. In contrast, both β7-expressing TK1-β7–null cells and WT TK1 cells bound MAdCAM-1, and binding was markedly increased by CXCL12 stimulation. The capacity of Mn2+ to stimulate MAdCAM-1 binding to β7(L721P) cells confirmed that this mutation did not disrupt the ability of the integrin to bind ligand, but only prevented the transmission of signaling events that led to activation.
Figure 5.

Suppressing talin-induced change in β7 TMD topology perturbed mouse lymphocyte homing to the gut. (A) Cell surface expression of α4β7 in TK1-β7 KO cells stably expressing WT β7 or mutant β7(L721P). (B) Binding of soluble mouse MAdCAM-1 to the cells shown in A with or without CXCL12 stimulation. The TK1-β7 KO cell was used as a negative control. Stimulated cells were compared with resting (None) for each cell line using one-way ANOVA. MFI, mean fluorescence intensity. (C and D) In vivo competitive homing of TK1-β7 KO cells that stably express WT β7(β7 WT) or proline mutant β7(β7[L721P]) to different lymphoid tissues. TK1-β7 WT or TK1-β7(L721P) cells were labeled with eFluor 670. TK1 parental (TK1) cells were labeled with CFSE as an input control. Equal numbers (2 × 107) of eFluor 670–labeled cells and CFSE-labeled cells were mixed and then intravenously injected into C57BL/6J mice. Lymphoid organs were isolated 2 h after injection. The eFluor 670– or CFSE-labeled cells that homed into different lymphoid organs were enumerated by flow cytometry. The total numbers of homed cells to different lymphoid organs are shown in C. MLN (per mouse), PP (per mouse), PLN (per lymph node), and SP (per mouse). The ratio of TK1 parental cells to TK1-KO cells reconstituted with β7 WT or β7(L721P) cells recovered from different lymphoid organs is shown in D. Error bars show means ± SD. n = 5 (A and B) or 24 (C and D). NS, P > 0.05; **, 0.001 < P < 0.01; ***, P < 0.001.
To examine the effect of the Pro mutation on lymphocyte homing, TK1-β7–null cells expressing either β7 WT or β7 proline mutation (β7[L721P]) cells were labeled with eFluor 670 or carboxyfluorescein succinimidyl ester (CFSE), respectively. Equal numbers (2 × 107) of eFluor 670–labeled cells and CFSE-labeled cells were mixed and then injected intravenously into C57BL/6J mice. The number of eFluor 670– or CFSE-labeled cells in mesenteric lymph node (MLN), Peyer's patch (PP), peripheral lymph node (PLN), and spleen (SP) were enumerated by flow cytometry. A homing index, defined as the ratio of TK1-β7 WT or TK1-β7(L721P) to TK1 cells, was determined. Compared with TK1 cells, TK1-β7 WT cells homed equally well to MLN, PP, PLN, and SP (Fig. 5, C and D). In contrast, TK1-β7(L721P) cells homing to PP were dramatically reduced. Homing to MLN was also significantly reduced, albeit to a lesser extent than PP. In contrast, TK1-β7(L721P) cells homing to PLN and SP were similar to TK1-β7 WT and TK1 cells (Fig. 5, C and D). Thus, blocking talin-induced change in β7 TMD topology impaired the capacity of α4β7 to mediate lymphocyte homing to GALT.
β7(L720P) mutation disrupts GALT development
To examine the role of the topology of the β7 TMD in development of GALT, we analyzed mice bearing β7(L720P) (homologous to human β7[L721P]) mutation. CRISPR/Cas9 mutagenesis was used to generate four male founder Itgb7L720P/L720P mice (Fig. 6, A and B) who were then crossed with C57BL/6J females. Itgb7L720P/WT male F1 mice were backcrossed to C57BL/6J females, and the Itgb7L720P/WT F2 mice were intercrossed to generate Itgb7L720P/L720P and WT littermates for comparison. Itgb7L720P/L720P mice were born at expected Mendelian ratios and were apparently healthy. Moreover, normal numbers of both T and B cells were present in the secondary lymphoid organs such as SP and PLNs (Fig. 6 C). In sharp contrast, there was a profound reduction in both cell types in the PPs and a lesser reduction in MLNs consistent with the defect in lymphocyte homing to these sites. The percentage of CD4 and CD8 cells in Itgb7L720P/L720P mice was similar to the WT mice (Fig. S5 A). Furthermore, the localization within lymphoid organs of CD4 T cells, CD8 T cells, B cells, macrophages, and dendritic cells in Itgb7L720P/L720P mice was not grossly different from WT mice (Fig. S5, B and C). Thus, this proline mutation in the central β7 TMD disrupts development of the GALT.
Figure 6.

Disruption of GALT development in Itgb7L720P/L720P mice. (A) Schematic of the Cas9/sgRNA-targeting sites in Itgb7. The sgRNA-targeting sequences are underlined in blue, and the protospacer-adjacent motif (PAM) sequence is labeled in blue in the WT sequence. The MaeI restriction site removed from the WT and ApaI site introduced in the mutant are labeled as are the Leu codon changed to Pro. (B) Sequencing analysis of WT and β7(L720P) knock-in mice. DNA sequencing confirmed a leucine to proline substitution at position 720 of the mouse β7 integrin gene (position 721 in human β7 integrin gene). The mutation is labeled in red. The silent mutations are labeled in green and with green asterisks. (C) The absolute number of CD3+ T cells and B220+ B cells isolated from Itgb7WT/WT mice and Itgb7WT/L720P, or Itgb7L720P/L720P littermates are shown. Error bars show means ± SD. n = 8. NS, P > 0.05; ***, P < 0.001.
