

Abstract
The inherited cardiomyopathies, hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) are relatively common, potentially life-threatening and currently untreatable. Mutations are often in the contractile proteins of cardiac muscle and cause abnormal Ca2+ regulation via troponin. HCM is usually linked to higher myofilament Ca2+-sensitivity whilst in both HCM and DCM mutant tissue there is often an uncoupling of the relationship between troponin I (TnI) phosphorylation by PKA and modulation of myofilament Ca2+-sensitivity, essential for normal responses to adrenaline. The adrenergic response is blunted, and this may predispose the heart to failure under stress. At present there are no compounds or interventions that can prevent or treat sarcomere cardiomyopathies. There is a need for novel therapies that act at a more fundamental level to affect the disease process. We demonstrated that epigallocatechin-3 gallate (EGCG) was found to be capable of restoring the coupled relationship between Ca2+-sensitivity and TnI phosphorylation in mutant thin filaments to normal in vitro, independent of the mutation (15 mutations tested). We have labeled this property “re-coupling.” The action of EGCG in vitro to reverse the abnormality caused by myopathic mutations would appear to be an ideal pharmaceutical profile for treatment of inherited HCM and DCM but EGCG is known to be promiscuous in vivo and is thus unsuitable as a therapeutic drug. We therefore investigated whether other structurally related compounds can re-couple myofilaments without these off-target effects. We used the quantitative in vitro motility assay to screen 40 compounds, related to C-terminal Hsp90 inhibitors, and found 23 that can re-couple mutant myofilaments. There is no correlation between re-couplers and Hsp90 inhibitors. The Ca2+-sensitivity shift due to TnI phosphorylation was restored to 2.2 ± 0.01-fold (n = 19) compared to 2.0 ± 0.24-fold (n = 7) in wild-type thin filaments. Many of these compounds were either pure re-couplers or pure desensitizers, indicating these properties are independent; moreover, re-coupling ability could be lost with small changes of compound structure, indicating the possibility of specificity. Small molecules that can re-couple may have therapeutic potential.
HIGHLIGHTS
- Inherited cardiomyopathies are common diseases that are currently untreatable at a fundamental level and therefore finding a small molecule treatment is highly desirable.
- We have identified a molecular level dysfunction common to nearly all mutations: uncoupling of the relationship between troponin I phosphorylation and modulation of myofilament Ca2+-sensitivity, essential for normal responses to adrenaline.
- We have identified a new class of drugs that are capable of both reducing Ca2+-sensitivity and/or recouping the relationship between troponin I phosphorylation and Ca2+-sensitivity.
- The re-coupling phenomenon can be explained on the basis of a single mechanism that is testable.
- Measurements with a wide range of small molecules of varying structures can indicate the critical molecular features required for recoupling and allows the prediction of other potential re-couplers.
Keywords: cardiomyopathy, sarcomeric protein mutations, troponin I phosphorylation, PKA, Ca2+ regulation, small molecule pharmacology, EGCG, silybin
Introduction
The contractile apparatus of cardiac muscle is controlled by the sarcoplasmic Ca2+-level acting upon the Ca2+ sensor, troponin. Troponin, together with tropomyosin and actin constitutes the thin filaments of muscle. In unstimulated muscle contraction is inhibited by the C-terminus of the troponin I subunit binding to actin-tropomyosin and blocking the interaction with myosin crossbridges. Upon stimulation intracellular Ca2+ rises and Ca2+ binds to the troponin C subunit (TnC), the C-terminus of TnI is released from its site on actin and crossbridge cycling can proceed (Gordon et al., 2000; MacLeod et al., 2010). Thus changes in Ca2+ level switch contraction on and off. However contractility in heart muscle is additionally modulated through the β-receptors. Activation of β1- receptors by adrenaline initiates formation of cAMP and activation of Protein Kinase A (PKA) leading to increased rate and magnitude of contraction. Lusitropy, an increased rate of relaxation allowing for shorter twitch duration, is a key component of adrenergic activation and involves TnI. Phosphorylation of ser22 and 23 of TnI by PKA leads to a 2-fold decrease in Ca2+-sensitivity and a corresponding increase in the rate of Ca2+ release from TnC and is essential for the lusitropic response (Robertson et al., 1982; Kentish et al., 2001; Layland et al., 2005).
Whilst the structural mechanism of the troponin Ca2+ switch is well-documented (Takeda et al., 2003), the structural basis of lusitropy is poorly understood since both the phosphorylated N-terminal peptide and the regulatory “switch peptide” of TnI are intrinsically disordered, however recent studies using molecular dynamics simulations have begun to explain this phenomenon (Papadaki and Marston, 2016). The study of this phosphorylation dependent regulation has been recently stimulated by the discovery that a primary effect of many mutations in thin filament proteins associated with cardiomyopathy is the uncoupling of this relationship (Bayliss et al., 2012; Memo et al., 2013; Messer and Marston, 2014).
Dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM) are often due to single mutations in a protein of the sarcomere and have been extensively studied in vitro. It is estimated that 1/250 of the population show symptoms of DCM, and that 40% of cases are inherited (Hershberger et al., 2013). Truncating mutations in the titin (TTN) gene are the most common but a substantial fraction (12%) of mutations are in the proteins of the thin filament. HCM is estimated to be present in 1/500 of the population; it is overwhelmingly an inherited disease and the majority of known disease causing mutations are in the contractile proteins of the heart muscle sarcomere (Walsh et al., 2017). It is a leading cause of sudden cardiac death in young adults, particularly athletes and if not controlled can lead to heart failure (Lopes and Elliott, 2014).
In HCM, mutations generally cause an increase of about 1.5- to 2-fold in myofilament Ca2+-sensitivity that is considered to be sufficient to induce the symptoms of HCM. The effect of DCM mutations in contractile proteins on Ca2+-sensitivity is variable with a trend toward deceased Ca2+-sensitivity (Marston, 2016). Importantly for this study it has been shown that HCM and DCM-related mutations in contractile proteins usually abolish the coupled relationship between Ca2+-sensitivity and TnI phosphorylation by PKA: we have called this phenomenon “uncoupling” (Memo et al., 2013; Messer and Marston, 2014; Messer et al., 2016). A blunting of the response to β-adrenergic activation is commonly observed in animal models with HCM or DCM mutations and it has been demonstrated in a DCM mouse model that this blunting is sufficient to induce heart failure under chronic stress (Messer and Marston, 2014; Wilkinson et al., 2015).
