

Abstract
PSMD7, a 19S proteasome subunit, is overexpressed in most carcinoma cells. It forms a dimer with PSMD14 that functions in the removal of attached ubiquitin chain. However, there is little knowledge about the cellular mechanism of PSMD7 and its exact biological function, especially in cancer cells. In this study, we explored the role of PSMD7 in proliferation, cell cycle, apoptosis, and proteasomal proteolysis in the esophageal squamous cell carcinoma (ESCC) cell line EC9706. Our results showed that PSMD7 was highly expressed in ESCC cells. Downregulation of PSMD7 by lentivirus‐mediated shRNA led to decreased proliferation, increased cell apoptosis, and reduced proteasomal function. Notably, lower expression level of mTOR and p70S6K and suppressed activity of mTOR/p70S6K pathway were detected after PSMD7 downregulation. By contrast, increased expression of p‐mTORSer2448 and p‐p70S6KThr421/Ser424 was discovered upon PSMD7 overexpression in Het‐1A cells. Furthermore, PSMD7 downregulation contributed to decelerated tumor growth, inhibition of proteasomal function, induced cell apoptosis and attenuated activity of mTOR/p70S6K pathway in vivo. These findings suggest that PSMD7 and the mTOR/p70S6K pathway may be a promising candidate for developing therapies for ESCC.
Keywords: apoptosis (ESCC), esophageal squamous cell carcinoma, mTOR/p70S6K pathway, PSMD7, tumorigenesis
Abbreviations
- ESCC
esophageal squamous cell carcinoma
- shRNA
short hairpin RNA
- TGI
tumor growth inhibition rate
- TV
tumor volume
Carcinoma of the esophagus is one of the most commonly diagnosed malignant cancers, ranking the eighth cancers worldwide with highly lethal rate and rapidly rising trend 1. In particular, the morbidity of esophageal squamous cell carcinoma (ESCC) remains high in northern China, which accounts for about 90% of the total esophageal cancer cases and possibly develops resistance to chemotherapeutic drugs 2. Chemical proteasomal inhibitors improve the survival rate of patients through strengthening the toxicity to cancer cells and increasing durable clinical response 3. It has been demonstrated that proteasome inhibitor bortezomib has anti‐proliferative effect not only to a large number of cancer cells, such as lung, breast, prostate, pancreas, and colon and melanoma cell lines, but also to xenograft models of solid tumors 4, 5, 6. However, disappointing results were yielded in clinical trials of solid tumors, no matter bortezomib used as single treatment or combined with other chemotherapy drugs, radiation therapy, immunotherapy, or other new targeting drugs in treatment 7, 8, 9. Thereafter, further investigation in the molecular mechanisms underlying the carcinogenesis and progression of ESCC and potential therapeutic targets becomes more and more urgent.
Accumulated studies in recent years have demonstrated that some deubiquitinases have abnormal expressions in tumors and participate in tumor development, which may be used as potential targets for drug therapy 10. The encoded deubiquitinases in human genome account for nearly 100 until now. It has been reported that four deubiquitinases (USP14, UCHL5/Uch37, PSMD7/RPN8, and PSMD14/RPN11) are related to proteasome function of removing or editing the ubiquitinated proteasome substrates 11. Among four deubiquitinases, PSMD7 and PSMD14 are non‐ATPase subunits of 19S lid subcomplex with Mpr1‐Pad1‐N‐terminal (MPN) domain, followed by long C‐terminal helices JAMM domain. PSMD14 has been reported to function in the removal of attached ubiquitin chain, which is a precondition for efficient substrate translocation through the narrow axial pore 12. Research on the proteasome holoenzyme revealed that PSMD14 and PSMD7 can form a dimer, which is locating directly above the central pore and leading the degraded substrate enter into the N‐terminal‐domain ring of the base ATPases 13.
In many types of tumor cells, in addition to the enhancement of the signaling pathways upstream to ubiquitin‐proteasome system, the activity of proteasome and expression of proteasomal subunits are abnormal 14, 15, 16. PSMD7 has been reported as a cancer‐associated signature in a variety of tumors, such as breast 17, 18, prostate 19, and bladder cancers 20. However, little knowledge about the cellular biological role of PSMD7 is known, and further research is needed to clarify its exact function in normal cells, particularly in cancer cells. Recent study has shown that inhibition of proteasomal activity induced cytotoxicity through the downregulation of ERK and Akt/mTOR signaling 21. Proteasome inhibitor MG132 caused the dephosphorylation of p70S6 kinase (p70S6K) and mTOR in a dose‐ and time‐dependent manner 22. The mTOR/p70S6K pathway is highly activated in ESCC, and its inhibition by rapamycin suppressed the phosphorylation of major downstream effectors, p70S6K and 4E‐BP1 23. Therefore, we supposed that PSMD7 was involved in the regulation of activity of mTOR/p70S6K pathway. Therefore, we hypothesized that PSMD7 maybe probably involved in the maintenance of proteasomal function in cancer cells.
In this study, the expression of PSMD7 in three ESCC cell lines was examined. In addition, its role on proliferation, cell cycle, cell apoptosis, and mTOR/p70S6K pathway was investigated using shRNA lentivirus‐mediated PSMD7 knockdown. Furthermore, we also found that PSMD7 knockdown in vivo suppressed the tumor growth using ESCC tumor models. Our results showed that PSMD7 is overexpressed in all three ESCC cells. Knockdown of PSMD7 showed inhibited proliferation and enhanced apoptosis, which is through mTOR/p70S6K signaling pathway. Taken together, PSMD7 exhibits pro‐cancer characteristics, which suggests PSMD7 could be a potential molecular target for ESCC therapy.
