

Abstract
Infection with Helicobacter pylori is known to decrease the level of glutathione in gastric epithelial cells and increase the production of reactive oxygen species (ROS), which can lead to DNA damage and the development of gastric cancer. Cation transport regulator 1 (CHAC1) has γ‐glutamylcyclotransferase activity that degrades glutathione. We found that cagA‐positive H. pylori infection triggered CHAC1 overexpression in human gastric epithelial (AGS) cells leading to glutathione degradation and the accumulation of ROS. Nucleotide alterations in the TP53 tumour suppressor gene were induced in AGS cells overexpressing CHAC1, whereas no mutations were detected in cells overexpressing a catalytically inactive mutant of CHAC1. A high frequency of TP53 mutations occurred in H. pylori‐infected AGS cells, but this was prevented in cells transfected with CHAC1 siRNA. These findings indicate that H. pylori‐mediated CHAC1 overexpression degrades intracellular glutathione, allowing the accumulation of ROS which subsequently causes mutations that could contribute to the development of gastric cancer.
Keywords: cagA, CHAC1, glutathione, H. pylori, p53, ROS
Abbreviations
- ACTB
β‐actin
- AGS cells
human gastric epithelial cells
- CHAC1
cation transport regulator 1
- ER
endoplasmic reticulum
- GSH
glutathione
- H. pylori
Helicobacter pylori
- ROS
reactive oxygen species
It is well established that Helicobacter pylori (H. pylori) infection can increase the production of reactive oxygen species (ROS) and decrease the level of glutathione (GSH) in gastric epithelial cells 1, 2, 3. These changes in redox balance and the elevation of oxidative stress lead to mutations in DNA that potentially contribute to the development of gastric carcinoma 4, 5. GSH provides the reducing equivalents used in the protection of cellular macromolecules against the oxidative damage caused by ROS 6, 7. However, it is not clear from prior studies whether the decrease in GSH associated with H. pylori infection results directly from its consumption following the excessive production of ROS, or the accumulation of ROS is exacerbated by a deficiency of GSH.
Recent studies have shown that a protein known as cation transport regulator 1 (CHAC1) is a novel member of the γ‐glutamylcyclotransferase family of enzymes that contribute to the γ‐glutamylcycle 8, 9. CHAC1 can catalyse the cleavage of GSH into 5‐oxoproline and the dipeptide cysteinylglycine and is one of the only cytosolic enzymes known to degrade GSH 8, 9. Overexpression of CHAC1 leads to GSH depletion and a change in the cellular redox balance 8, 9. CHAC1 has also been identified as a component of the unfolded protein response stress signalling pathway in the endoplasmic reticulum (ER) 10, 11, and its elevated mRNA expression level has been associated with a poor outcome in patients with breast and ovarian cancer 12, 13.
Given the role of CHAC1 in the degradation of GSH, we tested the hypothesis that the changes in GSH levels observed in H. pylori‐infected cells result from the induction of CHAC1. We report here that H. pylori infection causes increased expression of CHAC1 that leads to GSH depletion, elevated ROS accumulation and increased somatic DNA mutations in the TP53 tumour suppressor gene.
Materials and methods
Bacteria and infection
A standard strain (43 504) of H. pylori (cagA‐positive H. pylori) was purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). An isogenic cagA‐knockout H. pylori mutant (43 504) (cagA‐negative H. pylori) was kindly provided by one of the authors (HM). Prior to infection, H. pylori was grown in Brucella broth (Becton Dickinson, Sparks, MD, USA) with 10% fetal bovine serum (FBS) for 24 h.
For infection, H. pylori was added to human gastric epithelial (AGS) cells at a ratio of 100 bacteria per cell.
For the TP53 mutation analysis, AGS cells were infected with either cagA‐positive or cagA‐negative H. pylori every 3 days for 15 days postinfection. Before each reinfection, the cells were reseeded at a rate of 2 × 105 cells/well. Samples for CHAC1, GSH, ROS and mutation analysis were taken on day 16 after the first infection.
Cell culture and transfection
AGS cells obtained from ATCC were grown in RPMI‐1640 (Sigma‐Aldrich Co., St. Louis, MO, USA) supplemented with 10% FBS, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin. Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) was used for transfection of the plasmids and small interfering RNA (siRNA). A wild‐type CHAC1 cDNA clone in the expression vector pCMV6 (CHAC1‐WT) was obtained from OriGene Technologies (Rockville, MD, USA), and an inactive mutant (CHAC1‐MT) was created by the insertion of an E157Q substitution in the active site.
For CHAC1 knockdown, cells were treated with synthesized siRNA (5′‐AUCUUCAAGGAGCGUCACCAC‐3′). An unrelated scrambled siRNA (5′‐GUUAAAUAGCGAUAGGAAUUC‐3′) was used as a control for non‐sequence‐specific effects. AGS cells were transfected with a final working concentration of 50 nmol·L−1 siRNA in original RPMI‐1640 at 6 h before H. pylori infection.
