

Abstract
BACKGROUND
Blinatumomab, a bispecific monoclonal antibody construct that enables CD3-positive T cells to recognize and eliminate CD19-positive acute lymphoblastic leukemia (ALL) blasts, was approved for use in patients with relapsed or refractory B-cell precursor ALL on the basis of single-group trials that showed efficacy and manageable toxic effects.
METHODS
In this multi-institutional phase 3 trial, we randomly assigned adults with heavily pretreated B-cell precursor ALL, in a 2:1 ratio, to receive either blinatumomab or standard-of-care chemotherapy. The primary end point was overall survival.
RESULTS
Of the 405 patients who were randomly assigned to receive blinatumomab (271 patients) or chemotherapy (134 patients), 376 patients received at least one dose. Overall survival was significantly longer in the blinatumomab group than in the chemotherapy group. The median overall survival was 7.7 months in the blinatumomab group and 4.0 months in the chemotherapy group (hazard ratio for death with blinatumomab vs. chemotherapy, 0.71; 95% confidence interval [CI], 0.55 to 0.93; P = 0.01). Remission rates within 12 weeks after treatment initiation were significantly higher in the blinatumomab group than in the chemotherapy group, both with respect to complete remission with full hematologic recovery (34% vs. 16%, P<0.001) and with respect to complete remission with full, partial, or incomplete hematologic recovery (44% vs. 25%, P<0.001). Treatment with blinatumomab resulted in a higher rate of event-free survival than that with chemotherapy (6-month estimates, 31% vs. 12%; hazard ratio for an event of relapse after achieving a complete remission with full, partial, or incomplete hematologic recovery, or death, 0.55; 95% CI, 0.43 to 0.71; P<0.001), as well as a longer median duration of remission (7.3 vs. 4.6 months). A total of 24% of the patients in each treatment group underwent allogeneic stem-cell transplantation. Adverse events of grade 3 or higher were reported in 87% of the patients in the blinatumomab group and in 92% of the patients in the chemotherapy group.
CONCLUSIONS
Treatment with blinatumomab resulted in significantly longer overall survival than chemotherapy among adult patients with relapsed or refractory B-cell precursor ALL. (Funded by Amgen; TOWER ClinicalTrials.gov number, NCT02013167.)
The Prognosis for Adults with Newly diagnosed acute lymphoblastic leukemia (ALL) has improved over the past three decades. With the use of intensive chemotherapy regimens, complete remission rates are 85 to 90% and long-term survival rates are 30 to 50%.1–4 Still, most adults with B-cell precursor ALL will have a relapse and will die from complications of resistant disease or associated treatment. Among adults with relapsed or refractory ALL, remission rates are 18 to 44% with the use of standard salvage chemotherapy, but the duration of remission is typically short.5–10 A major goal in this population is to induce remission with sufficient duration to prepare for stem-cell transplantation.11 The median overall survival among patients with relapsed or refractory ALL ranges from 2 to 6 months, and 3-to-5-year survival rates are less than 10%.7–10,12 More effective treatment is needed for relapsed and refractory ALL in adults.
The B-lineage surface antigen CD-19 is expressed on the surface of more than 90% of B-cell precursor ALL blasts.13 Blinatumomab (Blincyto, Amgen) is a bispecific T-cell engager antibody construct. Blinatumomab binds simultaneously to CD3-positive cytotoxic T cells and to CD19-positive B cells, which allows the patient’s endogenous T cells to recognize and eliminate CD19-positive ALL blasts.14–16
Single-group trials have shown the efficacy and safety of blinatumomab in the treatment of heavily pretreated Philadelphia chromosome (Ph)–negative relapsed or refractory B-cell precursor ALL.17,18 In a pivotal, multicenter, single-group, phase 2 trial of blinatumomab, the rate of complete remission with complete or partial hematologic recovery was 43%, and the median overall survival was 6.1 months.18 We report here the results of a multinational, randomized, phase 3 trial that compared blinatumomab with standard chemotherapy in the treatment of patients with relapsed or refractory ALL.
METHODS
TRIAL DESIGN
In this prospective, randomized, phase 3 trial, investigators at 101 centers in 21 countries enrolled adults (18 years of age or older) with Ph-negative B-cell precursor ALL in any of the following stages: refractory to primary induction therapy or to salvage with intensive combination chemotherapy, first relapse with the first remission lasting less than 12 months, second or greater relapse, or relapse at any time after allogeneic stem-cell transplantation. Additional eligibility criteria included more than 5% blasts in the bone marrow and an Eastern Cooperative Oncology Group performance status of 2 or less (on a 5-point scale, with higher numbers indicating greater disability). Key exclusion criteria were other active cancers, a clinically relevant pathologic condition of the central nervous system, isolated extramedullary disease, autoimmune disease, acute graft-versus-host disease (GVHD) of grade 2 or higher or active chronic GVHD, allogeneic stem-cell transplantation within 12 weeks before randomization, autologous stem-cell transplantation within 6 weeks before randomization, chemotherapy or radiotherapy within 2 weeks before randomization, use of immunotherapy within 4 weeks before randomization, or ongoing use of investigational treatment.
TRIAL OVERSIGHT
All patients provided written informed consent. The trial was designed by Amgen in collaboration with the trial investigators. The trial protocol (including the statistical analysis plan), which is available with the full text of this article at NEJM.org, was approved by the investigational review board or independent ethics committee at each trial center. The first two authors and the last author prepared the first draft of the manuscript, with assistance from professional medical writers who were funded by Amgen. All the authors contributed revisions and had access to the data. All the authors vouch for the integrity and completeness of the data and for the fidelity of the trial to the protocol. Two authors who are employees of Amgen conducted the statistical analyses and contributed to the manuscript. An independent data and safety monitoring committee consisting of two clinicians and one biostatistician met regularly to review safety and efficacy data, which were provided by an independent statistician.
TREATMENTS
Eligible patients were randomly assigned, in a 2:1 ratio, with the use of an interactive voice-response system to receive open-label treatment with either blinatumomab or standard chemotherapy (Fig. S1 in the Supplementary Appendix, available at NEJM.org). Randomization was stratified according to age (<35 vs. ≥35 years), previous salvage therapy (yes vs. no), and previous allogeneic stem-cell transplantation (yes vs. no). Patients in each treatment group were to receive up to two cycles of induction therapy; in addition, in each group, patients in morphologic remission (≤5% bone marrow blasts) were to receive up to three cycles of consolidation therapy, and patients in continued morphologic remission were to receive up to 12 months of maintenance therapy.
