

Abstract
Background and Aims
Inflammatory bowel disease [IBD] is characterised by a disruption of immune homeostasis, which is tightly regulated to protect against harmful pathogens yet not react to commensal antigens. Animal studies indicate that regulatory T cells [Treg] modulate the immune response to prevent IBD development. Lactoferrin [LF] is an endogenous anti-inflammatory pleiotropic protein secreted at high concentrations in colostrum and at mucosal sites. However, the effect of LF on specific T lymphocyte populations has not been studied. Here, we identify a novel mechanism by which a recombinant human LF, VEN-120, regulates T cell populations in health and disease.
Methods
Two murine models of intestinal inflammation, the dextran sodium sulphate colitis model and the TNFΔARE/+ model of ileitis, were used to study the anti-inflammatory and T cell modulating ability of VEN-120. Flow cytometry was used to evaluate T cell populations within the lamina propria and mesenteric lymph nodes, and to evaluate the effect of VEN-120 on CD4+ T cells in vitro.
Results
VEN-120 reduced inflammation in both models of IBD, accompanied by increased Tregs in the intestinal lamina propria. Treatment of CD4+ T cells in vitro resulted in an upregulation of Treg genes and skewing towards a Treg population. This in vitro T cell skewing translated to an increase of Treg homing to the intestinal lamina propria and associated lymph tissue in healthy mice.
Conclusions
These data provide a novel immunological mechanism by which VEN-120 modulates T cells to restrict inflammatory T cell-driven disease.
Keywords: VEN-120, lactoferrin, colitis, regulatory T cells
1. Introduction
Although the aetiology of inflammatory bowel disease [IBD; comprising ulcerative colitis, UC; and Crohn’s disease, CD] is still poorly defined, current hypotheses suggest that it may develop through a dysregulated mucosal immune response toward the commensal enteric flora in genetically susceptible individuals.1–3 This immune dysregulation results in an overproduction of pro-inflammatory cytokines such as tumour necrosis factor [TNF] by monocytes, macrophages, and T cells, thereby allowing for persistence of a hyperactivated immune response.4,5 CD4+ T cells are critical drivers of pathogenesis in IBD, whereby an increase in activation and populations of CD4+ and CD8+ T cells are observed in patients with IBD.6 More recently, it has been appreciated that over-activation of Th1/Th17 T cell phenotypes drives much of the chronic pathology in IBD.7 On the other hand, it has been observed that the pro-inflammatory T cells can be tempered by the action of a subset of CD4+CD25+Foxp3+ cells, termed regulatory T cells [Treg], which exhibit suppressive effects on activated pro-inflammatory T cells, permitting resolution of inflammation and restoration of the homeostatic immune environment.
Lactoferrin [LF] is a single-chain iron-binding glycoprotein of ~ 80 kDa that belongs to the family of human transferrins.8 LF is present in myriad mucosal fluids,9–13 but is most predominant in human milk, particularly in the colostrum during early lactation, where it has been suggested to promote the healthy growth and development of the gastrointestinal [GI] tract,14 promote the growth of commensal bacteria, and deter the establishment of pathogenic bacteria and viruses.15–17 LF has been previously used for its beneficial effects in several models of human health including dermal inflammation,18,19 wound healing,20 and infectious diseases.19,21,22
Preclinical studies have shown the protective effects of orally administered human LF [hLF] in mice23 and bovine LF [bLF] in rats in chemically induced models of colitis.24,25 Though these studies have demonstrated the ability of LF to decrease the expression of cytokines that are associated with acute inflammation in these models, such as TNF and interleukin [IL]-6, the mechanism of action for LF has yet to be described.
In this study, we sought to determine the mechanism by which LF affects inflammation. To do this, we hypothesised that the beneficial effect of LF may be the result of its effect on T cell populations, specifically the Treg phenotype. Here we demonstrate the ability of LF [VEN-120] to induce a Treg phenotype in CD4+ cells in vitro, and in vivo, using two relevant models of disease, namely: the long-established DSS murine model of chemically induced colitis and the TNFΔARE/+ model, which recapitulates CD-like ileitis. With this model, we investigated the ability of VEN-120 to drive a Treg phenotype in vivo at the expense of Th17 populations and to ameliorate disease activity.
2. Materials and Methods
2.1. Chemicals and reagents
Recombinant human lactoferrin [rhLF, VEN-120] was obtained from Ventria Bioscience [Fort Collins, CO, USA]. Cytokines [IL-2, IL-6, IL12, TGF-β] were obtained from Peprotech [Rocky Hill, NJ, USA]. Phorbol 12-myristate 13-acetate [PMA], ionomycin, brefeldin A, and monensin were obtained from Sigma Aldrich [St Louis, MO, USA].
2.2. Animals
A colony of TNFΔARE/+ mice were bred in-house with age- and sex-matched wild type littermates used as controls. Faecal samples from these mice were consistently negative for Helicobacter, protozoa, and helminthes All animals were handled in accordance with procedures approved by the University of Colorado Institutional Animal Care and Use Committee.
2.3. Chemically induced acute murine colitis model
Eight-week old C57/BL6 mice [Jackson Laboratories, Bar Harbor, ME, USA] were treated with dextran sodium sulphate [DSS] ad libitum [3% w/v 36–50 kDa, MP Biomedicals, Santa Ana, CA, USA] in drinking water for 7 days. DSS groups received daily gavage of VEN-120 [500 mg/kg] or PBS. Body weight was assessed daily to assess disease severity. At the time of sacrifice, spleen, mesenteric lymph nodes, and segments of colonic tissue were excised for flow cytometric analysis of leukocyte subsets, colon lengths were assessed, and colon sections were processed for histology, RNA extraction, and cytokine analyses to assess disease activity. Histology was evaluated and scored by a trained pathologist blinded to the conditions of the experiments, according to a previously described scoring system.26 Colonic tissues were snap-frozen for polymerase chain reaction [PCR].