At present there are no compounds or interventions that can prevent or treat sarcomeric cardiomyopathies and current treatments available are directed at alleviating the symptoms. There is a need for novel therapies that act at a more fundamental level to affect the disease process (Tardiff et al., 2015). In our studies of the uncoupling effects of mutations we demonstrated that epigallocatechin-3 gallate (EGCG), originally studied as a Ca2+ desensitizer (Tadano et al., 2010; Friedrich et al., 2016), was also found to be capable of restoring the coupled relationship between Ca2+-sensitivity and TnI phosphorylation in mutant thin filaments to normal in vitro, a property we refer to as “re-coupling” (Papadaki et al., 2015; Messer et al., 2016, 2017). The action of EGCG in vitro to reverse the abnormality caused by myopathic mutations would appear to be an ideal pharmaceutical profile for treatment of inherited HCM and DCM. However, EGCG is known to have pleiotropic pharmaceutical properties in intact tissue, including inotropic activity, making it an unsuitable therapeutic drug (Singh et al., 2011; Feng et al., 2012; Baell and Walters, 2014; Ingólfsson et al., 2014). Moreover, this compound is not easily available in its pure state and it is not sufficiently stable under oxidative and hydrolytic conditions. Therefore, it is important to investigate whether other structurally related compounds can also re-couple myofilaments without these off-target effects.
EGCG and its analogs have been widely studied as Hsp90 inhibitors, so we began our investigations using analogs of EGCG and of Silybin, a natural product with structural similarity, originally studied as Hsp90 inhibitors (Hao et al., 2010; Zhao et al., 2011; Khandelwal et al., 2013). We used the quantitative in vitro motility assay to screen 40 compounds and found 23 that can re-couple mutant myofilaments. Many of these compounds were either pure re-couplers or pure desensitizers, unlike EGCG that has both properties. Moreover, re-coupling ability could be lost with minor modifications in the compound structure, indicating the possibility of specificity. Consideration of the molecular structures of re-coupling molecules (structure-activity relationships, SAR), compared with similar inactive molecules can provide considerable insight into the mechanism of re-coupling and may lead to the discovery of more potent re-coupling (lead) compounds with therapeutic potential.
Methods
Sources of contractile proteins
Troponin was isolated from donor heart tissue, supplied by Sydney Tissue Bank, University of Sydney, Australia. St Vincent's Hospital Sydney and Brompton, Harefield and NHLI Research Ethics Committees provided ethical approval for the collection of and experimentation with tissue samples (Lal et al., 2015). Donors had no previous history of heart disease and unremarkable ECG.
An anti-cardiac TnI monoclonal antibody affinity column was used to isolate troponin from 100 mg of donor heart tissue as previously described (Messer et al., 2007). This troponin has an intrinsically high level of TnI phosphorylation (1.6–2.2 mol Pi/mol). To reduce the phosphorylation level (<0.3 mol Pi/mol) troponin was treated with shrimp alkaline phosphatase (Memo et al., 2013). The level of TnI phosphorylation was measured by phosphate affinity SDS–PAGE as previously described (Messer et al., 2009).
Recombinant human sequence tropomyosin was a gift from Kristen Nowak and Elyshia MacNamara, University, Western Australia. Wild-type α-tropomyosin (Tpm1.1) and mutant E180G tropomyosin were expressed in a baculovirus/Sf9 system (Akkari et al., 2002; Marston et al., 2013). Skeletal muscle F-actin and myosin were isolated from rabbit fast-twitch skeletal muscle as previously described (Messer et al., 2007). TnC G159D mutant troponin was extracted from a mutant human heart sample (Dyer et al., 2009), E99K mutant cardiac actin was extracted from the E99K transgenic mouse heart (Song et al., 2013).
EGCG- and silybin-related compounds
The analogs of EGCG labeled 1–28 were prepared as described by Khandelwal and Blagg (Khandelwal et al., 2013). Table 1 identifies the compounds in that paper and the molecular structures are shown in Figure S1. Silybin diastereomereomers and silmarin-derived pure flavonolignanswere isolated as described by Kren et al. (Gažák et al., 2011; Krenek et al., 2014; Novotná et al., 2014) (for structures see Figure S1). All other compounds were purchased from Sigma-Aldrich. Dose was 100 μM from a 10 mM stock in DMSO unless otherwise stated.
Table 1.
Ability of small molecules to re-couple troponin in thin myofilaments measured by in vitro motility assay.
| Drug | Coupling Difference (Initial Screen E180G) | E180G EC50 % Motility Difference, μM | Other mutations | EC50 Hsp90 inhibition assay |
|---|---|---|---|---|
| EGCG | 23.0 | 59 μM | see Figure 1 | |
| Silybin(racemic mixture) | 12.0 | 47 μM | E54K, E99K | |
| 1 | 1.0 | |||
| 2 (23h) | 27.0 | 30 μM | 22 μM | |
| 3 (27a) | 8.0 | 14 μM | ||
| 4 (23p) | 27.0 | 34 μM | >100 μM | |
| 5 | 2.0 | |||
| 6 | 1.0 | |||
| 7 (27c) | −11.0 | 67 μM | E54K, E99K | 32 μM |
| 8 (27p) | No Effect | 38 μM | ||
| 9 (23q) | 20.0 | 35 μM | >100 μM | |
| 10 | No Effect | |||
| 11 (10c) | 7.0 | E54K | >100 μM | |
| 12 (23f) | −1.0 | No Effect | 63 μM | |
| 13 | No Effect | |||
| 14 | 17.0 | 36 μM | ||
| 15 (10j) | 13.0 | 19 μM | ||
| 16 (27h) | 6.0 | 31 μM | 8 μM | |
| 17 (23b) | 3.5 | 58 μM | ||
| 19 (27g) | 24.0 | 21 μM | ||
| 20 | 2.0 | |||
| 21 (11d) | 25.0 | 14 μM | ||
| 22 | 7.0 | |||
| 23 (23a) | 9.0 | 98 μM | ||
| 24 | 8.0 | 24.5 μM | ||
| 26 (7a) | 26.0 | E54K | ||
| 27 (31d) | 4.0 | 55 μM | ||
| 28 (10c) | 4.0 | >100 μM | ||
| Epicatechin (EC) | 69 | |||
| Epigallocatechin (EGC) | 62 | |||
| Gallocatechin (GC) | 68 | |||
| Epicatechin Gallate (ECG) | 8 | |||
| Silybin A | 5 | G159D | ||
| Silybin B | 55 | 39 μM | G159D | |
| Dehydrosilybin A | 3 | G159D | ||
| Dehydrosilybin B | 62 | 47 μM | G159D | |
| Isosilybin | 4 | |||
| Silychristin | 35 | |||
| Silydianin | 20 | |||
| Taxifolin | 60 | |||
| Quercetin | 51 | 8 μM | ||
| Resveratrol | 63 | |||
| Novobiocin | 46 | |||
| Pterostilbene | 50 | |||
| Sissotrin | 30 | |||
| W-7 | 50 | |||
| Omecamtiv Mecarbil | 0 |
Test system as shown in Figure 2. Compounds 1–28 were synthesized by Blagg and Khandelwal; their corresponding identification numbers in the original paper are given in brackets and the structures are shown in the supplement (Khandelwal et al., 2013). Difference between motility (%) in phosphorylated and unphosphorylated forms of thin filaments (and EC50, when measured) with TPM1 E180G HCM mutation are shown. Other mutations also used in this test are E54K, TPM1 E54K DCM mutation; E99K, ACTC E99K HCM mutation and G159D, TNNC1 G159D DCM mutation. Compounds highlighted in green are effective re-couplers. Compounds highlighted in red indicate reverse re-coupling where Ca2+-sensitivity of the phosphorylated Tn I is greater than that of the dephosphorylated Tn I. In orange is the reverse re-coupler #7. For comparison the last column shows the effectiveness of these compounds in an assay for Hsp90 inhibition; effective Hsp90 inhibitors are highlighted in green and EC50 is given when measured (Khandelwal et al., 2013).
