

Abstract
The epidermal growth factor (EGF)‐receptor ligand amphiregulin (AREG) is a potent growth factor implicated in proliferative skin diseases and in primary and metastatic epithelial cancers. AREG, synthesized as a propeptide, requires conversion to an active peptide by metalloproteases by a process known as ectodomain shedding. Although (ADAM17) a disintegrin and metalloprotease 17 is a key sheddase of AREG, ADAM8‐, ADAM15‐, and batimastat (broad metalloprotease inhibitor)‐sensitive metalloproteases have also been implicated in AREG shedding. In the present study, using a curly bare (Rhbdf2 cub) mouse model that shows loss‐of‐hair, enlarged sebaceous gland, and rapid cutaneous wound‐healing phenotypes mediated by enhanced Areg mRNA and protein levels, we sought to identify the principal ectodomain sheddase of AREG. To this end, we generated Rhbdf2 cub mice lacking ADAM17 specifically in the skin and examined the above phenotypes of Rhbdf2 cub mice. We find that ADAM17 deficiency in the skin of Rhbdf2 cub mice restores a full hair coat, prevents sebaceous gland enlargement, and impairs the rapid wound‐healing phenotype observed in Rhbdf2 cub mice. Furthermore, in vitro, stimulated shedding of AREG is abolished in Rhbdf2 cub mouse embryonic keratinocytes lacking ADAM17. Thus, our data support previous findings demonstrating that ADAM17 is the major ectodomain sheddase of AREG.
Keywords: ADAM17, amphiregulin, ectodomain shedding, EGFR, epithelial cancer, RHBDF2
Abbreviations
- ADAM17
a disintegrin and metalloprotease 17
- AREG
amphiregulin
- EGFR
epidermal growth factor receptor
- HB‐EGF
heparin‐binding EGF
- mEFs
mouse embryonic fibroblasts
- MEKs
mouse embryonic keratinocytes
- Rhbdf2cub
curly bare
- TGFα
transforming growth factor alpha
The epidermal growth factor receptor (EGFR) pathway plays a major role in normal development, and in multiple diseases including epithelial cancers and chronic obstructive pulmonary disease, and in liver diseases 1, 2, 3, 4, 5. A critical step in regulating this pathway is ectodomain shedding of type 1 transmembrane EGFR ligands from the cell surface by membrane‐anchored metalloproteases 6. For instance, type‐1 transmembrane EGFR ligands, including amphiregulin (AREG), transforming growth factor alpha (TGFα), epidermal growth factor (EGF), and heparin‐binding EGF (HB‐EGF), are produced as inactive propeptides. In the ectodomain shedding process, ADAMs (a disintegrin and metalloproteases) cleave propeptides to release soluble peptides, leading to activation of the EGFR signaling pathway 7, 8.
Among the multiple ADAMs studied (ADAM8, ADAM9, ADAM10, ADAM12, ADAM15, ADAM17, and ADAM19), ADAM10 and ADAM17 have emerged as key sheddases of the EGFR ligands EGF, betacellulin, HB‐EGF, and TGFA 9, 10. In culture, ectodomain shedding assays using mouse embryonic fibroblasts (mEFs) lacking ADAM10 or ADAM17 show impaired shedding of EGF and betacellulin, and HB‐EGF or TGFA, respectively 11. In line with these findings, ADAM17 knockout mice show defects in cardiac valve and eyelid development 8, 12, defects that are also observed in mice deficient in HB‐EGF (Hbegf −/− mice) and in TGFA (Tgfa −/− mice), respectively 12, 13, 14. Furthermore, using loss‐of‐function experiments in mEFs, Sahin et al. 9 demonstrated that both constitutive shedding and stimulated ectodomain shedding of EGFR ligands, including EGF, epiregulin, betacellulin, HB‐EGF, TGFA, and AREG, are unaltered in the absence of ADAM8, ADAM9, ADAM12, ADAM15, and ADAM19. Thus, substantial literature suggests that ADAM10 and ADAM17 have essential, but distinct, roles in shedding of EGFR ligands.
Results of in vivo studies implicate ADAM17 as the specific metalloprotease contributing to the ectodomain shedding of AREG 15, a potent growth factor implicated in proliferative skin diseases, and primary and metastatic epithelial cancers 16, 17, 18. Moreover, results of in vitro studies have suggested ADAM17 as a key sheddase 9, 10; nevertheless, ADAM8‐, ADAM15‐, and batimastat (broad metalloprotease inhibitor)‐sensitive metalloproteases have also been implicated in AREG shedding in vitro 11. Understanding of the sheddase mechanisms for AREG is critical for development of more effective therapies for diseases associated with this growth factor.
