

Abstract
In 1900, Adami speculated that a sequence of context‐independent energetic and structural changes governed the reversion of differentiated cells to a proliferative, regenerative state. Accordingly, we show here that differentiated cells in diverse organs become proliferative via a shared program. Metaplasia‐inducing injury caused both gastric chief and pancreatic acinar cells to decrease mTORC1 activity and massively upregulate lysosomes/autophagosomes; then increase damage associated metaplastic genes such as Sox9; and finally reactivate mTORC1 and re‐enter the cell cycle. Blocking mTORC1 permitted autophagy and metaplastic gene induction but blocked cell cycle re‐entry at S‐phase. In kidney and liver regeneration and in human gastric metaplasia, mTORC1 also correlated with proliferation. In lysosome‐defective Gnptab −/− mice, both metaplasia‐associated gene expression changes and mTORC1‐mediated proliferation were deficient in pancreas and stomach. Our findings indicate differentiated cells become proliferative using a sequential program with intervening checkpoints: (i) differentiated cell structure degradation; (ii) metaplasia‐ or progenitor‐associated gene induction; (iii) cell cycle re‐entry. We propose this program, which we term “paligenosis”, is a fundamental process, like apoptosis, available to differentiated cells to fuel regeneration following injury.
Keywords: dedifferentiation, regeneration, repair, reprogramming, transdifferentiation
Subject Categories: Autophagy & Cell Death, Cell Cycle, Development & Differentiation
Introduction
In 1900, George Adami wrote about the relationship between mitotic and differentiated cells, stating that he expected mitotic cells would generally devote energy toward replication and differentiated cells toward performing physiological functions (Adami, 1900). He also observed that upon injury, differentiated cells had the capacity to revert to a more primitive state, becoming mitotic again to promote tissue repair. Adami's observations on such cellular plasticity have largely been forgotten, as the focus in the 20th century was nearly exclusively on the unidirectional differentiation of stem cells into functional, “post‐mitotic” cells.
However, over the past decade or two, numerous examples have emerged to support plasticity in differentiated cells. First, it became clear that normal, somatic cells could be reprogrammed to pluripotency (Takahashi & Yamanaka, 2006). Furthermore, in tissues, injury can induce a repair process that recruits largely post‐mitotic, differentiated cells back into the cell cycle in most, if not all, organs and species, for example, glia (Boerboom et al, 2017; Mindos et al, 2017); lung (Logan & Desai, 2015); heart in mammals (Wang et al, 2017) and fish (Karra et al, 2015); in multiple gastrointestinal tract organs (Mills & Sansom, 2015). Each such example to date has been studied essentially in isolation within the context of a particular type of injury and a single organ; however, because the process is so widespread, we have postulated that it may be governed by a shared, evolutionarily conserved molecular and cellular program that is independent of tissue and species (Mills & Sansom, 2015).
It has long been known that the response of both the corpus of the stomach and the digestive‐enzyme‐secreting (exocrine) pancreas to certain types of injury involves phenotypical changes in cell differentiation and tissue architecture, known as metaplasia. In the acute setting, the metaplastic response appears to be a tissue repair mechanism and can be temporary, with full restoration of normal tissue architecture (Nomura et al, 2005; Huh et al, 2012). Chronically, however, ongoing damage and long‐term metaplasia are associated with and may fuel the majority of gastric and pancreatic adenocarcinomas (Mills & Sansom, 2015; Giroux & Rustgi, 2017; Storz, 2017). In both organs, the cells of origin for the metaplastic, proliferating epithelial cells are thought to be differentiated secretory cells (zymogenic chief cells in the stomach and acinar cells in the pancreas) that reprogram to re‐enter the cell cycle (Mills & Sansom, 2015; Murtaugh & Keefe, 2015; Mills & Goldenring, 2017; Radyk & Mills, 2017).
Here, we report that differentiated cells in both pancreas and stomach exhibit high levels of mTORC1 activity during homeostasis. Proliferation‐inducing injury caused rapid mTORC1 loss and a dramatic induction of autodegradative machinery (lysosomes and autophagy). As the functional and structural components were recycled, cells changed gene expression patterns (e.g., inducing the metaplastic marker Sox9); thereafter, they reactivated mTORC1 and re‐entered the cell cycle. Such changes in mTORC1 activity were corroborated in tissues from human patients. Also, established models of injury to differentiated cells in mouse liver (Espeillac et al, 2011) and kidney (Chang‐Panesso & Humphreys, 2017) correlate mTORC1 activity with the recruited proliferating cells. Blocking mTORC1 with rapamycin in murine pancreas and stomach impaired only cell cycle re‐entry but not earlier cellular changes. Differentiated cells in autophagy‐defective Gnptab −/− mice were blocked from both SOX9 expression and cell cycle re‐entry phases, consistent with the upstream autodegradative phase being necessary for downstream mTORC1‐mediated S‐phase entry.
Our results in the context of numerous previous reports on cellular reprogramming lead us now to propose that recruiting differentiated cells into a regenerative phenotype occurs via stepwise metabolic and molecular phases that constitute a conserved, fundamental, cellular program, akin to mitosis or apoptosis. This cellular program occurs during cell fate changes of various types (e.g., reversion, dedifferentiation, transdifferentiation, reprogramming). The lack of a standard term for the actual cellular process itself impedes finding shared features that transcend cell types, tissues, and model systems. We propose a new, unifying term: “paligenosis” from the Greek: pali/n/m (meaning backward or recurrence) + genea (born of, producing) + osis (an action or process).
Results
Diverse organs show similar changes in metabolic activity during acute injury
To induce injury in the stomach, we employed a high‐dose tamoxifen (“HD‐Tam”) injury model that has been used by us and others (Huh et al, 2012; Burkitt et al, 2017; Lee et al, 2017; Leushacke et al, 2017). HD‐Tam causes loss of nearly all acid‐secreting parietal cells in the body of the stomach (Figs EV1 and EV3) and induces mature, differentiated digestive‐enzyme‐secreting chief cells at the base of the unit to give rise to a proliferating cell population (Radyk et al, 2018). These former chief cells maintain low‐level expression of some mature chief cell markers and induce expression of wound repair‐associated genes like mucins and TFF2 (aka spasmolytic polypeptide). The pattern of parietal cell loss and abundant, proliferative cells co‐expressing TFF2 and chief cell markers has been called spasmolytic polypeptide‐expressing metaplasia (SPEM) or pseudopyloric metaplasia (Schmidt et al, 1999). Maximal parietal cell loss and proliferation stemming from chief cells occurs at 3 days after the first dose of tamoxifen (Schematized in Fig 1A). By 7 days, parietal cells have returned, and the entire stomach regenerates to pre‐treatment cell censuses within 14–21 days (Huh et al, 2012). HD‐Tam is a rapid, synchronous method to model, in a manner that lends itself to molecular analyses, the mechanisms of stomach repair that also occurs in human stomachs infected with the bacterium Helicobacter pylori.
Figure EV1. pS6 is an accurate proxy for rapamycin‐sensitive mTORC1 activity and shows that loss of mTORC1 does not affect parietal cell death or induction of metaplastic gene expression in reprogramming chief cells.
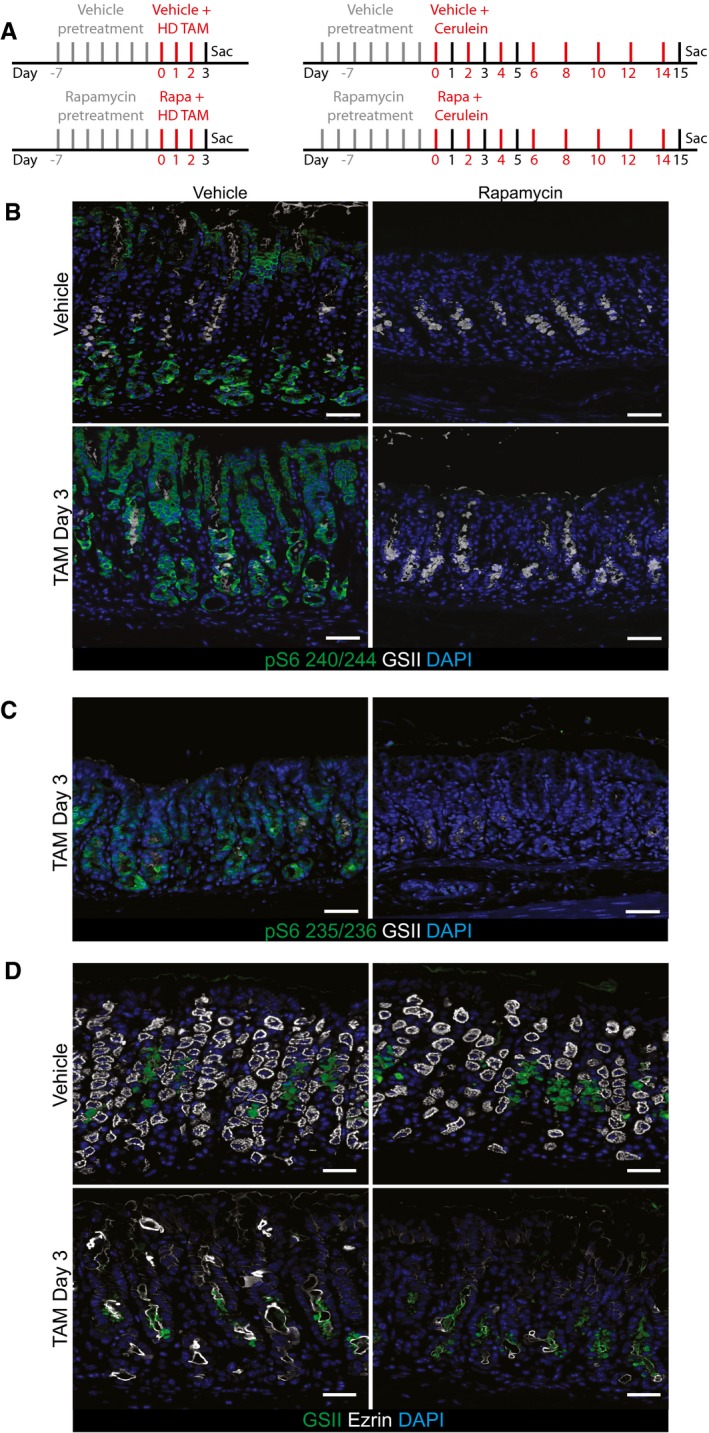
- Injection schemes for injury experiments with rapamycin in stomach (left) and pancreas (right).
- Representative epifluorescence images of the distribution of pS6 in the normal and injured stomach ±rapamycin treatment. pS6 is restricted to the chief cell zone (base) and pit zone of the normal corpus unit. At peak (HD‐Tam day 3) SPEM stages, it is located at high level throughout the unit. Upon rapamycin treatment, all pS6 staining is lost throughout the normal and injured corpus unit. The characteristic induction of GSII staining in reprogramming chief cells at the base of gastric units (indicating SPEM) occurs at least as markedly in the presence of rapamycin, indicating mTORC1 is not required for metaplastic gene induction. Green, pS6; white, GSII; blue, DAPI. Scale bars: 50 μm.
- At peak metaplasia stages, pS6 235/6 is upregulated in the stomach epithelium and rapamycin treatment at this stage abolishes all staining. Scale bars: 50 μm.
- Representative epifluorescence images of the loss parietal cells (marked by ezrin) upon injury and rapamycin treatment. Treatment with HD‐Tam caused the loss of the vast majority of parietal cells throughout the corpus. Rapamycin does not rescue that injury. Green, GSII; white, ezrin; blue, DAPI. Scale bars: 50 μm.
Figure EV3. Histological changes in the injured stomach and pancreas with and with rapamycin treatment.
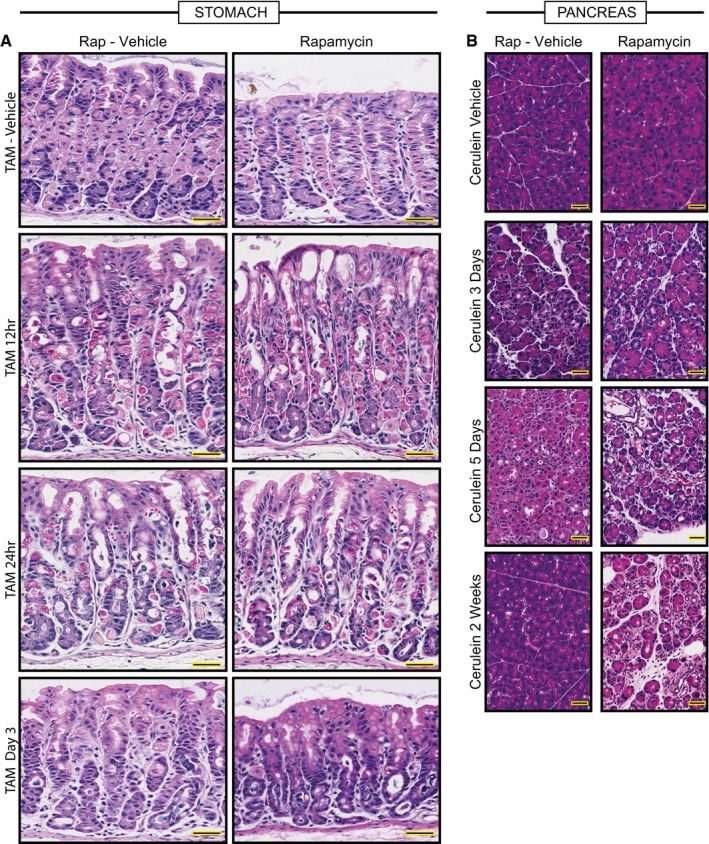
- Representative hematoxylin and eosin counterstained images of HD‐TAM stomach tissue ±rapamycin. Treatment with tamoxifen causes acute loss of parietal cells (large eosinophilic cells) by 12–24 h post‐injury. By 3 days, chief cells have reprogrammed into SPEM cells. The general pattern of loss of parietal cells and conversion of chief cells to metaplastic cells is not affected by rapamycin (right panels). Scale bars, 50 μm.