Induction and consolidation treatments with blinatumomab were administered in 6-week cycles; in each cycle, patients received treatment for 4 weeks (9 μg per day during week 1 of induction cycle 1 and 28 μg per day thereafter, by continuous infusion) and no treatment for 2 weeks. Maintenance treatment with blinatumomab was given as a 4-week continuous infusion every 12 weeks. Patients in the blinatumomab group who had high tumor load during screening were to receive dexamethasone before the start of treatment to prevent the cytokine release syndrome (Table S1 in the Supplementary Appendix). All patients in the blinatumomab group were to receive dexamethasone before their dose of blinatumomab to prevent infusion reactions (Table S1 in the Supplementary Appendix) and were to receive intrathecal prophylaxis for central nervous system disease according to institutional or national guidelines. Interruption or discontinuation of the dose of blinatumomab was required if neurologic events or other selected adverse events occurred (further details are provided in the Dose Modification section in the Supplementary Appendix); dose adjustment was permitted for patients receiving standard chemotherapy but was not required for specific events. Patients in the chemotherapy group received the investigator’s choice of one of the following four regimens: fludarabine, high-dose cytosine arabinoside, and granulocyte colony-stimulating factor with or without anthracycline; a high-dose cytosine arabinoside–based regimen; a high-dose methotrexate-based regimen; or a clofarabine-based regimen (Table S2 in the Supplementary Appendix). Protocol-specified therapy could be discontinued at any time after the first treatment cycle and the patient could subsequently undergo stem-cell transplantation if the investigator determined that such actions were in the patient’s best interest.
ASSESSMENTS
Complete remission was defined as 5% or less bone marrow blasts and no evidence of disease and was further characterized according to the extent of recovery of peripheral blood counts as follows: complete remission with full recovery (platelet count of >100,000 per microliter and absolute neutrophil count of >1000 per microliter), complete remission with partial recovery (platelet count of >50,000 per microliter and absolute neutrophil count of >500 per microliter), or complete remission with incomplete recovery (platelet count of >100,000 per microliter or absolute neutrophil count of >1000 per microliter). Minimal residual disease was assessed at one central laboratory for trial centers in the United States and Canada (64 patients) with the use of multicolor flow cytometry and at another central laboratory for other trial centers (341 patients) with the use of real-time quantitative polymerase chain reaction of clonal immunoglobulin or T-cell receptor gene rearrangements with an assay sensitivity of at least 0.0001.19,20 Lumbar puncture was performed during each cycle to evaluate leukemic involvement of the central nervous system. Adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), version 4.0. Serious adverse events included events that were fatal or life-threatening, required or prolonged hospitalization, resulted in disability or incapacity, or resulted in a congenital anomaly or birth defect or were other medically important serious events such as an overdose. Adverse events of interest were identified by a steering committee.
STATISTICAL ANALYSIS
The primary end point was overall survival, which was defined as the time from randomization to death from any cause. Key secondary end points included achievement of complete remission with full hematologic recovery within 12 weeks after initiation of treatment; achievement of complete remission with full, partial, or incomplete hematologic recovery within 12 weeks after initiation of treatment; and event-free survival (defined as the time from randomization until relapse after achieving a complete remission with full, partial, or incomplete hematologic recovery, or death). Other secondary end points included the duration of complete remission, minimal residual disease remission (defined as a minimal residual disease level below 0.0001), the rate of allogeneic stem-cell transplantation, and adverse event rates. Final results for key secondary end points were to be tested in a hierarchical manner if the results of the primary end point were found to be significant. We calculated that an enrollment target of 400 patients and a total of approximately 330 deaths would give the trial approximately 85% power to detect a hazard ratio for death of 0.70 in the blinatumomab group as compared with chemotherapy at a two-sided alpha level of 0.05; this calculation was based on an assumed median overall survival of 4.2 months in the chemotherapy group8,9 and a 10% dropout rate. Two interim analyses were planned to assess a survival benefit when 50% and 75% of the planned deaths for the final analysis had been observed. The O’Brien–Fleming stopping boundary at the time of each interim analysis was calculated with the use of a Lan–DeMets alpha-spending function.21,22
Efficacy analyses included all patients who underwent randomization (intention-to-treat population). Safety analyses and efficacy sensitivity analyses included all patients who received at least one dose of trial treatment (as-treated population). Time-to-event estimates were calculated with the use of the Kaplan–Meier method, and treatment groups were compared by means of two-sided stratified log-rank tests. The treatment effect was expressed as a hazard ratio with a 95% confidence interval, which was estimated with the use of a stratified Cox regression model. Remission rates were compared with the use of a stratified two-sided Cochran–Mantel–Haenszel test.
RESULTS
TRIAL POPULATION AND TREATMENT
Patients were enrolled from January 2014 through September 2015. After 75% of the total number of planned deaths for the final analysis had occurred, the independent data and safety monitoring committee recommended that the trial be stopped early because of benefit observed with blinatumomab therapy. The data cutoff date was January 4, 2016. Of the 405 patients who underwent randomization, 376 received open-label trial treatment (267 of 271 patients [99%] received blinatumomab and 109 of 134 patients [81%] received standard chemotherapy); less than 1% of the 271 patients in the blinatumomab group and 16% of the 134 patients in the chemotherapy group withdrew consent before receiving treatment (Fig. S2 in the Supplementary Appendix). A total of 22 patients (8%) in the blinatumomab group and no patients in the chemotherapy group were continuing trial treatment at the time of the analysis. The chemotherapy regimen was fludarabine, high-dose cytosine arabinoside, and granulocyte colony-stimulating factor with or without anthracycline for 49 patients (45%); a high-dose cytosine arabinoside–based regimen for 19 patients (17%); a high-dose methotrexate-based regimen for 22 patients (20%); and a clofarabine-based regimen for 19 patients (17%). The two treatment groups had similar demographic and disease characteristics at baseline when all patients who underwent randomization were assessed (Table 1) as well when patients who did not receive the trial treatment were excluded (Table S3 in the Supplementary Appendix).
Table 1.