2.4. TNFΔARE/+ murine model of CD-like ileitis
VEN-120 was solubilised in PBS, sterile filtered, and administered either orally by gavage or by micro-osmotic pump [Alzet #1002, flow rate:0.25uL/h, DURECT Corporation, Cupertino, CA, USA] implanted subcutaneously on the back, posterior to the scapulae, to 20-week old TNFΔARE/+ mice. Dosing was determined based on a DSS dose response curve [100 mg-1 g/kg/day], identifying 500 mg/kg/day as the optimum dose in mice [data not shown]. Following the course of VEN-120 administration, mice were sacrificed and ileums longitudinally transected and processed for standard haematoxylin-eosin staining after fixation in 10% formaldehyde. Histological severity of inflammation was determined based upon an established colitis recording system scored by a trained pathologist26 blinded to the treatment.
2.5. Intestinal barrier permeability assay
After 14 days’ treatment with VEN-120 or BSA control, TNFΔARE/+ mice were gavaged with 150 µl of 80 mg/ml FITC-labelled dextran [4 kDa, FD4] and blood was collected 4 h later by submandibular bleeding. Serum was separated from blood after clotting and fluorescence measured.27 Values were determined by interpolating from a standard curve of FD4.
2.6. Lymphocyte isolation
Single-cell suspensions were obtained by gently pressing the mesenteric lymph nodes [MLN] or spleen against a 70-μm cell strainer. Splenic red blood cells were lysed by 3-min incubation in ammonium chloride lysing reagent [ACK Lysis Buffer; Invitrogen, Carlsbad, CA, USA]. Intestinal segments were opened along the mesentery, and luminal content was rinsed off with phosphate-buffered saline solution before cutting into 1-cm sections in phosphate-buffered solution containing 15 mM HEPES and 1 mM EDTA with vigorous agitation on a vortex mixer. The tissue was then passed through a 70-μm tissue strainer, and the process was repeated until the wash remained clear. The remaining lamina propria was digested in 1 mg/mL collagenase type VIII [C9722; Sigma Aldrich, St Louis, MO, USA] for 10 min in an orbital shaker at 270 rpm and 37°C. Tissues were vortexed briefly and filtered to remove any remaining undigested material, and cells were counted before flow cytometric evaluation. CD4+ cells were then isolated by positive selection using EasySep™ Mouse CD4 Positive Selection [StemCell Technologies, Vancouver, BC, Canada].
2.7. Real-time PCR analysis
CD4+ cells were isolated from the spleens and mesenteric lymph nodes of healthy C57/BL6 mice. RNA was harvested from CD4+ lymphocytes obtained from the spleens of healthy C57/BL6 mice using Qiagen RNEasy kits. cDNA was subsequently synthesised from 0.5 μg total RNA with RT2 First Strand Kit [Qiagen, Valencia, CA, USA]. qPCR was carried out using primers provided with the Mouse T Helper Cell Differentiation RT2 Profiler™ PCR Array [Qiagen], per the manufacturer’s instruction. Data analysis was carried out using ΔΔCT method with normalisation of the raw data to endogenous control genes provided with the array.
2.8. Flow cytometry
Cells from indicated compartments were incubated with fluorescently labelled anti-mouse antibodies against: CD4 [GK1.5, BioLegend, San Diego, CA, USA]; IFN-γ [XMG1.2], IL-17a [FFA21], CD25 [PC61.5], FoxP3 [FJK-16s; BD Biosciences, San Jose, CA, USA]; or corresponding isotype controls. Intracellular staining was performed by use of the FoxP3 staining kit [eBioscience, San Diego, CA, USA], according to the manufacturer’s instructions. Intracellular cytokine staining was performed following 4 h of stimulation with PMA [50 ng/mL], ionomycin [1 μg/mL], and brefeldin and monensin. Flow cytometric analysis was performed using a BD FACSCanto II [BD Biosciences, San Jose, CA, USA]. FACS was performed by use of a BD FACSAria III [BD Biosciences]. Further analyses were performed by use of FlowJo software [Tree Star, Ashland, OR, USA].
2.9. Statistical analysis
All murine colitis and ileitis studies were performed using a minimum of six mice per group and were repeated a minimum of three times. Statistical analyses were performed using Student’s t test or repeated measures analysis of variance with Graphpad Prism Data Analysis software [GraphPad Software, La Jolla, CA]. Data were expressed as mean ± standard error of the mean [s.e.m]. Statistical significance was set at p < 0.05.
3. Results
3.1. VEN-120 ameliorates chemically-induced acute murine colitis
Previous studies have indicated that LF can ameliorate disease activity in chemically-induced models of colitis.23–25 We hypothesised that one result of the anti-inflammatory role of VEN120 would be to stimulate the regulatory T cells. Although not considered a T cell-based colitis model, there are nonetheless data suggesting that T cells and specifically Tregs can play a role in the protection from DSS colitis.28–30 We sought to investigate whether VEN-120 was efficacious in a similar model of colitis. For this purpose, 8-week old C57/BL6 mice were treated with either 500 mg/kg VEN-120 or vehicle [bovine serum albumin, 500 mg/kg] for 7 days while receiving 3% DSS ad libitum in drinking water. Treatment with VEN-120 significantly attenuated weight loss at Days 6 and 7 post induction of colitis compared with vehicle-treated animals [p < 0.01, Figure 1A]. The anti-inflammatory effect of VEN-120 administration was evident on gross examination of the colons from the DSS-treated mice, whereby the shortening of colons in vehicle-treated mice was significantly reversed in VEN-120 treated mice [5.0 ± 0.28 cm vs 5.9 ± 0.29 cm; p < 0.05, Figure 1B].
Figure 1.