- Representative hematoxylin and eosin counterstained images of pancreas tissue injured with cerulein at various stages ±rapamycin. Cerulein injury causes mosaic, asynchronous conversion of acinar cells into proliferative, acinar‐ductal metaplastic cells with maximal features of the process at day 5 in our protocol. By 2 weeks, the pancreas has compensated for the continuous injury and recovers a relatively normal morphology. Dual treatment with rapamycin and cerulein does not rescue the metaplastic response by day 5 and impedes normal tissue compensation by 2 weeks injury, with most of the tissue continuing to show abundant metaplastic forms. Scale bars, 50 μm.
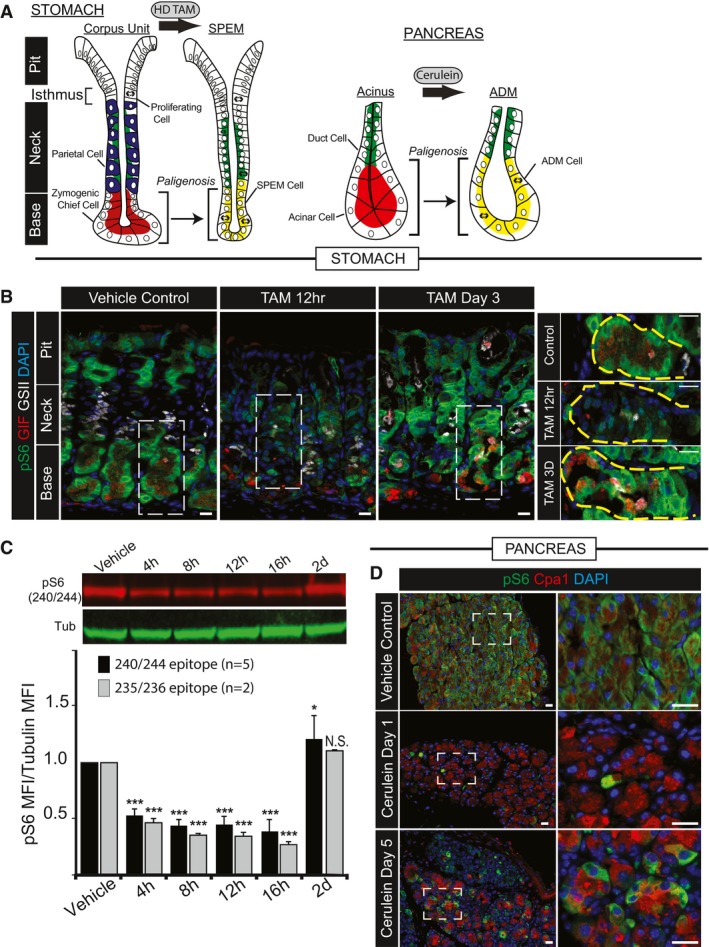
Figure 1. mTORC1 activity undergoes dramatic changes during stomach and pancreas metaplastic injury response.
-
A, BDigestive‐enzyme‐secreting (zymogenic) mature cell populations in the stomach (A) and pancreas (B) are recruited back into the cell cycle to fuel metaplasia in response to large‐scale injury. Digestive enzyme expression (red) decreases, and markers of mucous neck cells (green, stomach) or duct cells (green, pancreas) increase in metaplastic, proliferating cells (red + green = yellow). Stomach is further characterized by loss of acid‐secreting parietal cells (blue). (B) Representative epifluorescence images of mouse gastric corpus glands during homeostasis, early after injury (HD‐Tam 12 h) and at maximal metaplastic response (HD‐Tam Day 3), stained for mTORC1 activity using a downstream target, pS6 as a proxy. Green, pS6; red, GIF (gastric intrinsic factor, a chief cell marker); white, GSII (a mucous neck cell marker); blue, DAPI. Right—higher magnification images of boxed areas on left focus exclusively on the base of the unit where the digestive‐enzyme‐secreting cells are reprogramming. Yellow dashed area outlines the base of a single gastric unit. Scale bar, 20 μm; boxed area pull out, 10 μm.
-
CWestern blot of pS6 (red) and β‐tubulin control (green) from whole corpus protein extracts at various injury time points; pS6 (240/244 or 235/6) vs. tubulin fluorescent intensity from replicate blots quantified below (error bars = standard deviation). *P < 0.05, ***P < 0.001. Statistical analysis with both antibodies was done using ANOVA with a post hoc Dunnett's test.
-
DRepresentative epifluorescence images of pS6 staining of pancreas during homeostasis, acute injury (cerulein 12 h), and maximal injury (cerulein day 5). Green, pS6; red, amylase; blue, DAPI. Boxed areas on left depicted at higher magnification on right. Scale bar, 20 μm; boxed area pull out, 10 μm.
To induce injury in pancreas, we used a well‐described rapid method involving daily injection of the secretagogue cerulein. Cerulein injections cause large‐scale damage to the digestive‐enzyme‐secreting acinar cells of the exocrine pancreas (Adler et al, 1985; Niederau et al, 1985; Saluja et al, 1985). To repair the damage, acinar cells re‐enter the cell cycle, forming duct‐like structures called ADM (acinar‐to‐ductal metaplasia; schematic in Fig 1A). In our protocol, ADM peaks 5 days after commencement of cerulein. Thereafter, there is continued damage if cerulein administration is maintained, but the pancreas gradually adapts to the injury over 2 weeks. Similar to HD‐Tam injury in the stomach, cerulein injury models a metaplastic process that can also be a precursor for pancreatic ductal adenocarcinoma.
To determine whether the reversion from the differentiated to the replicative state involves conserved shifts in cellular energy use, we examined metabolic activity in both tissues using phosphorylated ribosomal S6 protein (pS6). The principal mediator of S6 phosphorylation is the S6 kinase enzyme via the cellular metabolism hub mTOR complex 1 (mTORC1). To confirm that S6 phosphorylation depends on mTORC1 activity, we treated mice with rapamycin, a specific inhibitor of the mTORC1‐mediated S6 kinase activity. We used an antibody against residues 240/244 of S6, because those sites are phosphorylated principally by pS6 kinase 1, whereas the 235/236 phosphorylation sites can have input from other signaling pathways. For example, 235/236 can be phosphorylated by p90 ribosomal S6 kinases that can be activated via ERK signaling (Roux et al, 2007). Figure EV1 shows that rapamycin, which is a specific inhibitor of mTORC1‐mediated S6 Kinase activity, abolished pS6 240/44 staining, which was normally abundant in gastric pit cells nearer the stomach lumen and in gastric chief cells. Rapamycin also blocked S6 phosphorylation efficiently during the HD‐Tam protocol (Fig EV1). Antibodies against 235/236 also showed strong phosphorylation at peak metaplasia as well as a similar abrogation of staining in the presence of rapamycin (Fig EV1). As anti‐240/244 antibodies have stronger signal in our experiments and are more specific for mTORC1‐mediated phosphorylation, we will use anti‐240/244 pS6 as a surrogate for mTORC1 activity for the remainder of the manuscript unless otherwise mentioned.
HD‐Tam or cerulein caused dramatic changes in pS6 expression. In stomach, pS6 was largely lost by 12 h. By 3 days, when SPEM is maximal in this system, the entire gastric unit expressed abundant pS6 (Fig 1B). Molecular and cellular changes in the stomach following HD‐Tam are sufficiently synchronous across the whole stomach that quantitative, molecular approaches can be used (Huh et al, 2012). Quantitatively, phosphorylation status of both pS6 240/244 and 235/236 in the corpus of the stomach was decreased by nearly half within the first 4 h and returned to at or above baseline by 48 h (Fig 1C). In pancreas, despite a slower and less synchronous time course, the same pattern of mTORC1 activity could be observed by immunofluorescence. pS6 was abundant in acinar cells at baseline, was nearly undetectable by 24 h, and recovered in many cells by day 5, when ADM is maximal (Fig 1D).
Thus, both tissues, when recruiting proliferative cells for repair, undergo a well‐defined pattern of changes in mTORC1 activity. During homeostasis, the organs are replete with differentiated secretory cells that are not dividing but are energetically active in synthesizing protein using their elaborate secretory apparatus (Mills & Taghert, 2012; Lo et al, 2017). When replicating cells must be recruited from those differentiated cells, the cells shut off mTORC1 temporarily, then re‐induce it at the time of maximal regenerative proliferation.
To further assess whether the upregulation of pS6 is a common feature during the recruitment of differentiated cells to regenerate damaged tissue, we examined liver (two‐thirds partial hepatectomy) and kidney (tunicamycin‐induced acute injury) for changes in S6 phosphorylation. Both injury models have previously been shown to involve recruitment of differentiated cells back into the cell cycle (Newberry et al, 2008). In kidney, as expected, tubules in the cortex and outer medulla are damaged as evinced by vacuolation (Fig EV2). Non‐damaged tubules show increased BrdU as cells re‐enter the cell cycle (Fig EV2). The proliferative tubules show marked increase in pS6. Similarly, the well‐known recruitment of hepatocytes into the cell cycle 48 h following partial hepatectomy is also accompanied by increased S6 phosphorylation (Fig EV2).
Figure EV2. Acute kidney injury and partial hepatectomy both cause upregulation of mTORC1 activity during proliferative phases.
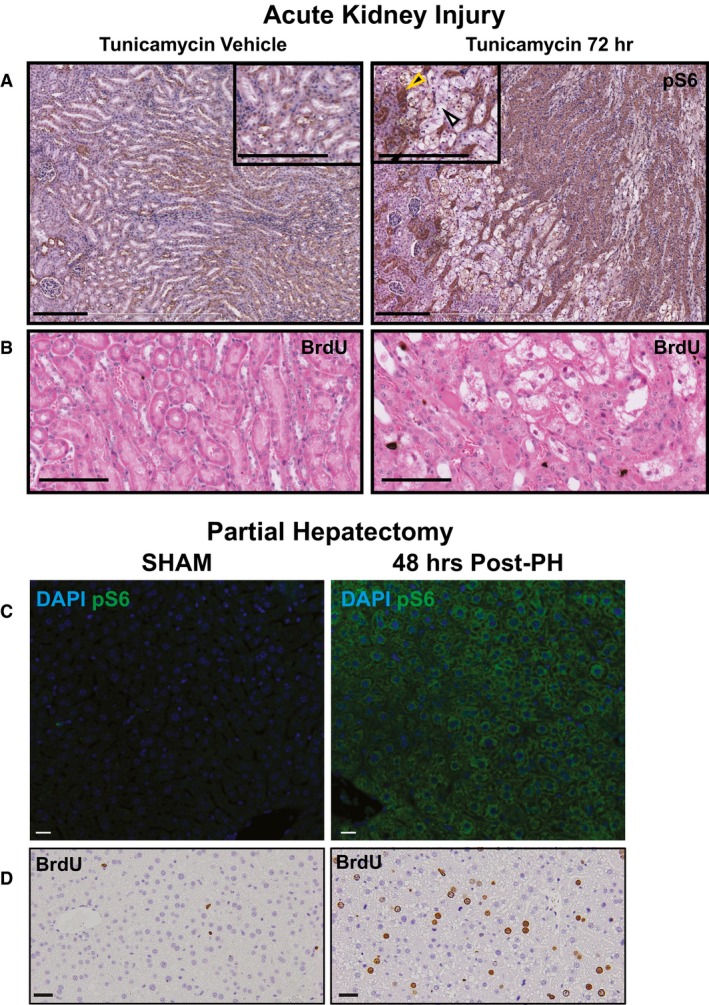
- Upon injury with tunicamycin, tubule cells in the kidney are damaged (white arrowhead) and surviving tubule cells (yellow arrowhead) upregulate pS6. Scale bars: 100 μm.
- Upregulation of the pS6 is associated with increased proliferation in this injury model as seen by BrdU+ nuclei. Scale bars: 100 μm.
- Two‐thirds partial hepatectomy causes a pronounced upregulation of pS6 in the remaining hepatocyte mass. Scale bars: 20 μm.
- The pS6+ hepatocytes are highly proliferative at this stage. Scale bars: 20 μm.
mTORC1 is required for injury‐induced proliferation
During SPEM and ADM, expression of genes encoding secretory cargo (like digestive enzymes) is scaled down, whereas wound repair and progenitor‐associated genes are scaled up (Capoccia et al, 2013). Many such scaled up genes (e.g., Cd44 and Sox9) are increased in both pancreas and stomach during the recruitment of differentiated cells back into the cell cycle. To determine whether blocking mTORC1 affected response to injury, we subjected mice to HD‐Tam or cerulein and examined the effects of blocking mTORC1 with rapamycin (Fig EV3). Figures EV1 and EV3 show that parietal cell death occurred in HD‐Tam even without mTORC1 activity. The gastric units also remodeled as expected with chief cells assuming the ductular morphology, characteristic of SPEM (Fig EV3).
We next examined the effects of rapamycin on induced proliferation. We noted that in control experiments, without HD‐Tam, proliferation of the cells in the isthmus (the narrow zone between pit and upper neck, Fig 1A), where there is active mitosis in homeostasis, was not affected markedly by rapamycin (Fig 2A and C). However, rapamycin decreased the injury‐induced proliferation by nearly half (P < 0.001; Fig 2C). The lack of proliferation did not affect SPEM induction, as defined by cells co‐stained with GSII and the chief cell marker, GIF (Fig 2A and B). Indeed, the SPEM marker SOX9 was induced in reprogramming chief cells to levels at least as high as those observed in HD‐Tam without rapamycin (Fig EV4). That cells expressing molecular features of metaplasia can arise in the absence of proliferation is consistent with multiple previous reports showing that chief cells can give rise directly to SPEM cells without contribution from the isthmal stem cell (Nam et al, 2010; Radyk et al, 2018).
Figure 2. Recruitment of proliferating cells during stomach and pancreas metaplastic injury depends on mTORC1.