Quantitative in vitro motility assay (IVMA)
Thin filaments were reconstituted with 10 nM rabbit skeletal or mouse cardiac muscle α-actin (labeled with TRITC phalloidin), tropomyosin (40–60 nM), and troponin (60 nM) to study Ca2+-regulation of filament motility by the quantitative in vitro motility assay (IVMA) (Fraser and Marston, 1995; Messer et al., 2007). Thin filament movement over a bed of immobilized rabbit fast skeletal muscle heavy meromyosin (100 μg/ml) was compared in paired motility cells, in which troponin varied by a single factor (mutation, phosphorylation state, or treatment with drug). The temperature was set to 29°C. Filament movement was recorded and analyzed as previously described (Marston et al., 1996), yielding two parameters, the fraction of filaments moving and the speed of moving filaments. Only the fraction of filaments motile is used in this study.
The fraction motile was measured over a range of Ca2+ concentrations to generate Ca2+-activation curves. The data were fitted to the 4-variable Hill equation to yield a value for EC50 and nH. Most assays of the effect of compounds are single measurements. When appropriate EC50 values from replicate experiments were analyzed by paired t-test since the distribution of EC50 has been shown to be normal. Error bars and n values are given in tables and figures.
For drug screening, motility was measured with paired phosphorylated and unphosphorylated tropomyosin E180G mutant-containing thin filaments at a fixed 0.037 μM Ca2+. Most assays were single measurements.
Results
EGCG can re-couple every uncoupling mutation tested
We measured the Ca2+ activation of contractility using an in vitro motility assay that measures the movement of reconstituted thin filaments over a bed of immobilized myosin heads. A series of measurements, some of which have been previously published (Papadaki et al., 2015; Messer et al., 2016, 2017), are summarized in Figure 1. These determinations of EC50 show that in every case where a mutation causes uncoupling of Ca2+-sensitivity from TnI phosphorylation, EGCG can restore the coupling (re-couple). Thus, the effect of EGCG is independent of the mutation that induced uncoupling; in addition, both in man and cat, HCM cases were found where no mutation in sarcomeric genes was detected yet the thin filaments were uncoupled and they could also be re-coupled by EGCG (Bayliss et al., 2012; Messer et al., 2016).
Figure 1.

EGCG recoupling of troponin. Ratio of EC50 for phosphorylated and unphosphorylated thin filaments is plotted for thin filaments that are uncoupled. EC50 was determined from Ca2+-concentration plots; see Figures 2A, 4A,C,D for examples. Raw EC50 data is given in Table S1. EC50 was measured by in vitro motility assay in the presence (red bars) and absence (blue bars) of 100 μM EGCG. Mutant troponin or tropomyosin was from tissue (mouse, cat or human) with mutations in TnC (TNNC1), TnI (TNNI3), TnT (TNNT2), Actin (ACTC), tropomyosin (TPM1), myosin binding protein C (MYBPC3), and from cat heart with HCM and Human heart material from myectomy operations to relieve HCM; in the latter two cases samples were genotyped but no causative mutation was found. EGCG restored coupling to all the uncoupled systems. Includes data from (Papadaki et al., 2015; Messer et al., 2016, 2017).
Rapid screening for re-coupling functionality
For the rapid screening of compounds, we devised a single Ca2+ concentration assay. This is illustrated in Figures 2A,B for quercetin. The test system contains skeletal muscle actin, human sequence recombinant tropomyosin 1.1 containing the E180G HCM-associated mutation and human cardiac troponin from donor hearts in the phosphorylated and unphosphorylated form. At 0.037 μM Ca2+ the filaments are about 40% motile irrespective of phosphorylation level whilst when quercetin is added the phosphorylated Ca2+ activity curve shifts to the right and % motility at 0.037 μM Ca2+ drops, whilst the unphosphorylated level is unchanged. We have set a difference between phosphorylated and unphosphorylated >10% as indicating re-coupling activity. This assay method can also be used to construct a dose-response curve as shown. Table 1 and Figures 2C,D show the results from screening 40 compounds in this assay. We investigated compounds in 4 categories.
Figure 2.

Single [Ca2+] screen for re-coupling compounds. (A) Ca2+-activation curves for phosphorylated and unphosphorylated thin filaments containing E180G tropomyosin in the presence and absence of 25 μM Quercetin, a typical pure recoupler. Solid lines, phosphorylated state; dotted lines, unphosphorylated state. Purple lines and points, E180G thin filaments; green lines and points, E180G thin filaments + Quercetin (B) Dose -response curve for quercetin recoupling activity measured at a constant concentration of 0.037 μM Ca2+. INSET, kinetic analysis of dose response curve; difference between phosphorylated and unphosphorylated states is plotted against quercetin concentration. The Hill equation is fitted to the data, EC50 = 8 μM. (C,D) The effect of 100 μM drug treatment on the difference between % of motility of phosphorylated and unphosphorylated tropomyosin E180G mutant-containing thin filaments at 0.037 μM Ca2+. (C) EGCG-related molecules (for structures 1–28 see Figure S1 and Table 1). (D) Silybin, related compounds and other HsP90 inhibitors and Ca2+ desensitizers. The threshold for positive re-coupling was set at a 10% difference (dotted line).
Variants of EGCG lacking the pyrogallol ring (Khandelwal et al., 2013).
Variants of EGCG lacking the galloyl moiety (Tadano et al., 2010).
Silybin, its derivatives and stereoisomers, since this has also been proposed as an Hsp90 C-terminal inhibitor (Gažák et al., 2011; Zhao et al., 2011).
Unrelated Hsp90 inhibitors and Ca2+-desensitizers (Hall et al., 2014).
For structures see Figure S1.
Re-couplers were clearly distinguishable from inactive compounds: 23/40 compounds were classed as re-couplers. A series of dose-response curves (Figure S2) indicate the magnitude of recoupling is approximately the same for all compounds measured together and the EC50 for EGCG and Silybin derivatives are similar, being in the range 30–60 μM (see Table 1). It should be noted that Ca2+-sensitisers and the myosin activator Omecamtiv Mercarbil do not re-couple (Figure 2).