RNA extraction and real‐time RT‐PCR
AGS cells were treated for RNA extraction with 1.0 mL of TRIzol reagent (Invitrogen) according to the manufacturer's instructions, and cDNA was synthesized with random primers using Superscript III Reverse Transcriptase (Invitrogen). Oligonucleotide primers and probes are listed in Table S1. Relative quantitation of mRNA was performed by real‐time reverse transcription (RT)‐PCR using TaqMan Universal PCR Master Mix (ABgene, Epsom, UK). Amplification and detection were performed with the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA).
Measurement of the intracellular GSH concentration
The intracellular GSH concentration was measured as indicated by the manufacturer's instructions (GSSG/GSH Quantification Kit; Sigma‐Aldrich Co.). A standard line was drawn using GSSG solution at various concentrations.
ROS measurement
The cells were washed twice with PBS, and the media were changed to original RPMI‐1640 containing a final working concentration of 20 μmol·L−1 carboxy‐H2DCFDA (Invitrogen) dissolved in dimethylsulfoxide (Wako Pure Chemical Industries, Ltd., Osaka, Japan). For a negative control, 20 μL of dimethylsulfoxide was added to the media. After incubation, the cells were trypsinized, washed and resuspended with PBS for examination by flow cytometry (FACSCanto II; Becton Dickinson).
Production of anti‐CHAC1 antibody
A full‐length human CHAC1 cDNA was subcloned in the pHUE vector, and recombinant protein was expressed in Escherichia coli and purified as described previously 14. A novel anti‐CHAC1 monoclonal antibody (IgM, κ) termed CHAC1‐mAb(v1v2) was made for the study by immunizing BALB/c mice (CLEA Japan, Tokyo, Japan) with the purified recombinant human CHAC1 protein, and hybridoma cell lines were prepared according to the previously described protocol 15. Hybridoma cell lines producing anti‐CHAC1 antibodies were verified by enzyme‐linked immunosorbent assay with the recombinant CHAC1 protein and were characterized further by immunohistochemistry and immunoblotting of CHAC1 expressed in transfected HEK293T cells purchased from ATCC (Fig. S1).
Western blotting
For protein extraction, AGS cell lysates were harvested with M‐PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific Inc., Kanagawa, Japan) supplemented with protease inhibitor cocktail (Sigma‐Aldrich Co.). Western blotting was performed as previously described 16 with minor modifications. The membranes were probed with the appropriately diluted primary antibodies (CHAC1‐mAb(v1v2) or anti‐β‐actin antibody [#4970; Cell Signaling Technology, Beverly, MA, USA]), followed by incubation with the appropriate secondary antibodies. Blots were developed with ECL (Bio‐Rad, Hercules, CA, USA). Finally, images were recorded and analysed with ChemiDoc MP Image System (Bio‐Rad).
Sequence analysis of TP53
AGS cells were treated with 1.0 mL of TRIzol reagent for RNA extraction, and cDNA was synthesized as described above. A total of 1179 bp of exome sequence between exons 2 and 11 of TP53 was amplified with PrimeSTAR Max DNA Polymerase (Takara Shuzo, Shiga, Japan) using the primers listed in Table S1. PCR products were subcloned using the TOPO TA Cloning Kit (Invitrogen) according to the manufacturer's instructions, followed by analysis of nucleotide sequences in randomly selected clones with ABI BigDye terminator ver. 3.1 (Applied Biosystems) and ABI Prism 3130xl Genetic Analyzer (Applied Biosystems).
Statistical analysis
GraphPad PRISM ver. 6 (GraphPad Software, Inc., CA, USA) was used for statistical analysis. The results are expressed as means ± standard errors of the means (SEM). A two‐sided P < 0.05 was regarded as statistically significant. A two‐way analysis of variance (ANOVA) and further analysis using Tukey's multiple comparisons test were used to test for statistical significance of the time‐independent mRNA expression of CHAC1. A one‐way analysis of variance (ANOVA) and further analysis using Holm–Sidak's multiple comparisons test and unpaired t‐test were used to test for comparison of intracellular levels of CHAC1, GSH and ROS. The TP53 mutation frequencies were analysed by Fisher's exact test.