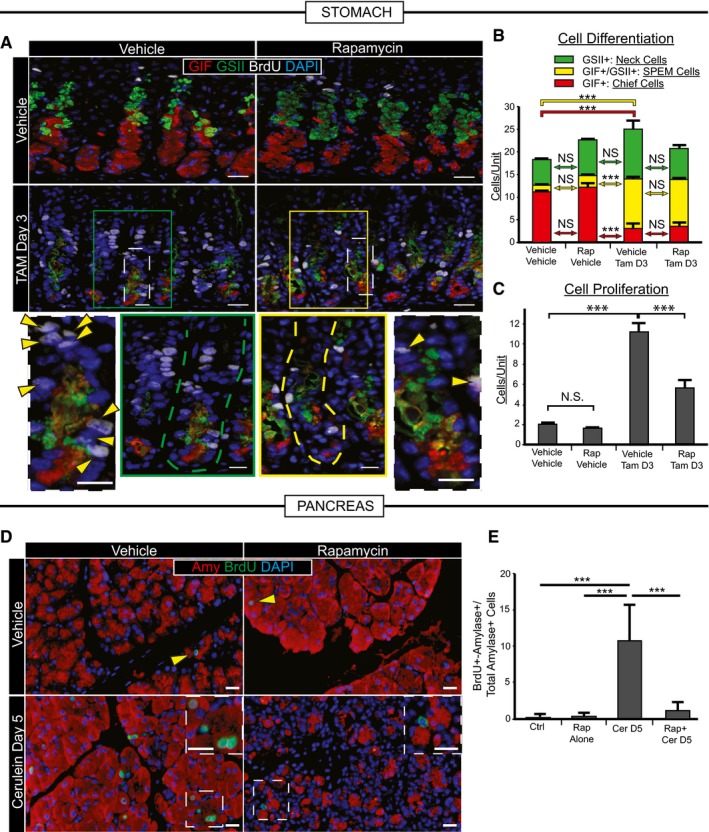
- Representative immunofluorescence images of stomach tissue ± metaplastic injury ± rapamycin treatment. Green, neck cells (GSII); red, chief cells (GIF); white, proliferating cells (BrdU); blue, nuclei (DAPI). Scale bars 20 μm; 10 μm for bottom images. Bottom—boxed areas from top pictures are shown at higher magnification with individual bases of gastric units (where reprogramming occurs) outlined by dashed lines and proliferating cells by arrowheads.
- Cells of each differentiation type, following scheme in Fig 1A, are quantified by scoring immunofluorescence images from multiple experiments. Metaplastic injury induces a massive accumulation of yellow (SPEM) cells and loss of red (Chief) cells (compare vehicle–vehicle with vehicle‐Tam, D3) that is not significantly affected by rapamycin treatment (compare vehicle‐Tam D3 with Rap‐Tam D3).
- Proliferative cells are quantified as for panel (B). Injury induces massive proliferation (compare vehicle–vehicle with vehicle‐Tam D3) significantly inhibited by rapamycin (compare vehicle‐Tam D3 with Rap‐Tam D3).
- Top panel arrowheads indicate rare proliferative acinar cells during homeostasis with or without rapamycin treatment. Cerulein induces proliferation of acinar cells recruited into the cell cycle that is inhibited by rapamycin. Boxed areas are magnified in insets. Note multiple BrdU+ cells (green) staining with amylase (red) a digestive enzyme, indicating acinar cell origin. BrdU+ cells following rapamycin + cerulein treatment are often not co‐stained with amylase. Blue, DAPI (nuclei). Scale bars 20 μm; 10 μm for insets.
- Quantification of multiple experiments with mice treated as in panel (D).
Figure EV4. mTORC1 is not required for increased SOX9 during metaplasia.
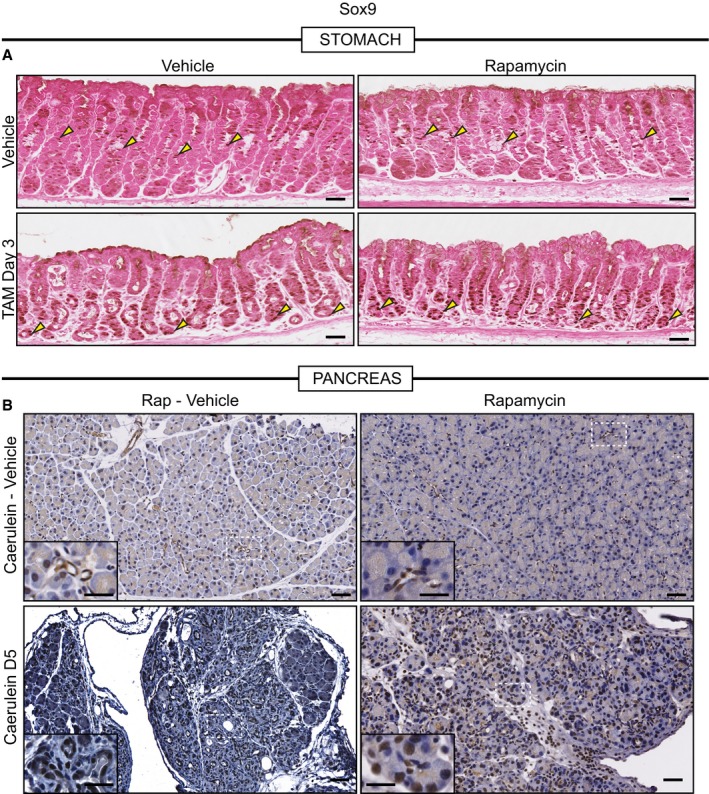
- Representative eosin counterstained IHC images of normal or metaplastic gastric tissue stained for SOX9. SOX9, in control tissue, stains the isthmal and mucous neck cells, which are proliferative progenitors (yellow arrowheads), of the corpus units and is generally excluded from the base of units. Upon injury with HD‐TAM, SOX9 expression is induced in the base of units (yellow arrowheads). Treatment with rapamycin does not alter either the normal or metaplasia distribution of SOX9 (yellow arrowheads). Scale bars, 50 μm.
- Representative hematoxylin counterstained IHC images of normal or metaplastic pancreatic tissue stained for SOX9. SOX9 expression in normal pancreatic tissue is restricted to the duct (see inset in top left panel which is a high magnification view of the boxed area). At peak metaplasia stages, SOX9 becomes expressed in dedifferentiating acinar cells (see bottom left inset). Treatment with rapamycin in normal (see top right inset) or injured (see bottom right inset) does not alter SOX9 expression. Scale bars 50 μm; inset 25 μm.
Rapamycin had equivalent effects on the pancreas. Metaplastic induction of SOX9 was not affected (Fig EV4); however, cell proliferation was even more substantially blocked than in the stomach (Fig 2D and E). This may be because the pancreas is entirely dependent on reprogramming acinar cells as a source for proliferation, whereas the stomach also has a constitutive stem cell that continues to proliferate even in the presence of rapamycin (Fig 1A). Continued HD‐Tam injections kill mice, so we cannot study adaptation of stomachs; however, we have maintained cerulein injections for up to 2 weeks by which point wild‐type pancreas usually adapts to the injury. Thus, we used the pancreas to determine whether mTORC1‐dependent proliferation was required for pancreatic repair. Figure EV3 shows that 2‐week cerulein with mTORC1 blocked led to tissue loss relative to cerulein treatment alone.
Changes in mTORC1 also characterize human metaplasia
To determine whether mTORC1 activity is modulated in human disease states, we first examined a database of stomach tissues from human patients exhibiting metaplastic response to H. pylori infection, previously compiled at Washington University (Lennerz et al, 2010; Radyk et al, 2018). A representative region from this dataset is shown in Fig 3A. As in mice, morphologically normal chief cells showed high pS6. In regions of SPEM, pS6 abundance varied. In lesions that had histological features of cells undergoing acute conversion to SPEM (what we have previously termed “hybrid SPEM” (Lennerz et al, 2010; Radyk et al, 2018) based on examination of a large dataset of SPEM lesions), pS6 levels were high (Fig 3A). In regions where basal cells showed more uniform metaplasia (“established SPEM”), pS6 levels were lower. In humans, SPEM is thought to be either transient and rapidly resolve (as in the mouse HD‐Tam model) or chronic and persist for decades, involving large patches of the stomach (Peterson, 2002). In the chronic case, SPEM is equivalent to the lesion pathologists call chronic atrophic gastritis (Rugge et al, 2008). In addition, SPEM is thought to progress to (or predate) another, proliferative, pre‐cancerous lesion, intestinal metaplasia (Yoshizawa et al, 2007; Correa & Piazuelo, 2012; Spechler et al, 2017) and to increase risk for progression to a cytologically atypical lesion, dysplasia, as well as to cancer itself.
Figure 3. mTORC1 activity correlates with stages of metaplasia during human gastric tumorigenesis.
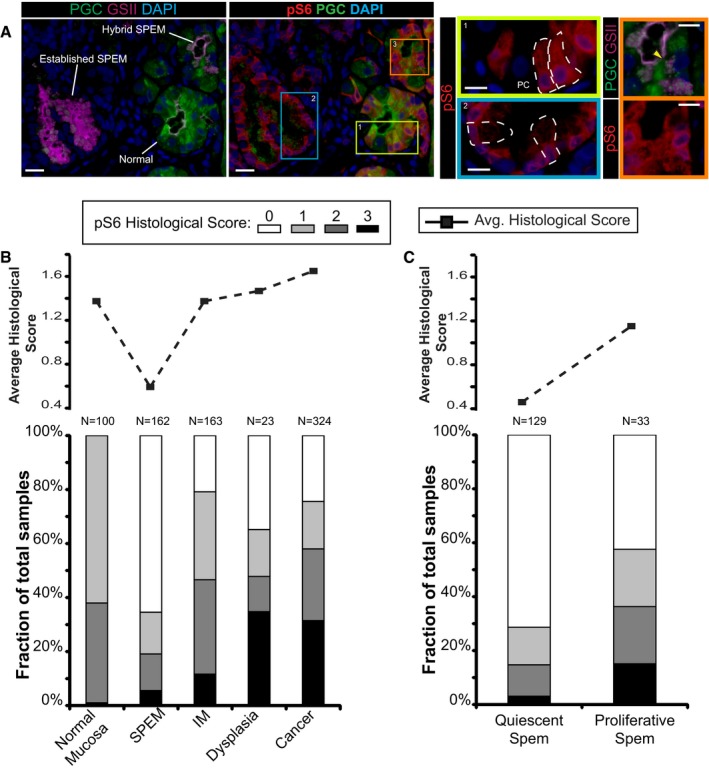
- Immunofluorescent images of human gastric tissue from a patient with intestinal‐type gastric adenocarcinoma elsewhere. In this non‐carcinoma containing region of the gastric corpus, various states of metaplasia can be observed that reflect mouse injury models. Extensive previous work (Lennerz et al, 2010) of a dataset of such resection specimens and of biopsies showing SPEM in a non‐cancer setting has indicated likely stages of progression of SPEM from essentially normal wherein large, pyramidal‐columnar cells at the base express only chief cell markers like pepsinogen C (PGC, green) to “hybrid SPEM” (yellow arrowhead, inset) where smaller, cuboidal columnar cells label with varying degrees of PGC and the neck/SPEM cell marker GSII (purple) to “established SPEM” characterized by cells that label extensively with GSII and have scant PGC; established SPEM cells are mucus‐stuffed, with peripheral, basal, flattened nuclei (blue, DAPI). Higher magnification of each cellular phenotype is shown by color‐coded box on right. As parietal cells are lost in SPEM, the remnant one in the yellow boxed area (labeled “PC”) is consistent with the normal chief cell phenotype (representative individual cells outlined by white dashed lines). Note that there is consistently high expression of pS6 (red) throughout the cytoplasm of such normal chief cells but that this pS6 varies in the hybrid SPEM lesion and is largely scaled down in the established SPEM region (note pS6 only around the nuclei of these cells). Scale bar, 20 μm; pullouts 10 μm.
- Analysis of a human gastric tissue microarray with normal, metaplastic, and cancer tissue all represented from patients with resections for gastric cancer. Serial tissues sections of the array were stained by immunohistochemistry with pS6 or Ki67, counterstained with hematoxylin, and visually graded by blinded observers, supervised by a human pathologist, for staining intensity (from score 0 meaning undetectable to 3 most intense). Top—average histological score is plotted for each phenotype. Bottom—the relative fraction of tissue cores with each score is plotted (total scores of each type provided at the top of each column.
- Given the biphasic nature of the SPEM histological score and given that established SPEM, as observed in panel (A), shows decreased pS6, we separated all the SPEM lesions into Ki‐67+ (“proliferative”) and Ki‐67− (“quiescent”) and replotted as for panel (B).
To further clarify the link between mTORC1 activity and metaplastic changes in humans, we analyzed pS6 levels in gastric tissue microarrays (Appendix Table S1) comprising tissue cores representing the following histological phenotypes: normal mucosa, SPEM, IM, dysplasia, and gastric adenocarcinoma. pS6 showed consistent, mid‐level expression in nearly all normal mucosal samples, in agreement with our smaller sample showing expression of pS6 in normal chief cells and with our mouse data (Fig 3B). Both cancer and dysplastic lesions showed higher average pS6 expression, though there was also more variability in that over a third of such lesions showed much stronger expression than normal tissue, while about a third showed lower expression (Fig 3B). On average, intestinal metaplasia pS6 levels were close to those of normal mucosa (Figs 3B and EV5). SPEM lesions showed a clear biphasic pattern with the majority like the “established SPEM” with low‐to‐no detectable pS6 (cf. Figs 3A and EV5) but with some SPEM lesions having much stronger pS6 (Figs 3 and EV5).
Figure EV5. Representative IHC images from human tissue microarray.
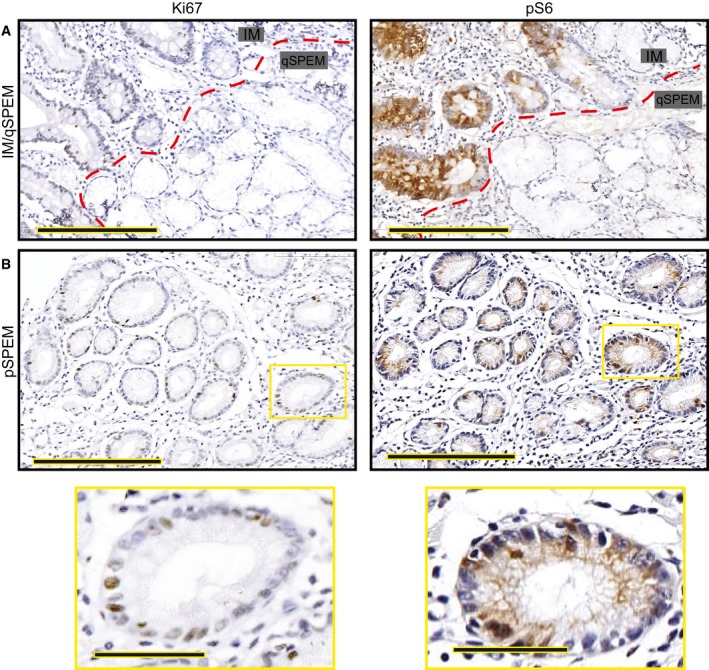
- Intestinal metaplasia (“IM” indicating the glands to upper left of red dashed line) is generally proliferative as evinced by frequent Ki‐67+ cells (left) and is strongly pS6 positive. Most SPEM has a quiescent phenotype (glands labeled on “qSPEM” side of panels) characterized by cells with abundant mucus, flattened basal nuclei, and a lack of both Ki‐67 and pS6 staining Scale bar, 200 μm.