Previous research demonstrated that integrin α4β7 is constitutively expressed on naive T and B cells at a relatively low level (Erle et al., 1994), and retinoic acid, produced by gut dendritic cells, enhances the expression of α4β7 on CD4+ T cells to imprint them with the gut tropic α4β7high phenotype (Gorfu et al., 2009). Thus, we speculated the β7 proline mutation, by preventing contact with gut dendritic cells during maturation, might reduce β7 expression in adult T cells. To avoid potential effects of postpartum lymphocyte cell education, we tested the surface expression of integrin αL, α4, β1, β2, and β7 as well as intracellular expression of kindlin 3 (FERMT3) and talin (TLN) in neonatal mice (Fig. 7 A). The expression of those integrins and integrin intracellular binding proteins was similar in both WT mice and Itgb7L720P/L720P mice. In contrast, lymphocyte α4β7 expression was decreased in adult Itgb7L720P/L720P mice (Fig. 7 B). To test the idea that the effector/memory T cells might be selectively affected, we compared β7 surface expression on naive T cells (CD62LhighCD44low) and effector T cells (CD62LlowCD44high) in adult WT and Itgb7L720P/L720P mice (Fig. 7 C). As expected, in WT mice, β7 expression was increased twofold in effector/memory compared with naive T cells. In sharp contrast, Itgb7L720P/L720P effector/memory T cells exhibited similar β7 expression to naive T cells.
Figure 7.

Itgb7L720P/L720Padult mice have reduced gut-tropic β7high effector/activated T cells. (A) Equivalent cell surface expression of integrin αL, α4, β1, β2, and β7 and intracellular expression of kindlin 3 and talin in neonatal Itgb7WT/WT mice and Itgb7L720P/L720P littermates are shown. (B) Cell surface expression of β7 is reduced in 8-wk-old Itgb7L720P/L720P compared with Itgb7WT/WT littermates. (C) The ratio of cell surface expression of integrin β7 on effector/activated T cells (CD62LlowCD44high) compared with naive T cells (CD62LhighCD44low) in 8-wk-old Itgb7WT/WT mice and Itgb7L720P/L720P littermates. Mean fluorescence intensities are displayed on the representative histograms. Error bars show means ± SD. **, 0.001 < P < 0.01. Teff, effector T cells.
Discussion
The concept of “inside-out signal transduction” was engendered by the capacity of signals generated within cells to regulate the affinity and functioning of integrin extracellular domains (Ginsberg et al., 1992), and research with platelet integrin αIIbβ3 has played a central role in a current model of the process. One model proposes that talin, by binding to the β3 integrin cytoplasmic domain, disrupts an electrostatic interaction that strengthens the inner membrane clasp that stabilizes the αIIbβ3 TMD association. Changes the topology of the β TMD disrupt an outer membrane clasp to complete the activation process (Kim et al., 2011). Recent research has indicated marked differences in the mechanism of inside-out activation of other classes of integrins (Lu et al., 2016). In this study, we have tested the talin-induced topology change model with a leukocyte integrin, α4β7. We find that introduction of proline, known to form a flexible kink in an integrin β TMD, blocks talin-induced inside-out integrin signaling and biological functions in development. These studies establish, in a physiological setting, the importance of transmission of β subunit TMD topology across the membrane.
Activation of integrin α4β7 is mediated by talin binding to the β7 cytoplasmic domain in a mechanism analogous to αIIbβ3. Talin deletion in T cells reduced but did not eliminate the α4β7-mediated constitutive binding of and adhesion to MAdCAM-1, whereas lack of talin virtually eliminated the PMA-induced increase in MAdCAM-1 interactions. Thus, as with β1, β2, and β3 integrins, talin is required for agonist-induced activation, defined as an increase of ligand binding affinity above a low constitutive level. These effects of talin deletion were mirrored by the effects of β7 mutations that disrupt talin binding, showing that the interaction of talin with β7 is required as it is with β1 and β3 integrins (Tadokoro et al., 2003). Similarly, talin mutants that block binding to the membrane proximal or membrane distal sites in β3 (Wegener et al., 2007) both impaired the capacity of THD to activate α4β7, indicating that talin binding activates α4β7 through a similar mechanism to αIIbβ3.
The integrin β TMD transmits talin-induced altered TMD topology to the extracellular domain to activate integrins and mediate their biological functions. Introduction of a proline residue in the β7 TMD impaired activation of α4β7. The effect of the proline cannot be ascribed to simple replacement of a hydrophobic leucine because substitution with other neutral residues, e.g., alanine, did not have this effect. Rather, it is the capacity to introduce a flexible kink in the TMD (von Heijne, 1991; Nilsson and von Heijne, 1998; Visiers et al., 2000; Kim et al., 2011) that accounts for this unique property of proline. The effect of this flexible kink is to reduce helix rigidity, thereby decoupling a physical displacement of the inner half of the TMD from the outer half (Kim et al., 2012), thus preserving the outer membrane clasp. The location of the proline was relevant in that placing the proline in place of Gly722 had a weaker effect. The increased flexibility provided by proline insertions can limit the transmission of talin-induced change in topology of the inner to the outer half of the β TMD domain, thereby stabilizing the α-β transmembrane helix association by preserving the helical packing required to form the outer membrane clasp.