Interestingly, one compound, #7, had a reverse effect—the phosphorylated troponin was more Ca2+-sensitive than the unphosphorylated troponin condition. The standard assay used E180G mutant tropomyosin as the uncoupling mutation but, as with EGCG, recoupling was independent of the mutation type when tested with uncoupled thin filaments containing TPM1 E54K, ACTC E99K, or TNNC1 G159D mutations as indicated in Table 1. It is interesting to note that when Ca2+ sensitivities of the phosphorylated and unphosphorylated troponin of re-coupled systems were measured, the shift in Ca2+-sensitivity was restored to a value close to the shift in Ca2+-sensitivity previously observed in PKA-phosphorylated native thin filaments. (see Figure 3 and Table S1). Following this preliminary screening, selected compounds were further investigated.
Figure 3.
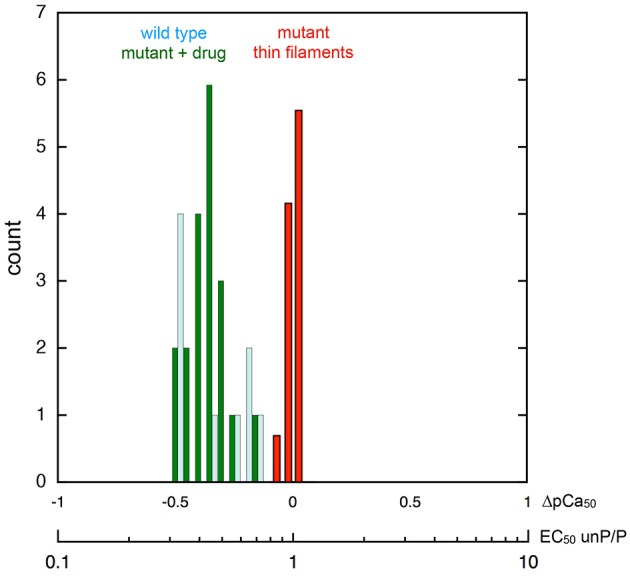
Phosphorylation-dependent change in thin filament Ca2+-sensitivity. Frequency histogram showing the distribution of the ratio of Ca2+-sensitivity in the phosphorylated and unphosphorylated state. The X-axes shows the ratio of EC50 unphosphorylated/phosphorylated and the corresponding shift in pCa50. Collected data from different mutations (Figure 1) and different recoupling compounds (Figure 2) is plotted. Red bars show distribution of shift in Ca2+ sensitivity on phosphorylation for mutant thin filament samples (n = 22 measurements by in vitro motility assay) showing no difference in Ca2+ sensitivity (uncoupling). Dark green bars show distribution of Ca2+ sensitivity shift from the same samples following the addition of small molecule recouplers. For comparison, the Ca2+-sensitivity shift on phosphorylation of native thin filaments and skinned muscle using a range of measurement systems is plotted (pale blue bars; n = 9, data from Marston, 2016, Table 5).
There is no correlation between re-couplers and Hsp90 inhibitors
Although many of the compounds investigated were selected from a study on Hsp90 inhibitors, there does not seem to be any correlation between their efficacy as re-couplers and Hsp90 inhibition. Thus, for the EGCG analogs tested (1–28) there were four compounds that were both Hsp90 inhibitors and re-couplers, four that were re-couplers only, four that were Hsp90 inhibitors only and seven that were inactive in both assays (Table 1).
The Ca2+-desensitizer and re-coupling activities may be separated
Although the lead compound, EGCG, was both a desensitizer and a re-coupler, we found that most of the compounds we studied did not have both properties. Some were desensitizers alone but the majority were re-couplers that did not influence myofilament Ca2+-sensitivity in WT muscle. Thus, the two activities of EGCG are not intrinsically linked.
Epicatechin gallate (ECG) is an example of a compound that is a pure desensitizer as illustrated in Figures 4A,B. When added to thin filaments that contain the uncoupling mutation E180G in tropomyosin, ECG equally reduces the Ca2+-sensitivity of both phosphorylated and unphosphorylated thin filaments so the system remains uncoupled. Silybin A also acts as a pure desensitizer (Figure 4C, Figure S2B).
Figure 4.

Separation of desensitization and recoupling activities (A,B) Epicatechin gallate (ECG) is a desensitizer but not a recoupler. (A) Ca2+-activation curves for phosphorylated and unphosphorylated thin filaments containing E180G tropomyosin in the presence and absence of 25 μM ECG. Solid lines, phosphorylated; dotted lines, unphosphorylated. Purple lines and points, E180G thin filaments; green lines and points, E180G thin filaments + ECG. (B) Dose-response curve for ECG activity measured at a constant Ca2+ concentration of 0.037. (C,D) Silybin A is a pure desensitizer whilst Silybin B is a pure recoupler Ca2+ activation curves are shown for phosphorylated and unphosphorylated thin filaments containing TnC G159D (DCM mutation) that are uncoupled. (C) In presence and absence of silybin A, (D) in presence and absence of Silybin B. Solid lines, phosphorylated; dotted lines, unphosphorylated. Purple lines and points, E180G thin filaments; green lines and points, E180G thin filaments + Silybin A or B. Structures of Silybin A and B are shown below the plots. (E,F) Compounds that recouple but are not Ca2+-desensitizers. (E) The effect of compounds on Ca2+-sensitivity of wild-type thin filaments. The ratio of EC50 in the presence and absence of tested compounds is plotted for the phosphorylated (dark green) and unphosphorylated (light green) states. Only EGCG is a significant desensitizer. DHSA/B, Dehydrosilybin A/B. (F) The effect of 100 μM drug treatment on the difference in fraction motile between phosphorylated and unphosphorylated states at 0.037μM Ca2+ in the phosphorylated and unphosphorylated states (see Figures 2C,D). Red bars, WT troponin; pink bars, WT troponin + compound. Dark green bars, E180G thin filaments, light green bars, E180G thin filaments + compound. Wild-type difference is unaffected by compound (not a desensitizer), whilst the value in E180G thin filaments is low indicating uncoupling, but restored by the compound indicating re-coupling.
Many of the compounds investigated were pure re-couplers. This is best demonstrated by their inability to influence the Ca2+-sensitivity of native thin filaments in either the phosphorylated or unphosphorylated state, in contrast to EGCG (Figure 4E). In mutant-uncoupled thin filaments, the pure recoupling ability is shown by the decrease in Ca2+-sensitivity of phosphorylated but not the unphosphorylated mutant thin filament in the presence of the compound as shown for Silybin B in Figure 4D, Quercetin in Figure 2A and Dehydrosilybin B in the Figure S3. In the rapid single Ca2+ concentration assay a large difference between % motility in the phosphorylated and unphosphorylated states is found in fully coupled systems. In wild-type thin filaments the difference is unaffected by the compounds indicating that the compound does not change wild-type Ca2+-sensitivity, whilst the value in E180G thin filaments is low, indicating uncoupling, but is restored by the compounds, indicating re-coupling.