Demographic and Baseline Clinical Characteristics of the Patients.*
| Characteristic | Blinatumomab Group (N=271) |
Chemotherapy Group (N = 134) |
|---|---|---|
| Age — yr | ||
| Mean | 40.8 ± 17.1 | 41.1 ± 17.3 |
| Range | 18–80 | 18–78 |
| Male sex — no. (%) | 162 (59.8) | 77 (57.5) |
| Race — no. (%)† | ||
| White | 228 (84.1) | 112 (83.6) |
| Asian | 19 (7.0) | 9 (6.7) |
| Black | 5 (1.8) | 3 (2.2) |
| Other | 19 (7.0) | 10 (7.5) |
| Hispanic ethnic group — no. (%) | 26 (9.6) | 11 (8.2) |
| Geographic region — no. (%) | ||
| Europe | 180 (66.4) | 85 (63.4) |
| United States or Canada | 41 (15.1) | 23 (17.2) |
| Rest of world | 50 (18.5) | 26 (19.4) |
| ECOG performance status — no. (%)‡ | ||
| 0 | 96 (35.4) | 52 (38.8) |
| 1 | 134 (49.4) | 61 (45.5) |
| 2 | 41 (15.1) | 20 (14.9) |
| Missing data | 0 | 1 (07) |
| Key trial inclusion criteria — no. (%) | ||
| Disease refractory to primary therapy or salvage therapy | 115 (42.4) | 54 (40.3) |
| First relapse, with duration of first remission <12 mo | 76 (28.0) | 37 (27.6) |
| Untreated second or greater relapse§ | 32 (11.8) | 16 (11.9) |
| Relapse after allogeneic stem-cell transplantation§ | 46 (17.0) | 27 (20.1) |
| Not specified | 2 (0.7) | 0 |
| Previous allogeneic stem-cell transplantation — no. (%) | ||
| Yes | 94 (34.7) | 46 (34.3) |
| No | 176 (64.9) | 87 (64.9) |
| Unknown | 1 (0.4) | 1 (07) |
| Salvage-treatment phase — no. (%) | ||
| First | 114 (42.1) | 65 (48.5) |
| Second | 91 (33.6) | 43 (32.1) |
| Third | 45 (16.6) | 16 (11.9) |
| Fourth | 14 (5.2) | 5 (3.7) |
| Fifth or later | 7 (2.6) | 5 (3.7) |
| Maximum central or local bone marrow blasts — no. (%) | ||
| >5 to <10% | 9 (3.3) | 7 (5.2) |
| 10 to <50% | 60 (22.1) | 23 (17.2) |
| ≥50% | 201 (74.2) | 104 (77.6) |
| Unknown | 1 (0.4) | 0 |
| Peripheral blast count in blood (x 10−9/liter) | 4.4 ± 15.5 | 5.0 ± 15.7 |
Plus–minus values are means ±SD. There were no significant between-group differences in the characteristics evaluated at baseline. A complete summary of disease characteristics at baseline is provided in Table S3 in the Supplementary Appendix. Data are summarized for all patients who underwent randomization (intention-to-treat population). Percentages may not total 100 because of rounding.
Race was determined by the investigator.
The Eastern Cooperative Oncology Group (ECOG) performance status scale ranges from 0 to 5, with higher numbers indicating greater disability.
Patients who met this inclusion criterion met none of the above inclusion criteria.
The median number of treatment cycles was 2 (range, 1 to 9) in the blinatumomab group and 1 (range, 1 to 4) in the chemotherapy group. Consolidation therapy with trial treatment was administered to 32% of treated patients in the blinatumomab group and to 3% of treated patients in the chemotherapy group.
OVERALL SURVIVAL
For this prespecified interim analysis, 251 deaths were recorded. Overall survival was significantly longer in the blinatumomab group than in the chemotherapy group. The median overall survival was 7.7 months (95% confidence interval [CI], 5.6 to 9.6) in the blinatumomab group versus 4.0 months (95% CI, 2.9 to 5.3) in the chemotherapy group (hazard ratio for death, 0.71; 95% CI, 0.55 to 0.93; P = 0.01, which crossed the prespecified stopping boundary) (Fig. 1A), with a median duration of follow-up of 11.7 and 11.8 months, respectively. Overall survival curves for the blinatumomab and chemotherapy groups separated within 3 months and converged again between 15 and 18 months. Similar results were seen after exclusion of the data from patients who did not receive trial treatment (Fig. S3 in the Supplementary Appendix). When data were censored at the time of allogeneic stem-cell transplantation, the median overall survival was 6.9 months (95% CI, 5.3 to 8.8) in the blinatumomab group and 3.9 months (95% CI, 2.8 to 4.9) in the chemotherapy group (hazard ratio for death, 0.66; 95% CI, 0.50 to 0.88; P = 0.004) (Fig. 1B). The transplantation-censored overall survival curves for the blinatumomab and chemotherapy groups separated within 3 months and did not converge again. Estimated survival at 6 months among all patients who underwent randomization was 54% in the blinatumomab group and 39% in the chemotherapy group. The treatment benefit with respect to overall survival was generally consistent across key subgroups (Fig. 2A).
Figure 1. Efficacy End Points.
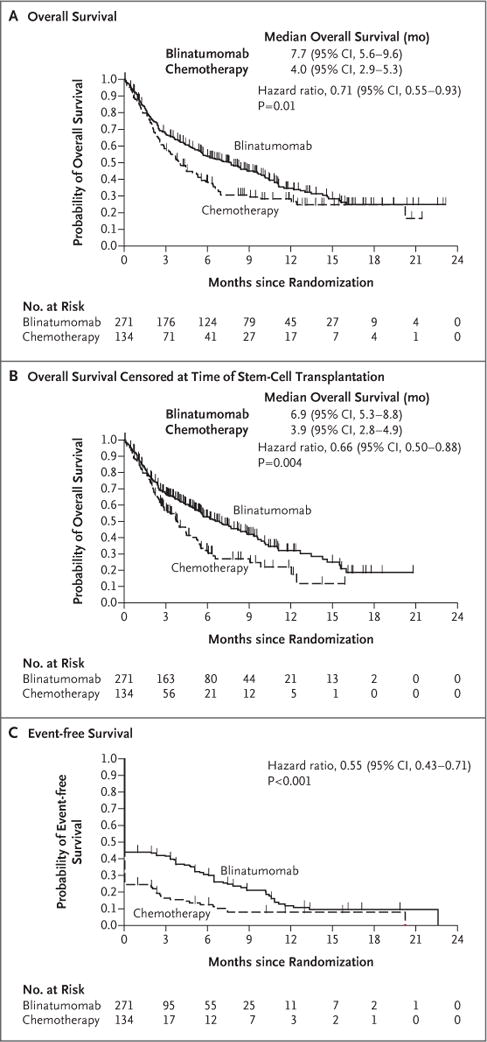
Panel A shows the probability of overall survival in the two groups. Overall survival was calculated as the time from randomization to death from any cause. The median duration of follow-up for overall survival was 11.7 months in the blinatumomab group and 11.8 months in the chemotherapy group. Panel B shows the probability of overall survival in the two groups (also calculated as the time from randomization to death from any cause) when data were censored at the time of allogeneic stem-cell transplantation. The median duration of follow-up for this analysis of overall survival was 7.0 months in the blinatumomab group and 6.0 months in the chemotherapy group. Panel C shows the probability of event-free survival, which was calculated as the time from randomization until relapse after complete remission with full, partial, or incomplete hematologic recovery, or death; patients who did not achieve a complete re-mission with full, partial, or incomplete hematologic recovery were assigned an event-free duration of 1 day. The median duration of follow-up for event-free survival was 7.8 months in the blinatumomab group and 10.2 months in the chemotherapy group. All three analyses were performed in the intention-to-treat population. P values were determined by means of stratified log-rank tests.