Our research provides new insight into how α4β7 mediates formation of the mucosal immune system and suggests new therapeutic approaches. The role of β7 integrins in assembly of the mucosal immune system has long been known (Wagner et al., 1996; Gorfu et al., 2009; Villablanca et al., 2011) as has the role of chemokine stimulation of those cells (Gorfu et al., 2009; Sun et al., 2014) that activates integrin α4β7; however, direct proof of an integrin activation requirement by itself for development is lacking. Our research is the first to establish the importance of integrin activation as the key effect of chemokine stimulation in homing and development. The pathogenesis of IBD involves a massive influx of immune cells into the gastrointestinal mucosa (Adams and Eksteen, 2006; Eksteen et al., 2008; Villablanca et al., 2011) with infiltration of inflammatory lymphocytes in the colonic lamina propria of IBD patients (Caradonna et al., 2000; Smith et al., 2005). Vedolizumab, a mAb that blocks α4β7 function, has been approved for the treatment of IBD in adults (McLean et al., 2012; Poole, 2014; Cherry et al., 2015); however, the extent to which it shares the risk of progressive multifocal leukoencephalopathy induced by anti-α4β1 antibodies is uncertain (Lam and Bressler, 2014). Moreover, aggravated colitis is observed in a small percentage of patients treated with high dose of vedolizumab (Feagan et al., 2005; Rutgeerts et al., 2013). As shown in this study, blockade of talin-induced activation preserves some α4β7 adhesive function, in contrast with full blockade with antibodies such as vedolizumab. Thus, blockade of activation may avert mechanism-based toxicities while preserving a therapeutic effect. In this study, we add to a deeper understanding of the signaling events that control α4β7 activation and development of the GALT, which may ultimately lead to new strategies for treating IBD or other α4β7 integrin-mediated pathologies such as HIV-AIDS.
Materials and methods
cDNAs and recombinant proteins
A modified pET-15b vector encoding a His6-AviTag at the N terminus of the integrin cytoplasmic tail was used as a recombinant model protein affinity matrix as previously described (Pfaff et al., 1998). cDNA encoding integrin β7 (730–779) cytoplasmic tails, WT, β7(L758A), and β7(Y759A) were cloned into this vector, replacing the αIIb tail sequence, and recombinant model proteins were produced as described previously (Gingras et al., 2012). The cDNAs encoding the mouse THD (residues 1–400) WT, L325R, and W359A were cloned into pEGFP-C1 vector. The cDNAs encoding the full-length human α4, β7, and mouse β7 proteins were amplified by RT-PCR and cloned into pCDNA3.1 or pLVX lentiviral expression vector. Cloning of β7 point mutants (L758A, Y759A, L721P, G722P, and L723P) was done by oligonucleotide-directed mutagenesis using the Infusion-HD Eco Dry cloning kit (Takara Bio Inc.). The cDNAs encoding the ectodomain of human and mouse MAdCAM-1 proteins were amplified by RT-PCR and fused to a cDNA encoding an IgG Fc fragment into pCDNA3.1. Recombinant MAdCAM-1 (Val1 to Pro315) fused to the Fc2/3 regions of human IgG1 was produced in pcDNA3.0 (Invitrogen) and purified by Protein A resin (GenScript) as described previously (Sun et al., 2011). Mouse β7 and human talin single guide RNAs (sgRNAs) were constructed in vector pSpCas9n(BB)-2A-Puro (48141; Addgene; Ran et al., 2013). The sgRNA sequences were as follows: β7-sgRNA1, 5′-GCATCGTGGCAGTGGGACTAGGG-3′; β7-sgRNA2, 5′-CGTGGCAGTGGGACTAGGGCTGG-3′; and talin-sgRNA, 5′-ACCTGACGGAGATGTCCCGTGGG-3′ (protospacer-adjacent motif sequences are underlined). All constructs were confirmed by DNA sequencing.
Cell culture experiments
293T, Jurkat, and TK1 cells were purchased from ATCC. 293T cells transiently expressing α4 and β7 WT and mutants were generated by Lipofectamine 3000 transfection (Invitrogen). Jurkat T cells that stably expressed WT and mutant β7 were generated by lentiviral transduction followed by selection with puromycin and cell sorting. WT or mutant β7-expressing lentiviral particles were prepared by cotransfection of pLVX-WT β7, β7(L721P), or β7(L723P) with pMDLg/pRRE, pRSV-Rev, and pMD2.G in 293T cells. Supernatants containing lentiviral particles were collected. For lentiviral delivery, Jurkat T cells were grown to 80% confluence and then infected with pLVX-WT β7–, β7(L721P)–, or β7(L723P)–containing lentiviral particles. Cells were then selected with puromycin (2 µg/ml) 72 h after infection.
To make TK1-β7 KO cells, TK1 cells were cotransfected with β7-sgRNA1 and 2 and Cas9-EGFP using an Amaxa electroporator (Lonza) followed by cell sorting of EGFP-positive cells. 5–7 d later, the TK1-β7 KO cells were incubated with anti-β7 antibody, and β7-null cells were isolated as unbound to anti–rabbit IgG MicroBeads (Miltenyi Biotech). The TK1-β7 KO cells were reconstituted with β7 or β7(L721P) by lentiviral transduction followed by selection with puromycin and FACS sorting for similar β7 expression. Cells were routinely tested for mycoplasma infection. To generate the Jurkat–talin KO cells, cells were transfected with pSpCas9(BB)-2A-EGFP vector expressing the sgRNA (5′-ACCTGACGGAGATGTCCCGTGGG-3′; protospacer-adjacent motifs are underlined) using an Amaxa electroporator (Lonza). 48 h later, EGFP-positive cells were single-cell cloned by automated cell deposition unit using a FACSAria (BD), and cells lacking talin 1 and talin 2 were selected by immunoblotting with an antibody that recognizes both forms of talin (Fig. 1 A).