- Rare SPEM lesions show cells with cuboidal columnar morphology. These lesions show Ki‐67 positivity usually associated with pS6 positivity. Boxed regions are shown at higher magnification below. Scale bar, 200 μm; pullout, 50 μm.
SPEM lesions with lower pS6 activity tended to express abundant mucin as well as epitope for the SPEM‐identifying lectin GSII (Fig 3A); nuclei tended to be flat and eccentric (Fig EV5). pS6‐expressing SPEM cells were more cuboidal columnar, resembling the SPEM cells in the acute, proliferative mouse SPEM that resolves in a few days after HD‐Tam. We hypothesized that SPEM with increased pS6 represented metaplastic cells that are actively proliferating (like D3 HD‐Tam in mice) to repair an injury, whereas the decreased pS6 lesions of established SPEM may be mitotically quiescent. Hence, we divided the SPEM lesions into mitotically active (“proliferative SPEM”) and inactive (“quiescent SPEM”) based on Ki‐67 staining of the same tissue core on another microtome section (Fig EV5) and then correlated those phenotypes to the previously scored pS6 expression for that lesion. Proliferative SPEM was far more likely to be associated with pS6 expression, whereas quiescent SPEM was largely negative for pS6 (Fig 3C, P < 0.001 by χ2). Thus, pS6 is low‐moderate in normal, physiologically active mucosa and high in most lesions that have increased proliferation (proliferative SPEM, IM, dysplasia, cancer). We conclude that metabolic activity correlates with differentiation state and recruitment into the cell cycle in humans as well as mice.
Loss of mTORC1 inhibits cell cycle progression at S‐phase
Because gastric chief cells respond to injury more synchronously than pancreatic acinar cells, we are able to perform molecular analyses based on changes of gene expression. We used this approach to determine specifically where the block in cell cycle re‐entry occurs when mTORC1 activity is inhibited. We analyzed Affymetrix GeneChips of whole gastric corpora ±HD‐Tam (3D) ±rapamycin by Gene Set Enrichment Analysis (GSEA) with a combination of both a publicly available and custom gene sets. In a control experiment to validate our approach, we dissociated gastric epithelial cells from Atp4b‐Cre; ROSA26 mTmG mouse stomachs and used flow cytometry to isolate parietal cells (GFP+) from other epithelial cells (Tomato+). Expression of isolated, amplified RNA applied to GeneChips was analyzed by Partek Genomics Suite, and the 94 genes whose expression was enriched ≥ eightfold in parietal cells vs. other epithelial cells was computed. As expected, GSEA showed that these PC‐enriched genes were highly preferentially expressed in control stomachs vs. HD‐Tam stomachs; the addition of rapamycin did not affect this pattern (Appendix Fig S1). Thus, global gene expression profiling with GSEA can detect the loss of parietal cells that epitomizes HD‐Tam‐induced metaplasia and also shows that parietal cell loss is independent of mTORC1, consistent with the histological data. In another control experiment, we performed GSEA of a published gene set of mature chief cell enriched genes (Capoccia et al, 2013) and contrasted HD‐Tam vs. HD‐Tam + rapamycin. There was no substantial effect of rapamycin, suggesting that the change in chief cell gene expression induced by injury is also not substantially affected by loss of mTORC1 (Appendix Fig S1).
On the other hand, although many transcripts from a previously published gene set of SPEM‐associated genes (Nozaki et al, 2008) did not show particular changes when rapamycin was administered in HD‐Tam, there was a cluster of genes enriched only when mTORC1 levels were normal (Appendix Fig S1). Injury that causes metaplasia induces both wound‐healing‐associated genes (e.g., Clu, Sox9, CD44v) and proliferation‐associated genes. Given that rapamycin blocks proliferation specifically in our histological analysis, we next examined the effects of rapamycin on the cell cycle using GSEA. Figure 4A shows that, indeed, rapamycin induces a marked de‐enrichment of cell cycle gene expression in HD‐Tam. The block appears specifically at the S‐phase and beyond, as gene sets for G1‐S, S, G2, and G2‐M showed that G1‐S genes were relatively similarly distributed regardless of mTORC1 activity, whereas genes expressed during the later stages in the cell cycle were skewed toward the HD‐Tam alone condition (Fig 4B–E). We used a slightly different approach to further investigate the interaction of mTORC1 with cell cycle stage by first determining the top 20 genes skewed most toward the HD‐Tam (vs. vehicle‐treated controls) in each cell cycle stage gene set. We then determined the average increased expression of those genes in both HD‐Tam and HD‐Tam + rapamycin vs. vehicle controls. Figure 4F shows that rapamycin decreased expression of the 20 top G1/S‐phase HD‐Tam‐enriched genes by only 16 ± 3%, whereas gene expression at other cell cycle stages was inhibited substantially more. Expression of G2/M‐phase genes was decreased by 49 ± 3% with rapamycin treatment (P < 0.001, HD‐Tam vs. HD‐Tam + rapamycin in G2‐M genes; P < 0.05 for G2‐M vs. G1‐S).
Figure 4. mTORC1 activity is required predominately for progression through S‐, G2‐, and M‐phases during metaplastic induction of proliferation.
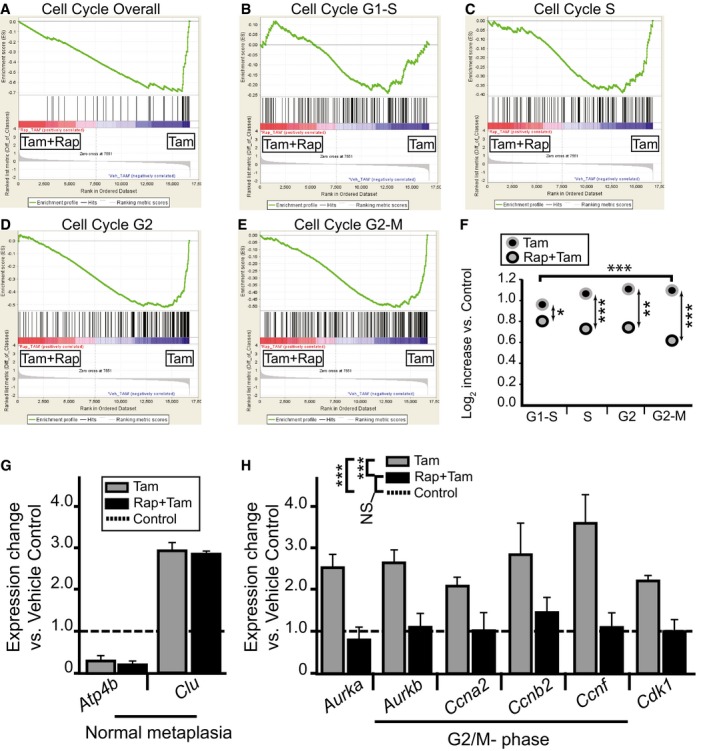
-
A–EMicroarrays of stomach corpora at D3 ± HD‐Tam ± rapamycin were analyzed using GSEA “Difference of Classes” function comparing rapamycin + HD‐Tam (“Tam+Rap”) vs. rapamycin vehicle + HD‐Tam (“Tam”). Whitfield Gene Sets specific for either overall cell cycle genes or specific phases of cell cycle are depicted. Note that rapamycin correlates with decreased cell cycle gene expression that is largely due to decreased S‐G2 phase gene expression.
-
FThe dot plots are of the actual average expression levels (in rapamycin + HD‐Tam and HD‐Tam alone Genechips) of the top 20 genes enriched in various Whitfield Gene Sets GSEA comparisons of HD‐Tam vs. vehicle controls (both without rapamycin). Expression levels of HD‐Tam and HD‐Tam + rapamycin for all genes were normalized to expression level in vehicle control Genechip to facilitate plotting and expressed as Log2 such that 1 = 2‐fold enriched vs. control. Note that average expression of G1‐phase genes is only somewhat reduced by rapamycin (by t‐test of Tam vs. Rap‐Tam, *P < 0.05, **P < 0.01; ***P < 0.001), whereas later phases of the cell cycle are substantially reduced (decrease in G2/M‐phase relative to G1‐phase by ANOVA with Dunnett's post hoc test is ***P < 0.001).
-
G, HqRT–PCR of select transcripts. Control genes known to be increased or decreased in SPEM (G) and genes associated with specifically with G2‐M cell cycle phase (H). Expression was normalized to housekeeping gene Tbp, then vehicle control samples for each gene were set at 1, and HD‐Tam and HD‐Tam + rapamycin expression was normalized to the control sample (statistics for the entire set of cell cycle genes among the different treatments are shown in legend, ***P < 0.001 by ANOVA with Tukey's post hoc test; data represented as mean ± SEM of the means from 3 replicates from a total of 3 independent experiments).
To independently validate the GeneChip findings, we performed qRT–PCR that showed that the expected decreases in a parietal cell (Atp4b) transcript and increase in a non‐cell‐cycle SPEM transcript (Clu) were not affected by rapamycin (Fig 4G). Also matching the GeneChip results, the G1 transcript, Ccnd1, was increased similarly regardless of mTORC1 status. As expected, a G2/M‐phase transcript cohort was uniformly increased in HD‐Tam but not in HD‐Tam + rapamycin (Fig 4H). Thus, molecular analysis indicates that inhibition of mTORC1 activity does not substantially affect chief cell G1‐phase entry from the quiescent, G0 state but slows S, G2, and M‐phase progression. BrdU uptake and incorporation into DNA occurs during S‐phase; thus, the block in BrdU seen histologically corroborates the molecular data suggesting that mTORC1 is required for G1 to S transition.
Autodegradative machinery is massively upregulated early following injury
We so far have observed that mTORC1 activity is rapidly extinguished within hours of inducing injury. Later, as cells re‐enter the cell cycle, mTORC1 is rekindled. Blocking re‐emergence of mTORC1 activity inhibits induced proliferation in both stomach and pancreas. In pancreas, where repair is entirely dependent on reprogramming, loss of mTORC1 activity blocks tissue regeneration. We hypothesized that the scaling down of mature cell architecture to “retool” a cell for more efficient proliferation would likely involve activation of lysosomes and autophagic machinery. The autodegradation of cellular structure could then liberate key macromolecules (nucleotides, amino acids, lipids) that would both stimulate mTORC1 reactivation and provide building blocks for replication. Figure 5 shows that there is a massive increase of lysosomes (by luminal marker Cathepsin D, Fig 5A) and autophagosomal puncta (by LC3‐GFP, Fig 5C) early following injury in gastric chief cells. Figure 5B quantifies a large spike in lysosomes, as a percentage of their PGC+ (pepsinogen C; chief cell marker) cell area, by 12–24 h of HD‐Tam that begins to resolve by later stages, when many cells have re‐entered the cell cycle. Increased lysosomes, autophagosomes, and autolysosomes can also be seen at the ultrastructure level (Fig 5D and E) on transmission electron microscopy (tEM). tEM analysis shows that rER, mitochondria, and secretory granules are all targeted for recycling during these early stages. The pancreas also shows an equivalent time course of changes in autodegradative machinery, with a spike in lysosome and autophagic puncta 8–24 h following cerulein, followed by decreasing, but still elevated levels, at D3 and near baseline levels at the time of maximal proliferation and pS6 activity (D5: Fig 5F, Appendix Fig S2).
Figure 5. Lysosomal and autophagic pathways are upregulated acutely following stomach and pancreas injury.
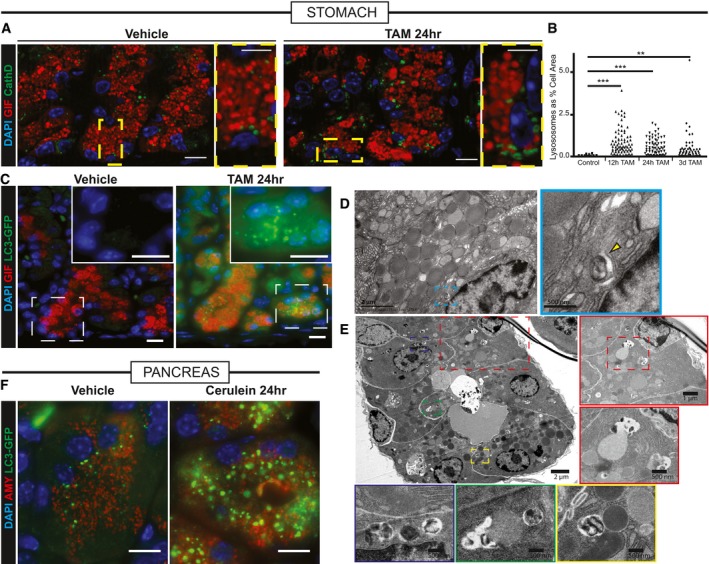
- Injured zymogenic cells upregulate Cathepsin D+ puncta (green) 24 h follow HD tamoxifen. Red, chief cells (GIF); blue, nuclei (DAPI). Boxed areas are shown at higher magnification at right of each panel. Scale bars 20 μm; 10 μm for pullouts.
- Quantification of Cathepsin D+ area in chief cells at various stages following injury. **P < 0.01; ***P < 0.001 by ANOVA with Dunnett's post hoc test. Each datapoint is an individual counted cell.
- LC3 puncta (detected by GFP fluorescence in Lc3‐gfp mice) shows increased autophagosomal puncta paralleling Cathepsin D+ results. Green, LC3‐GFP; red, GIF; blue, DAPI. Boxed areas are shown at higher magnification and differing fluorescence channels in insets. Scale bars 20 μm.