To determine whether ADAM17 is the key sheddase of AREG, we utilized the curly bare (Rhbdf2 cub) gain‐of‐function mouse mutation. Homozygosity for this spontaneous mutation in the Rhbdf2 gene augments Areg mRNA and protein levels and results in alopecia, sebaceous gland enlargement, and rapid wound‐healing phenotypes through enhanced secretion of AREG and subsequent hyperactivation of the EGFR pathway 19. Furthermore, AREG deficiency in Rhbdf2 cub/cub mice prevents the alopecia, sebaceous gland enlargement, and rapid wound‐healing phenotypes, suggesting that AREG is the primary mediator of the Rhbdf2 cub phenotype 19. Thus, the Rhbdf2 cub mouse mutation provides a powerful in vivo model system that allows us to examine the physiological role of ADAM17 in ectodomain shedding of AREG and in AREG‐mediated downstream events, including wound healing.
Here, we demonstrate that conditional deletion of ADAM17 in the skin of Rhbdf2 cub/cub mice impairs the AREG‐mediated hair, sebaceous gland, and wound‐healing phenotypes observed in these mice. We also demonstrate that ADAM17 deficiency significantly abolishes both stimulated and unstimulated shedding of AREG in Rhbdf2 cub/cub mouse embryonic keratinocytes (MEKs), suggesting that ADAM17 is indispensible for sheddase of AREG.
Materials and methods
Animals
All animal work conformed to regulations in the Guide for the Care And Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Research Council, National Academy of Sciences, 8th edition, 2011). Euthanasia was performed in a manner consistent with the 2013 recommendations of the American Veterinary Medical Association (AVMA) Guidelines on Euthanasia. All individuals working with animals in this project read and adhered to The Jackson Laboratory policy, POL.AWC.025 Euthanasia in Animal Experiments Involving Pain, Distress, or Illness. The Rhbdf2 cub/cub , Rhbdf2 −/−, and Rhbdf2 cub/cub Areg −/− mice are maintained on the C57BL/6J genetic background, and Adam17 flox/flox and Adam17 flox/flox K14‐Cre mice are of mixed genetic background 20. We generated Rhbdf2 cub/cub Adam17 flox/flox K14‐Cre mice by crossing female Rhbdf2 +/cub, Adam17 +/flox, K14‐Cre mice with male Rhbdf2 cub/cub, Adam17 flox/flox mice. Areg Mcub/Mcub mice are referred to as Areg −/− mice in this manuscript 19. Mice were maintained under modified barrier conditions on a 12‐h light and 12‐h dark cycle with constant temperature and humidity. The Animal Care and Use Committee at The Jackson Laboratory approved all of the experimental procedures.
Histology
Mice were euthanized by CO2 asphyxiation followed by open chest necropsy, a secondary method of euthanasia. Dorsal skin was removed, fixed in 10% neutral buffered formalin for 24 h, processed routinely, embedded in paraffin, sectioned and stained, with hematoxylin and eosin (H&E).
Isolation of primary keratinocytes
For isolation of MEKs, skin from embryonic day 18 mouse embryos was incubated overnight in neutral protease at 4 °C. Following separation of the epidermis from the dermis, the epidermis was placed in Petri dishes containing trypsin (#12563029; ThermoFisher Scientific, Waltham, MA, USA) and allowed to incubate for 30 min at room temperature. After blocking trypsin activity with soybean trypsin inhibitor (#R007100; ThermoFisher Scientific), cells were grown in KBM‐2 medium (#CC‐3107; Lonza, Fisher Scientific, Pittsburgh, PA, USA) supplemented with antibiotic/antimycotic.
Measurement of amphiregulin protein levels
AREG levels in the cell culture supernatant were measured via ELISA as described previously 19. Briefly, 100 μL of cell culture supernatant was added to capture antibody‐precoated plates and incubated for 2 h at room temperature (RT). After three washes, 100 μL of the detection antibody was added to each well and incubated for an additional 2 h at RT. Following three washes, 100 μL of streptavidin/HRP was added to each well and incubated at RT for 20 min, before adding 100 μL of substrate solution (20 min incubation) and 50 μL of stop solution. A spectrophotometer (SpectraMax 190; Molecular Devices, San Jose, CA, USA) was used to determine the optical density.
Statistical analysis
One‐way ANOVA and two‐way ANOVA were used for comparison of several groups using prism v7 software (GraphPad, La Jolla, CA, USA). A P < 0.05 was considered statistically significant. Data represent mean ± SD.