Antibodies and reagents
The following antibodies were purchased from BioLegend: CD4 (GK1.5), CD8 (53–6.7), CD44 (1M7), CD62L (MEL-14), B220 (RA3-6B2), CD11c (N418), and F4/80 (BM8). Monoclonal anti-GFP (1:5,000; Takara Bio Inc.), monoclonal anti–β-actin (1:5,000; Sigma-Aldrich), monoclonal anti–human β7 (EPR1357, 1:3,000; Abcam), monoclonal antitalin (8d4, 1:1,000; Sigma-Aldrich), and polyclonal anti–kindlin 3 antibody (1:500; Abgent; Ye et al., 2013) were used for immunoblotting. FIB504 against human/mouse β7 prepared by using hybridoma cells was used for immunofluorescence at 2 µg/ml. Act-1 against human α4β7 was used for immunoprecipitation as previously described (Sun et al., 2011). Secondary Alexa Fluor–labeled antibodies for Western blot analysis were from Rockland or Thermo Fisher Scientific. CFSE and eFluor 670 were purchased from Invitrogen and BioLegend, respectively. Human and mouse CXCL12 were purchased from R&D Systems. PMA was purchased from Sigma-Aldrich.
Mice
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of California, San Diego. All mice were housed in specific pathogen-free conditions before use. The Itgb7L720P/L720P knock-in mice were generated using the CRISPR/Cas9 approach by the Transgenic Mouse Facility of the University of California, Irvine. B6SJL female mice were used as embryo donors. Single-stranded oligodeoxynucleotide (ssODN) was used as a homology-directed repair template and purchased from Integrated DNA Technologies as Ultramer DNA oligonucleotides. ssODN was mixed with Cas9 mRNA and sgRNA directly without purification. The sequence of ssODN was 5′-GTCAGCCCTTAAACCTTGGCCCCACCAGAGGGAGTGGATCACACCCGTGCCATCATACTGGGCTGCACAGGGGGCATCGTGGCAGTGGGCCCAGGCCTCGTTCTGGCTTACCGGCTCTCTGTGGAAATC-3′. Mononuclear cells were isolated from SP, peripheral blood, PLN, MLN, and PP by tissue homogenizer (JXFSTPRP-24; ThunderSci) as previously described (Berlin et al., 1995; Sun et al., 2014). Cell counting with immunofluorescence cytometry was performed using Accuri C6 FACSCalibur (BD).
Flow cytometry
5 × 106 cells were washed and resuspended in HBSS containing 0.1% BSA and 1 mM Ca2+/Mg2+ and stained with allophycocyanin (APC) anti–human/mouse integrin β7 antibody (1:500; FIB504; BioLegend) for 30 min at 4°C. APC rat IgG2a was used as isotype control. Cells were washed twice before flow cytometry analysis using a FACSCalibur. Data were analyzed with FlowJo software.
For the soluble ligand binding assay, after treating with Fc receptor blocking (BioLegend) to reduce nonspecific binding, 5 × 106 cells were washed and resuspended in HBSS containing 0.1% BSA and 1 mM Ca2+/Mg2+. The MAdCAM-1/Fc fusion protein was added to the mixture and incubated for 30 min at room temperature with or without PMA (100 nM for 30 min) or CXCL12 (0.5 µg/ml for 2 min). Then cells were immediately fixed by 2% paraformaldehyde at room temperature for 15 min followed by two washes and then incubated with Alexa Fluor 647–conjugated anti–human IgG (1:1,000) for 30 min at 4°C.
Affinity chromatography with recombinant integrin cytoplasmic domains
Cells were lysed with cold cell lysis buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 10 mM MgCl2, 1% NP-40, and 10% glycerol) plus protease inhibitor cocktails (Roche) on ice for 30 min. Affinity chromatography was performed using recombinant His-Avi-integrin cytoplasmic domain model proteins bound to NeutrAvidin resin (Novagen) as previously described (Pfaff et al., 1998; Petrich et al., 2007a). Samples were separated on a 4–20% SDS polyacrylamide gel (Novex; Invitrogen). Bound THD was stained by monoclonal anti-GFP (1:5,000; Takara Bio Inc.) and detected and quantified using an Odyssey imaging system (LI-COR Biosciences).
Flow chamber assay
The assembly of the microfluidic devices used in this study has been described previously (Fan et al., 2016). In brief, coverslips were coated with MAdCAM-1/Fc (10 µg/ml) alone or with CXCL12 (2 µg/ml) for 1 h and then blocked for 1 h with casein (1%) at room temperature. After coating, coverslips were sealed to polydimethylsiloxane chips by magnetic clamps to create flow chamber channels ∼29 µm high and ∼300 µm across. By modulating the pressure between the inlet well and the outlet reservoir, 2 dyn/cm wall shear stress was applied in all experiments. Cells were diluted to 5 × 106 cells/ml in HBSS (with 1 mM Ca2+/Mg2+) and immediately perfused through the flow chamber. For the PMA stimulation, cells were prestimulated for 10 min with PMA (100 nM) at room temperature before perfusion.
Adhesive interactions between the flowing cells and the coated substrates were assessed by manually tracking the motions of individual cells for 1 min as previously described (Sun et al., 2014). The motion of each adherent cell was monitored for 10 s after the initial adhesion point, and two categories of cell adhesion were defined: rolling adhesion cells had rolling motions for >10 s with a velocity >1 µm/s, whereas cells that remained adherent and stationary for >10 s with a velocity <1 µm/s were defined as arrested adherent cells.