Results
H. pylori induces CHAC1 expression, GSH depletion and ROS accumulation in infected cells
AGS cells infected by cagA‐positive H. pylori significantly (P < 0.0001) expressed CHAC1 mRNA with peak expression at 24 h postinfection (Fig. 1A). In contrast, CHAC1 was not detected in uninfected AGS cells and was not strongly induced by cagA‐negative H. pylori. At 24 h postinfection, intracellular levels of CHAC1 (mRNA and protein), GSH and ROS were measured in cagA‐positive H. pylori‐infected AGS cells transfected with CHAC1 siRNA or scrambled control siRNA (Fig. 1B–E). Infection of AGS cells with cagA‐positive H. pylori induced a significant (P < 0.05) > 10‐fold increase in CHAC1 mRNA and protein expression (Fig. 1B,C). At the same time, there was a significant (P < 0.001) decrease in GSH (Fig. 1D) and a significant (P < 0.001) increase in ROS (Fig. 1E). Moreover, no changes in the GSH (Fig. 1D) and ROS (Fig. 1E) levels were observed in the cagA‐positive H. pylori‐infected AGS cells when the CHAC1 expression was suppressed by siRNA transfection to a level that was undetectable by western blotting (Fig. 1B). These experiments show that cagA‐positive H. pylori infection induces CHAC1 which degrades GSH thereby allowing ROS to accumulate.
Figure 1.
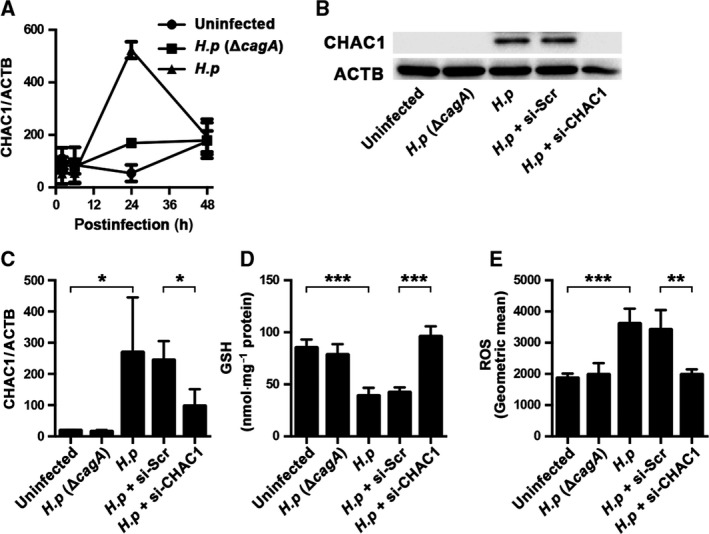
H. pylori infection and CHAC1 expression. (A) CHAC1 mRNA expression normalized with that of β‐actin (ACTB) in AGS cells was measured after cagA‐positive or cagA‐negative H. pylori infection. Total RNA was isolated at 2, 6, 24 and 48 h postinfection. (B–E) Untreated AGS cells were infected by either cagA‐positive or cagA‐negative H. pylori for 24 h. AGS cells transfected with CHAC1 siRNA or scrambled siRNA were also infected by cagA‐positive H. pylori. (B) The protein expression of CHAC1 and ACTB; (C) the mRNA expression of CHAC1 normalized with ACTB; (D) the level of intracellular GSH; (E) the level of intracellular ROS. H.p (ΔcagA) indicates AGS cells infected with cagA‐negative H. pylori; H.p indicates cells infected with cagA‐positive H. pylori; H.p + si‐Scr indicates cells infected with cagA‐positive H. pylori with transfection of scrambled siRNA; H.p + si‐CHAC1 indicates cells infected with cagA‐positive H. pylori with transfection of siRNA CHAC1. Data shown for A and C are the mean ± SE from triplicate measurements derived from three replicate experiments, and data for D and E are the mean ± SE from triplicate measurements derived from four replicate experiments. *P < 0.05, **P < 0.01 and ***P < 0.001.
Overexpression of catalytically active CHAC1 is required for the induction of TP53 mutations in the H. pylori‐infected cells
To confirm the observation that the effects of CHAC1 expression on GSH depletion and ROS accumulation are due to the enzymatic activity of CHAC1, we transfected AGS cells with wild‐type (CHAC1‐WT) and a catalytically inactive mutant CHAC1 (CHAC1‐MT). After 16 days, CHAC1‐WT and CHAC1‐MT (mRNA and protein) were expressed at similar levels (Fig. 2A,D). At this time, there was a decrease in GSH and an increase in ROS levels in cells expressing catalytically active CHAC1‐WT (Fig. 2B,C). In contrast, there was no change in the levels of GSH or ROS in transfected cells expressing inactive CHAC1‐MT (Fig. 2B,C).
Figure 2.