Results
The loss‐of‐hair, enlarged sebaceous gland, and rapid wound‐healing phenotypes of Rhbdf2 cub mice are mediated through ADAM17
To determine whether ADAM17 is essential for the loss‐of‐hair and enlarged sebaceous gland phenotypes exhibited by Rhbdf2 cub/cub mice, we generated Rhbdf2 cub/cub mice lacking ADAM17 in skin, by crossing Rhbdf2 cub/cub mice with Adam17 flox/flox K14‐Cre mice, and studied the phenotypes of second‐generation offspring. We noted that ADAM17 acts as a genetic modifier of Rhbdf2 cub/cub mice – Rhbdf2 cub/cub Adam17 flox/flox K14‐Cre mice display a full hair coat, in contrast to the complete hair loss in Rhbdf2 cub/cub mice (Fig. 1A). We next performed histopathological examination of truncal skin from Rhbdf2 cub/cub (Fig. 1B.a,b) and Rhbdf2 cub/cub Adam17 flox/flox K14‐Cre (Fig. 1B.c,d) mice at 3 weeks of age. Although, gross examination of Rhbdf2 cub/cub Adam17 flox/flox K14‐Cre mice showed a full hair coat (Fig. 1A, 3), histological examination revealed mild follicular dystrophy (arrowhead) in Rhbdf2 cub/cub Adam17 flox/flox K14‐Cre mice compared with extreme follicular dystrophy in Rhbdf2 cub/cub mice (arrows). Additionally, mice of both strains exhibited hyperkeratosis (H) and thickened epidermis (E), whereas enlargement of sebaceous glands (*) was observed only in Rhbdf2 cub/cub mice, suggesting that deletion of ADAM17 partially reverses the loss‐of‐hair and sebaceous gland phenotypes of Rhbdf2 cub/cub mice.
Figure 1.

(A) Conditional deletion of ADAM17 in the skin of Rhbdf2 cub/cub mice restores hair growth. Also, note the relatively smaller size of the Rhbdf2 cub/cub ADAM17 flox/flox K14‐Cre (3) and ADAM17 flox/flox K14‐Cre (1) mice compared with Rhbdf2 cub/cub (2) mice. (B) (a, b) Truncal skin sections of Rhbdf2 cub/cub mice (2) displaying extreme follicular dystrophy (arrow), thickened epidermis (E), enlarged sebaceous glands (*), and hyperkeratosis (H). (c, d) Although Rhbdf2 cub/cub ADAM17 flox/flox K14‐Cre mice exhibit a full hair coat, histological analysis of truncal skin sections of Rhbdf2 cub/cub ADAM17 flox/flox K14‐Cre mice (3) revealed mild follicular dystrophy (arrow heads); however, there was no evidence of sebaceous gland hyperplasia (★). Scale bars: 250 μm (a, c) and 50 μm (b, d). (C) Healing of ear tissue in 6‐ to 8‐week‐old female ADAM17 flox/flox K14‐Cre (1), Rhbdf2 cub ADAM17 flox/flox K14‐Cre (3), and Rhbdf2 cub/cub (2) mice (n = 3 per group; representative images are shown) at 0 and 14 days postwounding. Magnification = 4×; Scale bars = 1 mm; Quantification of ear‐hole closures on day 14; ***P < 0.001.
We next examined the rapid wound‐healing phenotype of Rhbdf2 cub/cub mice. The Rhbdf2 cub mutation induces a rapid wound‐healing phenotype through enhanced secretion of AREG 19, 21; when we punched 2‐mm through‐and‐through holes in the ear pinnae of Rhbdf2 cub/cub mice, within 2 weeks ear‐hole closure of more than 90% was observed in Rhbdf2 cub/cub mice, in contrast to approximately 20% ear‐hole closure in Rhbdf2 +/+ mice 19. Here, we wanted to determine whether the wound‐healing phenotype in Rhbdf2 cub/cub mice requires ADAM17. Using the above‐mentioned ear‐hole closure assay, we tested the wound‐healing phenotype of Rhbdf2 cub/cub Adam17 flox/flox K14‐Cre mice and compared it to those of Adam17 flox/flox K14‐Cre and Rhbdf2 cub/cub mice. Not surprisingly, impairment of wound healing was similar in Adam17 flox/flox K14‐Cre mice (Fig. 1C, left column) and Rhbdf2 cub/cub Adam17 flox/flox K14‐Cre mice (Fig. 1C, middle column), whereas Rhbdf2 cub/cub mice showed the rapid wound‐healing phenotype (Fig. 1C, right column). Collectively, these data suggest that loss of ADAM17 in the skin of Rhbdf2 cub/cub mice modifies the loss‐of‐hair phenotype and restores a full hair coat and diminishes the wound‐healing phenotype.