Static adhesion assay
Wells of a 96-well Immulon 2HB (Thermo Fisher Scientific) plate were coated overnight at 4°C with MAdCAM-1/Fc (10 µg/ml) with or without CXCL12 (2 µg/ml) in coating buffer (PBS and 10 mM NaHCO3, pH 9.0). After the plates were rinsed with PBS, free binding sites were blocked with 2% BSA in coating buffer for 1 h at 37°C. Serum-starved Jurkat T cells were then added to wells and were allowed to adhere for 2 h at 37°C. Plates were then washed with PBS, and bound cells were stained with crystal violet (1%) after fixation with 3.7% formaldehyde.
Immunoprecipitation and blotting
Cells were lysed with ice-cold cell lysis buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 10 mM MgCl2, 1% NP-40, and 10% glycerol) plus protease inhibitor cocktails (Roche) on ice for 30 min and immunoprecipitated with the α4β7 mAb, Act-1 (1 µg). Mouse IgG was used as a control. After overnight incubation with antibodies at 4°C, Protein G Sepharose (Invitrogen) was added to the reaction mixture and further incubated for 2–4 h at 4°C. After three washes with ice-cold lysis buffer, beads were mixed with sample buffer before separation on a 4–20% SDS polyacrylamide gel (Novex; Invitrogen). Western blots were stained with monoclonal anti–human β7 (EPR1357, 1:3,000; Abcam), monoclonal antitalin (8d4; 1:1,000; Sigma-Aldrich), and monoclonal anti–β-actin (1:5,000; Sigma-Aldrich). Signal was detected and results were quantified using an Odyssey imaging system (LI-COR Biosciences).
In vivo competitive lymphocyte homing
TK1-β7 WT or TK1-β7(L721P) cells were labeled with eFluor 670 (eBioscience), and TK1 cells were labeled with CFSE (BioLegend). Equal numbers (5 × 107) of eFluor 670–labeled cells and CFSE-labeled cells were mixed and then injected into the tail vein of recipient C57BL/6J mice. After 2 h, the recipient mice were sacrificed, and lymphocytes from MLN, PP, PLN, and SP were harvested. The absolute cell numbers were calculated by measuring the ratio of CFSE- and eFluor 670–labeled cells by flow cytometry and then adjusted to the total cell count (hemocytometer). All animal experiments were approved by the University of California, San Diego, Institutional Animal Care and Use Committee in adherence with National Institutes of Health guidelines and policies.
Immunofluorescence staining
Mice frozen lymphoid organ sections were fixed with 4% PFA for 10 min at room temperature and blocked with 5% FBS plus 5% rat serum for 1 h at room temperature. Then the section was stained with either 2.5 µg/ml of CD8 (53–6.7) Alexa Fluor 594, 2.5 µg/ml of CD4 (GK1.5) Alexa Fluor 647, and 2.5 µg/ml of B220 (RA3-6B2) Alexa Fluor 488, or 2.5 µg/ml of F4/80 (BM8) Alexa Fluor 647 and 2.5 µg/ml of CD11c (N418) Alexa Fluor 488 overnight at 4°C. All antibodies were purchased from BioLegend.
Images were acquired using a Keyence BZX-700 all-in-one fluorescence microscope with CFI Plan Apochromat λ4× fluorescent objective (Nikon Plan Apochromat; 0.2 NA) or CFI Plan Apochromat λ10× fluorescent objective (Nikon Plan Apochromat; 0.45 NA), which was operated with a 2/3-in, 2.83 million pixel monochrome charge-coupled device colorized with a liquid chromatography filter at 25°C. Fluorochromes used for these analyses were Alexa Fluor 594, Alexa Fluor 647, Alexa Fluor 488, and/or DAPI. Images were processed and analyzed by a BZ-II Analyzer (Keyence) and Photoshop Element 10 software (Adobe). Background was reduced using brightness and contrast adjustments applied to the whole image.
Statistical analysis
Statistical analysis was performed in PRISM software (6.00; GraphPad Software), and all datasets were checked for Gaussian normality distribution. Data analysis was performed using one-way ANOVA or two-way ANOVA with Bonferroni’s post hoc test as indicated in figure legends. The resulting p-values are indicated as follows: NS, P > 0.05; *, 0.01 < P < 0.05; **, 0.001 < P < 0.01; ***, P < 0.001. Data represent means ± SD or SEM of at least three independent experiments. Western blot results show representative images from at least three independent experiments.
Online supplemental material
Fig. S1 confirms that talin-mediated α4β7 activation required specific contacts between the β7 region and talin F3. Fig. S2 confirms that changed β7 TMD topology mediates α4β7 activation. Fig. S3 shows that introduction of Ala, Gly, or Arg residues instead Pro at the midpoint of the β7 TMD had no effects on α4β7 activation by THD. Fig. S4 confirms that disrupting talin-induced change in β7 TMD topology impaired CXCL12-mediated α4β7 activation. Fig. S5 shows the organization of lymph nodes and the distribution and localization of different cell types in 8-wk-old Itgb7WT/WT mice and Itgb7L720P/L720P littermates.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health grants HL 078784, R35 HL 139947, and NS 092521 and by an American Heart Association postdoctoral fellowship 17POST33660181.
The authors declare no competing financial interests.
Author contributions: H. Sun and M.H. Ginsberg conceived the project and designed experiments. H. Sun performed the experiments and analyzed data. A.R. Gingras made the structure models. F. Lagarrigue, Z. Fan, and K. Ley provided key reagents and devices and contributed expert advice to the design of experiments. H. Sun and M.H. Ginsberg wrote the manuscript with contributions from all authors.