- Transmission electron micrographs of a normal zymogenic cell. Yellow arrowhead indicates a rare lysosome seen during homeostatic conditions.
- Transmission electron micrographs of corpus units 24 h follow tamoxifen injury. Various selected pullouts highlight double membrane‐bound structures attacking cytosolic components in reprogramming chief cells.
- Acinar cells in pancreas have increased LC3‐GFP+ puncta following acute injury with cerulein. Green, LC3‐GFP; red, GIF; blue, DAPI. Scale bar 20 μm.
Autodegradative machinery is required for normal progression to later stages
We next sought to address whether autodegradative machinery activation is both upstream of and required for metaplasia formation and proliferation. To do this, we used mice defective in lysosomal hydrolase trafficking that have been shown previously to have defects in autodegradative function specifically in exocrine secretory cells like chief and acinar cells (Boonen et al, 2011). Gnptab −/− mice are deficient in an enzyme required for the addition of mannose‐6‐phosphate to lysosomal enzymes to ensure their proper trafficking. We treated Gnptab −/− and littermate controls (Gnptab −/+ and Gnptab +/+) with HD‐Tam or cerulein. HD‐Tam treatment in Gnptab −/− mice caused the expected loss of parietal cells; however, chief cell reprogramming was dramatically compromised (Fig EV6). Most units did not show loss of large chief cells with eccentric nuclei at all (red arrowhead, Fig EV6), suggesting reprogramming did not occur, whereas some gastric units showed complete loss of the base zone where chief cells normally reside (green arrowhead, Fig EV6), indicating chief cells were aberrantly lost instead of reprogrammed. Rarer gastric units seemed to complete the reprogramming (yellow arrowhead, Fig EV6). In pancreas, we detected almost no ADM in Gnptab −/− mice (Fig EV6) by D5. Rather, cells remained in an aberrant acinar morphology with considerable loss of eosinophilic cytoplasm but no decrease in size. By 2 weeks, whereas wild‐type controls had largely adapted to the cerulein injury, in Gnptab −/− mice, the exocrine pancreas comprised only scattered ducts and SOX9− acinar cells, still organized in typical lobules. Cytologically, these remnant cells were characterized by generous pale cytoplasms ranging from foamy to hyaline and lacking nearly all distinguishing features.
Figure EV6. Histological appearance of Gnptab −/− stomach and pancreas tissue at injury time points.
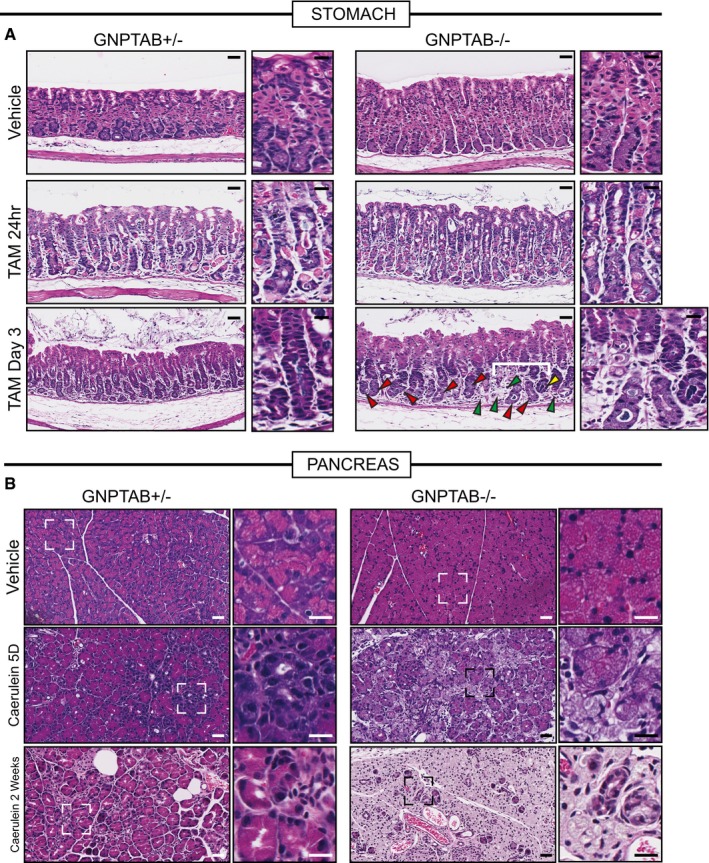
- Representative hematoxylin and eosin counterstained images of Gnptab +/− and Gnptab −/− stomach tissue. Gnptab −/− chief cell cytoplasms have a hypertrophic, frothy appearance compared to control zymogenic cells. Loss of parietal cells (fried‐egg appearing eosinophilic cells) following HD‐Tam is not affected by loss of GNPTAB; however, the base zones in Gnptab −/− mice at day 3 HD‐Tam are usually resistant to dedifferentiation (red arrowheads) with large, frothy chief cells remaining largely non‐reprogrammed. Another, less common phenotype is that all chief cells are lost such that most of the base of the unit disappears (green arrowheads). Rare units partially undergo morphological metaplastic changes, though usually those are also associated with loss of basal cells (yellow arrowheads). Higher magnification views are to right of each panel, with white bracket delineating particular region of interest in Gnptab −/− stomach Scale bar 50 μm; pullout, 25 μm.
- Representative hematoxylin and eosin counterstained images of Gnptab −/+ and Gnptab −/− pancreas. Similar to the stomach zymogenic cells, pancreatic acinar cells also have a hypertrophic, frothy appearance. Whereas control samples treated with cerulein show diffuse, asynchronous acinar‐to‐ductal metaplasia, Gnptab −/− mice have acinar cells that simply become less eosinophilic and foamy over time without undergoing ADM. By 2 weeks, wild‐type pancreas has largely adapted to cerulein, whereas Gnptab −/− pancreas parenchyma comprises only lobules of excessively pale (hyaline), frothy acinar cells and scattered reactive ducts. Scale bar 50 μm; pullout, 25 μm.
We next examined the molecular phenotype of the block in Gnptab −/− mice. In control stomachs in response to injury, reprogramming cells in the base showed the expected abundant increase in metaplastic genes like Sox9 (Fig 6A) and the epitope for GSII (Fig 6C). Proliferation in the base of the unit, where chief cells were reprogramming, was nearly equivalent to the rate of proliferation in the normal stem cell zone in the neck (Fig 6B and D). The bases of gastric units in Gnptab −/− mice were markedly compromised in both metaplastic changes and proliferation (Fig 6A–D). In Gnptab −/− mice, chief cells in the base remained both BrdU‐ and SOX9‐negative (Fig 6E and F). They also failed to reactivate mTORC1, as pS6 in these mice was largely not detectable in the base (Fig EV7).
Figure 6. Lysosomal function is required for metaplasia‐associated gene expression and increased proliferation.
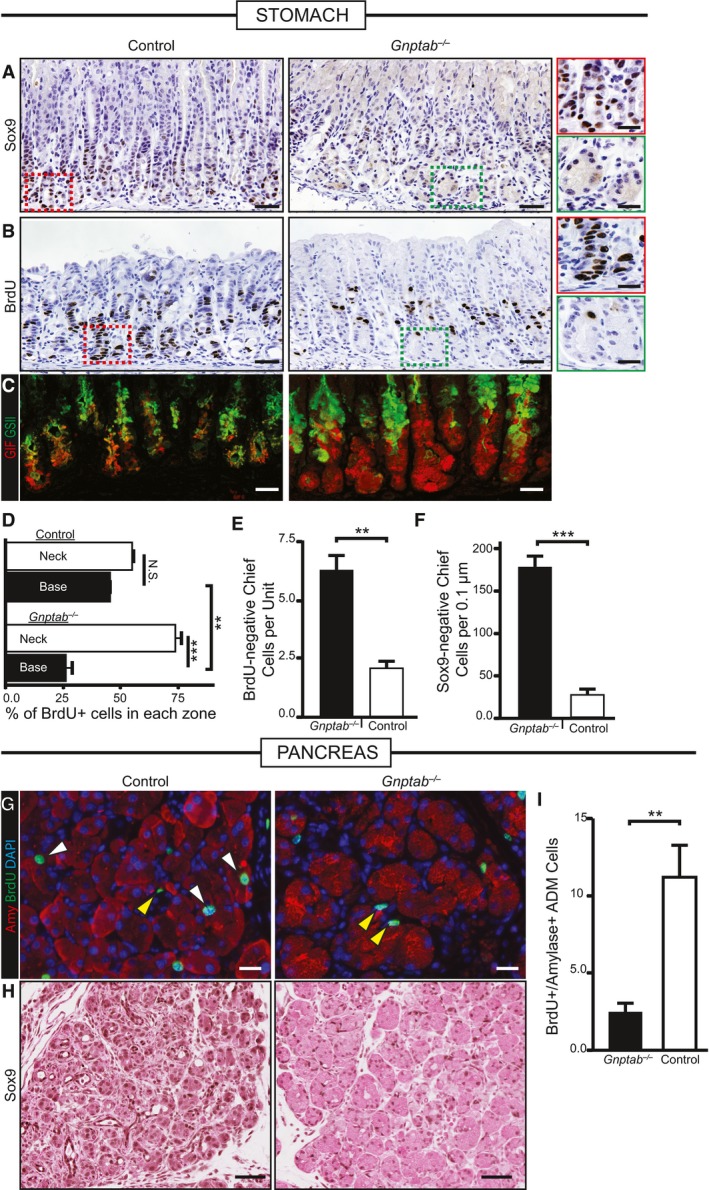
- Immunohistochemical analysis of SOX9 expression at peak SPEM stages following gastric injury. In control and Gnptab −/+ mice, SOX9 becomes expressed in reprogramming chief cells in the bases of the corpus at SPEM stages, but not in Gnptab −/− mice. Color‐coded boxes shown at higher magnification shown at right for panels (A and B). Scale bar, 50 μm; 25 μm pullout.
- S‐phase, cell cycle marker BrdU is incorporated throughout the gastric corpus unit at peak SPEM stages in control WT or Gnptab −/+ mice. In Gnptab −/− mice, the gastric unit bases, where proliferation is recruited from chief cells, show a marked relative deficit in BrdU+ cells. Scale bar, 50 μm; 25 μm pullout.
- Immunofluorescence analysis of injured gastric tissue from Gnptab −/− and control mice. GIF/GSII co‐expression is the hallmark of SPEM. In control mice, the vast majority of corpus unit bases are converted to GIF/GSII co‐expression state. In Gnptab −/− mice, bases are resistant to conversion and remain as GIF single positive cells. Red, GIF; green, GSII. Scale bar, 20 μm.
- Quantification of randomly sampled 20× fields stained with BrdU. Distribution of BrdU in neck region vs. base region (note total = 100%) is plotted. Note control mice have equivalent amounts of BrdU‐labeled cells in the neck and base (˜50% in each), whereas Gnptab −/− mice BrdU‐labeled cells substantially shifted away from the paligenotic base of units and into the isthmal‐neck region, where the constitutive stem cell is active.
- Quantification of randomly sampled 20× fields stained with BrdU in control and Gnptab −/− mice. Gnptab −/− mice have significantly more BrdU‐negative base cells compared to control animals.
- Quantification of control and Gnptab −/− corpus units stained for SOX9 scored for the amount of SOX9‐negative chief cells per unit at peak SPEM stages. Gnptab −/− mice have significantly more SOX9‐negative bases compared to control animals.
- Representative immunofluorescence images of injured control and Gnptab −/− pancreatic tissue at cerulein 5 days. Red, amylase; green, BrdU; blue, DAPI. White arrows show proliferating, amylase+ acinar‐derived cells (note these are not seen in Gnptab −/− mice). Yellow arrowheads show proliferating stromal cells that are not affected by loss of GNPTAB. Scale bar, 20 μm.
- Representative immunohistochemistry of SOX9 stained control and Gnptab −/− pancreatic tissue at cerulein 5 days. Gnptab −/− tissue has reduced metaplastic phenotype and reduced expression of SOX9. Scale bar, 50 μm.
- Quantification of amylase+BrdU+ cells of control and Gnptab −/− tissue in randomly sampled 20× fields at 5 days of cerulein injury.
Figure EV7. Lysosomal activity is required to re‐activate mTORC1 following HD tamoxifen injury.
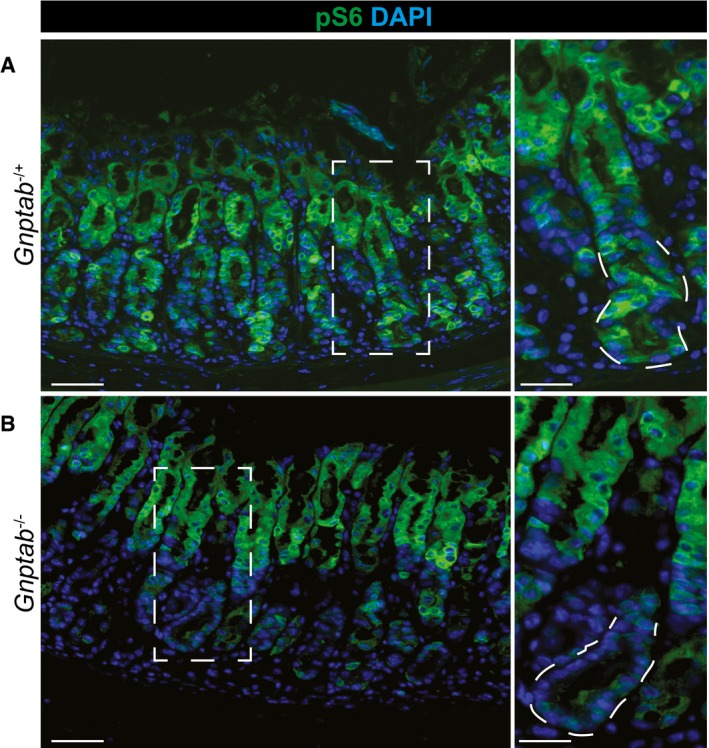
- At peak metaplasia stages in Gnptab −/+ tissue, pS6 is re‐expressed throughout the stomach epithelium, including intense staining within the pit and metaplastic base. Scale bars: 50 μm; pullout, 25 μm.