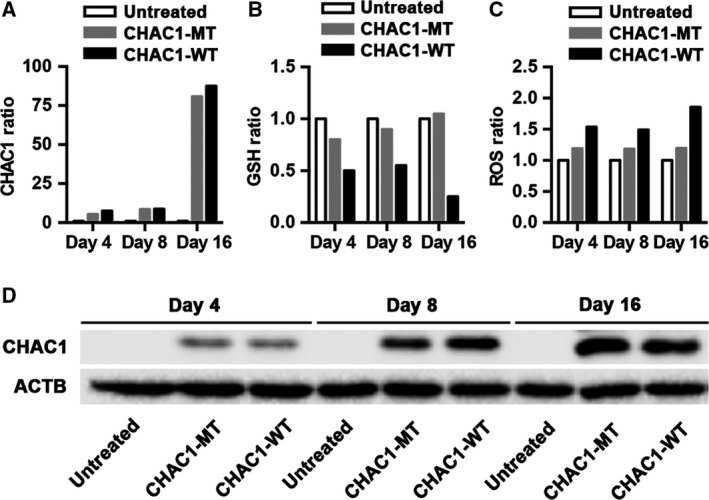
Levels of CHAC1 mRNA, GSH, ROS and CHAC1 protein in CHAC1‐transfected samples used for mutation analysis. (A–D) AGS cells expressing CHAC1‐WT or inactive CHAC1‐MT and untreated cells were cultured for 4, 8 and 16 days. The values of CHAC1 mRNA, GSH and ROS are the mean of two replicates used for mutation analysis and are expressed as the ratio of treated to untreated values.
To determine whether CHAC1 expression resulted in an increased mutation rate, a mutation analysis was undertaken on the untransfected and transfected cells after 4, 8 and 16 days. Nucleotide alterations in DNA recovered from these AGS cells were determined in the sequence between exons 2 and 11 of the tumour suppressor gene TP53. No mutations were detected in TP53 in untransfected control AGS cells or in cells transfected with catalytically inactive CHAC1‐MT (Table 1). In contrast, cells transfected with catalytically active CHAC1‐WT had multiple mutations that encoded amino acid substitutions in the TP53 gene. The frequency of these mutations increased in a time‐dependent manner (Table 1).
Table 1.
TP53 mutation in AGS cells related to CHAC1 overexpression
| Total mutated clones | Mutations causing amino acid substitutions | P value* | |
|---|---|---|---|
| AGS cells with CHAC1 overexpression by transfection | |||
| Day 4 CHAC1‐WT | 5/60 | 2/70 740 (0.28/104) | N.S. |
| Day 8 CHAC1‐WT | 10/59 | 7/69 561 (1.01/104) | 0.0074 |
| Day 16 CHAC1‐WT | 20/59 | 13/69 561 (1.87/104) | 0.0001 |
| Day 16 CHAC1‐MT | 0/50 | 0/58 950 (0.00/104) | N.S. |
| Day 16 control | 0/60 | 0/70 740 (0.00/104) | – |
| AGS cells with CHAC1 overexpression induced by H. pylori infection | |||
| Uninfected | 0/60 | 0/70 740 (0.00/104) | – |
| cagA (−) H. pylori | 0/59 | 0/69 561 (0.00/104) | N.S. |
| cagA (+) H. pylori | 14/59 | 8/69 561 (1.15/104) | 0.0037 |
| cagA (+) H. pylori + scrambled siRNA | 10/53 | 6/62 487 (0.96/104) | 0.0107 |
| cagA (+) H. pylori + CHAC1 siRNA | 0/58 | 0/68 382 (0.00/104) | N.S. |
*P values for the number of mutated bases causing amino acid substitutions were evaluated by Fisher's exact test.
Total mutated clones indicates the total number of mutations per total number of clones examined; Mutations causing amino acid substitutions indicates the number of mutated bases causing amino acid substitutions per total number of base pairs sequenced. Frequency of mutations causing amino acid substitutions per 104 base pairs is shown in parentheses. AGS cells were subjected to H. pylori infection for 16 days (see Methods). N.S., not significant.
To extend this study, we next performed the same TP53 mutation analysis on H. pylori‐infected AGS cells transfected with either CHAC1 siRNA or scrambled control siRNA. The levels of CHAC1 mRNA, GSH, ROS and CHAC1 protein in samples used for the mutation analysis are shown in Fig. S2A–D and had a similar phenotype to the infected cells shown in Fig. 1. As observed in the CHAC1‐transfected cells, cagA‐positive H. pylori‐infected cells and the infected cells transfected with scrambled control siRNA had a significantly higher frequency of nucleotide alterations in TP53 than uninfected cells. In contrast, there were no mutations in cagA‐positive H. pylori‐infected cells in which CHAC1 expression was knocked down by CHAC1 siRNA (Table 1). The results of the mutation analysis presented here are the combination of two independent experiments (Tables S2 and S3). Nucleotide alterations in TP53 causing amino acid substitutions are summarized in Table S4.
Discussion
CHAC1 is a novel ER stress‐inducible gene first identified in human aortic endothelial cells treated with oxidized phospholipids 10, and various stimuli that trigger ER stress, including infection, upregulate CHAC1 mRNA expression 11, 17. H. pylori‐triggered ER stress has been reported since 2013 18, 19, 20, and the present study is the first to demonstrate that CHAC1 overexpression is induced by the H. pylori‐triggered ER stress.