Materials and methods
Cell culture
Nonmalignant esophageal cell line Het‐1A was maintained in Dulbecco's modified Eagle's medium supplemented with 10% FBS (HyClone, USA). Human HeLa 229 cells and ESCC cell lines (EC9706, EC1 and Eca109 cells) were cultured in RPMI 1640 media (HyClone, USA) supplemented with 10% FBS and 1% L‐glutamine. All cells were grown at 37 °C humidified atmosphere with 5% CO2.
Western blot
Total proteins of Het‐1A, HeLa 229, EC9706, and Eca109 were isolated by Total Protein Extraction Kit (Solarbio, China), respectively, followed by the detection of PSMD7 protein expression. Protein samples were subjected to SDS/PAGE gels and transferred to polyvinylidene fluoride membrane (Millipore, USA), followed by incubation with anti‐PSMD7 antibody (Santa Cruz Biotechnology, USA), anti‐GADPH antibody (Abgent, USA), anti‐cleaved caspase‐3 antibody (Abgent, USA), anti‐PARP antibody (Abgent, USA), anti‐ubiquitin antibody (Santa Cruz Biotechnology, USA), anti‐mTOR (Cell Signaling Technology, USA), p‐mTOR (Cell Signaling Technology, USA), p70S6K (Cell Signaling Technology, USA), and p‐p70S6K (Cell Signaling Technology, USA). Membranes were incubated with horseradish peroxidase‐labeled secondary antibodies (Boster, China) and detected by Enhanced Chemiluminescent Kit (Santa Cruz Biotechnology, USA).
Immunofluorescence Staining
Cells were fixed by 4% paraformaldehyde and permeabilized by 0.1% saponin. Samples were blocked by 1% BSA and incubated with anti‐PSMD7 antibody (Santa Cruz Biotechnology, USA) and anti‐ubiquitin antibody (Santa Cruz Biotechnology, USA). Cell samples were visualized with the corresponding DyLight 649‐conjugated affinipure goat anti‐rabbit IgG (Earthox, USA). Cell nucleus was stained by incubation with 4′, 6‐diamidino‐2‐phenylindole (DAPI) for 10 min. Stained cells were imaged by IX71 inverted fluorescence microscope (Olympus Optical, Japan).
Proteasomal activity assay
The cellular 26S proteasomal activity was evaluated by an in vitro peptide hydrolysis method using β5‐selective fluorogenic substrate succinyl‐Leu‐Leu‐Val‐Tyr‐7‐amido‐4‐methylcoumarin (Merck, Germany) as described previously 24. For some experiments, EC9706 cells were transfected with negative control shRNA lentivirus or PSMD7 shRNA lentivirus, and then, stable transfected cell populations were selected and maintained in puromycin culture medium.
PSMD7 knockdown by lentivirus‐delivered RNA interference
Four short hairpin RNA (shRNA) sequences (Table 1) and one negative control scramble sequence (TTCTCCGAACGTGTCACGT) were designed and synthesized. The hairpin‐pGV112 vector was constructed containing a short hairpin RNA (shRNA) sequences. The lentivirus was packaged using the pHelper 1.0, pHelper 2.0, and pGV112 plasmids, as well as Lipofectamine 2000 (Invitrogen, USA), and then used to infect 293T cells. Lentivirus supernatant was collected 48 h later and used to infect EC9706 cells with polybrene (Sigma‐Aldrich, USA), followed by selection by puromycin (Thermo Scientific, USA).
Table 1.
Targeting sequence of PSMD7 RNAi
| NO. | Target Seq | CDS | GC% |
|---|---|---|---|
| LV‐PSMD7‐RNAi1 | TGCACAACCTCATCAACAA | 133…1107 | 42.11 |
| LV‐PSMD7‐RNAi2 | TGAGGAAGTTGGAGTTGAA | 133…1107 | 42.11 |
| LV‐PSMD7‐RNAi3 | CATCAACGAACTCATGAAA | 133…1107 | 36.84 |
| LV‐PSMD7‐RNAi4 | TGACAAAGACGATTCTGTA | 133…1107 | 36.84 |
Overexpression of PSMD7 in Het‐1A cells
The cDNA of PSMD7 was amplified by PCR and inserted into vector pEGFP‐N1 (Clontech, USA) to obtain a recombinated vector pEGFP‐N1‐PSMD7. EC9706 cells were seeded at a density of 2 × 106 cells/mL followed by transfection with 2.5 μg of pEGFP‐N1‐PSMD7 and 5 μL of Lipofectamine 2000 (Invitrogen, USA) according to the instructions. Meanwhile, empty vector pEGFP‐N1 was transfected to EC9706 cells using as negative control.
Cell proliferation and apoptosis analysis
Cells were seeded in 96‐well flat‐bottomed plate and cultured overnight, followed by detection of cell proliferation using CCK‐8 kit (Beyotime Inst Biotech, China). Cell apoptosis was detected using Annexin V‐APC/7‐AAD Apoptosis Detection Kit (KeyGEN Biotech, China) and analyzed by flow cytometry (Becton Dickson, USA). Annexin V+/7‐AAD− and Annexin V+/7‐AAD+ cells were, respectively, considered as early and late apoptotic cells.
Caspase activity assay
Caspase‐3 Fluorometric Assay Kit (BioVision, USA) was used to detect the caspase activity according to manufacturer's instructions. Cells were stained with Ac‐DEVD‐AFC substrate at 37 °C for 1 h. Tumor tissue sample was homogenized to generate tissue lysate and then followed the kit procedures. Fluorescence was generated from the cleavage of DEVD peptide and was measured by FACSCalibur flow cytometer (Becton Dickinson, USA).