Figure 2. Subgroup Analyses.
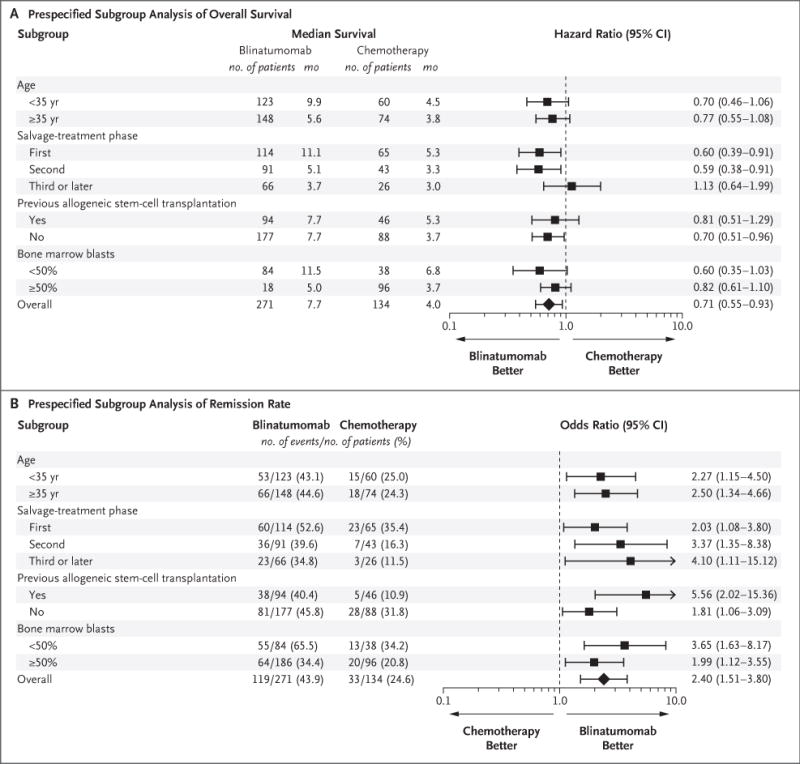
Panel A shows the results of an analysis of overall survival in prespecified subgroups of the intention-to-treat population that were defined according to baseline characteristics. Overall survival was calculated as the time from randomization to death from any cause. Panel B shows the results of an analysis of remission rates in prespecified subgroups of the intention-to-treat population that were defined according to baseline characteristics. The remission rate was defined as the percentage of patients who had complete hematologic remission with full, partial, or incomplete hematologic recovery by week 12. For both analyses, bone marrow blast data were from the central laboratory, if available; otherwise, data from the local laboratory were used. Central and local baseline results for bone marrow blasts were missing for one patient in the blinatumomab group.
REMISSION RATES
Remission rates within 12 weeks after treatment initiation were significantly higher in the blinatumomab group than in the chemotherapy group with respect to complete remission with full hematologic recovery (CR, 34% vs. 16%; P<0.001) and with respect to complete remission with full, partial, or incomplete hematologic recovery (CR, CRh, or CRi, 44% vs. 25%; P<0.001) (Table 2). Remission rates consistently favored blinatumomab over chemotherapy across key subgroups (Fig. 2B). Among the patients who had complete remission with full, partial, or incomplete hematologic recovery, 76% of the patients in the blinatumomab group and 48% in the chemotherapy group achieved a negative status (i.e., remission) for minimal residual disease (treatment difference, 28 percentage points; 95% CI, 9 to 47).
Table 2.
Best Hematologic Response Within 12 Weeks after Treatment Initiation.*
| Response Category | Blinatumomab Group (N = 271) |
Chemotherapy Group (N = 134) |
Treatment Difference (95% CI) |
P Value† | ||
|---|---|---|---|---|---|---|
| no. | % (95% CI) | no. | % (95% CI) | percentage points | ||
| Complete remission with full hematologic recovery | 91 | 33.6 (28.0–39.5) | 21 | 15.7 (10.0–23.0) | 17.9 (9.6–26.2) | <0.001 |
|
| ||||||
| Complete remission with full, partial, or incomplete hematologic recovery | 119 | 43.9 (37.9–50.0) | 33 | 24.6 (17.6–32.8) | 19.3 (9.9–28.7) | <0.001 |
|
| ||||||
| Complete remission with partial hematologic recovery | 24 | 8.9 (5.8–12.9) | 6 | 4.5 (1.7–9.5) | ||
|
| ||||||
| Complete remission with incomplete hematologic recovery | 4 | 1.5 (0.4–3.7) | 6 | 4.5 (1.7–9.5) | ||
Data are summarized for all patients who underwent randomization (intention-to-treat population). Complete remission was defined as 5% or less bone marrow blasts and no evidence of disease and was further characterized according to the extent of recovery of peripheral blood counts as follows: complete remission with full recovery (platelet count of >100,000 per microliter and absolute neutrophil count of >1000 per microliter), complete remission with partial recovery (platelet count of >50,000 per microliter and absolute neutrophil count of >500 per microliter), or complete remission with incomplete recovery (platelet count of >100,000 per microliter or absolute neutrophil count of >1000 per microliter).
Rates were compared with the use of a Cochran–Mantel–Haenszel test, with adjustment for the following stratification factors: age (<35 vs. ≥35 years), previous salvage therapy (yes vs. no), and previous allogeneic stem-cell transplantation (yes vs. no).
EVENT-FREE SURVIVAL AND DURATION OF REMISSION
Among the patients who had complete remission with full, partial, or incomplete hematologic recovery, the median duration of remission was 7.3 months (95% CI, 5.8 to 9.9) in the blinatumomab group and 4.6 months (95% CI, 1.8 to 19.0) in the chemotherapy group. For the key secondary efficacy end point of event-free survival, 6-month estimates were 31% in the blinatumomab and 12% in the chemotherapy group, and the hazard ratio for a relapse after achieving a complete remission with full, partial, or incomplete hematologic recovery, or death, was 0.55 (95% CI, 0.43 to 0.71; P<0.001) (Fig. 1C).