Figure 4F illustrates this behavior for 10 compounds. We did not identify any compound apart from EGCG that was both a desensitizer and a re-coupler and 15 out of 40 compounds had no desensitizing or recoupling effects.
Minimal effective structure
Since both EGCG and Silybin are quite large molecules, we investigated whether re-coupling could be achieved with smaller molecules. We found that removal of the pyrogallol ester moiety or the galloyl moiety, but not both, had no effect on re-coupling activity. Similarly, in Silybin it was possible to remove the lignan part without altering the compound's activity; thus, a substituted chromanone or chromane ring plus a phenolic ring seem to be sufficient for re-coupling (Figures 5A,B). We also observed that simpler structures such as the two-ring molecules, e.g. Resveratrol, Pterostilbene, compound #2 (Figure 5), and even the single ring calmodulin antagonist W-7 were effective re-couplers (Figure 2, Table 1). This suggests that it is not the chromane/chromanone ring system that is essential but rather the substitutions that define the charge structure in ring A.
Figure 5.

Structure-function relationships (A,B) Minimal structures that re-couple. (C,D) Small changes in A and D ring of EGCG determine recoupling activity (C), Dose-response curves for re-couplers #9 and #19, inactive compound #12 and reverse re-coupler #7. (D) Structures of related re-coupling, inactive, and reverse recoupling compounds.
Small differences change function
It is remarkable that the re-coupling function can be lost with small changes in key parts of the structure. For instance the change of an OH to H in the D ring of EGCG is sufficient to abolish recoupling whilst preserving desensitization (ECG, Figure 4A). The stereoisomers of Silybin and Dehydrosilybin, which have four or two chiral centers, respectively, have quite different activities. The B isoforms are good re-couplers contrary to the A isoforms (Figures 2, 4D, Figures S2B, S3).
A group of compounds with substitutions containing the core rings A, C, and D of EGCG shift function from good recoupling to no effect and even to reverse re-coupling. The structural differences are small, for instance compound 19 (re-coupler) and compound 7 that has a reverse effect differ only by the number and positions of O-methyl groups in the D ring whilst other substitutions in the D ring (compounds 8 and 16) render the compound inactive. In addition, a number of substitutions in the A ring are compatible with recoupling when combined with an appropriate substitution in the D ring (compounds 9, 19, and 2) (Figures 5C,D). Methylation can alter properties of compounds since the phenolic OH is a weak acid and it is thus available as H-bond acceptor; OMe is also an H-bond acceptor but rather weaker and the acidity is lost. The position of free OH on the phenolic ring is likely to be important; for instance there is a huge difference in the reactivity of OHs in the ortho(para) and meta positions.
Discussion
Mutations in contractile proteins can cause familial HCM or familial DCM. HCM has been linked to a higher myofilament Ca2+-sensitivity. In addition, we have identified a molecular level dysfunction common to both HCM and DCM-causing mutations (Papadaki et al., 2015; Messer et al., 2016). This is an uncoupling of the relationship between TnI phosphorylation and modulation of myofilament Ca2+-sensitivity, essential for normal responses to adrenaline. As a consequence, adrenergic response is blunted in vivo, which predisposes to heart failure under stress (Messer and Marston, 2014; Wilkinson et al., 2015). Thus uncoupling at the contractile protein level contributes to the symptoms of HCM and DCM and restoration of coupling is a significant target for therapy (Tardiff et al., 2015). We have identified compounds that can specifically reverse these abnormalities in vitro and therefore have potential for treatment that we call “re-couplers.” The first compound studied was Epigallocatechin-3-gallate (EGCG) (Tadano et al., 2010). EGCG is capable of both Ca2+ desensitization and re-coupling in mutant thin filaments (Papadaki et al., 2015). We have now identified a further 23 structural analogs that are capable of restoring coupling in vitro; many of these can re-couple independent of Ca2+-desensitization.
Mechanism of coupling, uncoupling, and re-coupling
The normal change in myofilament Ca2+-sensitivity due to TnI phosphorylation by PKA is reported to be 2.00 fold ± 0.24 (EC50 P/unP, see Marston, 2016). It is remarkable that our current measurements show the same Ca2+ sensitivity shift in phosphorylated and unphosphorylated mutant-containing thin filaments when re-coupled (mean EC50 P/unP 2.2 ± 0.1 (n = 19) (Figure 3). This indicates a complete restoration of Ca2+ sensitivity modulation by TnI phosphorylation that is independent of the nature of the re-coupling compound and independent of the cardiomyopathy-related mutation (Figure 1). Therefore, it is reasonable to propose that these perturbations act by changing the equilibrium between a small number of pre-existing states.
We propose that troponin exists in four functional states: phosphorylated form with high and low Ca2+-sensitivity and unphosphorylated form with high and low Ca2+ sensitivity (Figure 6). This model can account for the effects of TnI phosphorylation in wild-type and mutant (uncoupled) thin filaments and also the effects of re-coupling compounds. In wild-type muscle, phosphorylation results in a transition from the unphosphorylated, high Ca2+ sensitivity state to the phosphorylated low Ca2+ sensitivity state (red arrows). Mutations that produce uncoupling can only undergo transition from the unphosphorylated high Ca2+ sensitivity to phosphorylated high Ca2+ sensitivity state and the further transition to low Ca2+ sensitivity is blocked. Dvornikov et al. have proposed structural evidence that a normal phosphorylation-dependent structural transition is blocked in the TnI R145W HCM mutant troponin (Dvornikov et al., 2016). It is remarkable that the uncoupling phenomena can be induced by a wide range of mutations and other perturbations (Figure 1) and this suggests that the phosphorylated low affinity state is rather unstable and the high affinity state may be a default state.
Figure 6.
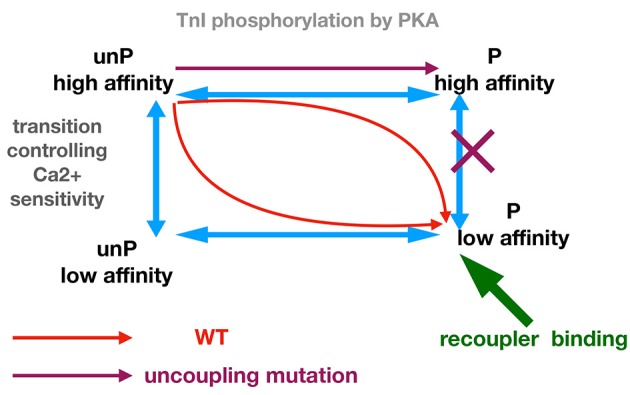
Allosteric model for modulation of Ca2+ -sensitivity by phosphorylation, mutations, and small molecule recouplers.