Animals and treatments
BALB/c nude mice (four‐ to six‐week‐old) were purchased from Hunan SJA Laboratory Animal Co., China, and raised under specific pathogen‐free (SPF) condition. All animal studies were carried out in compliance with the National Institutes of Health Guide for care and use of laboratory animals. Mice were housed four or five per cage in standard feeding condition.
EC9706 cells, negative shRNA control lentivirus, and PSMD7‐shRNA lentivirus‐transfected EC9706 cells were inoculated into the right flank of mice with 2 × 106 cells each. All animals were inspected daily, and tumor volume (TV) was measured every 2 days. Tumor volume was calculated in accordance with the formula: V = 0.5AB2, in which A and B, respectively, represent the long and short diameter of tumor measured with vernier caliper. Mice were sacrificed 2 weeks later. Tumor inhibition is calculated as [(tumor volume of control group ‐ tumor volume of experimental group)/tumor volume of control group] ×100%.
Tumor tissues were separated into two parts: One part was rapidly put into liquid nitrogen and frozen for western blot and caspase activity analysis, and the other part was fixed with 10% formaldehyde and embedded by paraffin for immunohistochemical analysis.
Immunohistochemistry
The paraffin‐embedded tumor tissue blocks were processed to obtain 5‐μm‐thick histologic sections. Samples were incubated with primary antibodies, anti‐cleaved caspase‐3 antibody (Abgent, USA), and anti‐ubiquitin antibody (Santa Cruz Biotechnology, USA) overnight at 4 °C, followed by staining with biotinylated antibody coupled to HRP‐linked streptavidin/biotin complex (Santa Cruz Biotechnology, USA). Then, the samples were visualized by DAB staining.
Statistical analysis
Data were shown as mean ± SD from three independent experiments with significance calculated by Students’ t‐test. The P value < 0.05 was considered as statistically significant.
Results
PSMD7 expression in ESCC cell lines
To explore PSMD7 expression level in cancer cells, the relative protein expression of PSMD7 was examined in nonmalignant esophageal cell line Het‐1A and ESCC cell lines. Results showed that PSMD7 was highly expressed in three ESCC cell lines and HeLa 299 cell line (Fig. 1A). The expression of PSMD7 showed statistical difference between the three ESCC cell lines and Het‐1A cells. The protein level of PSMD7 was higher changed by ~ 3.2 times in EC9706 cells (P < 0.05), ~ 1.96 times in EC1 cells (P < 0.05), and ~ 2.24 times in Eca109 cells (P < 0.05) than Het‐1A cells (Fig. 1A). The protein expression level of PSMD7 in EC9706 cells was high and was employed in our subsequent studies. Therefore, we found PSMD7 is differently expressed in cancer cell lines.
Figure 1.
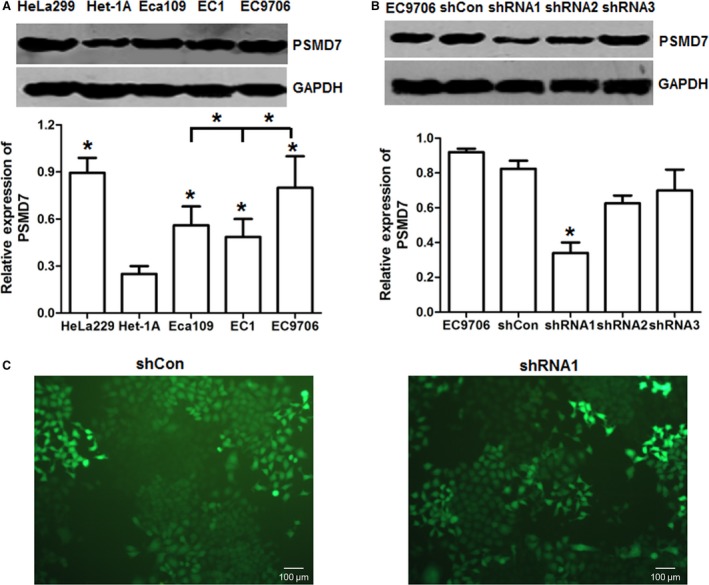
PSMD7 was overexpressed in esophageal squamous cell carcinoma (ESCC) cell lines. (A) Western blot results of PSMD7 in ESCC cell lines with GAPDH as internal control. (B) Lentivirus shRNA vectors targeting PSMD7 were constructed and used to infect EC9706 cells. The protein expression level of PSMD7 was examined by western blot 72 h after lentivirus transfection. *P < 0.05 by Student’ s t‐test. Error bars represent mean ± SD derived from three independent experiments. (C) The lentivirus‐transfected cells were selected by puromycin to obtain stable cell populations. The transfection efficiency was indicated by GFP fluorescent signal (×100).
PSMD7 knockdown inhibits proliferation and induces apoptosis in ESCC cell line
We have found that PSMD7 was highly expressed in EC9706 cells. To investigate affection by PSMD7 knockdown on malignant cell proliferation, lentivirus shRNA vectors targeting PSMD7 were constructed and used to infect EC9706 cells. The protein expression level of PSMD7 was decreased by 63% in shRNA1 lentivirus‐transfected EC9706 cells compared with the control (Fig. 1B), so PSMD7 shRNA1 lentivirus can inhibit the expression of PSMD7 in EC9706 cells. In the meantime, the lentivirus‐transfected cells were selected by puromycin to obtain stable knockdown cell populations (Fig. 1C). Together, we obtained transient and stable PSMD7 knockdown EC9706 cells.