VEN-120 reduces gross inflammatory indices and histopathological damage in a DSS model of colitis. A] Weight loss measurements demonstrated a protection of mice following administration of VEN-120 [*p < 0.05 two-way ANOVA, with Bonferoni’s post-test]. B] Administration of VEN-120 resulted in a protection of mucosal injury, as demonstrated by preservation of colon length [*p < 0.05, Student’s t test]. C] VEN-120 reduces overall injury and inflammation score in DSS colitis [***p < 0.001, Student’s t test]. D] H&E staining of the colon reveals a loss of epithelial integrity, loss of crypt architecture, absence of goblet cells, and immune cell infiltration upon insult with DSS alone, which is rescued by VEN-120 administration at 500 mg/kg/day, representative images, n = 7. DSS, dextran sodium sulphate; H&E, haematoxylin and eosin; ANOVA, analysis of variance.
Blinded histopathological analysis of the colons from vehicle-treated mice revealed a loss of epithelial integrity and crypt architecture, absence of goblet cells, immune cell infiltration, and hypertrophy of the mucosa muscularis, which was largely reversed in mice treated with VEN-120. Evaluation and scoring of injury and inflammation revealed a significant decrease in histopathological score from vehicle-treated animals [11.31 ± 0.54] compared with the VEN-120 treatment group [5.271 ± 0.69; p < 0.0001; Figure 1C]. This reduction in tissue injury, loss of crypt, architecture, and epithelial integrity, along with the reduction in inflammatory infiltrate, are represented by haematoxylin and eosin [H&E] micrographs [Figure 1D].
3.2. VEN-120 administration induces anti-inflammatory cytokines in CD4+ T cells during chemically induced colitis
In an effort to understand the potential for VEN-120 to affect the inflammatory environment in vivo, we assessed the cytokine profile of CD4+ T cells in the DSS mice treated with either vehicle or with VEN-120. Lymphocytes were isolated from the mesenteric lymph nodes [MLN] and lamina propria [LP] and evaluated by flow cytometry. Figure 2 demonstrates that treatment with VEN-120 resulted in a decrease in the frequency of CD4+ T cells that express pro-inflammatory cytokines, IL-17 [LP: 11.8 ± 0.17% vs 8.7 ± 0.69%, MLN: 18.34 ± 1.3% vs 12.0 ± 0.01%, Figure 2A, B] and IFNγ [LP: 14.78 ± 1.61% vs 6.47 ± 0.34%, Figure 2C], as well as a concomitant increase in CD4+ cells that produce the anti-inflammatory cytokine IL-10 [LP: 8.3 ± 0.09% vs 13.39 ± 1.11% and MLN: 2.33 ± 0.72% vs 4.7 ± 0.75%], which was evident in the LP, but not the MLN of VEN-120 treated mice [Figure 2E, F].
Figure 2.

VEN-120 promotes a shift in balance of CD4+ T cells in a DSS model colitis. Flow cytometric analysis demonstrated that VEN-120 reduced expression of the pro-inflammatory cytokine IL-17 in: A] LP CD4+ cells; and B] MLN CD4+ cells with concomitant reduction in the pro-inflammatory cytokine IFNγ in: C] LP CD4+ cells, with no effect demonstrated at: D] the MLN. Conversely, IL-10 producing CD4+ cells were demonstrated to be increased in: E] MLN of mice administered VEN-120. However, in CD4+ cells isolated from: F] colonic LP, no significant effect on IL-10 production was demonstrated, although an increased trend was identified. [*p < 0.05; **p < 0.01, one-way ANOVA, n = 7]. MLN, mesenteric lymph nodes; LP, lamina propria; ANOVA, analysis of variance.
To investigate the role of VEN-120 in the regulation of Treg in the DSS model of colitis, cell populations were evaluated from both DSS-treated groups to assess for the presence of CD4+Foxp3+ cells. By investigating the burden of all CD4+ T cell subtypes, we were able to demonstrate that VEN-120 reduced cellularity at the draining lymph nodes [MLN: 28.4 ± 2.39% vs 20.3 ± 0.5%], but not at the local site of inflammation [LP: 13.28 ± 0.88% vs 13.41 ± 1.2%], indicative of an overall decrease in inflammatory infiltrates following VEN-120 administration [Figure 3A, B]. However, of the populations of CD4+ T cells that are present at both tissue sites, we demonstrate an increase in Foxp3+ CD4+ T cells [LP: 19 ± 0.83% vs 28.7 ± 2.2% and MLN: 8.8 ± 0.5% vs 12 ± 0.49%], indicative of an induced population of Tregs in response to VEN-120 administration [Figure 3C, D]. Taken together, these data indicate an overall decrease in inflammation and subsequent decrease in inflammatory cytokine output at the source of inflammation in the DSS-mediated model of colitis.
Figure 3.

VEN-120 decreases overall T cell burden and promotes Treg phenotype in a DSS model of colitis. Flow cytometric analysis demonstrated CD4+ cell infiltration to be reduced in the VEN-120 treated mice versus the vehicle control at the B] MLN but not A] LP. Of the T cell populations present, an increased frequency in Tregs [CD4+Foxp3+] was observed in mice treated with VEN-120 at both the C] LP and D] MLN. [*p < 0.05; **p < 0.01, one-way ANOVA, n = 7]. DSS, dextran sodium suphate; MLN, mesenteric lymph nodes; LP, lamina propria; ANOVA, analysis of variance.