The functions of small molecules can be accounted for by their preferential binding to, and hence stabilizing of, one or more troponin states. Thus, a small molecule could re-couple by preferentially binding to the phosphorylated low affinity state, which would overcome the blocked transition from phosphorylated high affinity state to low affinity phosphorylated state. In such a mechanism the small molecule would have no effect upon the transitions of wild-type troponin, as observed. Molecules that desensitize mutant thin filaments without re-coupling (ECG, Silybin A) could be accounted for if they bound equally to the phosphorylated and unphosphorylated low affinity states. The reverse re-coupler, compound #7, could be incorporated into this scheme if it is preferentially bound to the unphosphorylated low Ca2+ affinity state. Such predictions could be tested if ligand binding to wild-type and mutant troponin in the different states could be measured; in the absence of such techniques, molecular dynamics simulations of the ligands bound in phosphorylated and unphosphorylated states would be a suitable approach (Zamora et al., 2017).
Troponin structural correlations
Although structural determinations of ligand binding (EGCG, W-7) to troponin subunits have been made, most studies have involved binary complexes (TnI and TnC) and partial sequences (Robertson et al., 2010; Botten et al., 2013; Hwang and Sykes, 2015). Recent molecular dynamics simulations have emphasized the importance of studying the intact troponin core with TnI, TnC, and TnT present (Papadaki and Marston, 2016; Zamora et al., 2016). Moreover, studies on the docking of EGCG, Silybin A and Silybin B by molecular dynamics simulations suggest that these compounds bind and crosslink the N-terminal peptide of TnI in the vicinity of the phosphorylatable serines22 and−23 to the N terminal domain of TnC and that the three compounds have different dynamics. In this position the re-coupling compound could influence the phosphorylation-dependent modulation of Ca2+-sensitivity of TnC compatible with our four state model (Zamora et al., 2017).
It is notable that the phosphorylatable N-terminus and the “switch peptide” of TnI, critical for Ca2+-regulation, are intrinsically disordered peptides (Hwang et al., 2014; Papadaki and Marston, 2016). By analogy with similar disordered peptides containing regulatory phosphorylation sites it is suggested that there may be negligible differences in time-averaged structures between phosphorylated and unphosphorylated states, but rather phosphorylation induces a change in protein dynamics from an ordered to a less ordered state (Colson et al., 2012).
More precise structure-function analysis requires further extensive docking modeling, however, it appears that the substituted chromane (in EGCG) or chromanone (in Silybin) are fundamental. The way in which the substitutions define the structure, charge distribution, hydrogen bonding potential or acidity of the A ring, apparently involved in its intercalation between TnI and TnC, is likely to be critical for the compound effect. In the EGCG analogs it seems that either the B ring (pyrogallol) or the D ring (galloyl) are sufficient to confer re-coupling activity, so one ring may be able to assume the configuration of the other when one of them is absent. In Silybin and Dehydrosilybin, the lignan part is not fully needed for function (taxifolin, quercetin) and only the B isomers are recouplers, whilst the A stereoisomers of Silybin and Dehydrosilybin are inactive as recouplers. In this case it seems likely that the A isomers have an unfavorable conformation that prevents the A and D rings binding in the same way as the B isoform. It is noteworthy that the B isomer is more active than the A isomer is several different bio-assays (Kren and Walterová, 2005; Biedermann et al., 2014).
Physiological correlates
The main effect of uncoupling of the modulation of Ca2+ sensitivity by TnI phosphorylation is the loss of the lusitropic response to beta adrenergic stimulation, which is usually accompanied by blunting of the whole physiological adrenergic response. This is demonstrated most directly by a lack of response to dobutamine stimulation observed at the whole animal, papillary muscle and single cardiomyocyte levels (Song et al., 2010, 2011; Wilkinson et al., 2015). Tadano showed that EGCG restored the impaired pump function due to diastolic dysfunction in a transgenic HCM mouse model (Tadano et al., 2010). Our preliminary experiments in isolated cardiomyocytes from ACTC E99K HCM transgenic and non-transgenic mice (Marston et al., 2018) show that the lusitropic effect of dobutamine is blunted in ACTC E99K mouse but could be restored by Resveratrol and Silybin B, whilst the effect of EGCG is anomalous, reflecting the known off target effects, especially positive inotropy (Feng et al., 2012). Further structural and functional investigations of these compounds could produce a new lead compound to direct our search for an effective and specific re-coupler with a practical potential in the treatment of inherited cardiomyopathies at the fundamental level.
Author contributions
AS, AM, MP, AC performed the experiments reported here; BB, AK, VK, DB prepared and supplied EGCG and silybin analogs; SM obtained funding, directed research and wrote the paper. All the authors contributed to editing and completing the paper.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the Sydney Heart Bank (Prof. Cristobal Dos Remedios, Dr. Sean Lal, and Dr. Amy Li) for the human heart samples used in this study.
This work was supported by grants from the British Heart Foundation (RG/ 11/20/29266 and FS/12/24/29568). AS has a Ph.D. studentship funded by the BHF Centre of Research Excellence (RE/13/4/30184). VK and DB acknowledge grant from the Czech Ministry of Health (AZV 16-27317A).