Loss of ADAM17 specifically in the skin causes dermatitis and myeloproliferative disease in Rhbdf2 cub/cub mice
A previous study showed that sustained deficiency of ADAM17 in the epidermis of wild‐type mice results in epidermal barrier defects, and subsequently dermatitis and myeloproliferative disease; that is, a significant increase in the myeloid‐cell infiltration 20. Thus, to determine whether ADAM17 deficiency also causes dermatitis in Rhbdf2 cub/cub mice, we examined the skin phenotype of Rhbdf2 cub/cub ADAM17 flox/flox K14‐Cre mice in comparison with that of ADAM17 flox/flox K14‐Cre and ADAM17 flox/flox control mice. The skin of Rhbdf2 cub/cub ADAM17 flox/flox K14‐Cre mice displayed noticeable scaling (Fig. 2A, left), indistinguishable from the phenotype of ADAM17 flox/flox K14‐Cre mice (Fig. 2A, right). Furthermore, histological examination of H&E sections revealed a thicker hypodermis (H) and a thinner dermis (D) in ADAM17 flox/flox control mice (Fig. 2B.a) in contrast to a thinner hypodermis and a thicker dermis in both ADAM17 flox/flox K14‐Cre (Fig. 2B.b) and Rhbdf2 cub/cub ADAM17 flox/flox K14‐Cre (Fig. 2B.c) mice. Additionally, we observed epidermal thickening (asterisk), hyperkeratosis (arrow head), and considerable infiltration of inflammatory cells, including macrophages and neutrophils (arrows), in the dermis of ADAM17 flox/flox K14‐Cre mice (Fig. 2B.e) and Rhbdf2 cub/cub ADAM17 flox/flox K14‐Cre mice (Fig. 2B.f), in contrast to ADAM17 flox/flox control mice (Fig. 2B.d), which did not manifest any indication of skin disease.
Figure 2.
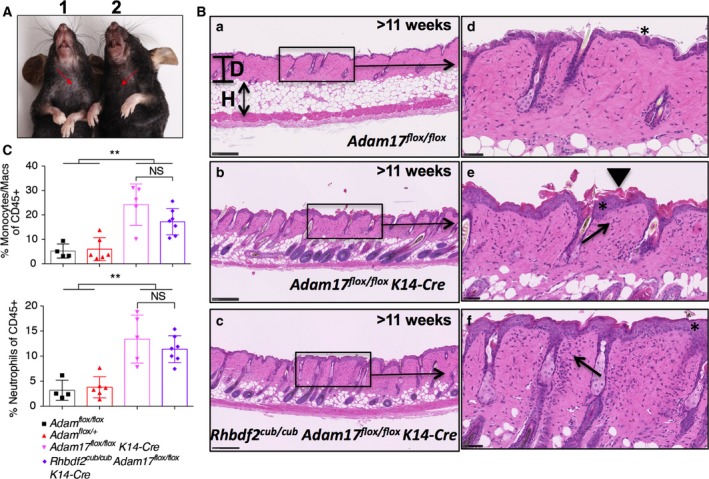
(A) The role of ADAM17 in regulating the skin barrier has been established previously 20 – deletion of ADAM17 in the skin results in epidermal defects and dermatitis. A similar phenotype – dry scaly skin (arrows) – was observed in both Adam17 flox/flox K14‐Cre (1) and Rhbdf2 cub/cub Adam17 flox/flox K14‐Cre (2) mice. (B) Deletion of ADAM17 in the skin results in epidermal defects and dermatitis. A similar dermatitis‐like phenotype, including epidermal thickening (asterisk), hyperkeratosis (arrow head), and considerable infiltration of inflammatory cells (arrows), was observed in both Adam17 flox/flox K14‐Cre (b, e) and Rhbdf2 cub/cub Adam17 flox/flox K14‐Cre (c, f) mice, compared to the normal skin in Adam17 flox/flox control mice (a, d). Note the relatively thicker dermis (D) and thinner hypodermis (H) in both Adam17 flox/flox K14‐Cre (b) and Rhbdf2 cub/cub Adam17 flox/flox K14‐Cre (c) mice, compared to the thinner dermis and thicker hypodermis in Adam17 flox/flox control mice (a). Scale bars: 250 μm (low magnification) and 50 μm (high magnification). (C) Both ADAM17 flox/flox K14‐Cre and Rhbdf2 cub/cub ADAM17 flox/flox K14‐Cre mice develop myeloproliferative disease, evidenced by the increased percentage of macrophages (top) and neutrophils (bottom) in the spleens of these mice compared with control mice (ADAM17 flox/flox and ADAM17 flox/+ mice).
Next, using flow cytometry analyses we determined whether there was any indication of myeloproliferative disease in Rhbdf2 cub/cub Adam17 flox/flox K14‐Cre mice by quantifying the differences in the percentages of splenic macrophages (Fig. 2C, top panel) and neutrophils (Fig. 2C, bottom panel) between ADAM17 flox/flox K14‐Cre and Rhbdf2 cub/cub ADAM17 flox/flox K14‐Cre mice. Compared with control mice (ADAM17 flox/flox and ADAM17 flox/+ mice), we observed significantly higher percentages of macrophages and neutrophils in both of ADAM17 flox/flox K14‐Cre and Rhbdf2 cub/cub Adam17 flox/flox K14‐Cre mice, suggesting that loss of ADAM17 specifically in the skin results in considerable myeloproliferation in Rhbdf2 cub/cub mice. Taken together, our results indicate that lack of ADAM17 in the skin results in dermatitis and myeloproliferative disease, which validates previous findings by Franzke et al. 20 that ADAM17 maintains the skin barrier. Moreover, our results showing development of a similar overt skin phenotype observed by Franzke et al. and restoration of hair growth in Rhbdf2 cub/cub mice lacking ADAM17 implicate ADAM17 as a participant in ectodomain shedding of AREG.