3.3. VEN-120 drives a Treg cell phenotype in vitro
To ascertain the potential for VEN-120 to differentially modulate the phenotype of naïve murine Th cells, we evaluated its effects on skewing conditions of Treg and Th1 cells in vitro. Murine naïve CD4+CD25- T cells were isolated and placed in naïve T cell conditions, or Treg conversion cultures, for 3 days as described in the methods. During this incubation time, T cells were incubated in the absence or presence of 10 nM VEN-120. Cells were then restimulated with PMA/ionomycin in the presence of intracellular transport inhibitor brefeldin A and assayed by flow cytometric intracellular cytokine staining to identify IL-10+ cells. VEN-120 significantly induced an increase in IL-10 expression in both Th0 and Treg skewing conditions [Figure 4A]. In order to investigate the ability for VEN-120 to drive the canonical transcription factor associated with Treg, we placed CD4+ cells in Treg conversion culture in the presence or absence of VEN-120. Following nuclear staining and subsequent flow cytometric analysis, we were able to identify a significant increase in Foxp3+ cells upon treatment with VEN-120 [Figure 4B]. This finding was further supported by the finding that the majority of genes positively regulated by VEN-120 treatment were those thought to drive Treg, notably foxp3 and nuclear factor of activated T cell genes [nfatc1, nfatc2]. Furthermore, another subset of genes were decreased which are associated with the development of pro-inflammatory T cell subtypes, most notably Il-17, as assessed using a T helper cell differentiation array with pre-determined primer sets corresponding to specific T cell subsets [Supplementary Figure 1, available at as Supplementary data at ECCO-JCC online].
Figure 4.
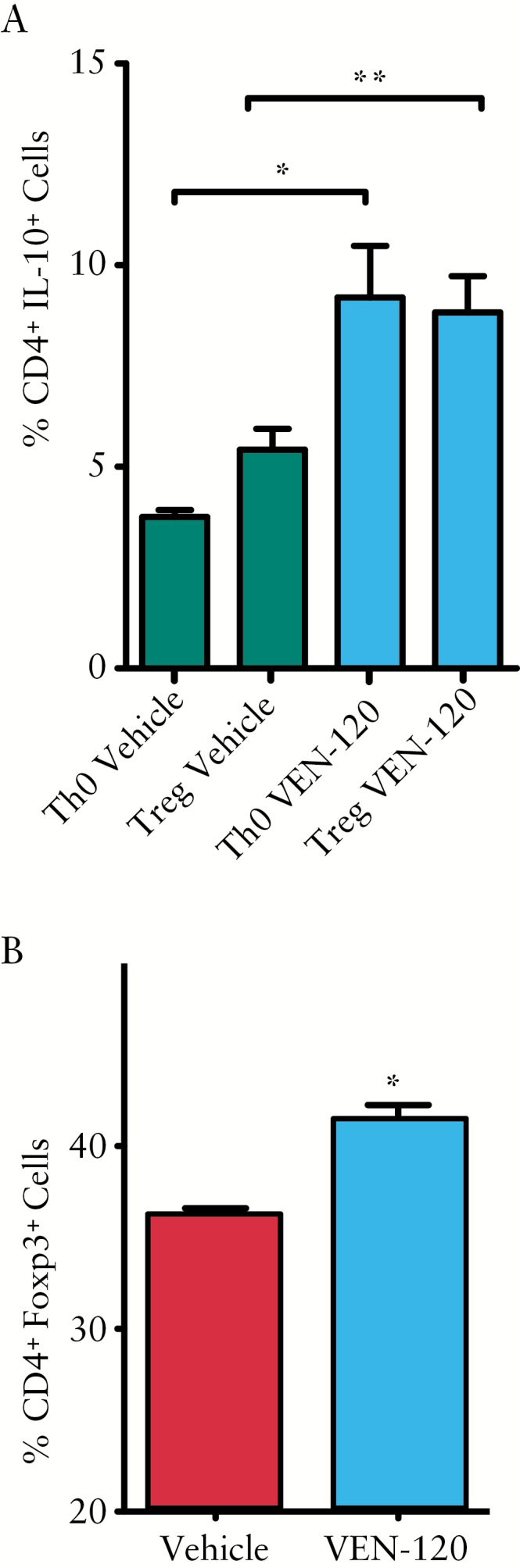
Conversion of CD4+CD25Neg T cells under polarising conditions is skewed by VEN-120 to a pro-regulatory phenotype. A] Isolated CD4+CD25Neg T cells were cultured in either naïve [Th0] or Treg polarising conditions in the presence of 10 nM VEN-120 or vehicle control. VEN-120 significantly induced an increase in IL-10 expression in both Th0 and Treg skewing conditions. B] VEN-120 drives the expression of the Treg transcription factor, Foxp3, under Treg skewing conditions [*p < 0.05; **p < 0.01, one-way ANOVA n = 7]. ANOVA, analysis of variance.
3.4. VEN-120 promotes Treg homing to the gut and associated lymph nodes
Following the observation that VEN-120 can affect the fate of naïve T cells in vitro, treatment effects on the T cell repertoire in vivo were subsequently determined. Healthy C57/BL6 mice were treated either with sham injection [PBS], VEN-120 delivered subcutaneously by osmotic pump [200 mg/kg/day], or by gavage at a dose of 500 mg/kg/day for 14 days. Upon termination of the experiment, tissues were collected [intestinal lamina propria, LP; mesenteric lymph nodes, MLN; axillary lymph nodes, AxLN; peripheral blood mononuclear cells, PBMC; and spleen], and evaluated by flow cytometry for expression of CD4, CD25, and Foxp3. Figure 5 demonstrates that either route of administration of VEN-120 resulted in a marked increase in Treg populations at the intestinal LP, followed by at the MLN and at the spleen. Treg induction did not appear to occur in the lymph tissue adjacent to the site of insertion of the osmotic pump [AxLN], nor within the circulating blood. This suggests that VEN-120 modulates Treg cell populations and directs them to the intestinal tissues in the absence of disease, regardless of route of administration.
Figure 5.

S.Q. and oral administration of VEN-120 results in Treg homing to intestinal tissues and associated lymphoid organs. Healthy, 8-week old C56/BL6 mice were treated with VEN-120 S.Q. by osmotic pump [200 mg/kg/day] or gavage [500 mg/kg/day] for a 14-day period. Flow cytometry revealed the ability of VEN-120 to induce accumulation of Treg at the LP, the MLN, and the spleen. No significant induction of Treg was noted at the AxLN or PBMC. [**p> 0.01, one-way ANOVA, n = 7]. S.Q., subcutaneous; MLN, mesenteric lymph nodes; LP, lamina propria; ANOVA, analysis of variance; AxLN, axillary lymph nodes; PBMC, peripheral blood mononuclear cells.