Next, cell proliferation capability of parental EC9706, negative control shRNA‐transfected (shCon), and PSMD7‐targeted shRNA‐transfected (shPSMD7) EC9706 was explored by CCK‐8 assay. Knockdown of PSMD7 considerably decreased the cell viability with ~ 47% of shPSMD7 group compared to shCon group (P < 0.05) (Fig. 2A), suggesting that inhibition of PSMD7 decreased the survival of EC9706 cells. Whether PSMD7 knockdown in EC9706 cells had impacts on apoptosis and cell cycle distribution was investigated by flow cytometry. Results showed that inhibition of PSMD7 in EC9706 cells caused cell apoptosis with significantly increased numbers of early apoptosis cells and late apoptosis cells (P < 0.05) (Fig. 2B,C), but made no difference on cell cycle distribution (P > 0.05) (Fig. 2D). The caspase‐3 activity was quantified through detecting the fluorogenic response as a result of DEVD peptide cleavage (Fig. 2E). PSMD7 knockdown raised caspase‐3 activity compared with the control (P < 0.05), which was further confirmed by the increased cleavage of caspase‐3 in shPSMD7 group (Fig. 2F). As a marker for cell apoptosis, PARP is cleaved by caspase‐3 and facilitates disassembling of the cellular components. A prominent cleaved PARP band was detected in PSMD7 knockdown tumor cells, whereas no PARP cleavage band was observed in control groups (Fig. 2F). In conclusion, PSMD7 knockdown inhibits proliferation and induces apoptosis in ESCC cells.
Figure 2.
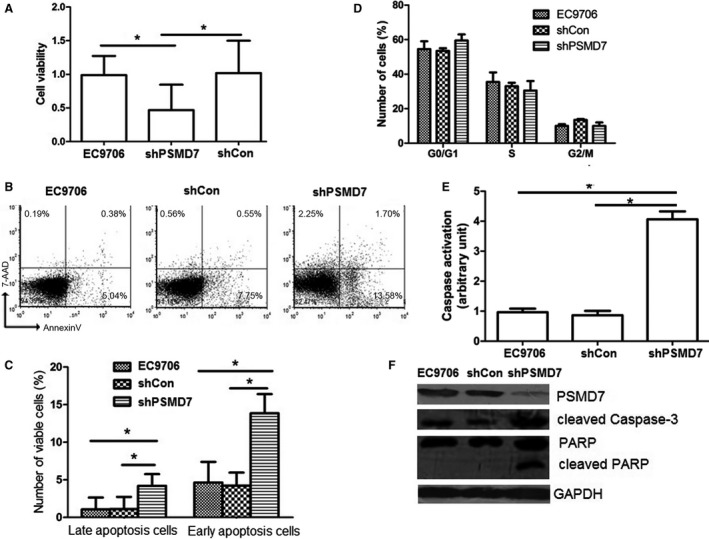
Effect of PSMD7 knockdown on proliferation, cell cycle, and apoptosis in EC9706 cells. (A) Cells were subjected to CCK‐8 assay for the determination of cell proliferation. shCon: EC9706 cells transfected with control lentivirus; shPSMD7: EC9706 cells transfected with shRNA1 lentivirus targeting PSMD7. *P < 0.05 by Student’ s t‐test. Error bars represent mean ± SD derived from three independent experiments. (B) One representative of flow cytometry scatter plots showed Annexin V‐FITC staining and 7‐AAD from three independent experiments. (C) The numbers represent the percentage of early apoptotic cells (lower right quadrant) and late apoptotic cells (upper right quadrant) are shown. (D) Cell cycle distribution data were shown in PSMD7 knockdown in C9706 cells. (E–F) Caspase activation assay (E) and immunoblot assay of cleaved by caspase‐3 and PARP (F) in EC9706 cells were performed. Results are presented as the mean ± SD based on three independent experiments. *P < 0.05 by Student’ s t‐test.
Decreased proteasomal activity after PSMD7 knockdown
As ubiquitin‐proteasome system plays an important role in apoptosis, western blot of ubiquitin from cell lysate showed that PSMD7 knockdown resulted in a remarkable increased level of ubiquitinated protein (Fig. 3A). Also, stronger fluorescence of ubiquitin and reduced proteasomal activity (P < 0.05) were observed in PSMD7 knockdown cells in comparison with control (Fig. 3B,C). So, the increased total level of ubiquitinated protein and decreased proteasomal activity supported that knockdown of PSMD7 was involved in the disruption of the ubiquitin‐proteasome pathway.
Figure 3.
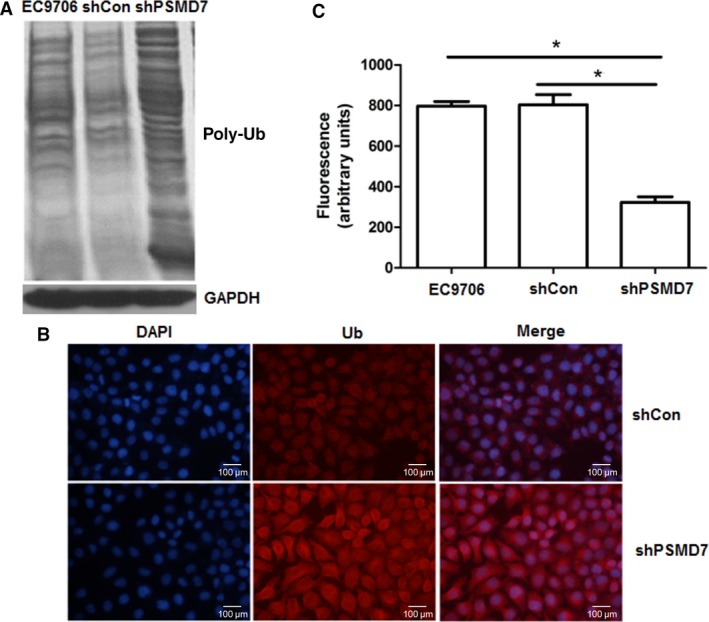
Knockdown of PSMD7 affected the ubiquitin‐proteasome pathway. (A) Western blots of ubiquitin from cell lysate showed the level of ubiquitinated protein in PSMD7 normal and knockdown condition. (B) Immunofluorescence of ubiquitin conjugates after lentivirus transfection (×200). (C) The proteolytic capability of 26S proteasome was measured by an in vitro peptide hydrolysis method using the β5‐selective fluorogenic substrate succinyl‐Leu‐Leu‐Val‐Tyr‐7‐amido‐4‐methylcoumarin. *P < 0.05 by Student’ s t‐test. Error bars represent mean ± SD derived from three independent experiments.