Ectodomain shedding of AREG requires ADAM17
To determine whether ADAM17 is required for AREG ectodomain shedding, we asked whether loss of ADAM17 in Rhbdf2 cub/cub keratinocytes alters AREG secretion with or without stimulation with 100 nm phorbol‐12‐myristate‐13‐acetate (PMA). We also examined AREG‐stimulated and AREG‐unstimulated secretion in Rhbdf2 +/+, Rhbdf2 cub/cub Areg −/−, and Rhbdf2 −/− keratinocytes, for comparison. First, in line with our previous findings 19, we found that both the stimulated secretion (red column) and unstimulated (blue column) secretion of AREG are significantly increased in Rhbdf2 cub/cub keratinocytes compared with control keratinocytes (Fig. 3A; 1, Rhbdf2 +/+; 2, Rhbdf2 cub/cub). Additionally, because Rhbdf2 cub/cub Areg −/− mice do not produce a functional AREG protein 19, as expected, there was no detectable AREG in the culture supernatants of Rhbdf2 cub/cub Areg −/− keratinocytes (Fig. 3A; 3). Furthermore, there was a significant reduction in AREG levels in the culture supernatants of Rhbdf2 −/− keratinocytes with or without stimulation in comparison with those of Rhbdf2 +/+ and Rhbdf2 cub/cub keratinocytes (Fig. 3A; 4), suggesting that RHBDF2 is required for both stimulated and unstimulated shedding of AREG.
Figure 3.
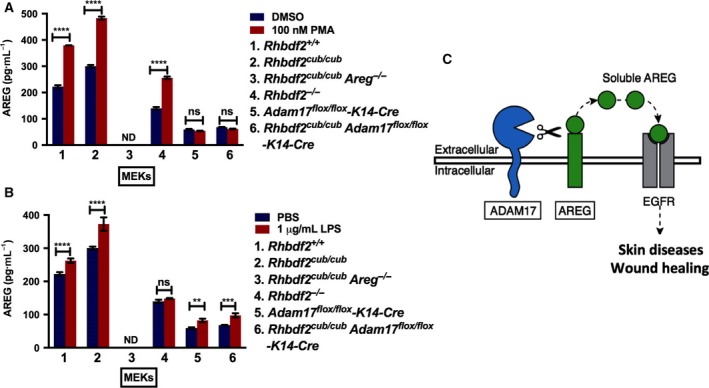
(A) ELISA quantitation of cleaved AREG from the supernatant of MEKs after overnight stimulation with either DMSO or 100 nm PMA. ND, not detected; Data represent mean ± SD. ****P < 0.0001; ns, not significant. (B) ELISA quantitation of cleaved AREG from the supernatant of MEKs after overnight stimulation with either PBS or 1 μg·mL−1 LPS. ND, not detected; Data represent mean ± SD. ****P < 0.0001; ***P < 0.001; **P < 0.01; ns, not significant. (C) Loss of ADAM17 modifies the Rhbdf2 cub/cub hair loss and ear‐punch closure phenotypes and in vitro deficiency of ADAM17 in Rhbdf2 cub/cub keratinocytes prevents stimulated secretion of AREG, together suggesting that ADAM17 is indispensible for sheddase of AREG.
Second, we observed that, compared to control Rhbdf2 +/+ keratinocytes, keratinocytes lacking ADAM17 showed approximately 85% and 75% lower AREG levels in stimulated and unstimulated conditions, respectively (Fig. 3A; 5, Adam17 flox/flox K14‐Cre). Moreover, there was no significant difference in AREG levels between stimulated and unstimulated conditions in Adam17 flox/flox K14‐Cre keratinocytes (Fig. 3A; 5). Third, consistent with the restoration of hair growth and loss of rapid wound‐healing phenotypes in Rhbdf2 cub/cub Adam17 flox/flox K14‐Cre mice, we observed that Rhbdf2 cub/cub Adam17 flox/flox K14‐Cre keratinocytes failed to secrete AREG even after stimulation with PMA (Fig. 3A; 6), in contrast to a significant increase in both stimulated and unstimulated Rhbdf2 cub/cub keratinocytes (Fig. 3A; 2), suggesting that the Rhbdf2 cub mutation fails to promote AREG secretion in the absence of ADAM17. Notably, the levels of AREG observed in keratinocytes lacking ADAM17 (Fig. 3A; 5 and 6) can be attributed to constitutive shedding, which does not require ectodomain shedding by metalloproteases 9.