3.5. VEN-120 administration reduces severity of disease in a TNFΔARE/+ model of ileitis
In an effort to demonstrate that the protective effects of VEN-120 are not restricted to a single model, and to interrogate the broad spectrum of disease phenotypes that IBD encompasses, the TNFΔARE/+ murine ileitis model was used. TNFΔARE/+ mice represent a relevant chronic model of Crohns disease [CD] generated by deletion within the AU-rich element [ARE] of the TNF gene in mice31 [ieTNFΔARE/+]. In order to test the immunoregulatory effect of VEN-120 in these mice, and to remove any potential confounding effects that VEN-120 may have on the gut microbiome and direct effects on the intestinal epithelium, 20-week old TNFΔARE/+ mice were treated for 14 days by subcutaneous delivery with VEN-120 or vehicle [BSA, 200 mg/kg/day] using a 2-week Alzet osmotic pump. As a monitor of disease activity, the effect of VEN-120 treatment on the intestinal barrier function was assessed as described in the Methods section. Figure 6A demonstrates a significant reduction [p < 0.05] in permeability of FD4 by roughly 50% from the intestine, suggesting a protective effect of VEN-120 at the site of inflammatory injury. To further demonstrate the protective effects of VEN-120, mice were sacrificed at the end of the 14-day treatment period and tissues harvested for analysis. Herein, we were able to demonstrate that the moderate-to-severe inflammation observed in the sham-treated mice was reversed in the VEN-120 treated mice [Figure 6B, C]. Although VEN-120 did not completely reverse the tissue inflammation over the 14-day treatment period, it significantly reduced inflammatory scoring across all parameters [chronic and active inflammation and preservation of villous architecture]. As an indicator of the anti-inflammatory effects of VEN-120, we harvested various lymphoid tissues [lamina propria, LP; mesenteric lymph nodes, MLN; and spleen, SP] and analysed the infiltration of naïve T cells [CD4+CD44LowCD62LHigh] at these sites by flow cytometry. Figure 6D demonstrates the ability of VEN-120 to significantly decrease the number of naïve T cells in all tissues analysed [p < 0.01]. Furthermore, in an analysis of unsorted lymphocytes, there was a significant decrease in cellularity at the LP, with an observed increase in cellularity at the MLN and spleen, which may be due to a temperospatial phenomenon of T cells migrating away from the local site of injury to the draining lymph nodes following resolution of inflammation [Figure 6E].
Figure 6.

Subcutaneous administration with VEN-120 attenuates chronic murine ileitis. A] VEN-120 treatment of TNFΔARE/+ mice resulted in a significant decrease in FD4 transport across the intestinal epithelial barrier. B] Two-week treatment of TNFΔARE/+ mice with VEN-120 results in a significant improvement in tissue architecture and a significant decrease in histological indices of inflammation. C] Representative H&E staining demonstrates improved architectural appearance and decreased chronic and acute inflammation. D] Flow cytometric analysis demonstrating a significant reduction in the influx of naïve CD4+ cells known to perpetuate inflammation in this model at the ileal LP, MLN and at the spleen. [*p < 0.05; **p < 0.01, one-way ANOVA, n = 6]. MLN, mesenteric lymph nodes; LP, lamina propria; ANOVA, analysis of variance; H&E, haematoxylin and eosin.
3.6. Oral VEN-120 reduces severity of disease in a TNFΔARE/+ model of ileitis
Twelve-week-old TNFΔARE/+ mice were given 200 µl by mouth [p.o.] of 500 mg/kg/day of VEN-120 for 14 days. As a positive control, 5 mg/kg anti-TNF monoclonal antibody [IFX; infliximab, Janssen Biotec, Inc.] was given intraperitoneally twice weekly. Upon termination of the experiment, mice were euthanised and tissues collected for further analysis. Mice treated with VEN120 had a significant reversal of pathology, as analysed by a pathologist in a blinded manner. Administration of VEN-120 resulted in improved histopathological scores, to a similar extent as those that were observed with the administration of IFX [Figure 7A]. Most notable was the preservation of normal tissue architecture, with a clear restoration of villus height and the presence of mucus-secreting goblet cells [unstained cells embedded within the mucosal epithelium, Figure 7B].
Figure 7.

Oral administration with VEN-120 attenuates chronic murine ileitis. A, B] H&E staining demonstrates improved architectural appearance and decreased chronic and acute inflammation following oral administration of VEN-120 or subcutaneous anti-TNF antibody, infliximab [IFX] [*p < 0.05; ***p < 0.0001, Student’s t test]. Flow cytometric analysis demonstrating that VEN-120 administration significantly decreases CD4+ burden at C] the LP, and D] at the MLN in TNFΔARE/+ mice. Further interrogation of these T cells identified a significant increase in IL-10 at the E] LP but not the F] MLN of TNFΔARE/+ mice treated with VEN-120. A significant reduction in IL-17 expressing cells was demonstrated at G] the LP but not H] the MLN of TNFΔARE/+ mice treated with VEN-120 [*p < 0.05; **p < 0.01, one-way ANOVA, n = 6]. MLN, mesenteric lymph nodes; LP, lamina propria; ANOVA, analysis of variance; H&E, haematoxylin and eosin; TNF, tumour necrosis factor.