References
- Adams D.H., and Eksteen B.. 2006. Aberrant homing of mucosal T cells and extra-intestinal manifestations of inflammatory bowel disease. Nat. Rev. Immunol. 6:244–251. 10.1038/nri1784 [DOI] [PubMed] [Google Scholar]
- Agace W.W. 2008. T-cell recruitment to the intestinal mucosa. Trends Immunol. 29:514–522. 10.1016/j.it.2008.08.003 [DOI] [PubMed] [Google Scholar]
- Anthis N.J., Wegener K.L., Ye F., Kim C., Goult B.T., Lowe E.D., Vakonakis I., Bate N., Critchley D.R., Ginsberg M.H., and Campbell I.D.. 2009. The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. EMBO J. 28:3623–3632. 10.1038/emboj.2009.287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcario M.J., and Tajkhorshid E.. 2014. Membrane-induced structural rearrangement and identification of a novel membrane anchor in talin F2F3. Biophys. J. 107:2059–2069. 10.1016/j.bpj.2014.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthos J., Cicala C., Martinelli E., Macleod K., Van Ryk D., Wei D., Xiao Z., Veenstra T.D., Conrad T.P., Lempicki R.A., et al. . 2008. HIV-1 envelope protein binds to and signals through integrin alpha4beta7, the gut mucosal homing receptor for peripheral T cells. Nat. Immunol. 9:301–309. 10.1038/ni1566 [DOI] [PubMed] [Google Scholar]
- Berlin C., Berg E.L., Briskin M.J., Andrew D.P., Kilshaw P.J., Holzmann B., Weissman I.L., Hamann A., and Butcher E.C.. 1993. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. 74:185–195. 10.1016/0092-8674(93)90305-A [DOI] [PubMed] [Google Scholar]
- Berlin C., Bargatze R.F., Campbell J.J., von Andrian U.H., Szabo M.C., Hasslen S.R., Nelson R.D., Berg E.L., Erlandsen S.L., and Butcher E.C.. 1995. alpha 4 integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell. 80:413–422. 10.1016/0092-8674(95)90491-3 [DOI] [PubMed] [Google Scholar]
- Byrareddy S.N., Arthos J., Cicala C., Villinger F., Ortiz K.T., Little D., Sidell N., Kane M.A., Yu J., Jones J.W., et al. . 2016. Sustained virologic control in SIV+ macaques after antiretroviral and α4β7 antibody therapy. Science. 354:197–202. 10.1126/science.aag1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caradonna L., Amati L., Lella P., Jirillo E., and Caccavo D.. 2000. Phagocytosis, killing, lymphocyte-mediated antibacterial activity, serum autoantibodies, and plasma endotoxins in inflammatory bowel disease. Am. J. Gastroenterol. 95:1495–1502. 10.1111/j.1572-0241.2000.02085.x [DOI] [PubMed] [Google Scholar]
- Cherry L.N., Yunker N.S., Lambert E.R., Vaughan D., and Lowe D.K.. 2015. Vedolizumab: an α4β7 integrin antagonist for ulcerative colitis and Crohn’s disease. Ther. Adv. Chronic Dis. 6:224–233. 10.1177/2040622315586970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe D.T., Chiu H., Fong S., and Weissman I.L.. 1994. Regulation of the avidity of integrin alpha 4 beta 7 by the beta 7 cytoplasmic domain. J. Biol. Chem. 269:14411–14418. [PubMed] [Google Scholar]
- Eksteen B., Liaskou E., and Adams D.H.. 2008. Lymphocyte homing and its role in the pathogenesis of IBD. Inflamm. Bowel Dis. 14:1298–1312. 10.1002/ibd.20453 [DOI] [PubMed] [Google Scholar]
- Erle D.J., Briskin M.J., Butcher E.C., Garcia-Pardo A., Lazarovits A.I., and Tidswell M.. 1994. Expression and function of the MAdCAM-1 receptor, integrin alpha 4 beta 7, on human leukocytes. J. Immunol. 153:517–528. [PubMed] [Google Scholar]
- Fan Z., McArdle S., Marki A., Mikulski Z., Gutierrez E., Engelhardt B., Deutsch U., Ginsberg M., Groisman A., and Ley K.. 2016. Neutrophil recruitment limited by high-affinity bent β2 integrin binding ligand in cis. Nat. Commun. 7:12658 10.1038/ncomms12658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feagan B.G., Greenberg G.R., Wild G., Fedorak R.N., Paré P., McDonald J.W., Dubé R., Cohen A., Steinhart A.H., Landau S., et al. . 2005. Treatment of ulcerative colitis with a humanized antibody to the alpha4beta7 integrin. N. Engl. J. Med. 352:2499–2507. 10.1056/NEJMoa042982 [DOI] [PubMed] [Google Scholar]
- Feagan B.G., Rutgeerts P., Sands B.E., Hanauer S., Colombel J.F., Sandborn W.J., Van Assche G., Axler J., Kim H.J., Danese S., et al. GEMINI 1 Study Group . 2013. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 369:699–710. 10.1056/NEJMoa1215734 [DOI] [PubMed] [Google Scholar]
- García-Alvarez B., de Pereda J.M., Calderwood D.A., Ulmer T.S., Critchley D., Campbell I.D., Ginsberg M.H., and Liddington R.C.. 2003. Structural determinants of integrin recognition by talin. Mol. Cell. 11:49–58. 10.1016/S1097-2765(02)00823-7 [DOI] [PubMed] [Google Scholar]
- Gingras A.R., Liu J.J., and Ginsberg M.H.. 2012. Structural basis of the junctional anchorage of the cerebral cavernous malformations complex. J. Cell Biol. 199:39–48. 10.1083/jcb.201205109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg M.H., O’Toole T.E., Loftus J.C., and Plow E.F.. 1992. Ligand binding to integrins: dynamic regulation and common mechanisms. Cold Spring Harb. Symp. Quant. Biol. 57:221–231. 10.1101/SQB.1992.057.01.027 [DOI] [PubMed] [Google Scholar]