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2018.00243/full#supplementary-material
References
- Akkari P. A., Song Y., Hitchcock-DeGregori S., Blechynden L., Laing N. (2002). Expression and biological activity of Baculovirus generated wild-type human slow alpha tropomyosin and the Met9Arg mutant responsible for a dominant form of nemaline myopathy. Biochem. Biophys. Res. Commun. 296, 300–304. 10.1016/S0006-291X(02)00852-5 [DOI] [PubMed] [Google Scholar]
- Baell J., Walters M. A. (2014). Chemistry: chemical con artists foil drug discovery. Nature 513, 481–483. 10.1038/513481a [DOI] [PubMed] [Google Scholar]
- Bayliss C. R., Jacques A. M., Leung M.-C., Ward D. G., Redwood C. S., Gallon C. E., et al. (2012). Myofibrillar Ca2+-sensitivity is uncoupled from troponin i phosphorylation in hypertrophic obstructive cardiomyopathy due to abnormal troponin T. Cardiovasc. Res. 97, 500–508. 10.1093/cvr/cvs322 [DOI] [PubMed] [Google Scholar]
- Biedermann D., Vavrikova E., Cvak L., Kren V. (2014). Chemistry of silybin. Nat. Prod. Rep. 31, 1138–1157. 10.1039/C3NP70122K [DOI] [PubMed] [Google Scholar]
- Botten D., Fugallo G., Fraternali F., Molteni C. (2013). A computational exploration of the interactions of the green tea polyphenol (−)-epigallocatechin 3-gallate with cardiac muscle troponin C. PLoS ONE 8:e70556. 10.1371/annotation/23964dab-0e50-4644-b3b1-7c9e4ee09958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colson B. A., Gruber S. J., Thomas D. D. (2012). Structural dynamics of muscle protein phosphorylation. J. Muscle Res. Cell Motil. 33, 419–429. 10.1007/s10974-012-9317-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvornikov A. V., Smolin N., Zhang M., Martin J. L., Robia S. L., de Tombe P. P. (2016). Restrictive cardiomyopathy Troponin-I R145W mutation does not perturb myofilament length dependent activation in human cardiac sarcomeres. J Biol. Chem. 291, 21817–21828. 10.1074/jbc.M116.746172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer E., Jacques A., Hoskins A., Ward D., Gallon C., Messer A., et al. (2009). Functional analysis of a unique troponin C mutation, Gly159Asp that causes familial dilated cardiomyopathy, studied in explanted heart muscle. Circ. Heart Fail. 2, 456–464. 10.1161/CIRCHEARTFAILURE.108.818237 [DOI] [PubMed] [Google Scholar]
- Feng W., Hwang H. S., Kryshtal D. O., Yang T., Padilla I. T., Tiwary A. K., et al. (2012). Coordinated regulation of murine cardiomyocyte contractility by Nanomolar (−)-Epigallocatechin-3-Gallate, the Major Green Tea Catechin. Mol. Pharmacol. 82, 993–1000. 10.1124/mol.112.079707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser I. D. C., Marston S. B. (1995). In vitro motility analysis of actin-tropomyosin regulation by troponin and Ca2+: the thin filament is switched as a single cooperative unit. J. Biol. Chem. 270, 7836–7841. 10.1074/jbc.270.14.7836 [DOI] [PubMed] [Google Scholar]
- Friedrich F. W., Flenner F., Nasib M., Eschenhagen T., Carrier L. (2016). Epigallocatechin-3-Gallate Accelerates Relaxation and Ca2+ transient decay and desensitizes myofilaments in healthy and Mybpc3-targeted knock-in cardiomyopathic mice. Front. Physiol. 7:607. 10.3389/fphys.2016.00607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gažák R., Valentová K., Fuksová K., Marhol P., Kuzma M., Medina M. Á., et al. (2011). Synthesis and antiangiogenic activity of new silybin galloyl esters. J. Med. Chem. 54, 7397–7407. 10.1021/jm201034h [DOI] [PubMed] [Google Scholar]
- Gordon A. M., Homsher E., Regnier M. (2000). Regulation of contraction in striated muscle. Physiol. Rev. 80, 853–924. 10.1152/physrev.2000.80.2.853 [DOI] [PubMed] [Google Scholar]
- Hall J. A., Forsberg L. K., Blagg B. S. (2014). Alternative approaches to Hsp90 modulation for the treatment of cancer. Future Med. Chem. 6, 1587–1605. 10.4155/fmc.14.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao H., Naomoto Y., Bao X., Watanabe N., Sakurama K., Noma K., et al. (2010). HSP90 and its inhibitors (Review). Oncol. Rep. 23, 1483–1492. 10.1158/1541-7786.MCR-15-0234 [DOI] [PubMed] [Google Scholar]
- Hershberger R. E., Hedges D. J., Morales A. (2013). Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 10, 531–547. 10.1038/nrcardio.2013.105 [DOI] [PubMed] [Google Scholar]
- Hwang P. M., Cai F., Pineda-Sanabria S. E., Corson D. C., Sykes B. D. (2014). The cardiac-specific N-terminal region of troponin I positions the regulatory domain of troponin C. Proc. Natl. Acad. Sci. U.S.A. 111, 14412–14417. 10.1073/pnas.1410775111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang P. M., Sykes B. D. (2015). Targeting the sarcomere to correct muscle function. Nat. Rev. Drug Discov. 14, 313–328. 10.1038/nrd4554 [DOI] [PubMed] [Google Scholar]
- Ingólfsson H. I., Thakur P., Herold K. F., Hobart E. A., Ramsey N. B., Periole X., et al. (2014). Phytochemicals perturb membranes and promiscuously alter protein function. ACS Chem. Biol. 9, 1788–1798. 10.1021/cb500086e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kentish J. C., McCloskey D. T., Layland J., Palmer S., Leiden J. M., Martin A. F., et al. (2001). Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ. Res. 88, 1059–1065. 10.1161/hh1001.091640 [DOI] [PubMed] [Google Scholar]
- Khandelwal A., Hall J. A., Blagg B. S. J. (2013). Synthesis and Structure–Activity Relationships of EGCG Analogues, a Recently Identified Hsp90 Inhibitor. J. Org. Chem. 78, 7859–7884. 10.1021/jo401027r [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kren V., Walterová D. (2005). Silybin and silymarin–new effects and applications. Biomed. Pap. Med. Fac. Univ. Palacký Olomouc Czech Repub. 149, 29–41. 10.5507/bp.2005.002 [DOI] [PubMed] [Google Scholar]
- Krenek K., Marhol P., Peikerov,á Ž., V K., Biedermann D. (2014). Preparatory separation of the silymarin flavonolignans by Sephadex LH-20 gel. Food Res. Int. 65, 115–120. 10.1016/j.foodres.2014.02.001 [DOI] [Google Scholar]
- Lal S., Li A., Allen D., Allen P. D., Bannon P., Cartmill T., et al. (2015). Best practice biobanking of human heart tissue. Biophys. Rev. 7, 399–406. 10.1007/s12551-015-0182-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layland J., Solaro R. J., Shah A. M. (2005). Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc. Res. 66, 12–21. 10.1016/j.cardiores.2004.12.022 [DOI] [PubMed] [Google Scholar]
- Lopes L. R., Elliott P. M. (2014). A straightforward guide to the sarcomeric basis of cardiomyopathies. Heart 100, 1916–1923. 10.1136/heartjnl-2014-305645 [DOI] [PubMed] [Google Scholar]
- MacLeod K. T., Marston S. B., Poole-Wilson P. A., Severs N. J., Sugden P. H. (2010). Cardiac myocytes and the cardiac action potential, in Oxford Textbook of Medicine 5th edn, eds Warrel D. A., Cox T. M., Firth J. D. (Oxford: Oxford University Press; ), 2603-2617. [Google Scholar]
- Marston S. (2016). Why is there a limit to the changes in myofilament Ca2+-sensitivity associated with myopathy causing mutations? Front. Physiol. 7:415. 10.3389/fphys.2016.00415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston S. B., Fraser I. D. C., Wu B., Roper G. (1996). A simple method for automatic tracking of actin filaments in the motility assay. J. Musc. Res. Cell Motil. 17, 497–506. 10.1007/BF00123365 [DOI] [PubMed] [Google Scholar]