3.7. VEN-120 drives Treg phenotype in TNFΔARE/+ model of ileitis
To gain further insights into the role of CD4+ T cells, we isolated T cells from the LP and MLN, and assessed their phenotypes by flow cytometry. By determining the burden of all CD4+ T cell subtypes, we were able to demonstrate that VEN-120 reduced cellularity both locally at the LP and at the MLN [Figure 7C and D, respectively]. This indicates an overall decrease in inflammatory activity in response to VEN-120 administration. In order to characterise the inflammatory profile of T cells at the site of inflammation, we stained for the cytokine markers of Treg and Th17 cells, IL-10 and IL-17, respectively. Here, we demonstrated that VEN-120 significantly induced IL-10 expression with a concomitant decrease in IL-17 in LP CD4+ cells [Figure 7E and G, respectively]. However, we were unable to demonsrate a similar effect on IL-10 and IL-17 at the MLN [Figure 7F and H, respectively]. Together, these data demonstrate that in a mouse model of IBD, oral administration of VEN-120 decreases the severity of TNF-driven disease as evidenced by a protection of tissue at the histopathological level.
4. Discussion
IBD affects an estimated 1.4 million people in the USA,32,33 and is associated with high morbidity and decreased quality of life.34–36,37 Corticosteroids are often used in the short term to achieve symptomatic relief and decreased inflammation, yet are ineffective in mucosal healing and not appropriate for long-term maintenance therapy.38 Over the past two decades, monoclonal antibodies against TNF have revolutionised the treatment and management of IBD.39–41 However, more than one-third of patients show no response to induction therapy and, for up to 50% of responders, anti-TNF therapy becomes ineffective over time.42–46 Additionally, anti-TNF therapy has multiple concerning side effects including increased risk of infection, anaphylaxis, and increased incidence of malignancies.47 Therefore, the development of a novel therapeutic approach, with minimal side effects, for the long-term treatment of IBD represents a significant unmet medical need. The anti-inflammatory potential of LF has been extensively studied in various models of disease. In experimental models of sepsis and rheumatoid arthritis, LF has been demonstrated to exert protection by inhibiting the production of pro-inflammatory cytokines [TNF, IL-6, and IL-1β], and stimulating anti-inflammatory/pro-restitution cytokines [IL-10 and IL-4].48–51
Moreover, studies have demonstrated the protective effects of orally administered human LF [hLF] in mice23 and bovine LF [bLF] in rats in chemically induced models of colitis.24,25 In each of these studies, administration of LF was associated with a decrease in TNF at the site of tissue injury. Interestingly, in both the DSS24 and TNBS25 models of colitis in rats, the Treg-associated cytokine, IL-10, was observed to be increased in colonic tissue following LF treatment. However, the scope of these studies did not ascertain the source of the cytokine in the whole-colon lysates. Although DSS colitis is not a classical T cell-driven model of disease, the induction of IL-10 by LF indicates a potential involvement of Treg as an important mediator of Treg’s suppressive function,52 and Treg has been shown to supress inflammation in this model.28–30 In a clinical study, bLF was shown to reduce the incidence of necrotising enterocolitis [NEC] in preterm infants. This study identified an increase in Treg cells in the group of patients that received the bLF, although no experimental evidence was generated in order to directly link bLF adminstration to the increase in Tregs─as Treg populations were noted to rise with age.53 It is possible that the increase in Treg numbers was simply due to decreased inflammation in the bLF-treated patients. Collectively, these past studies failed to address the direct effect of LF treatment on specific immune cells known to modulate inflammatory pathology.
This current study is the first to describe a novel mechanism by which LF regulates inflammation. Here we interrogate the T cell populations at various peripheral lymphoid tissues in both a chemically induced model of colitis and a TNF-driven model of CD-like ileitis. Our initial experiments demonstrated that VEN-120 can ameliorate DSS-induced colitis in mice, as has been demonstrated in previous studies with native forms of bovine or human lactoferrin.23 However, our suggested mechanism of action has demonstrated direct effects of VEN-120 on the immune compartment, with decreased infiltrating CD4+ cells demonstrated at the draining lymph nodes of VEN-120 treated animals, although not evident at the site of local inflammation [LP]. This may be indicative of the late-stage response of accumulation of CD4+ cells at the LP in the DSS model, which may be evident in a more chronic disease or at a later time point sampling. Furthermore, when phenotyping the cells present at the MLN and LP of VEN-120 treated mice, we were able to demonstrate significant increases in Foxp3+ cells, indicative of VEN-120’s ability to drive the expression of the canonical Treg transcription factor. VEN-120 was found to induce Tregs at the expense of pro-inflammatory Th17, with a concomitant amelioration of histopathological manifestations in both models. Interestingly, this effect of VEN-120 was found to be similar in magnitude to the administration of the anti-TNF monoclonal antibody (anti-TNF clone CA2, [infliximab]) in the TNF-driven CD-like ileitis model.
Subsequent experiments aimed at further elucidating this novel mechanism noted the significant increase in Treg frequency in the intestinal LP and the draining MLNs in healthy mice treated orally or via osmotic pump with VEN-120. However, no significant increase in Treg was noted in the peripheral blood or in the axillary lymph nodes adjacent to the site of implantation of the pump. Collectively, these data suggest the ability of VEN-120 to activate a tolerogenic phenotype, which is likely a function of LF’s importance in the infant’s early stages of development of a healthy gut immune system which can accommodate commensal bacteria.54 Furthermore, these studies agree with other previously published reports that Treg homing to the gut is required for immunological tolerance,55 and that specific effect on Tregs by factors such as retinoic acid affects their ability to preferentially home to distinct tissues and increase their protective effects in a model of T-cell induced murine colitis.56–58
Although LF may act to alter the gut microbiome in health and disease, we show that in vitro T cell experiments and subcutaneous administration of LF result in direct skewing of T cell populations and less severe disease in vivo. Together, these data suggest that LF may act without direct action on the gut microbiome. However, given the importance of specific microbial species in the context of IBD, further investigations are required to assess the role LF may play in influencing the microbial landscape.