Fourth, similar results were obtained when keratinocytes isolated from the aforementioned strains (1 through 6) of mice were exposed to bacterial endotoxin lipopolysaccharide (LPS) and assayed for AREG levels in the culture supernatants (Fig. 3B). Interestingly, there was a subtle but significant increase in the levels of AREG in Adam17 flox/flox K14‐Cre and Rhbdf2 cub/cub Adam17 flox/flox K14‐Cre keratinocytes upon stimulation with LPS (Fig. 3B; 5 and 6); however, this could be due to differential regulation of AREG constitutive shedding by PMA versus LPS. Taken together, because loss of ADAM17 significantly abolished stimulated secretion of AREG in Rhbdf2 cub/cub keratinocytes, these data strongly suggest that ADAM17 is essential for ectodomain shedding of AREG (Fig. 3C).
Discussion
Amphiregulin plays an important role in pathological processes, including psoriasis induction 22, 23, cancer progression, and resistance to chemotherapy and anti‐EGFR therapies 16, 24. For example, AREG has been characterized as a multicrine – autocrine, paracrine, and endocrine (systemic) – growth factor in primary and metastatic epithelial cancers 25, 26, 27. AREG induces its own expression to enable self‐sufficiency of growth signals acting through EGFR, via an extracellular autocrine loop 28, suggesting that dysregulation of this loop could lead to overexpression of AREG. Additionally, cancer cells overexpressing AREG can induce neoplastic transformation of neighboring cells through paracrine or endocrine activity 15. Also, more recently, we showed in mice that AREG underlies the hyperproliferative skin disease tylosis and that loss of AREG restores the normal skin phenotype in a mouse model of human tylosis 29. Together, these studies highlight the key role of AREG in several pathological processes, and the potential of AREG depletion as a therapeutic approach in multiple diseases. To develop effective therapeutic strategies targeting AREG, it is important to understand how AREG secretion is regulated in vivo.
Amphiregulin synthesized as pro‐AREG is converted to an active form by metalloproteases. Although several ADAMs have been implicated, a study by Sahin et al. 9 showed that Adam17 −/− MEFs exhibit impaired shedding of AREG, indicating that ADAM17 may be a major sheddase. Comparison of the phenotype of Adam17 −/− mice with that of Areg −/− mice is a potential means of providing support for a role of ADAM17 as the major sheddase of AREG. However, literature suggests that in contrast to Hbegf −/− and Tgfa −/− mice 13, 14, Areg −/− mice are viable and do not present with an overt phenotype, except for defects in mammary gland development during puberty and in nursing 30. Thus, it remains to be determined whether the phenotype of mice with Areg depletion resembles any aspects of the Adam17 −/− phenotype. Adam17 −/− mice exhibit perinatal lethality, limiting the ability to examine mammary gland development and nursing competence phenotypes. Furthermore, although at birth Adam17 −/− pups exhibit stunted growth and development 31, including defective mammary branching, suggesting a role for ADAM17 in shedding of AREG, there is a lack of direct evidence. In the present study, using mouse genetics and in vitro ectodomain shedding assays, we sought to determine whether loss of ADAM17 abolishes shedding of AREG in vivo. We demonstrate that loss of ADAM17 impairs the AREG‐mediated loss‐of‐hair, enlarged sebaceous gland, and rapid wound‐healing phenotypes observed in Rhbdf2 cub/cub mice. Moreover, we find that conditional deletion of ADAM17 in the skin of Rhbdf2 cub/cub mice significantly inhibits stimulated secretion of AREG in keratinocytes, suggesting that ADAM17 is necessary for ectodomain shedding of AREG in keratinocytes. Notably, in macrophages, RHBDF2 also regulates stimulated secretion of pro‐inflammatory cytokine tumor necrosis factor alpha (TNFα) through ADAM17 32, 33, implicating that, similar to ectodomain shedding of AREG, ADAM17 might be key for shedding of RHBDF2‐mediated secretion of TNFα. Consistently, RHBDF2 has recently been suggested to be an essential regulator of stimulated growth factor and cytokine signaling via ADAM17 34, 35.
We previously observed that Rhbdf2 gain‐of‐function alleles, including Rhbdf2 cub, could induce secretion of AREG in the presence of a potent ADAM17 inhibitor marimastat 19. Moreover, both our group 19 and Siggs et al. 36 observed that ADAM17 activity is reduced in Rhbdf2 cub/cub mice. Based on these findings, we postulated that RHBDF2 might regulate secretion of AREG independent of ADAM17 activity 19. However, results from the present study showing that loss of ADAM17 reverses the phenotype of Rhbdf2 cub/cub mice suggest that ADAM17 is indispensible for sheddase of AREG and that RHBDF2 does not act as a sheddase of AREG.
Author contributions
VH, LDS, and MVW designed research; VH, LMB, and MLF performed experiments; VH, LMB, and MLF acquired data; VH, LDS, and MVW analyzed data; and VH, LDS, and MVW wrote the paper.