ALLOGENEIC STEM-CELL TRANSPLANTATION
A total of 24% of the patients in the blinatumomab group and 24% of the patients in the chemotherapy group underwent allogeneic stem-cell transplantation, including 14% and 9% of patients, respectively, who achieved remission without the use of another treatment; 3% and 4% of patients who achieved remission with the use of an intervening anticancer therapy; 1% of patients in each group who had a relapse after having achieved complete remission with full, partial, or incomplete hematologic recovery; and 6% and 10% of patients who did not achieve complete remission with full, partial, or incomplete hematologic recovery. Among the patients who achieved complete remission with full, partial, or incomplete hematologic recovery and who underwent allogeneic stem-cell transplantation, 10 of 38 patients (26%) in the blinatumomab group and 3 of 12 patients (25%) in the chemotherapy group died during a median follow-up period of 206 and 279 days, respectively.
ADVERSE EVENTS
Adverse events were reported in 99% of the patients in each treatment group (Table 3, and Table S4 in the Supplementary Appendix). Serious adverse events were reported in 62% of the patients in the blinatumomab group and in 45% in the chemotherapy group (Table 3, and Table S5 in the Supplementary Appendix). After adjustment for treatment exposure, the event rate for serious adverse events was 349.4 per 100 patient-years in the blinatumomab group and 641.9 per 100 patient-years in the chemotherapy group. Fatal adverse events were reported in 19% of the patients in the blinatumomab group and in 17% of the patients in the chemotherapy group (Table 3). Fatal adverse events that occurred in at least 1% of the patients in either treatment group were sepsis (eight patients [3%] in the blinatumomab group and four patients [4%] in the chemotherapy group), septic shock (six patients [2%] and no patients, respectively), multiorgan failure (three patients [1%] and no patients), respiratory failure (one patient [<1%] and two patients [2%]), and bacteremia (no patients and two patients [2%]). Investigators considered fatal adverse events to be related to treatment with blinatumomab or chemotherapy in eight patients (3%) and eight patients (7%), respectively.
Table 3.
Adverse Events.*
| Event | Blinatumomab Group (N = 267) |
Chemotherapy Group (N = 109) |
|---|---|---|
| no. of patients (%) | ||
| Any adverse event | 263 (98.5) | 108 (99.1) |
|
| ||
| Event leading to premature discontinuation of trial treatment | 33 (12.4) | 9 (8.3) |
|
| ||
| Serious adverse event | 165 (61.8) | 49 (45.0) |
|
| ||
| Fatal serious adverse event | 51 (19.1) | 19 (17.4) |
|
| ||
| Any adverse event of grade ≥3 | 231 (86.5) | 100 (91.7) |
|
| ||
| Grade ≥3 adverse event of interest reported in at least 3% of patients in either group | ||
|
| ||
| Neutropenia | 101 (37.8) | 63 (57.8) |
|
| ||
| Infection | 91 (34.1) | 57 (52.3) |
|
| ||
| Elevated liver enzyme | 34 (12.7) | 16 (14.7) |
|
| ||
| Neurologic event | 25 (9.4) | 9 (8.3) |
|
| ||
| Cytokine release syndrome | 13 (4.9) | 0 |
|
| ||
| Infusion reaction | 9 (3.4) | 1 (09) |
|
| ||
| Lymphopenia | 4 (1.5) | 4 (3.7) |
|
| ||
| Any decrease in platelet count | 17 (6.4) | 13 (11.9) |
|
| ||
| Any decrease in white-cell count | 14 (5.2) | 6 (5.5) |
Data are summarized for all patients who received at least one dose of trial treatment.
Adverse events of grade 3 or higher were reported in 87% of the patients in the blinatumomab group and in 92% of the patients in the chemotherapy group (Table 3). The incidence of grade 3 or higher events of interest that were categorized as neutropenia or infection was lower with blinatumomab than with chemotherapy; in contrast, neurologic events of grade 3 or higher occurred at a similar rate in the two groups (Table 3, and Table S6 in the Supplementary Appendix). The rates of treatment discontinuation due to any adverse event were 12% in the blinatumomab group and 8% in the chemotherapy group, including 4% and 1%, respectively, due to a neurologic event and 1% and 0% due to the cytokine release syndrome. In the blinatumomab group, treatment interruptions were required, as specified in the protocol, for 32% of patients overall, including 7% for infections, 6% for neurologic events, 5% for the cytokine release syndrome, 3% for infusion reactions, and 3% for neutropenia. In the chemotherapy group, treatment interruptions were not required by the protocol but were reported in 6% of patients. In the blinatumomab group, events of interest in the category of the cytokine release syndrome were reported as serious adverse events in 4% of the patients and as events of grade 3 or higher in 5% of the patients.
DISCUSSION
In this randomized phase 3 trial involving adults with Ph-negative relapsed or refractory B-cell precursor ALL, blinatumomab resulted in significantly longer overall survival than standard chemotherapy; the risk of death was 29% lower and the median duration of survival was 3.7 months longer in the blinatumomab group than in the chemotherapy group. Rates of complete remission with full hematologic recovery and complete remission with full, partial, or incomplete hematologic recovery were significantly higher with blinatumomab therapy than with chemotherapy, and the median duration of remission was longer. Adverse events that occurred in the blinatumomab group were consistent with those reported in previous trials. In single-group trials of blinatumomab,17,18 neurologic events and the cytokine release syndrome were identified as two adverse events of interest. In the current controlled trial, blinatumomab was associated with lower incidences of myelosuppression and associated complications than chemotherapy and was associated with higher incidences of serious adverse events, including neurologic events, the cytokine release syndrome, administration-site conditions, and procedural complications (Table S5 in the Supplementary Appendix). After adjustment for differences in treatment exposure between the two groups, the event rate for serious adverse events was lower overall in the blinatumomab group than in the chemotherapy group. Rates of neurologic events of grade 3 or higher were similar in the two groups. The cytokine release syndrome was reported in the blinatumomab group but usually did not require discontinuation of blinatumomab treatment. On the basis of efficacy and safety results from the planned interim analysis, the independent data and safety monitoring committee recommended that the trial be stopped early because of the overall survival benefit observed with blinatumomab as compared with chemotherapy. In an analysis of patient-reported outcomes in this trial that was reported separately,23 health-related quality of life, patient function, and symptoms associated with blinatumomab as compared with chemotherapy were examined. In that analysis, the global health status and quality-of-life score improved in the blinatumomab group and worsened in the chemotherapy group, with a hazard ratio for deterioration in a time-to-event analysis of 0.67 (95% CI, 0.52 to 0.87). Hazard ratios for other quality-of-life outcomes ranged from 0.59 to 0.80 in favor of blinatumomab, with upper boundaries of the 95% confidence intervals that were less than 1.0 across all subscales and single items except for insomnia (95% CI, 0.62 to 1.02).