- In Gnptab −/− tissue, pS6 is not reactivated in the base, indicating lysosomal activity is required for mTORC1 re‐activation at later stages following injury. Boxed regions are shown at higher magnification at right with a representative base (in which pS6 remains inactive without lysosomal activity) outlined by dotted line. Lysosomal activity appears dispensable for pit cells (at top of gastric unit) mTORC1 activity. Scale bars: 50 μm; pullout, 25 μm.
In the pancreas, there was a similar defect in both BrdU (Fig 6G and I) and Sox9 (Fig 6H). The remnant acinar cells that remained in Gnptab −/− mice following 2 weeks of cerulein treatment expressed E‐cadherin and low levels of amylase but were not positive for other mature acinar nuclear markers like GATA4 or metaplasia markers like CK8/18 (Appendix Fig S3).
Finally, to determine whether the dropout of gastric bases was due to increased cell death in the absence of lysosomal hydrolase activity, we examined tissue for cleaved caspase 3. In wild‐type mice (either with or without rapamycin), we did not detect substantial apoptotic death of the chief cells, consistent with our previous observations that death in HD‐Tam is essentially confined to parietal cells (Huh et al, 2012; Radyk et al, 2018; Fig EV1). In Gnptab −/− mice, however, we frequently observed multiple cells in some bases of gastric units that were undergoing apoptosis (Appendix Fig S4). Thus, in stomach, aberrant autodegradative function leads either to stalling of the chief cell reprogramming process or cell death. In pancreas, we observed a pattern of scattered apoptosis of acinar cells in wild‐type mice ±rapamycin following cerulein treatment. Loss of GNPTAB did not seem to affect this basal rate of death, which is consistent with the survival of many acinar cell remnants out to 2 weeks, as discussed above.
Discussion
There has been a recent burgeoning of examples of cellular plasticity in tissue in response to injury, not to mention a growing, already large literature on in vitro systems for reprogramming cells back to progenitors. The instances of such plasticity span numerous species and nearly all tissues. Despite the breadth of examples of cellular reprogramming, studies focusing on the specific molecular mechanisms responsible for the process are still relatively scant. This is particularly true in studies of cells in tissue, likely because investigators have focused more on the outcome of cellular reprogramming—regeneration or tumorigenesis—than on the stepwise mechanisms differentiated cells use to contribute to those outcomes. Here, we have speculated that there could be a shared cellular program that governs the many diverse examples of differentiated cells changing their fate to facilitate repair. There have been many terms that either focus on the outcome of the program or are overly broad: “dedifferentiation”, “transdifferentiation”, “reversion”, “reprogramming”. We now propose “paligenosis” as a specific term describing the cellular process differentiated cells use to re‐acquire regenerative capacity. We highlight that paligenosis may be a conserved cellular process with shared molecular and cellular regulation akin to other basic cellular processes like mitosis and apoptosis.
To support our assertion that there may be a shared program for recruiting differentiated cells, we have analyzed the cellular and molecular changes that occur during injury‐induced reprogramming in two distinct organs. Upon injury, both the stomach and pancreas have the capacity to repair tissue damage through the recruitment of fully differentiated cells into a less differentiated, proliferative state to replenish cell numbers. This pattern of change in cell phenotype is known to pathologists as metaplasia. We find that the cellular and molecular changes that characterize cells undergoing such metaplastic injury response in either stomach or pancreas are remarkably similar. Specifically, we found that acutely following injury, autodegradative pathways increase alongside a decrease in mTORC1 activity (Fig 7). As the injury progresses, we observed the induction of genes that are known to occur during metaplasia followed by the rise of mTORC1 activity and increased proliferation (Fig 7). A similar pattern of changes in mTORC1 activity relative to metaplasia and the differentiated vs. proliferative phenotype was observed in human patients. We found that mTORC1 activity was specifically required for progression through S‐phase. Previous literature has also shown that mTORC1 activity is critical for S‐phase progression of cancer cells following DNA damage, as mTORC1 is needed to generate pyrimidines in a nutrient‐poor environment (Robitaille et al, 2013; Silvera et al, 2017; Zhou et al, 2017). mTORC1 activation is also needed for yeast to pass through G1 into S‐phase as they emerge from quiescence (Dhawan & Laxman, 2015; Moreno‐Torres et al, 2015). Using an animal model of lysosomal dysfunction, we uncovered that normal lysosomal function after injury is required for cell phenotype and gene expression changes associated with metaplasia. In pancreas, where constitutive stem cells are not available for regeneration, loss of either autodegradative function or mTORC1 activity compromised eventual organ repair.
Figure 7. Schematic model of shared program: paligenosis.
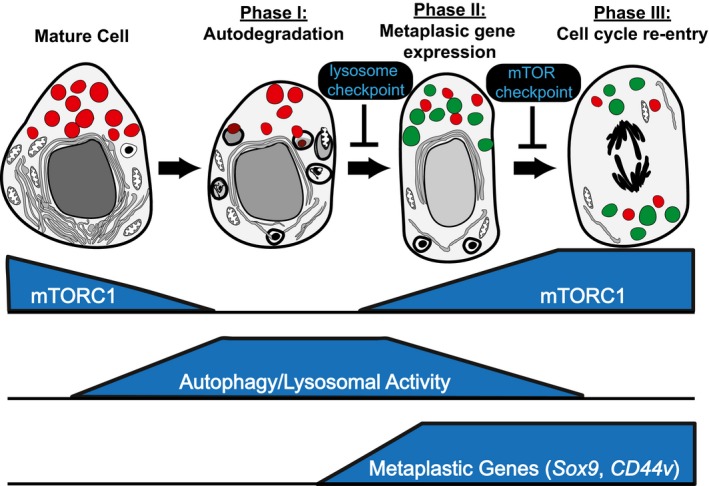
Data presented in the paper suggest that differentiated cells revert to a regenerative/proliferative state via a program involving stepwise progression through three stages. Progression can be blocked at intervening checkpoints. The potential context‐independent nature of this sequence of structural‐energetic changes suggests that it is available to differentiated cells in multiple organs and species. We have termed this general program of differentiated cells acquiring regenerative potential “paligenosis”.
Recent advances in the understanding of how mTORC1 is controlled have described a role for the lysosome as an activator of the pathway through the release of nutrients like key amino acids (Zoncu et al, 2011). Thus, our current working model is that due to injury‐induced stress, autodegradative pathways are upregulated, and flux increases. The activation of autodegradative pathways appears to act in parallel with loss of the mature gene regulatory network, as forcing expression of key mature‐cell‐promoting transcription factors like MIST1 (BHLHA15) impairs the injury/repair process (Direnzo et al, 2012; Lo et al, 2017). MIST1 controls a cassette of genes that help direct a cell's energy toward secretion and away from lysosomal activation and autophagy (Mills & Taghert, 2012). We reason, as did Adami over a century ago, that to convert from the differentiated state (structurally complex, energetically active) to the replicative state (structurally simple, energetically active), cellular energy use must be repurposed as an autodegradative program is activated to convert differentiated cell structure into building blocks for replication. The release of nutrients through the lysosome is sensed in cells during the autodegradative phase, resulting in reactivation of mTORC1, which, once the cell has reached sufficient energy levels, subsequently facilitates cell cycle progression and growth to replace cells lost during the injury.
Pancreatic adenocarcinoma and—to a lesser extent—gastric adenocarcinoma are commonly driven by oncogenic mutations in Kras. In mouse models in both the pancreas (Hingorani et al, 2005) and stomach (Choi et al, 2016), Kras G12D mutations, in concert with tissue inflammation, promote changes in gene expression and cell phenotypes resembling injury‐induced metaplasia. In the pancreas, genetically disabling autophagy in the context of K‐Ras mutations prevents K‐Ras from driving high‐grade lesions (Rosenfeldt et al, 2013). Furthermore, cells unable to phosphorylate S6 in the context of activating K‐Ras mutations also exhibit less pancreatic cancer progression (Khalaileh et al, 2013). A similar critical role for mTORC1 downstream of another key driver oncogene pathway, Wnt activation mediated by APC mutation, has been described in intestinal carcinogenesis (Morran et al, 2014). Thus, tumorigenesis in diverse tissues may also involve modulating lysosomal activity and mTORC1, similar to what we observe in our injury models here. Other pathways downstream of K‐Ras, such as PI3K/Rac1 signaling (Heid et al, 2011; Wu et al, 2014), also play similar roles in injury‐induced metaplasia.
If there truly is a shared cellular program, paligenosis, underlying the process of recruiting mature cells to become regenerative cells, we would expect the general features we have described here in stomach and pancreas to be recapitulated in many other tissues and species. Obviously, it will be important to conduct new studies in other systems to begin to support that assertion; however, we can at this point re‐examine the extant literature to determine whether roles for lysosomes/autophagy and/or mTORC1 in the process of cellular reprogramming to a regenerative state have previously been described. One such previous study, using a different injury protocol, with the endpoint to determine the role of mTORC1 and autophagy in severity of pancreatitis, similarly showed a pattern of early autodegradation followed by mTORC1 activation (Hu et al, 2015). The authors also found that rapamycin worsened severity of pancreatitis. In liver, it has long been known that the earliest phase of hepatocyte response to partial hepatectomy is massive activation of autophagy/lysosomes (Becker & Lane, 1965). mTORC1 is required for the later stages of the process, when proliferation is maximal, consistent with observations we make in the current manuscript (Jiang et al, 2001; Nelsen et al, 2003; Buitrago‐Molina et al, 2009; Espeillac et al, 2011). In kidney, the reprogramming process involves mTORC1 (Kato et al, 2012), and we show here that mTORC1 activity is increased specifically in the tubular cells, which are the cell population called back into the cell cycle to regenerate damaged tissue. To our knowledge, lysosomes/autophagy has not been examined in regenerating kidney. In mature glial cells that dedifferentiate following axonal injury, activation of autophagy/lysosomes is a well‐established early event (Jessen & Mirsky, 2016). To our knowledge, mTORC1 activity has not been examined in the process. Furthermore, in tissue culture cellular reprogramming models to generate induced pluripotent stem cells, there is an emerging literature that an early autophagy phase is followed eventually by mTORC1 activation. Inhibition of either autophagy or prolonged inhibition of mTORC1 reduces reprogramming efficiency (He et al, 2012; Wang et al, 2013; Wu et al, 2015). Hence, the stages and checkpoints appear to be the same as the ones we examine in the current manuscript.
Thus, there are numerous reports indicating that the pattern we show here systematically of autodegradation first, then mTORC1 activation may be universal. Moreover, teleologically, it makes sense that a mature cell would first recycle cellular components required for physiological function to use them as substrates for subsequent synthesis of components needed for proliferation. In organs like the vertebrate pancreas or liver, where there are no constitutively active stem cells, repair would likely depend in large part on paligenosis. In tissues with constitutive stem cells, like stomach and intestines, the tissues would have the choice of regenerating with either constitutive stem cells or paligenotic cells, depending potentially on type, extent, and location of injury.
Not all differentiated cells are likely to be able to undergo paligenosis. In the stomach, for example, we have never observed this phenomenon in mature parietal cells (Huh et al, 2012; Mills & Sansom, 2015). Cells that are constitutively undifferentiated and replicative like those of the isthmus of the stomach or LGR5+ crypt‐base columnar cells should not need any stage of paligenosis (Fig 7). They may acquire the building block nucleotides and amino acids from the blood and/or extracellular environment, given that, by definition, their lack of differentiation means they contain limited non‐nuclear components to recycle. Other cells, such as mucous neck cells in the stomach or +4 cells in the intestine (van Es et al, 2012; Roth et al, 2012; Buczacki et al, 2013), may be able to respond to injury but are less well differentiated and thus may be able to skip the autodegradative phase and go directly to the activating mTORC1 and cell division phase of paligenosis.
Paligenosis may be beneficial for its potential to provide lifelong tissue repair in adult organs, but this capacity also seems inherently tied to increased risk for tumorigenesis. Chronic injury of the type that repetitively induces paligenotic/metaplastic events has long been known to increase risk for acquisition of mutations and progression to neoplasm. We have proposed that the reason that risk increases with age is that cycles of paligenosis and subsequent redifferentiation allow accumulation of mutations that may be stored in long‐lived, differentiated cells. Eventually, a critical mutation may be unmasked during paligenosis, and a clone of cells that is unable to redifferentiate arises. We have termed this the “cyclical hit” model of tumorigenesis (Mills & Sansom, 2015; Saenz & Mills, 2018).
There are numerous questions that our current study prompts. What molecular events underlie the competence to pass through each stage of paligenosis? What is the relationship between paligenosis and chronic injury, and what causes the increased risk for cancer? Why are some cells able to undergo paligenosis, whereas others are not? We expect that the framework of sequential phases of paligenosis that we introduce here, along with the potential checkpoints that serve as molecular barriers between each stage of the process, can serve as a starting point for future questions.
Materials and Methods
Animal studies and reagents
All experiments using animals followed protocols were approved by the Washington University School of Medicine Animal Studies Committee. WT C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME). Tg(Atp4b‐cre)1Jig/JcmiJ (Atp4b‐Cre) (Syder et al, 2004), Gt(ROSA)26Sortm4(ACTB‐tdTomato,‐EGFP)Luo/J (ROSA26mtmg) (Muzumdar et al, 2007), Gnptab (Gelfman et al, 2007 ),and LC3‐GFP (Mizushima et al, 2004) mice were previously described. Gnptab mice were a kind gift from Dr. Stuart Kornfeld of Washington University. Tamoxifen (5 mg/20 g body weight; Toronto Research Chemicals) was injected intraperitoneally (IP) daily for 2–3 days to induce maximal gastric injury (Huh et al, 2012; Saenz et al, 2016). Tamoxifen was prepared by first dispersing in 100% ethanol by sonication and then emulsifying in sunflower oil (Sigma‐Aldrich) 9:1 (oil:ethanol). Pancreatitis was induced by 6 hourly IP injections of 50 μg/kg (in 0.9% saline) cerulein (Sigma‐Aldrich) given every other day for up to 2 weeks. Mice were sacrificed 24 h after the final cerulein injection. Rapamycin (60 μg/20 g body weight; LC Laboratories) was injected IP in 0.25% Tween‐20, 0.25% polyethylene glycol in PBS for 3–7 days prior to starting and throughout injury time course. Tunicamycin (Carlisle et al, 2014) and two‐thirds partial hepatectomy (Blanc et al, 2010) injuries were performed as previously described. Mice were given an IP injection containing 5‐bromo‐2′‐deoxyuridine (BrdU; 120 mg/kg) and 5‐fluoro‐2′‐deoxyuridine (12 mg/kg) in sterile water 90 min before sacrifice for all BrdU labeling experiments.