Effect of PSMD7 downregulation on the mTOR/p70S6K pathway
To explore the potential mechanisms underlying the inhibited proliferation and increased cell apoptosis due to PSMD7 inhibition, the protein expression levels of mTOR and p70S6K were investigated by western blot (Fig. 4A). Compared with the control groups, the expression levels of mTOR and p70S6K were reduced in shPSMD7 group, ~ 52.5% (P < 0.05) and ~ 71.3% (P < 0.05), respectively (Fig. 4B,C). Moreover, the protein level of p‐mTORSer2448 and p‐p70S6KThr421/Ser424 was reduced by ~ 60.0% (P < 0.05) and ~ 73.7% (P < 0.05), respectively (Fig. 4B,C), suggesting the suppressed activity of mTOR/p70S6K pathway in PSMD7 knockdown cells. Moreover, PSMD7 was overexpressed in cells of Het‐1A by transfection with the constructed vector pEGFP‐N1‐PSMD7 (Fig. 4D). Results showed that the protein level of PSMD7 was increased to ~ 1.45 times compared with control (P < 0.05) (Fig. 4E,F). The protein level of p‐mTORSer2448 and p‐p70S6KThr421/Ser424 in PSMD7 overexpressing Het‐1A cells was raised to ~ 1.26 times (P < 0.05) and ~ 1.84 times (P < 0.05), respectively, compared with controls (Fig. 4G).
Figure 4.
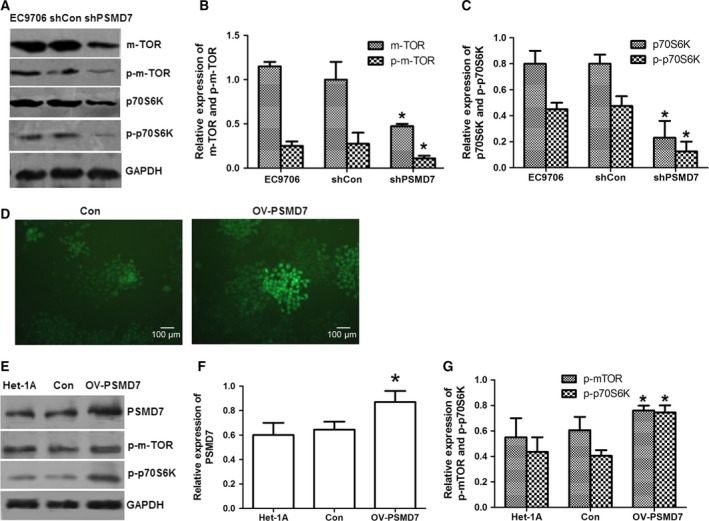
Effects of PSMD7 on the activity of mTOR/p70S6K pathway in EC9706 cells and Het‐1A cells. (A) Western blot results of mTOR, p‐mTOR, p70S6K, and p‐p70S6K with GAPDH as internal control. (B and C) The expression of mTOR, p‐mTOR, p70S6K, and p‐p70S6K was analyzed by densitometry using ImageJ software and plotted from three independent experiments. (D) PSMD7 was overexpressed in Het‐1A cells by transfection with the constructed vector pEGFP‐N1‐PSMD7 (OV‐PSMD7) using empty vector as control (Con). The green fluorescence indicated the transfected cells (×100). (E) Western blot results of PSMD7, p‐mTOR, and p‐p70S6K with GAPDH as internal control. (F and G) The expression of PSMD7, p‐mTOR, and p‐p70S6K was analyzed by densitometry using ImageJ software and plotted from three independent experiments. *P < 0.05 by Student’ s t‐test.
Inhibited tumor growth in vivo by PSMD7 knockdown
As the apoptosis was enhanced and the proteasomal activity was inhibited in the in vitro experiments, the potential effect of PSMD7 knockdown on the inhibition of ESCC tumorigenesis in vivo was subsequently tested. The tumor‐bearing mice models were established using EC9706 cells and PSMD7 shRNA lentivirus infected EC9706 cells, and tumor volumes were monitored every 2 days. Tumors were excised at the end of the experiment (Fig. 5A). Tumor volumes were calculated and plotted (Fig. 5B). Our results showed that the growth rate of xenograft in shPSMD7 group was decreased by comparison with control groups (P < 0.05), with tumor growth inhibition rate (TGI) of shPSMD7/shCon as 58.3% at day 14 after inoculation (Fig. 5B). Summary, PSMD7 knockdown induces impaired tumor growth in vivo.
Figure 5.