Several surface antigens are expressed on B-cell precursor ALL blasts, which allows for the use of targeted therapies.24–26 CD20 is expressed on B-cell precursor leukemic cells in up to 50% of patients.13,27 Rituximab, an unconjugated CD20 monoclonal antibody, had minimal efficacy in the treatment of B-cell precursor ALL, but its addition to intensive chemotherapy was associated with longer survival in both Burkitt’s leukemia and CD20-positive B-cell precursor ALL than was intensive chemotherapy alone.28–32 CD22 is expressed in more than 90% of patients with B-cell ALL. Among patients with relapsed or refractory ALL, treatment with inotuzumab ozogamicin (a CD22 monoclonal antibody conjugated to calicheamicin) resulted in higher rates of complete remission and higher rates of negative minimal residual disease than the rates seen with chemotherapy.33–35 Overall survival, a primary end point of the trial, was not significantly longer with inotuzumab than with chemotherapy at the prespecified boundary of P = 0.0208. Treatment with chimeric antigen receptor T cells that target CD19 was associated with complete remission rates of 70 to 90% and durable remissions in single-center trials involving patients who had minimal residual disease or overt relapse.36–38 Combinations of targeted therapies have not been investigated.
This trial of blinatumomab versus chemotherapy differs in several ways from a recently reported trial of inotuzumab versus chemotherapy in relapsed or refractory B-cell precursor ALL.35 The median overall survival was 7.7 months in the experimental group in both trials, but when similar chemotherapy regimens were administered, the median survival was 4.0 months in the current trial and 6.7 months in the inotuzumab trial, which suggests that the patient population in this trial had more aggressive disease than the population in the inotuzumab trial. Indeed, one in four patients in our trial received blinatumomab as third or later salvage therapy, whereas such patients were excluded from participation in the inotuzumab trial. No patient in the blinatumomab trial and 43% of inotuzumab-treated patients had a late first relapse (≥12 months after initial remission). Patients with high peripheral blasts (>10,000 per microliter) at baseline were permitted to receive blinatumomab in this trial but could not receive inotuzumab in the other trial. The percentage of patients who had undergone previous allogeneic stem-cell transplantation was 35% in this trial and 16% for inotuzumab-treated patients. The inotuzumab trial included patients with Ph-positive ALL, whereas this trial did not. Collectively, these differences suggest that patients were at greater risk of death and complications in this trial than in the inotuzumab trial. Thus, the efficacy results for blinatumomab and inotuzumab should not be compared across trials. However, both trials highlight the potential for directed therapies for patients with relapsed or refractory B-cell precursor ALL to reduce the need for prolonged, intensive chemotherapy.
Patients in the blinatumomab group who achieved complete remission with full, partial, or incomplete hematologic recovery had a higher incidence of response with respect to minimal residual disease than did patients in the chemotherapy group, which highlighted the depth and quality of remissions achieved with blinatumomab. Although minimal residual disease status during a patient’s first complete remission has emerged as the most important prognostic factor in the initial treatment of ALL in children and adults,39,40 its value as a prognostic factor in relapsed or refractory ALL is less well established. In this trial, the use of two central laboratories with different methods for assessing minimal residual disease may have introduced a variable that could limit interpretation of the trial. Nevertheless, the introduction of targeted immunotherapy for patients who remain positive for minimal residual disease during their first complete remission may improve the prognosis of such patients.
In summary, this large, randomized trial of single-agent immunotherapy with blinatumomab shows a significant survival benefit as compared with chemotherapy in adults with Ph-negative relapsed or refractory ALL. Blinatumomab resulted in significantly higher rates of hematologic remission and longer survival than chemotherapy. Given the previous exposure of these patients to myelosuppressive and immunosuppressive treatments, the activity of an immune-based therapy such as blinatumomab, which depends on functioning T cells for its activity, provides encouragement that responses may be further enhanced and made durable with additional immune activation strategies.
Supplementary Material
Acknowledgments
Supported by Amgen.
Dr. Kantarjian reports receiving grant support from Amgen, Pfizer, Bristol-Myers Squibb, Novartis Pharmaceuticals, and Ariad Pharmaceuticals; Dr. Stein, receiving lecture fees from Amgen; Dr. Gökbuget, serving on an advisory board for Kite Pharma (uncompensated), receiving fees for serving on advisory boards from Amgen, Pfizer, and Celgene/Juno Therapeutics, travel support from Amgen and Pfizer, and grant support from Amgen; Dr. Fielding, receiving fees for lectures and serving on an advisory board from Amgen; Dr. Schuh, receiving fees for serving on an advisory board from Amgen; Dr. Wei, receiving fees for serving on an advisory board and grant support from Amgen; Dr. Dombret, receiving honoraria from Roche/Genentech, Amgen, Pfizer, Ariad Pharmaceuticals, Novartis Pharmaceuticals, Celgene, Jazz Pharmaceuticals, Agios Pharmaceuticals, Sunesis Pharmaceuticals, Ambit Biosciences (now Daiichi-Sankyo), Karyopharm Therapeutics, Kite Pharma, Menarini, Astellas, Janssen, Servier, and Seattle Genetics, consulting fees from Amgen and Celgene, and grant support from Roche/Genentech, Amgen, Ariad Pharmaceuticals, Jazz Pharmaceuticals, and Kite Pharma; Dr. Foà, receiving fees for serving on advisory boards from Roche, Genentech, Janssen, Celgene, Ariad Pharmaceuticals, and Amgen, and lecture fees from Roche, Genentech, Janssen, Gilead Sciences, Celgene, Novartis Pharmaceuticals, and Sandoz; Dr. Bassan, receiving fees for consulting and serving on advisory boards from Amgen; Dr. Arslan, receiving fees for serving on an advisory board from Amgen; Dr. Rambaldi, receiving lecture fees and travel support from Amgen; Dr. Horst, receiving study fees from Sunesis Pharmaceuticals and Boehringer Ingelheim; Dr. Brüggemann, receiving fees for serving on advisory boards from Incyte and Amgen, lecture fees from Roche and Amgen, grant support from Amgen, and financial support for reference diagnostics from Affimed, Regeneron Pharmaceuticals, and Amgen; Dr. Klapper, receiving grant support to his institution from Roche, Novartis Pharmaceuticals, Regeneron Pharmaceuticals, and Janssen; Dr. Wood, receiving honoraria from Amgen and Seattle Genetics and holding laboratory service agreements with Pfizer, Juno Therapeutics, and Seattle Genetics; Mr. Fleishman, Dr. Nagorsen, Mr. Holland, and Dr. Zimmerman, being employees of and holding stock in Amgen; Mr. Holland, also holding stock in MacroGenics; and Dr. Topp, receiving fees for serving on advisory boards from Amgen, Regeneron Pharmaceuticals, Affimed, Jazz Pharmaceuticals, Gilead Sciences, and Pfizer and travel support from Amgen, Roche, Regeneron Pharmaceuticals, and Affimed.