For parietal cell isolation, stomachs were harvested and washed several times with PBS. The forestomach and antrum were carefully removed and the remaining corpus minced with a razor blade. The tissue was mechanically dissociated using a 50 μm Medicon (Beckman) for two 30‐s pulses. Chunks of tissue were further dissociated by incubating in 10 ml HBSS with 5 mM EDTA and 1 mM DTT with vigorous shaking for 1 h at 37°C, and then, the solution was run through a 100‐μm filter. Single cells were allowed to rest at 37°C, while filtered chunks were incubated in 10 ml RPMI 1640 with 5% BSA (Sigma) and 1.5 mg/ml Dispase II (Stem Cell Technologies) with vigorous shaking for 1.5 h at 37°C and then filtered again. Dissociated cells were pelleted and washed with cold HBSS three times and then resuspended in PBS with 1% BSA and 5 mM EDTA. Cells were sorted into a parietal cell population (GFP) and all remaining cells (tdTomato) using a MoFlo FACS machine (Dako/Cytomation).
Imaging and tissue analysis
Mouse tissues were immediately excised and flushed with phosphate‐buffered saline and fixed overnight in 4% paraformaldehyde in PBS. Tissues were washed, embedded in 3% agar, and then underwent routine paraffin processing. Sections prepared for immunofluorescence or immunohistochemistry underwent standard deparaffinization and rehydration protocols, were blocked in 5% normal serum, and left overnight with primary antibodies. Sections were washed in phosphate‐buffered saline and incubated for 1 h with secondary antibodies and then washed prior to mounting. For antibodies used in this study, see Appendix Table S2. Immunofluorescence images were taken on a Zeiss Apotome or LSM710 confocal (Zeiss). Bright field images were taken on a Nanozoomer (Hamamatsu) whole slide scanner or DP70 microscope (Olympus). Counting of stomach cell populations and proliferation was done as previously described (Burclaff et al, 2017), except for analysis of Gnptab −/− mice. To account for frequent gland loss in the base of these mice, a different approach was taken. For chief cell quantification (SOX9+ and BrdU+), 10 random, 20× fields were chosen in three Gnptab −/− and three control animals, and chief cells scored in slides from SOX9 or BrdU immunostained sections. For BrdU, distribution, the 10 fields were further subdivided into two rectangular regions: a basal one 100 μm perpendicular and 450 μm parallel to the muscularis mucosa and a region of the same size immediately adjacent and encompassing the neck of the gastric unit. All BrdU+ cells were scored and the proportion in each zone calculated. Quantification of proliferation in the pancreas was done by counting 10 randomly sampled whole 20× fields per condition. Cathepsin D+ area was calculated by generating a region of interest around PGC+ zymogenic cell cytoplasms and using particle counting analysis in ImageJ (NIH) to calculate Cathepsin D+ area relative to total cytoplasmic area. Tissue preparation and imaging for electron microscopy was done as previously described (Ramsey et al, 2007).
Human tissue studies
Human gastric pathological tissue specimens were obtained with approval by the Institutional Review Board of Washington University School of Medicine. Figure 3A is a representative image from a qualitative analysis of 44 separate curated gastric clinical samples that have been previously described (Lennerz et al, 2010; Radyk et al, 2018). The study of tissue microarray cases included in this paper was also approved by the China Medical University First Hospital Institutional Review Board and Ethics Committee. This patient cohort was initially treated at the China First Medical University, and routine standard of care specimens was obtained from patients treated between 2005 and 2009. Tumor, metaplastic, and uninvolved normal tissue from each patient was formalin‐fixed and paraffin‐embedded. Staining was scored on the following scale: 0, no staining; 1, minimal staining; 2, moderate to strong staining in at least 20% of cells; 3, strong staining in at least 50% of cells. The scoring system was designed, and independently verified, by a human pathologist.
Bioinformatics, microarray, qRT–PCR, and statistical analyses
For qRT–PCR and microarray analyses of mouse stomach ±rapamycin, two independent experiments were run and a total of two to three separate mice and corresponding microarrays were generated for each condition. All mice were harvested 3 days after first injection and treated as per protocol in (Fig EV1). Conditions were Veh‐Veh (rapamycin vehicle regimen + 3 days of tamoxifen vehicle), Veh‐Tam (3 days of rapamycin vehicle regimen + 3 days of HD‐Tam), Rap‐Veh (rapamycin regimen, 3 days of tamoxifen vehicle); Rap‐Tam (rapamycin regimen + 3 days of HD‐Tam). RNA for microarray and qRT–PCR analysis was isolated as previously described (Lo et al, 2017). For microarray, samples were processed and hybridized to Affymetrix Mouse Gene 2.0 ST per the manufacturer's instructions by the Washington University Genome Technology Access Core (GTAC). GeneChips were analyzed with Partek Genomic Suite 6.6 (Partek, Inc.) analysis software using default settings (Lo et al, 2017). Mapping to Gene Symbols was done either via GSEA (Subramanian et al, 2005) or GenePattern software (Reich et al, 2006). GSEA was done using default 3.0 settings. GMX files were made using previously published microarray data in the case of laser‐capture micro‐dissected chief cells (Capoccia et al, 2013), generated de novo or acquired from GSEA molecular signatures database. For the list of parietal cell‐specific genes generated de novo for the current manuscript, flow cytometry was used to sort parietal cells and control cells into 500 μl RNA protect reagent (Qiagen). RNA was isolated using the RNeasy Micro Kit (Qiagen) following the manufacturer's instructions. Mouse Gene 2.0 ST Array (Affymetrix) was used to analyze gene expression, and the gene set whose expression was enhanced at least eightfold (96 separate genes) in parietal cells vs. control was determined by Partek. For primers used in qRT–PCR, see Appendix Table S3. Statistics for cell counts and qRT–PCR were done by Student's t‐test (in the case of pair‐wise analysis of significance) or ANOVA (if multiple conditions were compared). For determining statistically significant differences among various conditions in ANOVA, the post hoc tests were either Tukey's (for multiple crosswise comparisons of means) or Dunnett's (for comparisons of multiple experimental samples to a single control). For the tissue microarray, a χ2 analysis was performed.
Western blot
Approximately 100 mg mouse corpus stomach tissue was lysed in urea buffer (8 M urea, 1% SDS, 150 mM Tris–HCl, pH = 7.0) with 1× protease/phosphatase inhibitor cocktail (Thermo). Protein concentration was determined using the DC protein assay (Bio‐Rad). Protein (30 μg) was separated using a 10% SDS–PAGE gel and transferred to PVDF membranes (Millipore). Membranes were incubated overnight at 4°C with Rabbit polyclonal pS6 240/244 or 235/236 (1:1,000 diluted, CST) and Rabbit polyclonal beta‐tubulin antibody (1:1,000 diluted, CST) and then incubated with infrared fluorescent dye‐conjugated secondary antibodies (LI‐COR Biosciences). Protein signal intensities were normalized against a tubulin loading control for each sample. Fluorescent intensity values were determined and quantified on Western blots at non‐saturating exposures using the ImageJ software. Statistical analysis with both antibodies was done using ANOVA with a post hoc Dunnett's test.
Data availability
All analyzed microarray data have been deposited in NCBI GEO under accession GSE103570.
Author contributions
Study concept and design: SGW, MAL, JCM, GS, and DL. Data acquisition and analysis: SGW, MAL, Z‐FM, JCM, RLC, MDR, GS, and H‐YGL. Drafting of manuscript: SGW and JCM. Key manuscript revisions and figure preparation: SGW, JCM, MAL, JB, MDR, Z‐FM, NOD, and VB. Histopathological interpretation: SGW, MAL, Z‐FM, and JCM. Bioinformatics and statistics: JCM and Z‐FM. Supplied crucial reagents and funding: JCM, Z‐FM, Z‐NW, and DL.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
This study was supported by the NIDDK R01s (DK094989, DK105129, DK110406), Alvin J. Siteman Cancer Center/Barnes Jewish Hospital Foundation Cancer Frontier Fund, NIH NCI P30 CA091842, The Barnard Trust, and DeNardo Education & Research Foundation grants to J.C.M., NCI training grant (CA00954731) to S.G.W., NIH training grant (5T32GM007067‐43) to M.D.R., NIH training grant (GM007067) and the Philip and Sima Needleman Student Fellowship in Regenerative Medicine to J.B., National Natural Science Foundation of China Grant (81371688, 81673089) to D.L. HL38180, DK112378, and DK56260 to N.O.D. Histology was performed by the Digestive Disease Research Core Center (P30DK052574).
Conflicts of interest
The authors declare that they have no conflicts of interest.
The EMBO Journal (2018) 37: e98311
See also: https://doi.org/10.15252/embj.201899206 (April 2018)
Contributor Information
Zhen‐Ning Wang, Email: josieon826@sina.cn.
Jason C Mills, Email: jmills@wustl.edu.
References
- Adami JG (1900) On growth and overgrowth In “Festschrift” in honor of Abraham Jacobi, MD, LLD: to commemorate the seventieth anniversary of his birth, May sixth, 1900, Huber F, Sondern FE. (eds), pp 422–432. New Rochelle, NY: Knickerbocker Press; [Google Scholar]
- Adler G, Hahn C, Kern HF, Rao KN (1985) Cerulein‐induced pancreatitis in rats: increased lysosomal enzyme activity and autophagocytosis. Digestion 32: 10–18 [DOI] [PubMed] [Google Scholar]
- Becker FF, Lane BP (1965) Regeneration of the mammalian liver. I. Auto‐phagocytosis during dedifferentiation of the liver cell in preparation for cell division. Am J Pathol 47: 783–801 [PMC free article] [PubMed] [Google Scholar]
- Blanc V, Sessa KJ, Kennedy S, Luo J, Davidson NO (2010) Apobec‐1 complementation factor modulates liver regeneration by post‐transcriptional regulation of interleukin‐6 mRNA stability. J Biol Chem 285: 19184–19192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerboom A, Reusch C, Pieltain A, Chariot A, Franzen R (2017) KIAA1199: a novel regulator of MEK/ERK‐induced Schwann cell dedifferentiation. Glia 65: 1682–1696 [DOI] [PubMed] [Google Scholar]
- Boonen M, van Meel E, Oorschot V, Klumperman J, Kornfeld S (2011) Vacuolization of mucolipidosis type II mouse exocrine gland cells represents accumulation of autolysosomes. Mol Biol Cell 22: 1135–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczacki SJ, Zecchini HI, Nicholson AM, Russell R, Vermeulen L, Kemp R, Winton DJ (2013) Intestinal label‐retaining cells are secretory precursors expressing Lgr5. Nature 495: 65–69 [DOI] [PubMed] [Google Scholar]
- Buitrago‐Molina LE, Pothiraju D, Lamle J, Marhenke S, Kossatz U, Breuhahn K, Manns MP, Malek N, Vogel A (2009) Rapamycin delays tumor development in murine livers by inhibiting proliferation of hepatocytes with DNA damage. Hepatology 50: 500–509 [DOI] [PubMed] [Google Scholar]
- Burclaff J, Osaki LH, Liu D, Goldenring JR, Mills JC (2017) Targeted apoptosis of parietal cells is insufficient to induce metaplasia in stomach. Gastroenterology 152: 762–766 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkitt MD, Williams JM, Townsend T, Hough R, Pritchard DM (2017) Mice lacking NF‐kappaB1 exhibit marked DNA damage responses and more severe gastric pathology in response to intraperitoneal tamoxifen administration. Cell Death Dis 8: e2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capoccia BJ, Jin RU, Kong YY, Peek RM Jr, Fassan M, Rugge M, Mills JC (2013) The ubiquitin ligase Mindbomb 1 coordinates gastrointestinal secretory cell maturation. J Clin Invest 123: 1475–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlisle RE, Brimble E, Werner KE, Cruz GL, Ask K, Ingram AJ, Dickhout JG (2014) 4‐Phenylbutyrate inhibits tunicamycin‐induced acute kidney injury via CHOP/GADD153 repression. PLoS One 9: e84663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang‐Panesso M, Humphreys BD (2017) Cellular plasticity in kidney injury and repair. Nat Rev Nephrol 13: 39–46 [DOI] [PubMed] [Google Scholar]
- Choi E, Hendley AM, Bailey JM, Leach SD, Goldenring JR (2016) Expression of activated ras in gastric chief cells of mice leads to the full spectrum of metaplastic lineage transitions. Gastroenterology 150: 918–930 e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa P, Piazuelo MB (2012) The gastric precancerous cascade. J Dig Dis 13: 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan J, Laxman S (2015) Decoding the stem cell quiescence cycle–lessons from yeast for regenerative biology. J Cell Sci 128: 4467–4474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Direnzo D, Hess DA, Damsz B, Hallett JE, Marshall B, Goswami C, Liu Y, Deering T, Macdonald RJ, Konieczny SF (2012) Induced Mist1 expression promotes remodeling of mouse pancreatic acinar cells. Gastroenterology 143: 469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Es JH, Sato T, van de Wetering M, Lyubimova A, Yee Nee AN, Gregorieff A, Sasaki N, Zeinstra L, van den Born M, Korving J, Martens ACM, Barker N, van Oudenaarden A, Clevers H (2012) Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol 14: 1099–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espeillac C, Mitchell C, Celton‐Morizur S, Chauvin C, Koka V, Gillet C, Albrecht JH, Desdouets C, Pende M (2011) S6 kinase 1 is required for rapamycin‐sensitive liver proliferation after mouse hepatectomy. J Clin Invest 121: 2821–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfman CM, Vogel P, Issa TM, Turner CA, Lee WS, Kornfeld S, Rice DS (2007) Mice lacking alpha/beta subunits of GlcNAc‐1‐phosphotransferase exhibit growth retardation, retinal degeneration, and secretory cell lesions. Invest Ophthalmol Vis Sci 48: 5221–5228 [DOI] [PubMed] [Google Scholar]