H. pylori infection is known to cause the depletion of cellular GSH levels with a concomitant accumulation of ROS 1, 2, 3, 21, 22, 23, 24, 25, 26. The mechanism of H. pylori‐induced GSH depletion, however, remains unknown. Because CHAC1 was reported in 2012 to be a novel member of the γ‐glutamylcyclotransferase family that degrades GSH 8, we proposed that the decreased level of GSH in H. pylori‐infected cells may be caused by CHAC1 expression. The experiments in the present study confirmed that cagA‐positive but not cagA‐negative H. pylori infection induced CHAC1 overexpression. The depletion of GSH in cagA‐positive H. pylori‐infected cells was found to be due to the expression of CHAC1 as the GSH depletion did not occur in infected cells when the expression of CHAC1 was knocked down by specific CHAC1 siRNA. In addition, the expression of CHAC1 with an inactivating mutation (CHAC1‐MT) also prevented the depletion of GSH. The present results also support the view that the accumulation of ROS occurs as a result of the depletion of GSH as ROS did not accumulate in H. pylori‐infected cells that were transfected with CHAC1 siRNA that blocked the expression of CHAC1 protein. This result confirms the importance of GSH in the maintenance of the cells redox balance and its role in the protection of the cell against the deleterious effects of oxidative stress.
ROS interact directly with macromolecules, including genomic DNA, thereby causing damage to specific genes responsible for cell proliferation 27, 28 and tumour suppression that can lead to tumorigenesis 29. An accumulation of ROS in gastric epithelial cells infected by H. pylori may lead to somatic cell DNA mutations that interfere with both the expression and function of tumour‐suppressing genes such as TP53, and contribute to the development of gastric cancer 4, 22, 30, 31, 32. Thus, our finding that H. pylori‐induced CHAC1 expression is responsible for the depletion of GSH and the accumulation of intracellular ROS led us to investigate whether elevated CHAC1 expression is associated with an increased frequency of DNA mutations in infected gastric epithelial cells. Although we would expect that all DNA would be subject to oxidative damage, we focussed on the TP53 gene because of its well‐established role as a tumour suppressor.
As expected, TP53 mutations causing amino acid substitutions were found to increase in a time‐dependent manner in AGS cells overexpressing wild‐type CHAC1. However, no mutations were detected in untreated control cells or in cells overexpressing catalytically inactive CHAC1‐MT. The levels of CHAC1, GSH and ROS in the cells used for the mutation analysis clearly demonstrated that independent of H. pylori infection, CHAC1‐induced depletion of GSH and the accumulation of intracellular ROS are required for the induction of TP53 mutations. In subsequent experiments with H. pylori infection, TP53 mutations were observed in the untreated or scrambled siRNA‐transfected control cells infected by cagA‐positive H. pylori, whereas TP53 mutations were not found in similarly infected cells transfected with CHAC1 siRNA. These experiments indicated that CHAC1 overexpression is a significant cause of TP53 mutations in gastric epithelial cells infected by cagA‐positive H. pylori. Our view that the increase in ROS in cagA‐positive H. pylori‐infected cells leads to oxidative DNA modification is further supported by the prior observations that the occurrence of mutations and carcinogenic transformation in H. pylori infection are negatively correlated with the concentration of the ROS scavenger vitamin C 33.
The frequency of TP53 mutations with amino acid substitutions in H. pylori‐infected AGS cells was reported in a previous study in connection with aberrant expression of activation‐induced cytidine deaminase (AID) 32. The mean mutation frequency of AGS cells caused by H. pylori was higher in the previous study than in the present study (2.23 vs 1.15 per 104 nucleotides). This may be due to differences in the strains of H. pylori used for infection (a clinical isolate vs ATCC 43504) and the DNA polymerase used for sequence analysis of TP53 (High Fidelity DNA Polymerase from Finnzymes vs PrimeSTAR Max DNA Polymerase). In the previous study, TP53 mutations were observed even in the AID knockdown cells infected by cag pathogenicity island (PAI)‐positive H. pylori at a still high frequency (2.08 per 104 nucleotides), suggesting that H. pylori‐mediated molecular events other than aberrant AID expression contributed to induce TP53 mutations. Thus, the TP53 mutation frequencies in the two different studies performed with AID and CHAC1, respectively, suggest that potential AID‐mediated TP53 mutations added to the basal level of CHAC1‐mediated TP53 mutations, and both are induced by cagA‐positive H. pylori infection and may contribute to gastric carcinogenesis independently via different pathways.