- Gorfu G., Rivera-Nieves J., and Ley K.. 2009. Role of beta7 integrins in intestinal lymphocyte homing and retention. Curr. Mol. Med. 9:836–850. 10.2174/156652409789105525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haling J.R., Monkley S.J., Critchley D.R., and Petrich B.G.. 2011. Talin-dependent integrin activation is required for fibrin clot retraction by platelets. Blood. 117:1719–1722. 10.1182/blood-2010-09-305433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes P.E., Diaz-Gonzalez F., Leong L., Wu C., McDonald J.A., Shattil S.J., and Ginsberg M.H.. 1996. Breaking the integrin hinge. A defined structural constraint regulates integrin signaling. J. Biol. Chem. 271:6571–6574. 10.1074/jbc.271.12.6571 [DOI] [PubMed] [Google Scholar]
- Kalli A.C., Campbell I.D., and Sansom M.S.. 2011. Multiscale simulations suggest a mechanism for integrin inside-out activation. Proc. Natl. Acad. Sci. USA. 108:11890–11895. 10.1073/pnas.1104505108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C., Lau T.L., Ulmer T.S., and Ginsberg M.H.. 2009. Interactions of platelet integrin alphaIIb and beta3 transmembrane domains in mammalian cell membranes and their role in integrin activation. Blood. 113:4747–4753. 10.1182/blood-2008-10-186551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C., Schmidt T., Cho E.G., Ye F., Ulmer T.S., and Ginsberg M.H.. 2011. Basic amino-acid side chains regulate transmembrane integrin signalling. Nature. 481:209–213. 10.1038/nature10697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C., Ye F., Hu X., and Ginsberg M.H.. 2012. Talin activates integrins by altering the topology of the β transmembrane domain. J. Cell Biol. 197:605–611. 10.1083/jcb.201112141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam M.C., and Bressler B.. 2014. Vedolizumab for ulcerative colitis and Crohn’s disease: results and implications of GEMINI studies. Immunotherapy. 6:963–971. 10.2217/imt.14.66 [DOI] [PubMed] [Google Scholar]
- Lau T.L., Partridge A.W., Ginsberg M.H., and Ulmer T.S.. 2008. Structure of the integrin beta3 transmembrane segment in phospholipid bicelles and detergent micelles. Biochemistry. 47:4008–4016. 10.1021/bi800107a [DOI] [PubMed] [Google Scholar]
- Lau T.L., Kim C., Ginsberg M.H., and Ulmer T.S.. 2009. The structure of the integrin alphaIIbbeta3 transmembrane complex explains integrin transmembrane signalling. EMBO J. 28:1351–1361. 10.1038/emboj.2009.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R., Babu C.R., Valentine K., Lear J.D., Wand A.J., Bennett J.S., and DeGrado W.F.. 2002. Characterization of the monomeric form of the transmembrane and cytoplasmic domains of the integrin beta 3 subunit by NMR spectroscopy. Biochemistry. 41:15618–15624. 10.1021/bi026822l [DOI] [PubMed] [Google Scholar]
- Lu Z., Mathew S., Chen J., Hadziselimovic A., Palamuttam R., Hudson B.G., Fässler R., Pozzi A., Sanders C.R., and Zent R.. 2016. Implications of the differing roles of the β1 and β3 transmembrane and cytoplasmic domains for integrin function. eLife. 5:e18633 10.7554/eLife.18633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B.H., Springer T.A., and Takagi J.. 2004. A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biol. 2:e153 10.1371/journal.pbio.0020153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean L.P., Shea-Donohue T., and Cross R.K.. 2012. Vedolizumab for the treatment of ulcerative colitis and Crohn’s disease. Immunotherapy. 4:883–898. 10.2217/imt.12.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora J.R., and Von Andrian U.H.. 2006. Specificity and plasticity of memory lymphocyte migration. Curr. Top. Microbiol. Immunol. 308:83–116. [PubMed] [Google Scholar]
- Nieswandt B., Moser M., Pleines I., Varga-Szabo D., Monkley S., Critchley D., and Fässler R.. 2007. Loss of talin1 in platelets abrogates integrin activation, platelet aggregation, and thrombus formation in vitro and in vivo. J. Exp. Med. 204:3113–3118. 10.1084/jem.20071827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson I., and von Heijne G.. 1998. Breaking the camel’s back: proline-induced turns in a model transmembrane helix. J. Mol. Biol. 284:1185–1189. 10.1006/jmbi.1998.2219 [DOI] [PubMed] [Google Scholar]
- Partridge A.W., Liu S., Kim S., Bowie J.U., and Ginsberg M.H.. 2005. Transmembrane domain helix packing stabilizes integrin alphaIIbbeta3 in the low affinity state. J. Biol. Chem. 280:7294–7300. 10.1074/jbc.M412701200 [DOI] [PubMed] [Google Scholar]