- Giroux V, Rustgi AK (2017) Metaplasia: tissue injury adaptation and a precursor to the dysplasia‐cancer sequence. Nat Rev Cancer 17: 594–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Kang L, Wu T, Zhang J, Wang H, Gao H, Zhang Y, Huang B, Liu W, Kou Z, Zhang H, Gao S (2012) An elaborate regulation of Mammalian target of rapamycin activity is required for somatic cell reprogramming induced by defined transcription factors. Stem Cells Dev 21: 2630–2641 [DOI] [PubMed] [Google Scholar]
- Heid I, Lubeseder‐Martellato C, Sipos B, Mazur PK, Lesina M, Schmid RM, Siveke JT (2011) Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterology 141: 719–730, 730.e1–7 [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA (2005) Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7: 469–483 [DOI] [PubMed] [Google Scholar]
- Hu YY, Zhou CH, Dou WH, Tang W, Hu CY, Hu DM, Feng H, Wang JZ, Qian MJ, Cheng GL, Wang SF (2015) Improved autophagic flux is correlated with mTOR activation in the later recovery stage of experimental acute pancreatitis. Pancreatology 15: 470–477 [DOI] [PubMed] [Google Scholar]
- Huh WJ, Khurana SS, Geahlen JH, Kohli K, Waller RA, Mills JC (2012) Tamoxifen induces rapid, reversible atrophy, and metaplasia in mouse stomach. Gastroenterology 142: 21–24 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R (2016) The repair Schwann cell and its function in regenerating nerves. J Physiol 594: 3521–3531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YP, Ballou LM, Lin RZ (2001) Rapamycin‐insensitive regulation of 4e‐BP1 in regenerating rat liver. J Biol Chem 276: 10943–10951 [DOI] [PubMed] [Google Scholar]
- Karra R, Knecht AK, Kikuchi K, Poss KD (2015) Myocardial NF‐kappaB activation is essential for zebrafish heart regeneration. Proc Natl Acad Sci USA 112: 13255–13260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Nakajima S, Saito Y, Takahashi S, Katoh R, Kitamura M (2012) mTORC1 serves ER stress‐triggered apoptosis via selective activation of the IRE1‐JNK pathway. Cell Death Differ 19: 310–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalaileh A, Dreazen A, Khatib A, Apel R, Swisa A, Kidess‐Bassir N, Maitra A, Meyuhas O, Dor Y, Zamir G (2013) Phosphorylation of ribosomal protein S6 attenuates DNA damage and tumor suppression during development of pancreatic cancer. Cancer Res 73: 1811–1820 [DOI] [PubMed] [Google Scholar]
- Lee C, Lee H, Hwang SY, Moon CM (2017) Hong SN (2017) IL‐10 plays a pivotal role in tamoxifen‐induced spasmolytic polypeptide‐expressing metaplasia in gastric mucosa. Gut Liv 11: 789–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennerz JK, Kim SH, Oates EL, Huh WJ, Doherty JM, Tian X, Bredemeyer AJ, Goldenring JR, Lauwers GY, Shin YK, Mills JC (2010) The transcription factor MIST1 is a novel human gastric chief cell marker whose expression is lost in metaplasia, dysplasia, and carcinoma. Am J Pathol 177: 1514–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leushacke M, Tan SH, Wong A, Swathi Y, Hajamohideen A, Tan LT, Goh J, Wong E, Denil S, Murakami K, Barker N (2017) Lgr5‐expressing chief cells drive epithelial regeneration and cancer in the oxyntic stomach. Nat Cell Biol 19: 774–786 [DOI] [PubMed] [Google Scholar]
- Lo HG, Jin RU, Sibbel G, Liu D, Karki A, Joens MS, Madison BB, Zhang B, Blanc V, Fitzpatrick JA, Davidson NO, Konieczny SF, Mills JC (2017) A single transcription factor is sufficient to induce and maintain secretory cell architecture. Genes Dev 31: 154–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan CY, Desai TJ (2015) Keeping it together: pulmonary alveoli are maintained by a hierarchy of cellular programs. BioEssays 37: 1028–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JC, Taghert PH (2012) Scaling factors: transcription factors regulating subcellular domains. BioEssays 34: 10–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JC, Sansom OJ (2015) Reserve stem cells: differentiated cells reprogram to fuel repair, metaplasia, and neoplasia in the adult gastrointestinal tract. Sci Signal 8: re8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JC, Goldenring JR (2017) Metaplasia in the stomach arises from gastric chief cells. Cell Mol Gastroenterol Hepatol 4: 85–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindos T, Dun XP, North K, Doddrell RD, Schulz A, Edwards P, Russell J, Gray B, Roberts SL, Shivane A, Mortimer G, Pirie M, Zhang N, Pan D, Morrison H, Parkinson DB (2017) Merlin controls the repair capacity of Schwann cells after injury by regulating Hippo/YAP activity. J Cell Biol 216: 495–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y (2004) In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15: 1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Torres M, Jaquenoud M, De Virgilio C (2015) TORC1 controls G1‐S cell cycle transition in yeast via Mpk1 and the greatwall kinase pathway. Nat Commun 6: 8256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morran DC, Wu J, Jamieson NB, Mrowinska A, Kalna G, Karim SA, Au AY, Scarlett CJ, Chang DK, Pajak MZ, Australian Pancreatic Cancer Genome I , Oien KA, McKay CJ, Carter CR, Gillen G, Champion S, Pimlott SL, Anderson KI, Evans TR, Grimmond SM et al (2014) Targeting mTOR dependency in pancreatic cancer. Gut 63: 1481–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaugh LC, Keefe MD (2015) Regeneration and repair of the exocrine pancreas. Annu Rev Physiol 77: 229–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L (2007) A global double‐fluorescent Cre reporter mouse. Genesis 45: 593–605 [DOI] [PubMed] [Google Scholar]
- Nam KT, Lee HJ, Sousa JF, Weis VG, O'Neal RL, Finke PE, Romero‐Gallo J, Shi G, Mills JC, Peek RM Jr, Konieczny SF, Goldenring JR (2010) Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology 139: 2028–2037 e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelsen CJ, Rickheim DG, Tucker MM, Hansen LK, Albrecht JH (2003) Evidence that cyclin D1 mediates both growth and proliferation downstream of TOR in hepatocytes. J Biol Chem 278: 3656–3663 [DOI] [PubMed] [Google Scholar]
- Newberry EP, Kennedy SM, Xie Y, Luo J, Stanley SE, Semenkovich CF, Crooke RM, Graham MJ, Davidson NO (2008) Altered hepatic triglyceride content after partial hepatectomy without impaired liver regeneration in multiple murine genetic models. Hepatology 48: 1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederau C, Ferrell LD, Grendell JH (1985) Caerulein‐induced acute necrotizing pancreatitis in mice: protective effects of proglumide, benzotript, and secretin. Gastroenterology 88: 1192–1204 [DOI] [PubMed] [Google Scholar]
- Nomura S, Yamaguchi H, Ogawa M, Wang TC, Lee JR, Goldenring JR (2005) Alterations in gastric mucosal lineages induced by acute oxyntic atrophy in wild‐type and gastrin‐deficient mice. Am J Physiol Gastrointest Liver Physiol 288: G362–G375 [DOI] [PubMed] [Google Scholar]
- Nozaki K, Ogawa M, Williams JA, Lafleur BJ, Ng V, Drapkin RI, Mills JC, Konieczny SF, Nomura S, Goldenring JR (2008) A molecular signature of gastric metaplasia arising in response to acute parietal cell loss. Gastroenterology 134: 511–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson WL (2002) Review article: Helicobacter pylori and gastric adenocarcinoma. Aliment Pharmacol Ther 16(Suppl 1): 40–46 [DOI] [PubMed] [Google Scholar]
- Radyk MD, Burclaff J, Willet SG, Mills JC (2018) Metaplastic cells in the stomach arise, independently of stem cells, via dedifferentiation or transdifferentiation of chief cells. Gastroenterology 154: 839–843.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radyk MD, Mills JC (2017) A chief source of cancer and repair in stomachs. EMBO J 36: 2318–2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey VG, Doherty JM, Chen CC, Stappenbeck TS, Konieczny SF, Mills JC (2007) The maturation of mucus‐secreting gastric epithelial progenitors into digestive‐enzyme secreting zymogenic cells requires Mist1. Development 134: 211–222 [DOI] [PubMed] [Google Scholar]
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP (2006) GenePattern 2.0. Nat Genet 38: 500–501 [DOI] [PubMed] [Google Scholar]
- Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, Prescianotto‐Baschong C, Sauer U, Jenoe P, Hall MN (2013) Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 339: 1320–1323 [DOI] [PubMed] [Google Scholar]
- Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, Adams PD, Anderson KI, Gottlieb E, Sansom OJ, Ryan KM (2013) p53 status determines the role of autophagy in pancreatic tumour development. Nature 504: 296–300 [DOI] [PubMed] [Google Scholar]
- Roth S, Franken P, Sacchetti A, Kremer A, Anderson K, Sansom O, Fodde R (2012) Paneth cells in intestinal homeostasis and tissue injury. PLoS One 7: e38965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, Sonenberg N, Blenis J (2007) RAS/ERK signaling promotes site‐specific ribosomal protein S6 phosphorylation via RSK and stimulates cap‐dependent translation. J Biol Chem 282: 14056–14064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugge M, Correa P, Di Mario F, El‐Omar E, Fiocca R, Geboes K, Genta RM, Graham DY, Hattori T, Malfertheiner P, Nakajima S, Sipponen P, Sung J, Weinstein W, Vieth M (2008) OLGA staging for gastritis: a tutorial. Dig Liver Dis 40: 650–658 [DOI] [PubMed] [Google Scholar]
- Saenz JB, Burclaff J, Mills JC (2016) Modeling murine gastric metaplasia through tamoxifen‐induced acute parietal cell loss. Methods Mol Biol 1422: 329–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenz JB, Mills JC (2018) Acid and the basis for cellular plasticity and reprogramming in gastric repair and cancer. Nat Rev Gastroenterol Hepatol https://doi.org/10.1038/nrgastro.2018.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saluja A, Saito I, Saluja M, Houlihan MJ, Powers RE, Meldolesi J, Steer M (1985) In vivo rat pancreatic acinar cell function during supramaximal stimulation with caerulein. Am J Physiol 249: G702–G710 [DOI] [PubMed] [Google Scholar]
- Schmidt PH, Lee JR, Joshi V, Playford RJ, Poulsom R, Wright NA, Goldenring JR (1999) Identification of a metaplastic cell lineage associated with human gastric adenocarcinoma. Lab Invest 79: 639–646 [PubMed] [Google Scholar]
- Silvera D, Ernlund A, Arju R, Connolly E, Volta V, Wang J, Schneider RJ (2017) mTORC1 and ‐2 coordinate transcriptional and translational reprogramming in resistance to DNA damage and replicative stress in breast cancer cells. Mol Cell Biol 37: e00577–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spechler SJ, Merchant JL, Wang TC, Chandrasoma P, Fox JG, Genta RM, Goldenring JR, Hayakawa Y, Kuipers EJ, Lund PK, McKeon F, Mills JC, Odze RD, Peek RM Jr, Pham T, Que J, Rustgi AK, Shaheen NJ, Shivdasani RA, Souza RF et al (2017) A summary of the 2016 James W. Freston conference of the American gastroenterological association: intestinal metaplasia in the esophagus and stomach: origins, differences, similarities and significance. Gastroenterology 153: e6–e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz P (2017) Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat Rev Gastroenterol Hepatol 14: 296–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syder AJ, Karam SM, Mills JC, Ippolito JE, Ansari HR, Farook V, Gordon JI (2004) A transgenic mouse model of metastatic carcinoma involving transdifferentiation of a gastric epithelial lineage progenitor to a neuroendocrine phenotype. Proc Natl Acad Sci USA 101: 4471–4476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: 663–676 [DOI] [PubMed] [Google Scholar]
- Wang S, Xia P, Ye B, Huang G, Liu J, Fan Z (2013) Transient activation of autophagy via Sox2‐mediated suppression of mTOR is an important early step in reprogramming to pluripotency. Cell Stem Cell 13: 617–625 [DOI] [PubMed] [Google Scholar]
- Wang WE, Li L, Xia X, Fu W, Liao Q, Lan C, Yang D, Chen H, Yue R, Zeng C, Zhou L, Zhou B, Duan DD, Chen X, Houser SR, Zeng C (2017) Dedifferentiation, proliferation, and redifferentiation of adult mammalian cardiomyocytes after ischemic injury. Circulation 136: 834–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CY, Carpenter ES, Takeuchi KK, Halbrook CJ, Peverley LV, Bien H, Hall JC, DelGiorno KE, Pal D, Song Y, Shi C, Lin RZ, Crawford HC (2014) PI3K regulation of RAC1 is required for KRAS‐induced pancreatic tumorigenesis in mice. Gastroenterology 147: 1405–1416 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Li Y, Zhang H, Huang Y, Zhao P, Tang Y, Qiu X, Ying Y, Li W, Ni S, Zhang M, Liu L, Xu Y, Zhuang Q, Luo Z, Benda C, Song H, Liu B, Lai L, Liu X et al (2015) Autophagy and mTORC1 regulate the stochastic phase of somatic cell reprogramming. Nat Cell Biol 17: 715–725 [DOI] [PubMed] [Google Scholar]
- Yoshizawa N, Takenaka Y, Yamaguchi H, Tetsuya T, Tanaka H, Tatematsu M, Nomura S, Goldenring JR, Kaminishi M (2007) Emergence of spasmolytic polypeptide‐expressing metaplasia in Mongolian gerbils infected with Helicobacter pylori . Lab Invest 87: 1265–1276 [DOI] [PubMed] [Google Scholar]
- Zhou X, Liu W, Hu X, Dorrance A, Garzon R, Houghton PJ, Shen C (2017) Regulation of CHK1 by mTOR contributes to the evasion of DNA damage barrier of cancer cells. Sci Rep 7: 1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Bar‐Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM (2011) mTORC1 senses lysosomal amino acids through an inside‐out mechanism that requires the vacuolar H(+)‐ATPase. Science 334: 678–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
All analyzed microarray data have been deposited in NCBI GEO under accession GSE103570.