Evaluation of TP53 mutations in human cancer cases indicates that the majority are G : C > T:A transversions and G : C > A : T transitions 34 that are considered to result from oxidative stress and the ROS‐mediated DNA damage 35, 36. In the present study, we also noted the majority of mutations (63%, 58%) are G : C > T : A transversions and G : C > A : T transitions (Figs 3 and 4). This further supports our contention that CHAC1 expression leads ultimately to oxidative damage to DNA.
Figure 3.
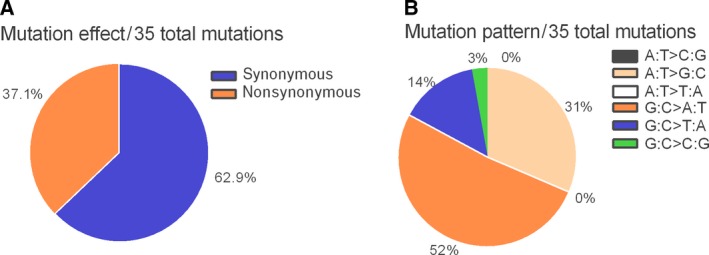
Type of somatic TP53 mutations found in AGS cells with CHAC1 overexpression by transfection. (A) Pie charts showing the proportion of nonsynonymous and synonymous mutations. (B) The proportion of different mutation patterns of 35 total mutations.
Figure 4.
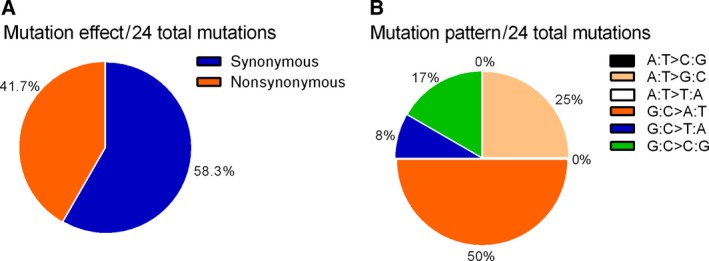
Type of somatic TP53 mutations found in AGS cells with CHAC1 overexpression induced by H. pylori infection. (A) Pie charts showing the proportion of nonsynonymous and synonymous mutations. (B) The proportion of different mutation patterns of 24 total mutations.
In a review of all cancer‐associated TP53 mutations, 86% were found between codons 125 and 300, a region that corresponds to the DNA binding domain. In the present study, 47% of the mutations causing amino acid substitutions occurred within this region and are therefore more likely to be deleterious 34. Substitutions at codons 175, 245, 248, 249, 273 and 282 account for approximately 30% of cancer‐associated TP53 mutations 34. In the present study, we detected mutations at codons 245 and 248. Together, these substitutions account for approximately 11% of previously described TP53 cancer‐associated mutations. In addition, several mutations identified in this study at codons 176, 177, 244 and 270 are in close proximity to high‐frequency cancer‐associated codons.
In conclusion, this study has shown that cagA‐positive H. pylori‐mediated CHAC1 overexpression causes GSH depletion and the accumulation of ROS that subsequently leads to nucleotide alterations in the DNA of the infected cells. Oxidative DNA damage caused by H. pylori‐induced CHAC1 overexpression in infected gastric epithelial cells may directly contribute to the development of gastric cancer.
Author contributions
YW designed the study design, performed most of the experiments, analysed and interpreted the data and wrote the manuscript. KT performed the analysis of CHAC1 expression induced by H. pylori infection and contributed to the study design. PT, KU, KK, AF, YI and TI provided the study material and technical support. YH conducted the mutation analysis. TS conducted the experiments. HM kindly provided the isogenic cagA‐knockout H. pylori mutant. PGB and YE supervised and directed the project and contributed to the manuscript preparation.
Supporting information
Fig. S1. Reactivity of the novel monoclonal antibody to CHAC1.
Fig. S2. The levels of CHAC1 mRNA, GSH, ROS and CHAC1 protein in H. pylori‐infected samples used for mutation analysis.
Table S1. Primers and probes used for this study.
Table S2. CHAC1 overexpression leading to TP53 mutation in AGS cells (results from each of the two experiments).
Table S3. CHAC1 expression induced by H. pylori infection leading to TP53 mutation in AGS cells (results from each of the two experiments).
Table S4. Distribution of nucleotide alterations in the TP53 sequence and resultant amino acid substitutions.
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science KAKENHI (16K19077) and Project Grant 525458 from the Australian National Health and Medical Research Council.
P.G.B. and Y.E. made equal senior author contributions to the article.