- Petrich B.G., Fogelstrand P., Partridge A.W., Yousefi N., Ablooglu A.J., Shattil S.J., and Ginsberg M.H.. 2007a The antithrombotic potential of selective blockade of talin-dependent integrin alpha IIb beta 3 (platelet GPIIb-IIIa) activation. J. Clin. Invest. 117:2250–2259. 10.1172/JCI31024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrich B.G., Marchese P., Ruggeri Z.M., Spiess S., Weichert R.A., Ye F., Tiedt R., Skoda R.C., Monkley S.J., Critchley D.R., and Ginsberg M.H.. 2007b Talin is required for integrin-mediated platelet function in hemostasis and thrombosis. J. Exp. Med. 204:3103–3111. 10.1084/jem.20071800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaff M., Liu S., Erle D.J., and Ginsberg M.H.. 1998. Integrin beta cytoplasmic domains differentially bind to cytoskeletal proteins. J. Biol. Chem. 273:6104–6109. 10.1074/jbc.273.11.6104 [DOI] [PubMed] [Google Scholar]
- Poole R.M. 2014. Vedolizumab: first global approval. Drugs. 74:1293–1303. 10.1007/s40265-014-0253-1 [DOI] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., and Zhang F.. 2013. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8:2281–2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüegg C., Postigo A.A., Sikorski E.E., Butcher E.C., Pytela R., and Erle D.J.. 1992. Role of integrin α 4 β 7/α 4 β P in lymphocyte adherence to fibronectin and VCAM-1 and in homotypic cell clustering. J. Cell Biol. 117:179–189. 10.1083/jcb.117.1.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutgeerts P.J., Fedorak R.N., Hommes D.W., Sturm A., Baumgart D.C., Bressler B., Schreiber S., Mansfield J.C., Williams M., Tang M., et al. . 2013. A randomised phase I study of etrolizumab (rhuMAb β7) in moderate to severe ulcerative colitis. Gut. 62:1122–1130. 10.1136/gutjnl-2011-301769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandborn W.J., Feagan B.G., Rutgeerts P., Hanauer S., Colombel J.F., Sands B.E., Lukas M., Fedorak R.N., Lee S., Bressler B., et al. GEMINI 2 Study Group . 2013. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N. Engl. J. Med. 369:711–721. 10.1056/NEJMoa1215739 [DOI] [PubMed] [Google Scholar]
- Shattil S.J., Kim C., and Ginsberg M.H.. 2010. The final steps of integrin activation: the end game. Nat. Rev. Mol. Cell Biol. 11:288–300. 10.1038/nrm2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson W.T., Franco S.J., and Huttenlocher A.. 2006. Talin1 regulates TCR-mediated LFA-1 function. J. Immunol. 177:7707–7714. 10.4049/jimmunol.177.11.7707 [DOI] [PubMed] [Google Scholar]
- Smith P.D., Ochsenbauer-Jambor C., and Smythies L.E.. 2005. Intestinal macrophages: unique effector cells of the innate immune system. Immunol. Rev. 206:149–159. 10.1111/j.0105-2896.2005.00288.x [DOI] [PubMed] [Google Scholar]
- Sun H., Wu Y., Qi J., Pan Y., Ge G., and Chen J.. 2011. The CC’ and DE loops in Ig domains 1 and 2 of MAdCAM-1 play different roles in MAdCAM-1 binding to low- and high-affinity integrin alpha4beta7. J. Biol. Chem. 286:12086–12092. 10.1074/jbc.M110.208900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H., Liu J., Zheng Y., Pan Y., Zhang K., and Chen J.. 2014. Distinct chemokine signaling regulates integrin ligand specificity to dictate tissue-specific lymphocyte homing. Dev. Cell. 30:61–70. 10.1016/j.devcel.2014.05.002 [DOI] [PubMed] [Google Scholar]
- Tadokoro S., Shattil S.J., Eto K., Tai V., Liddington R.C., de Pereda J.M., Ginsberg M.H., and Calderwood D.A.. 2003. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 302:103–106. 10.1126/science.1086652 [DOI] [PubMed] [Google Scholar]
- Villablanca E.J., Cassani B., von Andrian U.H., and Mora J.R.. 2011. Blocking lymphocyte localization to the gastrointestinal mucosa as a therapeutic strategy for inflammatory bowel diseases. Gastroenterology. 140:1776–1784. 10.1053/j.gastro.2011.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visiers I., Braunheim B.B., and Weinstein H.. 2000. Prokink: a protocol for numerical evaluation of helix distortions by proline. Protein Eng. 13:603–606. 10.1093/protein/13.9.603 [DOI] [PubMed] [Google Scholar]
- von Heijne G. 1991. Proline kinks in transmembrane alpha-helices. J. Mol. Biol. 218:499–503. 10.1016/0022-2836(91)90695-3 [DOI] [PubMed] [Google Scholar]
- Wagner N., Löhler J., Kunkel E.J., Ley K., Leung E., Krissansen G., Rajewsky K., and Müller W.. 1996. Critical role for beta7 integrins in formation of the gut-associated lymphoid tissue. Nature. 382:366–370. 10.1038/382366a0 [DOI] [PubMed] [Google Scholar]
- Wegener K.L., Partridge A.W., Han J., Pickford A.R., Liddington R.C., Ginsberg M.H., and Campbell I.D.. 2007. Structural basis of integrin activation by talin. Cell. 128:171–182. 10.1016/j.cell.2006.10.048 [DOI] [PubMed] [Google Scholar]
- Woolfson D.N., and Williams D.H.. 1990. The influence of proline residues on alpha-helical structure. FEBS Lett. 277:185–188. 10.1016/0014-5793(90)80839-B [DOI] [PubMed] [Google Scholar]
- Ye F., Petrich B.G., Anekal P., Lefort C.T., Kasirer-Friede A., Shattil S.J., Ruppert R., Moser M., Fassler R., and Ginsberg M.H.. 2013. The Mechanism of Kindlin-Mediated Activation of Integrin αIIbβ3. Curr. Biol. 23:2288–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.