- Marston S. B., Messer A. E., Eiros-Zamora J., Gould I., Papadaki M., Choudry A., et al. (2018). The molecular defects in Ca2+ regulation due to mutations that cause hypertrophic cardiomyopathy can be reversed by small molecules that bind to Troponin. Biophys. J. 114:37a 10.1016/j.bpj.2017.11.253 [DOI] [Google Scholar]
- Marston S., Memo M., Messer A., Papadaki M., Nowak K., McNamara E., et al. (2013). Mutations in repeating structural motifs of tropomyosin cause gain of function in skeletal muscle myopathy patients. Hum. Mol. Genet. 22, 4978–4987. 10.1093/hmg/ddt345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memo M., Leung M.-C., Ward D. G., dos Remedios C., Morimoto S., Zhang L., et al. (2013). Mutations in thin filament proteins that cause familial dilated cardiomyopathy uncouple Troponin I phosphorylation from changes in Myofibrillar Ca2+-sensitivity. Cardiovasc. Res. 99, 65–73. 10.1093/cvr/cvt071 [DOI] [PubMed] [Google Scholar]
- Messer A., Bayliss C., El-Mezgueldi M., Redwood C., Ward D. G., Leung M.-C., et al. (2016). Mutations in troponin T associated with Hypertrophic Cardiomyopathy increase Ca2+-sensitivity and suppress the modulation of Ca2+-sensitivity by troponin I phosphorylation. Arch. Biochem. Biophys. 601, 113–120. 10.1016/j.abb.2016.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer A. E., Chan J., Daley A., Copeland O., Marston S. B., Connolly D. J. (2017). Investigations into the sarcomeric protein and Ca2+-regulation abnormalities underlying hypertrophic cardiomyopathy in cats (Felix catus). Front. Physiol. 8:348. 10.3389/fphys.2017.00348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer A. E., Jacques A. M., Marston S. B. (2007). Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J. Mol. Cell. Cardiol. 42, 247–259. 10.1016/j.yjmcc.2006.08.017 [DOI] [PubMed] [Google Scholar]
- Messer A., Gallon C., McKenna W., Elliott P., Dos Remedios C., Marston S. (2009). The use of phosphate-affinity SDS-PAGE to measure the troponin I phosphorylation site distribution in human heart muscle. Proteomics Clin. Appl. 3, 1371–1382. 10.1002/prca.200900071 [DOI] [PubMed] [Google Scholar]
- Messer A., Marston S. (2014). Investigating the role of uncoupling of Troponin I phosphorylation from changes in myofibrillar Ca2+-sensitivity in the pathogenesis of Cardiomyopathy. Front. Physiol. 5:315 10.3389/fphys.2014.00315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotná M., Gažák R., Biedermann D., Di Meo F., Marhol P., Kuzma M., et al. (2014). cis– transIsomerization of silybins A and B. Beilstein J. Org. Chem. 10, 1047–1063. 10.3762/bjoc.10.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadaki M., Marston S. B. (2016). The importance of intrinsically disordered segments of cardiac troponin in modulating function by phosphorylation and disease-causing mutations. Front. Physiol. 7:735. 10.3389/fphys.2016.00508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadaki M., Vikhorev P. G., Marston S. B., Messer A. E. (2015). Uncoupling of myofilament Ca2+ sensitivity from troponin I phosphorylation by mutations can be reversed by epigallocatechin-3-gallate. Cardiovasc. Res. 108, 99–110. 10.1093/cvr/cvv181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson I. M., Sun Y.-B., Li M. X., Sykes B. D. (2010). A structural and functional perspective into the mechanism of Ca2+-sensitizers that target the cardiac troponin complex. J. Mol. Cell. Cardiol. 49, 1031–1041. 10.1016/j.yjmcc.2010.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson S. P., Johnson J. D., Holroyde M. J., Kranias E. G., Potter J. D., Solaro R. J. (1982). The effect of troponin I phosphorylation on the Ca2+-binding properties of the Ca2+-regulatory site of bovine cardiac troponin. J. Biol. Chem. 257, 260–263. [PubMed] [Google Scholar]
- Singh B. N., Shankar S., Srivastava R. K. (2011). Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochem. Pharmacol. 82, 1807–1821. 10.1016/j.bcp.2011.07.093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W., Dyer E., Stuckey D., Copeland O., Leung M., Bayliss C., et al. (2011). Molecular mechanism of the Glu99lys mutation in cardiac actin (ACTC gene) that causes apical hypertrophy in man and mouse. J. Biol. Chem. 286, 27582–27593. 10.1074/jbc.M111.252320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W., Dyer E., Stuckey D., Leung M.-C., Memo M., Mansfield C., et al. (2010). Investigation of a transgenic mouse model of familial dilated cardiomyopathy. J. Mol. Cell. Cardiol. 49, 380–389. 10.1016/j.yjmcc.2010.05.009 [DOI] [PubMed] [Google Scholar]
- Song W., Vikhorev P. G., Kashyap M. N., Rowlands C., Ferenczi M. A., Woledge R. C., et al. (2013). Mechanical and energetic properties of papillary muscle from ACTC E99K transgenic mouse models of hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 304, H1513–H1524. 10.1152/ajpheart.00951.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadano N., Du C., Yumoto F., Morimoto S., Ohta M., Xie M., et al. (2010). Biological actions of green tea catechins on cardiac troponin C. Br. J. Pharmacol. 161, 1034–1043. 10.1111/j.1476-5381.2010.00942.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N., Yamashita A., Maeda K., Maeda Y. (2003). Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature 424, 35–41. 10.1038/nature01780 [DOI] [PubMed] [Google Scholar]
- Tardiff J. C., Carrier L., Bers D. M., Poggesi C., Ferrantini C., Coppini R., et al. (2015). Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc. Res. 105, 457–470. 10.1093/cvr/cvv023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh R., Thomson K. L., Ware J. S., Funke B. H., Woodley J., McGuire K. J., et al. (2017). Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 19, 192–203. 10.1038/gim.2016.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson R., Song W., Smoktunowicz N., Marston S. (2015). A dilated cardiomyopathy mutation blunts adrenergic response and induces contractile dysfunction under chronic angiotensin II stress. Am. J. Physiol. Heart Circ. Physiol. 309, H1936–H1946. 10.1152/ajpheart.00327.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamora J. E., Hoben G., Sheehan A., Messer A., Chaudry A., Biedermann D., et al. (2017). EGCG and Silybin as treatment for inherited cardiomyopathies: binding simulations to cardiac troponin, in: 253rd, Conference: American Chemical Society National Meeting & Exposition. [Google Scholar]
- Zamora J. E., Papadaki M., Messer A. E., Marston S. B., Gould I. R. (2016). Troponin structure: its modulation by Ca2+ and phosphorylation studied by molecular dynamics simulations. Phys. Chem. Chem. Phys. 18, 20691–20707. 10.1039/C6CP02610A [DOI] [PubMed] [Google Scholar]
- Zhao H., Brandt G. E., Galam L., Matts R. L., Blagg B. S. J. (2011). Identification and initial SAR of silybin: an Hsp90 inhibitor. Bioorg. Med. Chem. Lett. 21, 2659–2664. 10.1016/j.bmcl.2010.12.088 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.