References
- 1. Tsugawa H, Suzuki H, Saya H, Hatakeyama M, Hirayama T, Hirata K, Nagano O, Matsuzaki J and Hibi T (2012) Reactive oxygen species‐induced autophagic degradation of helicobacter pylori CagA is specifically suppressed in cancer stem‐like cells. Cell Host Microbe 12, 764–777. [DOI] [PubMed] [Google Scholar]
- 2. Ding S‐Z, Minohara Y, Fan XJ, Wang J, Reyes VE, Patel J, Dirden‐Kramer B, Boldogh I, Ernst PB and Crowe SE (2007) Helicobacter pylori infection induces oxidative stress and programmed cell death in human gastric epithelial cells. Infect Immun 75, 4030–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Obst B, Wagner S, Sewing KF, Beil W and Hannover MH (2000) Helicobacter pylori causes DNA damage in gastric epithelial cells. Carcinogenesis 21, 1111–1115. [PubMed] [Google Scholar]
- 4. Farinati F, Cardin R, Degan P, Rugge M, Di Mario F, Bonvicini P and Naccarato R (1998) Oxidative DNA damage accumulation in gastric carcinogenesis. Gut 42, 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baik S‐C, Youn H‐S, Chung M‐H, Lee W‐K, Cho M‐J, Ko G‐H, Park C‐K, Kasai H and Rhee K‐H (1996) Increased oxidative DNA damage in Helicobacter pylori‐infected human gastric mucosa. Cancer Res 56, 1279–1282. [PubMed] [Google Scholar]
- 6. Oakley AJ, Yamada T, Liu D, Coggan M, Clark AG and Board PG (2008) The identification and structural characterization of C7orf24 as γ‐glutamyl cyclotransferase. J Biol Chem 283, 22031–22042. [DOI] [PubMed] [Google Scholar]
- 7. Oakley AJ, Coggan M and Board PG (2010) Identification and characterization of γ‐glutamylamine cyclotransferase, an enzyme responsible for γ‐glutamyl‐ε‐lysine catabolism. J Biol Chem 285, 9642–9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kumar A, Tikoo S, Maity S, Sengupta S, Sengupta S, Kaur A and Bachhawat AK (2012) Mammalian proapoptotic factor ChaC1 and its homologues function as γ‐glutamyl cyclotransferases acting specifically on glutathione. EMBO Rep 13, 1095–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crawford RR, Prescott ET, Sylvester CF, Higdon AN, Shan J, Kilberg MS and Mungrue IN (2015) Human CHAC1 protein degrades glutathione, and mRNA induction is regulated by the transcription factors ATF4 and ATF3 and a bipartite ATF/CRE regulatory element. J Biol Chem 290, 15878–15891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gargalovic PS, Imura M, Zhang B, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Patel S et al (2006) Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci USA 103, 12741–12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mungrue IN, Pagnon J, Kohannim O, Gargalovic PS and Lusis AJ (2008) CHAC1/MGC4504 is a novel proapoptotic component of the unfolded protein response, downstream of the ATF4‐ATF3‐CHOP cascade. J Immunol 182, 466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goebel G, Berger R, Strasak A, Egle D, Muller‐Holzner E, Schmidt S, Rainer J, Presul E, Parson W, Lang S et al (2012) Elevated mRNA expression of CHAC1 splicing variants is associated with poor outcome for breast and ovarian cancer patients. Br J Cancer 106, 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jahn B, Arvandi M, Rochau U, Fiegl H and Goebel G (2017) Development of a novel prognostic score for breast cancer patients using mRNA expression of CHAC1. J Comp Eff Res 6, 563–574. [DOI] [PubMed] [Google Scholar]
- 14. Baker RT, Catanzariti AM, Karunasekara Y, Soboleva TA, Sharwood R, Whitney S and Board PG (2005) Using deubiquitylating enzymes as research tools. Methods Enzymol 398, 540–554. [DOI] [PubMed] [Google Scholar]
- 15. Harlow E and Lane D (1988) Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, New York. [Google Scholar]
- 16. Takemura K, Kawachi H, Eishi Y, Kitagaki K, Negi M, Kobayashi M, Uchida K, Inoue J, Inazawa J, Kawano T et al (2014) γ‐Glutamylcyclotransferase as a novel immunohistochemical biomarker for the malignancy of esophageal squamous tumors. Hum Pathol 45, 331–341. [DOI] [PubMed] [Google Scholar]
- 17. Tattoli I, Sorbara MT, Vuckovic D, Ling A, Soares F, Carneiro LAM, Yang C, Emili A, Philpott DJ and Girardin SE (2012) Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 11, 563–575. [DOI] [PubMed] [Google Scholar]
- 18. Baird M, Ang PW, Clark I, Bishop D, Oshima M, Cook MC, Hemmings C, Takeishi S, Worthley D, Boussioutas A et al (2013) The unfolded protein response is activated in Helicobacter‐induced gastric carcinogenesis in a non‐cell autonomous manner. Lab Investig 93, 112–122. [DOI] [PubMed] [Google Scholar]