The induction of immune tolerance is currently a topic of intense interest in several disease areas. For this reason, several groups are investigating the therapeutic potential of cell therapy using Tregs to ameliorate autoimmune disorders such as acute graft-versus-host disease [GVHD]59,60 and type I diabetes.61 The use of an oral therapy consisting of an endogenous protein with a proven safety profile19,62 to induce Tregs may be a more attractive approach to treating autoimmune disease and may be associated with lower cost. Furthermore, LF has been shown to exhibit resistance to proteolytic breakdown and loss of iron at low pH, thereby allowing it to retain its native structure and function.63
Whereas VEN-120 was shown to significantly influence the phenotypic skewing of naïve T cells towards Treg and away from pro-inflammatory Th17 cells, studies have not addressed the upstream mechanisms responsible for this event, either receptor engagement or intracellular signalling events. The molecular mechanisms through which LF exerts its anti-inflammatory effects are not completely understood but appear in part to occur through the modulation of transcription factors, such as the inhibition of nuclear factor kappa B [NFκB] signalling pathways.64–68 These events may be downstream of a number of putative LF receptors such as Toll-like receptor 4 [TLR4],67 low-density lipoprotein receptor-related protein-1 [LRP-1],69 intelectin-1 [also known as omentin-1],70 and CXCR4.71 As an extension of these potential signalling events, VEN-120 was found to induce the transcription of various genes that are associated with the development of Treg. Most notable were the induction of foxp3, nfatc1, nfatc2, and il2ra genes. Interestingly, this is in agreement with the observation that VEN-120 acts to drive Treg in vivo and attenuate disease pathology in models whose activity is largely influenced by the overabundance of pro-inflammatory T cell subtypes and in which the induction of Treg could result in amelioration of disease. Although the scope of this current trial did not investigate the events from receptor binding to gene and protein regulation, future efforts are warranted to more clearly delineate the mechanism by which LF drives an anti-inflammatory response.
Although we demonstrate that LF acts through the promotion of Tregs to restrict disease, it cannot be ignored that LF may act to influence diverse cell types to achieve normal immune function and homeostasis. It has also been reported that bLF acts as a potent anti-inflammatory agent on monocytes by triggering a tolerogenic-like programme during their differentiation into dendritic cells [DC].62 Other groups have suggested that LF may act to preserve barrier function in epithelial cells in vitro; however, we have been unable to demonstrate an ability of VEN-120 to prevent decreases in transepithelial resistance in Caco-2 and T84 transwell following inflammatory insult [data not shown]. These studies identify a complex role for LF in immunomodulation, which may represent the requirement for initial activation of the inflammatory response in an orchestrated manner, together with the requirement for a dampening of inflammation to prevent a sustained pathophysiological outcome. Numerous studies have identified an increase in faecal LF in patients with active IBD,72,73 and it has been suggested as a potential biomarker to monitor disease progression. Although the levels of LF are elevated in these patients, there are many possible explanations as to the lack of benefit from this secreted LF. This increased LF output could be due to increased neutrophil degranulation at the site, or distal to the area of inflammation, where it may not be exerting an effect due to mechanical washout in the setting of diarrhoea. Additionally, it is conceivable that there is an increased loss of LF into the luminal contents during flareup of inflammation which results in increased faecal LF levels. Thus, augmenting LF by either systemic or topical administration may result in a reconstitution of sufficient concentrations of LF and thereby augment its activity, as has been suggested with other proteins elevated in inflammation such as alpha-1-anti-trypsin,74 grehlin,75 and adrenomedullin.76
Regardless of additional mechanisms that may be important for the anti-inflammatory abilities of LF, the importance of VEN-120 and LF to induce Treg populations may be of particular interest when designing future human trials. In light of our current findings, we speculate that the use of LF and various related peptides and isoforms may be deleterious in the treatment of diseases whose aetiology is driven in part by immune evasion as may be seen in certain neoplasms and sepsis. The role of Treg signalling has been demonstrated to result in higher rates of proliferation and increased disease progression in models of breast,77 pancreatic,78 and lung cancer.79 Indeed, this may in part explain the failure of LF- based therapies in several clinical trials. When patients were administered talactoferrin alpha, a recombinant form of hLF to treat severe sepsis, the 28-day mortality rate was higher in talactoferrin-treated patients, although not statistically significant. However, in-hospital mortality rates were significantly higher in talactoferrin-treated patients than in patients in the placebo group.80 Furthermore, talactoferrin conferred no advantages in 6-month survival, progression-free survival, or disease control rate in patients with advanced non-small cell lung cancer.81
The data presented here demonstrate for the first time that lactoferrin acts directly to modulate the immune response in a TNF-driven model of Crohn’s-like ileitis and DSS-induced colitis, with rescue of normal intestinal physiology as demonstrated by enhanced gut barrier function. Furthermore, VEN-120 reverses the severe chronic-stage pathology seen in both TNFΔARE/+ and DSS mice, with reversal of tissue damage and a decrease in associated T cell infiltration. Taken together, these data suggest that LF acts by skewing the phenotype of CD4+ cells away from a pro-inflammatory Th17 phenotype and towards a tolerogenic Treg phenotype to modulate inflammation. This mechanism of action through which rhLF can be used to target inflammation is relevant not only to models of IBD, but potentially to other diseases in which excessive inflammation drives chronic pathologies such as rheumatoid arthritis and multiple sclerosis.
Funding
This work was supported in part by NIH grant AI106278 from the National Institute of Allergy and Infectious Diseases.
Conflict of Interest
CM and RA are employees of Ventria Bioscience. CC, TN, and EdZ report no conflict of interest relating to the submitted work.
Author Contributions
The authors listed herein had significant input into the concept and design of the study [CM, CC, RA, EdZ], acquisition of data [CM, TN], analysis and interpretation of data [CM, CC, PJ, EdZ], drafting the article or revising it critically for important intellectual content [CM, CC, RA, EdZ], and final approval of the version to be submitted [CM, RA, EdZ].
Supplementary Data
Supplementary data are available at ECCO-JCC online.