Acknowledgements
We are grateful to Stephen B. Sampson for critical reading and for providing valuable comments on the manuscript. We also thank Scientific Services at The Jackson Laboratory for assistance with histology (Elaine Bechtel) and flow cytometry (Will Schott and Ted Duffy). Research reported in this publication was partially supported by the National Cancer Institute of the National Institutes of Health under Award Number P30CA034196, and by the Director's Innovation Fund at The Jackson Laboratory (VH).
For a preprint version of this article, see 37 (https://doi.org/10.1101/218891).
References
- 1. Sasaki T, Hiroki K and Yamashita Y (2013) The role of epidermal growth factor receptor in cancer metastasis and microenvironment. Biomed Res Int 2013, 546318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Werner S and Grose R (2003) Regulation of wound healing by growth factors and cytokines. Physiol Rev 83, 835–870. [DOI] [PubMed] [Google Scholar]
- 3. Singh B, Carpenter G and Coffey RJ (2016) EGF receptor ligands: recent advances. F1000Res 5, 2270 https://doi.org/10.12688/f1000research.9025.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burgel PR and Nadel JA (2008) Epidermal growth factor receptor‐mediated innate immune responses and their roles in airway diseases. Eur Respir J 32, 1068–1081. [DOI] [PubMed] [Google Scholar]
- 5. Komposch K and Sibilia M (2015) EGFR signaling in liver diseases. Int J Mol Sci 17 https://doi.org/10.3390/ijms17010030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Singh B and Coffey RJ (2014) Trafficking of epidermal growth factor receptor ligands in polarized epithelial cells. Annu Rev Physiol 76, 275–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blobel CP (2005) ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol 6, 32–43. [DOI] [PubMed] [Google Scholar]
- 8. Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN et al (1998) An essential role for ectodomain shedding in mammalian development. Science 282, 1281–1284. [DOI] [PubMed] [Google Scholar]
- 9. Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P and Blobel CP (2004) Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol 164, 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sunnarborg SW, Hinkle CL, Stevenson M, Russell WE, Raska CS, Peschon JJ, Castner BJ, Gerhart MJ, Paxton RJ, Black RA et al (2002) Tumor necrosis factor‐alpha converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J Biol Chem 277, 12838–12845. [DOI] [PubMed] [Google Scholar]
- 11. Horiuchi K, Le Gall S, Schulte M, Yamaguchi T, Reiss K, Murphy G, Toyama Y, Hartmann D, Saftig P and Blobel CP (2007) Substrate selectivity of epidermal growth factor‐receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol Biol Cell 18, 176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jackson LF, Qiu TH, Sunnarborg SW, Chang A, Zhang C, Patterson C and Lee DC (2003) Defective valvulogenesis in HB‐EGF and TACE‐null mice is associated with aberrant BMP signaling. EMBO J 22, 2704–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luetteke NC, Qiu TH, Peiffer RL, Oliver P, Smithies O and Lee DC (1993) TGF alpha deficiency results in hair follicle and eye abnormalities in targeted and waved‐1 mice. Cell 73, 263–278. [DOI] [PubMed] [Google Scholar]
- 14. Iwamoto R, Yamazaki S, Asakura M, Takashima S, Hasuwa H, Miyado K, Adachi S, Kitakaze M, Hashimoto K, Raab G et al (2003) Heparin‐binding EGF‐like growth factor and ErbB signaling is essential for heart function. Proc Natl Acad Sci USA 100, 3221–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sternlicht MD, Sunnarborg SW, Kouros‐Mehr H, Yu Y, Lee DC and Werb Z (2005) Mammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17‐dependent shedding of epithelial amphiregulin. Development 132, 3923–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nam SO, Yotsumoto F, Miyata K, Fukagawa S, Yamada H, Kuroki M and Miyamoto S (2015) Warburg effect regulated by amphiregulin in the development of colorectal cancer. Cancer Med 4, 575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu Q, Chiao P and Sun Y (2016) Amphiregulin in cancer: new insights for translational medicine. Trends Cancer 2, 111–113. [DOI] [PubMed] [Google Scholar]
- 18. Chung E, Cook PW, Parkos CA, Park YK, Pittelkow MR and Coffey RJ (2005) Amphiregulin causes functional downregulation of adherens junctions in psoriasis. J Invest Dermatol 124, 1134–1140. [DOI] [PubMed] [Google Scholar]