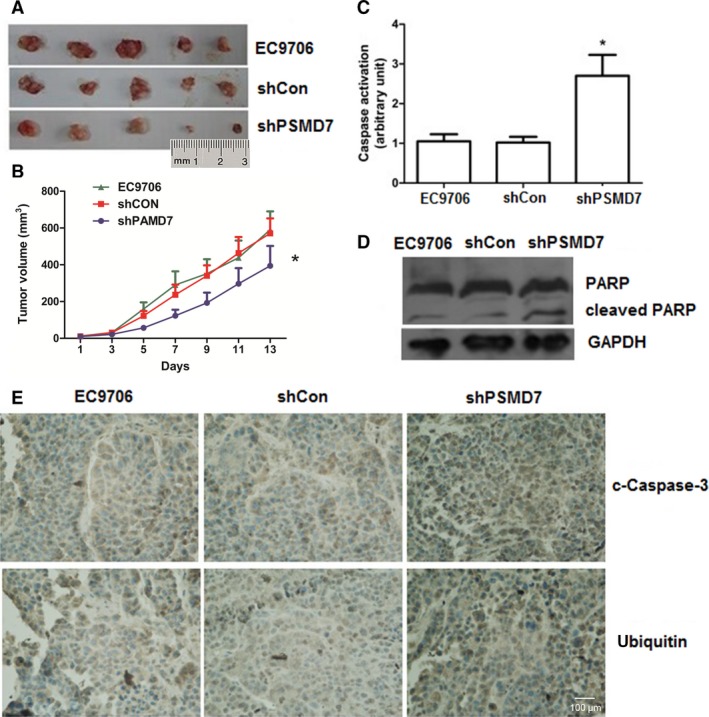
PSMD7 knockdown regulates tumor growth in an ESCC mouse model. (A) Tumor tissue retrieved from ESCC xenografts (n = 5). (B) The mean tumor volume of ESCC xenografts was calculated and plotted. (C–E) Caspase activation (C), western blot for PARP cleavage (D), and immunohistochemical staining (E) of tumor section obtained from mice are shown. Error bars represent mean ± SD derived from three independent experiments. **P < 0.05.
As shown in Fig. 5C, the activity of caspase‐3 in shPSMD7 group was higher than that in control groups (P < 0.05). Meanwhile, it was associated with increased expression of cleaved PARP and stronger staining of cleaved caspase‐3 after PSMD7 inhibition (Fig. 5D), suggesting that cell apoptosis was induced by PSMD7 knockdown in vivo. The expression intensity of ubiquitin in xenograft of nude mice was analyzed by immunohistochemical assay. High density of ubiquitinated protein was found in shPSMD7 group (Fig. 5E), which confirmed the inhibitory role of PSMD7 in ubiquitin‐proteasome system in vivo.
Based on the inhibited activity of the mTOR pathway in PSMD7 knockdown ESCC cell line, whether the anti‐cancer effect of PSMD7 knockdown in vivo through mTOR/p70S6K inhibition was evaluated. The expression of mTOR and p70S6K, and the phosphorylation status of them in xenograft were examined (Fig. 6A,B). The expression of PSMD7 is significantly reduced in shPSMD7 group (P < 0.05). PSMD7 knockdown blocked the expression levels of mTOR and p70S6K in vivo, reduced by 33% and 37%, respectively (P < 0.05) (Fig. 6C,D). The protein levels of p‐mTORSer2448 and p‐p70S6KThr421/Ser424 were also decreased by 18% (P < 0.05) and 36% (P < 0.05), respectively (Fig. 6C,D). These data suggest that inhibition of PSMD7 suppressed the activity of mTOR/p70S6K in vivo.
Figure 6.
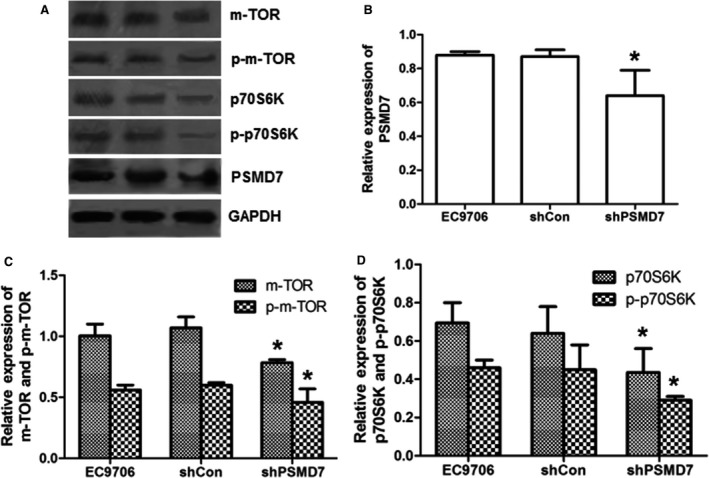
Effect of PSMD7 knockdown on the activity of mTOR/p70S6K pathway in vivo. (A) Western blot results of PSMD7, mTOR, p‐mTOR, p70S6K, and p‐p70S6K normalized with GAPDH expression. (B–D) The expression of PSMD7, mTOR, p‐mTOR, p70S6K, and p‐p70S6K was analyzed by densitometry using ImageJ software and plotted from three independent experiments. *P < 0.05 by Student’ s t‐test.