We thank Robert Dawson of CACTUS Communications for graphics support, funded by Amgen; and Geoffrey Smith of Amgen and Jonathan Latham for medical writing support, funded by Amgen.
APPENDIX
The authors’ affiliations are as follows: the Department of Leukemia, University of Texas M.D. Anderson Cancer Center, Houston (H.K.); City of Hope National Medical Center, Duarte (A.S.), and Amgen, Thousand Oaks (A.F., D.N., Z.Z.) — both in California; Goethe University, University Hospital, Department of Medicine II, Frankfurt am Main (N.G.), Medical Department II (H.-A.H., M.B.) and Hematopathology Section and Lymph Node Registry (W.K.), University Hospital Schleswig Holstein, Campus Kiel, Kiel, and Medizinische Klinik und Poliklinik II, Universitätsklinikums Würzburg, Würzburg (M.S.T.) — all in Germany; Royal Free Hospital and University College London Cancer Institute, London (A.K.F.); Princess Margaret Cancer Centre, Toronto (A.C.S.), and Centre Intégré Universitaire de Santé et de Services Sociaux de l’est de l’île de Montréal, Hôpital Maisonneuve-Rosemont, Montreal (J.B.) — all in Canada; ICO-Hospital Universitari Germans Trias i Pujol, Jose Carreras Research Institute, Universitat Autonoma de Barcelona, Barcelona (J.-M.R.), the Department of Medicine, Hospital Universitari i Politecnic La Fe, University of Valencia, Valencia, and Centro de Investigación Biomédica en Red de Cáncer, Instituto Carlos III, Madrid (M.A.S.) — all in Spain; Alfred Hospital and Monash University, Melbourne, VIC, Australia (A.W.); Institut Universitaire d’Hématologie, Hôpital Saint-Louis (Assistance Publique – Hôpitaux de Paris), Paris (H.D.), and Centre Hospitalier Lyon Sud, Pierre-Benite (X.T.) — both in France; Ematologia, Dipartimento di Biotecnologie Cellulari ed Ematologia, Azienda Ospedaliera Policlinico Umberto I, Università Sapienza di Roma, Rome (R.F.), Azienda Unità Locale Socio Sanitaria 12 Veneziana Ospedale Dell Angelo, Venice (R.B.), and Azienda Socio Sanitaria Territoriale Papa Giovanni XXIII, Bergamo, Università Statale di Milano, Milan (A.R.) — all in Italy; Ankara Universitesi, Tip Fakültesi, Cebeci Arastirma ve Uygulama Hastanesi, Ankara (Ö.A.), and Dokuz Eylül Üniversitesi Tıp Fakültesi, İzmir (F.D.) — both in Turkey; Instytut Hematologii i Transfuzjologii and Centrum Medyczne Kształcenia Podyplomowego, Warsaw, Poland (E.L.-M.); University of Washington Medical Center, Seattle (B.L.W.); and Amgen, Washington, DC (C.H.).
Footnotes
No other potential conflict of interest relevant to this article was reported.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Jabbour E, O’Brien S, Konopleva M, Kantarjian H. New insights into the pathophysiology and therapy of adult acute lymphoblastic leukemia. Cancer. 2015;121:2517–28. doi: 10.1002/cncr.29383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bassan R, Hoelzer D. Modern therapy of acute lymphoblastic leukemia. J Clin Oncol. 2011;29:532–43. doi: 10.1200/JCO.2010.30.1382. [DOI] [PubMed] [Google Scholar]
- 3.Kantarjian H, Thomas D, O’Brien S, et al. Long-term follow-up results of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (Hyper-CVAD), a dose-intensive regimen, in adult acute lymphocytic leukemia. Cancer. 2004;101:2788–801. doi: 10.1002/cncr.20668. [DOI] [PubMed] [Google Scholar]
- 4.Gökbuget N, Hoelzer D, Arnold R, et al. Treatment of adult ALL according to protocols of the German Multicenter Study Group for Adult ALL (GMALL) Hematol Oncol Clin North Am. 2000;14:1307–25. doi: 10.1016/s0889-8588(05)70188-x. [DOI] [PubMed] [Google Scholar]
- 5.Tavernier E, Boiron JM, Huguet F, et al. Outcome of treatment after first relapse in adults with acute lymphoblastic leukemia initially treated by the LALA-94 trial. Leukemia. 2007;21:1907–14. doi: 10.1038/sj.leu.2404824. [DOI] [PubMed] [Google Scholar]
- 6.Oriol A, Vives S, Hernández-Rivas JM, et al. Outcome after relapse of acute lymphoblastic leukemia in adult patients included in four consecutive risk-adapted trials by the PETHEMA Study Group. Haematologica. 2010;95:589–96. doi: 10.3324/haematol.2009.014274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas DA, Kantarjian H, Smith TL, et al. Primary refractory and relapsed adult acute lymphoblastic leukemia: characteristics, treatment results, and prognosis with salvage therapy. Cancer. 1999;86:1216–30. doi: 10.1002/(sici)1097-0142(19991001)86:7<1216::aid-cncr17>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 8.Kantarjian HM, Thomas D, Ravandi F, et al. Defining the course and prognosis of adults with acute lymphocytic leukemia in first salvage after induction failure or short first remission duration. Cancer. 2010;116:5568–74. doi: 10.1002/cncr.25354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Brien S, Thomas D, Ravandi F, et al. Outcome of adults with acute lymphocytic leukemia after second salvage therapy. Cancer. 2008;113:3186–91. doi: 10.1002/cncr.23919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gökbuget N, Stanze D, Beck J, et al. Outcome of relapsed adult lymphoblastic leukemia depends on response to salvage chemotherapy, prognostic factors, and performance of stem cell transplantation. Blood. 2012;120:2032–41. doi: 10.1182/blood-2011-12-399287. [DOI] [PubMed] [Google Scholar]
- 11.Gökbuget N, Hoelzer D. Salvage therapy of adult acute lymphoblastic leukemia. In: Faderl S, Kantarjian H, editors. Leukemias: principles and practice of therapy. Oxford, United Kingdom: Wiley-Black-well; 2011. pp. 217–27. [Google Scholar]
- 12.Fielding AK, Richards SM, Chopra R, et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood. 2007;109:944–50. doi: 10.1182/blood-2006-05-018192. [DOI] [PubMed] [Google Scholar]