- 19. Hosur V, Johnson KR, Burzenski LM, Stearns TM, Maser RS and Shultz LD (2014) Rhbdf2 mutations increase its protein stability and drive EGFR hyperactivation through enhanced secretion of amphiregulin. Proc Natl Acad Sci USA 111, E2200–E2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Franzke CW, Cobzaru C, Triantafyllopoulou A, Loffek S, Horiuchi K, Threadgill DW, Kurz T, van Rooijen N, Bruckner‐Tuderman L and Blobel CP (2012) Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand‐dependent terminal keratinocyte differentiation. J Exp Med 209, 1105–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hosur V, Burzenski LM, Stearns TM, Farley ML, Sundberg JP, Wiles MV and Shultz LD (2017) Early induction of NRF2 antioxidant pathway by RHBDF2 mediates rapid cutaneous wound healing. Exp Mol Pathol 102, 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bhagavathula N, Nerusu KC, Fisher GJ, Liu G, Thakur AB, Gemmell L, Kumar S, Xu ZH, Hinton P, Tsurushita N et al (2005) Amphiregulin and epidermal hyperplasia: amphiregulin is required to maintain the psoriatic phenotype of human skin grafts on severe combined immunodeficient mice. Am J Pathol 166, 1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cook PW, Brown JR, Cornell KA and Pittelkow MR (2004) Suprabasal expression of human amphiregulin in the epidermis of transgenic mice induces a severe, early‐onset, psoriasis‐like skin pathology: expression of amphiregulin in the basal epidermis is also associated with synovitis. Exp Dermatol 13, 347–356. [DOI] [PubMed] [Google Scholar]
- 24. Busser B, Sancey L, Brambilla E, Coll JL and Hurbin A (2011) The multiple roles of amphiregulin in human cancer. Biochem Biophys Acta 1816, 119–131. [DOI] [PubMed] [Google Scholar]
- 25. Ciardiello F, Kim N, Saeki T, Dono R, Persico MG, Plowman GD, Garrigues J, Radke S, Todaro GJ and Salomon DS (1991) Differential expression of epidermal growth factor‐related proteins in human colorectal tumors. Proc Natl Acad Sci USA 88, 7792–7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Picihard V, Berthois Y, Roccabianca M, Prevot C, Sarrazin M, Portugal H, Kumar S, Kumar P and Rognoni JB (2006) Concomitant cell growth and differentiation are dependent on erbB1 and integrin activation in an autonomously surviving colon adenocarcinoma: involvement of autocrine amphiregulin secretion. Anticancer Res 26, 2769–2783. [PubMed] [Google Scholar]
- 27. Yamada M, Ichikawa Y, Yamagishi S, Momiyama N, Ota M, Fujii S, Tanaka K, Togo S, Ohki S and Shimada H (2008) Amphiregulin is a promising prognostic marker for liver metastases of colorectal cancer. Clin Cancer Res 14, 2351–2356. [DOI] [PubMed] [Google Scholar]
- 28. Johnson GR, Saeki T, Gordon AW, Shoyab M, Salomon DS and Stromberg K (1992) Autocrine action of amphiregulin in a colon carcinoma cell line and immunocytochemical localization of amphiregulin in human colon. J Cell Biol 118, 741–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hosur V, Low BE, Shultz LD and Wiles MV (2017) Genetic deletion of amphiregulin restores the normal skin phenotype in a mouse model of the human skin disease tylosis. Biol Open 6, 1174–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luetteke NC, Qiu TH, Fenton SE, Troyer KL, Riedel RF, Chang A and Lee DC (1999) Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development 126, 2739–2750. [DOI] [PubMed] [Google Scholar]
- 31. Sternlicht MD and Sunnarborg SW (2008) The ADAM17‐amphiregulin‐EGFR axis in mammary development and cancer. J Mammary Gland Biol Neoplasia 13, 181–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adrain C, Zettl M, Christova Y, Taylor N and Freeman M (2012) Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 335, 225–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McIlwain DR, Lang PA, Maretzky T, Hamada K, Ohishi K, Maney SK, Berger T, Murthy A, Duncan G, Xu HC et al (2012) iRhom2 regulation of TACE controls TNF‐mediated protection against Listeria and responses to LPS. Science 335, 229–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grieve AG, Xu H, Kunzel U, Bambrough P, Sieber B and Freeman M (2017) Phosphorylation of iRhom2 at the plasma membrane controls mammalian TACE‐dependent inflammatory and growth factor signalling. Elife 6, e23968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cavadas M, Oikonomidi I, Gaspar CJ, Burbridge E, Badenes M, Felix I, Bolado A, Hu T, Bileck A, Gerner C et al (2017) Phosphorylation of iRhom2 controls stimulated proteolytic shedding by the metalloprotease ADAM17/TACE. Cell Rep 21, 745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Siggs OM, Grieve A, Xu H, Bambrough P, Christova Y and Freeman M (2014) Genetic interaction implicates iRhom2 in the regulation of EGF receptor signalling in mice. Biol Open 3, 1151–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hosur V, Farley ML, Burzenski LM, Shultz LD and Wiles MV (2017) ADAM17 is the principal ectodomain sheddase of the EGF‐receptor ligand amphiregulin. bioRxiv [PREPRINT]. [DOI] [PMC free article] [PubMed] [Google Scholar]