Discussion
In this study, we found the high expression of PSMD7 and elevated proteasomal activity in ESCC cells. Recent studies have shown that siRNA targeting PSMD7 increased fluorescence level of an ubiquitin‐dependent cytoplasmic GFP reporter similar to proteasome inhibitor MG132 treatment leading to cell death 25. In our study, we found that PSMD7 have the similar cellular effect similar with proteasome inhibitor. It is interesting that we, at the first time, reported that PSMD7 knockdown resulted in decreased proliferation and enhanced apoptosis in ESCC cell lines. Moreover, PSMD7 knockdown cells had higher caspase‐3 activity and strengthened the cleavage of caspase‐3 compared with control cells (ineffective shRNA). Also, the cleavage of PARP band was detected in PSMD7 knockdown tumor cells, but not in control groups. These data provided strong evidences that PSMD7 impairment induced apoptosis of ESCC cell lines.
Structural research on PSMD7 revealed that the JAMM domain is a metal‐free structure without isopeptidase activity 26, suggesting that it may not incise the poly‐ubiquitinated protein chain directly. However, PSMD7 was not detected as an individual subunit but presented in heteropolymer, in which it occupied a central position in an independent subcomplex isolated from 19S proteasome 27. The depletion of PSMD7 may affect the assembly of 19S lid subcomplex and eventually decrease the ability of recognition to poly‐ubiquitinated chain and cleavage of the degraded protein from ubiquitinated chain. Here, we showed that a remarkable increase in ubiquitinated protein levels, elevated fluorescence for ubiquitin, and reduced proteasomal activity were observed in PSMD7 knockdown ESCC cells, suggesting that PSMD7 participated in the maintenance of normal proteasome function. It may also suggest that the decreased proteasome activity was due to the abnormal function of 19S proteasome caused by PSMD7 knockdown.
The mTOR/p70S6K pathway exerts critical function in cell proliferation and is regulated by other protein 28, 29. In ESCC cell lines, mTOR/p70S6K pathway is activated via phosphorylation at multiple serine and threonine residues 30. MG132 has been shown to cause the dephosphorylation of p70S6K/mTOR and induce cell autophagy 22. Inhibition of PSMD7 not only reduced the expression of mTOR and p70S6K, but also decreased the expression of p‐mTORSer2448 and p‐p70S6KThr421/Ser424, whereas the augmented expression of phosphorylation of mTOR and p70S6K was detected after PSMD7 overexpression in Het‐1A cells. Our results showed that the effect of PSMD7 depletion in ESCC cell lines was similar to MG132 treatment, but the decreased phosphorylation residue of p70S6K was different, which are Thr421/Ser424 in PSMD7 knockdown and Thr389 in MG132 treatment. Our results indicated that the cellular function exhibited by PSMD7 inhibition in ESCC cell lines possibly had close relationship to the mTOR/p70S6K pathway.
In ESCC xenografts bearing mice with repression of PSMD7, we also proved that in vivo ability of PSMD7 inhibition suppresses tumor growth and induces apoptosis. Knockdown of PSMD7 deferred tumor growth in comparison with controls. The in vivo experimental results were consistent with those of the in vitro experiments, as the caspase activity and the level of cleaved PARP and ubiquitinated protein were significantly elevated after PSMD7 inhibition. Furthermore, PSMD7 inhibition restrained the mTOR pathway by reducing the phosphorylation of mTOR at Ser2448 and p70S6K at Thr421/Ser424 in vivo. Thus, we concluded that the suppression impact of PSMD7 on ESCC xenograft model may be via impeding the mTOR/p70S6K pathway.
From the above discussion, conclusion can be reached that PSMD7 downregulation contributes to inhibited proteasomal function and induced apoptosis in ESCC cell lines, which is likely to tightly associate with the mTOR/p70S6K pathway. Hence, operation of PSMD7 and mTOR/p70S6K pathway is a promising candidate target for therapy of ESCC.
Author contributions
KS and DG conceived and designed the project, R‐lZ, KS, and LY acquired the data, KS and J‐zZ analyzed and interpreted the data, as well as wrote the article.
Acknowledgements
Our work was supported by grants from the College and University Major Scientific Research Program of Henan Educational Committee, China (no. 16A310008), and Scientific and Technological Project of Henan Province, China (nos. 162102310047 and 201601027).