- 13.Raponi S, De Propris MS, Intoppa S, et al. Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody-based immunotherapy in acute lymphoblastic leukemia: analysis of 552 cases. Leuk Lymphoma. 2011;52:1098–107. doi: 10.3109/10428194.2011.559668. [DOI] [PubMed] [Google Scholar]
- 14.Löffler A, Gruen M, Wuchter C, et al. Efficient elimination of chronic lymphocytic leukaemia B cells by autologous T cells with a bispecific anti-CD19/anti-CD3 single-chain antibody construct. Leukemia. 2003;17:900–9. doi: 10.1038/sj.leu.2402890. [DOI] [PubMed] [Google Scholar]
- 15.Dreier T, Lorenczewski G, Brandl C, et al. Extremely potent, rapid and co-stimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int J Cancer. 2002;100:690–7. doi: 10.1002/ijc.10557. [DOI] [PubMed] [Google Scholar]
- 16.Hoffmann P, Hofmeister R, Brischwein K, et al. Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int J Cancer. 2005;115:98–104. doi: 10.1002/ijc.20908. [DOI] [PubMed] [Google Scholar]
- 17.Topp MS, Gökbuget N, Zugmaier G, et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol. 2014;32:4134–40. doi: 10.1200/JCO.2014.56.3247. [DOI] [PubMed] [Google Scholar]
- 18.Topp MS, Gökbuget N, Stein AS, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16:57–66. doi: 10.1016/S1470-2045(14)71170-2. [DOI] [PubMed] [Google Scholar]
- 19.Brüggemann M, Schrauder A, Raff T, et al. Standardized MRD quantification in European ALL trials: proceedings of the Second International Symposium on MRD assessment in Kiel, Germany, 18–20 September 2008. Leukemia. 2010;24:521–35. doi: 10.1038/leu.2009.268. [DOI] [PubMed] [Google Scholar]
- 20.Borowitz MJ, Wood BL, Devidas M, et al. Prognostic significance of minimal residual disease in high risk B-ALL: a report from Children’s Oncology Group study AALL0232. Blood. 2015;126:964–71. doi: 10.1182/blood-2015-03-633685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Brien PC, Fleming TR. A multiple testing procedure for clinical trials. Biometrics. 1979;35:549–56. [PubMed] [Google Scholar]
- 22.Lan KKG, DeMets DL. Discrete sequential boundaries for clinical trials. Biometrika. 1983;70:659–63. [Google Scholar]
- 23.Topp M, Zimmerman Z, Cannell P, et al. Health-related quality of life (HRQoL) of blinatumomab versus standard of care (SOC) chemotherapy in patients with relapsed or refractory Philadelphia negative B-cell precursor acute lymphoblastic leukemia in a randomized, open-label phase 3 study (TOWER) Blood. 2016;128:222. abstract. [Google Scholar]
- 24.Jabbour E, O’Brien S, Ravandi F, Kantarjian H. Monoclonal antibodies in acute lymphoblastic leukemia. Blood. 2015;125:4010–6. doi: 10.1182/blood-2014-08-596403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoelzer D, Gökbuget N. Chemoimmunotherapy in acute lymphoblastic leukemia. Blood Rev. 2012;26:25–32. doi: 10.1016/j.blre.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 26.Kantarjian H, Thomas D, Wayne AS, O’Brien S. Monoclonal antibody-based therapies: a new dawn in the treatment of acute lymphoblastic leukemia. J Clin Oncol. 2012;30:3876–83. doi: 10.1200/JCO.2012.41.6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas DA, O’Brien S, Jorgensen JL, et al. Prognostic significance of CD20 expression in adults with de novo precursor B-lineage acute lymphoblastic leukemia. Blood. 2009;113:6330–7. doi: 10.1182/blood-2008-04-151860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas DA, Faderl S, O’Brien S, et al. Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer. 2006;106:1569–80. doi: 10.1002/cncr.21776. [DOI] [PubMed] [Google Scholar]
- 29.Thomas DA, O’Brien S, Faderl S, et al. Chemoimmunotherapy with a modified hyper-CVAD and rituximab regimen improves outcome in de novo Philadelphia chromosome-negative precursor B-lineage acute lymphoblastic leukemia. J Clin Oncol. 2010;28:3880–9. doi: 10.1200/JCO.2009.26.9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ribrag V, Koscielny S, Bosq J, et al. Rituximab and dose-dense chemotherapy for adults with Burkitt’s lymphoma: a randomised, controlled, open-label, phase 3 trial. Lancet. 2016;387:2402–11. doi: 10.1016/S0140-6736(15)01317-3. [DOI] [PubMed] [Google Scholar]
- 31.Maury S, Chevret S, Thomas X, et al. Rituximab in B-lineage adult acute lymphoblastic leukemia. N Engl J Med. 2016;375:1044–53. doi: 10.1056/NEJMoa1605085. [DOI] [PubMed] [Google Scholar]
- 32.Jabbour E, Kantarjian H. Chemoimmunotherapy as a new standard of care for Burkitt leukaemia/lymphoma. Lancet. 2016;387:2360–1. doi: 10.1016/S0140-6736(16)00164-1. [DOI] [PubMed] [Google Scholar]
- 33.Kantarjian H, Thomas D, Jorgensen J, et al. Inotuzumab ozogamicin, an anti-CD22-calecheamicin conjugate, for refractory and relapsed acute lymphocytic leukaemia: a phase 2 study. Lancet Oncol. 2012;13:403–11. doi: 10.1016/S1470-2045(11)70386-2. [DOI] [PubMed] [Google Scholar]
- 34.Kantarjian H, Thomas D, Jorgensen J, et al. Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic leukemia. Cancer. 2013;119:2728–36. doi: 10.1002/cncr.28136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375:740–53. doi: 10.1056/NEJMoa1509277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–28. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gökbuget N, Kneba M, Raff T, et al. Adult patients with acute lymphoblastic leukemia and molecular failure display a poor prognosis and are candidates for stem cell transplantation and targeted therapies. Blood. 2012;120:1868–76. doi: 10.1182/blood-2011-09-377713. [DOI] [PubMed] [Google Scholar]
- 40.Goekbuget N, Dombret H, Bonifacio M, et al. BLAST: A confirmatory, single-arm, phase 2 study of blinatumomab, a bispecific T-cell engager (BiTE®) antibody construct, in patients with minimal residual disease B-precursor acute lymphoblastic leukemia (ALL) Blood. 2014;124:379. abstract. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.