- 19. Akazawa Y, Isomoto H, Matsushima K, Kanda T, Minami H, Yamaghchi N, Taura N, Shiozawa K, Ohnita K, Takeshima F et al (2013) Endoplasmic reticulum stress contributes to Helicobacter pylori VacA‐induced apoptosis. PLoS One 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim JM, Kim JS, Kim N, Ko SH, Jeon JI and Kim Y (2015) Helicobacter pylori vacuolating cytotoxin induces apoptosis via activation of endoplasmic reticulum stress in dendritic cells. Gastroenterology 30, 99–108. [DOI] [PubMed] [Google Scholar]
- 21. Wang F, Meng W, Wang B and Qiao L (2014) Helicobacter pylori‐induced gastric inflammation and gastric cancer. Cancer Lett 345, 196–202. [DOI] [PubMed] [Google Scholar]
- 22. Handa O, Naito Y, Yoshikawa T, Handa O, Naito Y and Yoshikawa T (2011) Redox biology and gastric carcinogenesis : the role of Helicobacter pylori. Redox Rep 16, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nathan C and Cunningham‐Bussel A (2013) Beyond oxidative stress: an immunologist's guide to reactive oxygen species. Nat Rev Immunol 13, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beil W, Obst B, Sewing KF and Wagner S (2000) Helicobacter pylori reduces intracellular glutathione in gastric epithelial cells. Dig Dis Sci 45, 1769–1773. [DOI] [PubMed] [Google Scholar]
- 25. Ray PD, Huang BW and Tsuji Y (2012) Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24, 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Raza Y, Khan A, Farooqui A, Mubarak M and Facista A (2014) Oxidative DNA damage as a potential early biomarker of Helicobacter pylori associated carcinogenesis. Pathol Oncol Res 20, 839–846. [DOI] [PubMed] [Google Scholar]
- 27. Forney R (2010) Oxidative stress and oxidative damage in carcinogenesis. Toxicol Pathol 38, 96–109. [DOI] [PubMed] [Google Scholar]
- 28. Auten RL and Davis JM (2009) The role of oxygen in health and disease ‐ A series of reviews oxygen toxicity and reactive oxygen species : the devil is in the details. Pediatr Res 66, 121–127. [DOI] [PubMed] [Google Scholar]
- 29. Inokuma T, Haraguchi M, Fujita F, Tajima Y and Kanematsu T (2009) Oxidative stress and tumor progression in colorectal cancer. Hepatogastroenterology 56, 343–347. [PubMed] [Google Scholar]
- 30. Farinati F, Cardin R, Russo VM, Busatto G, Franco M and Rugge M (2003) Helicobacter pylori CagA status, mucosal oxidative damage and gastritis phenotype: a potential pathway to cancer? Helicobacter 8, 227–234. [DOI] [PubMed] [Google Scholar]
- 31. Varon C, Mosnier J‐F, Lehours P, Matysiak‐Budnik T and Mégraud F (2009) Gastric carcinogenesis and Helicobacter pylori infection In Methods in Molecular Biology (Clifton NJ, ed), pp. 237–265. Inflammation and Cancer, New York City. [DOI] [PubMed] [Google Scholar]
- 32. Matsumoto Y, Marusawa H, Kinoshita K, Endo Y, Kou T, Morisawa T, Azuma T, Okazaki I‐M, Honjo T and Chiba T (2007) Helicobacter pylori infection triggers aberrant expression of activation‐induced cytidine deaminase in gastric epithelium. Nat Med 13, 470–476. [DOI] [PubMed] [Google Scholar]
- 33. Feiz HR and Mobarhan S (2002) Does Vitamin C intake slow the progression of gastric cancer in Helicobacter pylori‐infected populations. Nutrition Rev 60, 34–36. [DOI] [PubMed] [Google Scholar]
- 34. Olivier M, Hollstein M and Hainaut P (2010) TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hagen TM, Huang S, Curnutte J, Fowler P, Martinez V, Wehr CM, Ames BN and Chisari FV (1994) Extensive oxidative DNA damage in hepatocytes of transgenic mice with chronic active hepatitis destined to develop hepatocellular carcinoma. Proc Natl Acad Sci USA 91, 12808–12812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Varatharasa T, Anoma S, David EV, Tapas KH, Mand Sankar and David GG (2008) Base‐pairing properties of the oxidized cytosine derivative, 5‐hydroxy uracil. Biochem Biophys Res Commun 366, 752–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Reactivity of the novel monoclonal antibody to CHAC1.
Fig. S2. The levels of CHAC1 mRNA, GSH, ROS and CHAC1 protein in H. pylori‐infected samples used for mutation analysis.
Table S1. Primers and probes used for this study.
Table S2. CHAC1 overexpression leading to TP53 mutation in AGS cells (results from each of the two experiments).
Table S3. CHAC1 expression induced by H. pylori infection leading to TP53 mutation in AGS cells (results from each of the two experiments).
Table S4. Distribution of nucleotide alterations in the TP53 sequence and resultant amino acid substitutions.