References
- 1. van Hagen P, Hulshof MC, van Lanschot JJ, Steyerberg EW, van Berge Henegouwen MI, Wijnhoven BP, Richel DJ, Nieuwenhuijzen GA, Hospers GA, Bonenkamp JJ et al (2012) Preoperative chemoradiotherapy for esophageal or junctional cancer. N Engl J Med 366, 2074–2084. [DOI] [PubMed] [Google Scholar]
- 2. Li M, Wan X, Wang Y, Sun Y, Yang G and Wang L (2017) Time trends of esophageal and gastric cancer mortality in China, 1991‐2009: an age‐period‐cohort analysis. Sci Rep 7, 6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goldberg AL (2012) Development of proteasome inhibitors as research tools and cancer drugs. J Cell Biol 199, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen D and Dou QP (2010) The ubiquitin‐proteasome system as a prospective molecular target for cancer treatment and prevention. Curr Protein Pept Sci 11, 459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Orlowski RZ and Kuhn DJ (2008) Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res 14, 1649–1657. [DOI] [PubMed] [Google Scholar]
- 6. Williams S, Pettaway C and Song R (2003) Differential effects of the proteasome inhibitor bortezomib on apoptosis and angiogenesis in human prostate tumor xenografts. Mol Cancer Ther 2, 835–843. [PubMed] [Google Scholar]
- 7. Caravita T, De Fabritiis P and Palumbo A (2006) Bortezomib: efficacy comparisons in solid tumors and hematologic malignancies. Nat Clin Pract Oncol 3, 374–387. [DOI] [PubMed] [Google Scholar]
- 8. Wright JJ (2010) Combination therapy of bortezomib with novel targeted agents: an emerging treatment strategy. Clin Cancer Res 16, 4094–40104. [DOI] [PubMed] [Google Scholar]
- 9. Milano A, Perri F and Caponigro F (2009) The ubiquitin‐proteasome system as a molecular target in solid tumors: an update on bortezomib. Onco Targets Ther 2, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Daviet L and Colland F (2008) Targeting ubiquitin specific proteases for drug discovery. Biochimie 90, 270–283. [DOI] [PubMed] [Google Scholar]
- 11. D'Arcy P and Linder S (2012) Proteasome deubiquitinases as novel targets for cancer therapy. Int J Biochem Cell Biol 44, 1729–1738. [DOI] [PubMed] [Google Scholar]
- 12. Verma R, Aravind L, Oania R, McDonald WH, Yates JR III, Koonin EV and Deshaies RJ (2002) Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 298, 611–615. [DOI] [PubMed] [Google Scholar]
- 13. Matyskiela ME, Lander GC and Martin A (2013) Conformational switching of the 26S proteasome enables substrate degradation. Nat Struct Mol Biol 20, 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen L and Madura K (2005) Increased proteasome activity, ubiquitin‐conjugating enzymes, and eEF1A translation factor detected in breast cancer tissue. Cancer Res 65, 5599–5606. [DOI] [PubMed] [Google Scholar]
- 15. Arlt A, Bauer I, Schafmayer C, Tepel J, Müerköster SS, Brosch M, Röder C, Kalthoff H, Hampe J, Moyer MP et al (2009) Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2‐related factor 2 (Nrf2). Oncogene 28, 3983–3996. [DOI] [PubMed] [Google Scholar]
- 16. Rho JH, Qin S, Wang JY and Roehrl MH (2008) Proteomic expression analysis of surgical human colorectal cancer tissues: up‐regulation of PSB7, PRDX1, and SRP9 and hypoxic adaptation in cancer. J Proteome Res 7, 2959–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shu XO, Long J, Lu W, Li C, Chen WY, Delahanty R, Cheng J, Cai H, Zheng Y, Shi J et al (2012) Novel genetic markers of breast cancer survival identified by a genome‐wide association study. Cancer Res 72, 1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luise C, Capra M, Donzelli M, Mazzarol G, Jodice MG, Nuciforo P, Viale G, Di Fiore PP and Confalonieri S (2011) An atlas of altered expression of deubiquitinating enzymes in human cancer. PLoS One 6, e15891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang CN, Huang SP, Pao JB, Chang TY, Lan YH, Lu TL, Lee HZ, Juang SH, Wu PP, Pu YS et al (2012) Genetic polymorphisms in androgen receptor‐binding sites predict survival in prostate cancer patients receiving androgen‐deprivation therapy. Ann Oncol 23, 707–713. [DOI] [PubMed] [Google Scholar]
- 20. Niu H, Jiang H, Cheng B, Li X, Dong Q, Shao L, Liu S and Wang X (2012) Stromal proteome expression profile and muscle‐invasive bladder cancer research. Cancer Cell Int 12, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Klappan AK, Hones S, Mylonas I and Brüning A (2012) Proteasome inhibition by quercetin triggers macroautophagy and blocks mTOR activity. Histochem Cell Biol 137, 25–36. [DOI] [PubMed] [Google Scholar]
- 22. Choi CH, Lee BH, Ahn SG and Oh SH (2012) Proteasome inhibition‐induced p38 MAPK/ERK signaling regulates autophagy and apoptosis through the dual phosphorylation of glycogen synthase kinase 3β. Biochem Biophys Res Commun 418, 759–764. [DOI] [PubMed] [Google Scholar]
- 23. Hou G, Xue L, Lu Z, Fan T, Tian F and Xue Y (2007) An activated mTOR/p70S6K signaling pathway in esophageal squamous cell carcinoma cell lines and inhibition of the pathway by rapamycin and siRNA against mTOR. Cancer Lett 253, 236–248. [DOI] [PubMed] [Google Scholar]
- 24. Gorbea C, Pratt G, Ustrell V, Bell R, Sahasrabudhe S, Hughes RE and Rechsteiner M (2010) A protein interaction network for Ecm29 links the 26 S proteasome to molecular motors and endosomal components. Biol Chem 285, 31616–31633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sowa ME, Bennett EJ, Gygi SP and Harper JW (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sanches M, Alves BS, Zanchin NI and Guimarães BG (2007) The crystal structure of the human Mov34 MPN domain reveals a metal‐free dimer. J Mol Biol 370, 846–855. [DOI] [PubMed] [Google Scholar]
- 27. Sharon M, Taverner T, Ambroggio XI, Deshaies RJ and Robinson CV (2006) Structural organization of the 19S proteasome lid: insights from MS of intact complexes. PLoS Biol 4, e267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fenton TR and Gout IT (2011) Functions and regulation of the 70 kDa ribosomal S6 kinases. Int J Biochem Cell Biol 43, 47–59. [DOI] [PubMed] [Google Scholar]
- 29. Gong F, Hou G, Liu H and Zhang M (2015) Peroxiredoxin 1 promotes tumorigenesis through regulating the activity of mTOR/p70S6K pathway in esophageal squamous cell carcinoma. Med Oncol 32, 455. [DOI] [PubMed] [Google Scholar]
- 30. Ragan TJ, Ross DB, Keshwani MM and Harris TK (2008) Expression, purification, and characterization of a structurally disordered and functional C‐terminal autoinhibitory domain (AID) of the 70 kDa 40S ribosomal protein S6 kinase‐1 (S6K1). Protein Expr Purif 57, 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]