Supplementary Material
References
- 1. Cho JH, Brant SR. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology 2011;140:1704–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dessein R, Chamaillard M, Danese S. Innate immunity in Crohn’s disease: the reverse side of the medal. J Clin Gastroenterol 2008;42[Suppl 3 Pt 1]:S144–7. [DOI] [PubMed] [Google Scholar]
- 3. Stefanelli T, Malesci A, Repici A, Vetrano S, Danese S. New insights into inflammatory bowel disease pathophysiology: paving the way for novel therapeutic targets. Curr Drug Targets 2008;9:413–8. [DOI] [PubMed] [Google Scholar]
- 4. Naser SA, Romero C, Urbina P, Naser N, Valentine J. Cellular infiltration and cytokine expression correlate with fistulizing state in Crohn’s disease. Clin Vaccine Immunol 2011;18:1416–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Van Deventer SJ. Tumour necrosis factor and Crohn’s disease. Gut 1997;40:443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Funderburg NT, Stubblefield Park SR, Sung HC, et al. Circulating CD4[+] and CD8[+] T cells are activated in inflammatory bowel disease and are associated with plasma markers of inflammation. Immunology 2013;140:87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zenewicz LA, Antov A, Flavell RA. CD4 T-cell differentiation and inflammatory bowel disease. Trends Mol Med 2009;15:199–207. [DOI] [PubMed] [Google Scholar]
- 8. Brock JH. The physiology of lactoferrin. Biochem Cell Biol 2002;80:1–6. [DOI] [PubMed] [Google Scholar]
- 9. Laibe S, Bard E, Biichlé S, et al. New sensitive method for the measurement of lysozyme and lactoferrin to explore mucosal innate immunity. Part II: time-resolved immunofluorometric assay used in HIV patients with oral candidiasis. Clin Chem Lab Med 2003;41:134–8. [DOI] [PubMed] [Google Scholar]
- 10. Ohashi Y, Ishida R, Kojima T, et al. Abnormal protein profiles in tears with dry eye syndrome. Am J Ophthalmol 2003;136:291–9. [DOI] [PubMed] [Google Scholar]
- 11. Niemelä A, Kulomaa M, Vija P, Tuohimaa P, Saarikoski S. Lactoferrin in human amniotic fluid. Hum Reprod 1989;4:99–101. [DOI] [PubMed] [Google Scholar]
- 12. Lin AL, Johnson DA, Patterson TF, et al. Salivary anticandidal activity and saliva composition in an HIV-infected cohort. Oral Microbiol Immunol 2001;16:270–8. [DOI] [PubMed] [Google Scholar]
- 13. Caccavo D, Sebastiani GD, Di Monaco C, et al. Increased levels of lactoferrin in synovial fluid but not in serum from patients with rheumatoid arthritis. Int J Clin Lab Res 1999;29:30–5. [DOI] [PubMed] [Google Scholar]
- 14. Zhang P, Sawicki V, Lewis A, Hanson L, Nuijens JH, Neville MC. Human lactoferrin in the milk of transgenic mice increases intestinal growth in ten-day-old suckling neonates. Adv Exp Med Biol 2001;501:107–13. [DOI] [PubMed] [Google Scholar]
- 15. Barboza M, Pinzon J, Wickramasinghe S, et al. Glycosylation of human milk lactoferrin exhibits dynamic changes during early lactation enhancing its role in pathogenic bacteria-host interactions. Mol Cell Proteomics 2012;11:M111.015248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ochoa TJ, Cleary TG. Effect of lactoferrin on enteric pathogens. Biochimie 2009;91:30–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ammendolia MG, Agamennone M, Pietrantoni A, et al. Bovine lactoferrin-derived peptides as novel broad-spectrum inhibitors of influenza virus. Pathog Glob Health 2012;106:12–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mueller EA, Trapp S, Frentzel A, Kirch W, Brantl V. Efficacy and tolerability of oral lactoferrin supplementation in mild to moderate acne vulgaris: an exploratory study. Curr Med Res Opin 2011;27:793–7. [DOI] [PubMed] [Google Scholar]
- 19. Zavaleta N, Figueroa D, Rivera J, Sánchez J, Alfaro S, Lönnerdal B. Efficacy of rice-based oral rehydration solution containing recombinant human lactoferrin and lysozyme in Peruvian children with acute diarrhea. J Pediatr Gastroenterol Nutr 2007;44:258–64. [DOI] [PubMed] [Google Scholar]
- 20. Lyons TE, Miller MS, Serena T, et al. Talactoferrin alfa, a recombinant human lactoferrin promotes healing of diabetic neuropathic ulcers: a phase ½ clinical study. Am J Surg 2007;193:49–54. [DOI] [PubMed] [Google Scholar]
- 21. King JC, Jr, Cummings GE, Guo N, et al. A double-blind, placebo-controlled, pilot study of bovine lactoferrin supplementation in bottle-fed infants. J Pediatr Gastroenterol Nutr 2007;44:245–51. [DOI] [PubMed] [Google Scholar]
- 22. Ochoa TJ, Chea-Woo E, Campos M, et al. Impact of lactoferrin supplementation on growth and prevalence of Giardia colonization in children. Clin Infect Dis 2008;46:1881–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Håversen LA, Baltzer L, Dolphin G, Hanson LA, Mattsby-Baltzer I. Anti-inflammatory activities of human lactoferrin in acute dextran sulphate-induced colitis in mice. Scand J Immunol 2003;57:2–10. [DOI] [PubMed] [Google Scholar]
- 24. Togawa J, Nagase H, Tanaka K, et al. Oral administration of lactoferrin reduces colitis in rats via modulation of the immune system and correction of cytokine imbalance. J Gastroenterol Hepatol 2002;17:1291–8. [DOI] [PubMed] [Google Scholar]
- 25. Togawa J, Nagase H, Tanaka K, et al. Lactoferrin reduces colitis in rats via modulation of the immune system and correction of cytokine imbalance. Am J Physiol Gastrointest Liver Physiol 2002;283:G187–95. [DOI] [PubMed] [Google Scholar]
- 26. Burns RC, Rivera-Nieves J, Moskaluk CA, Matsumoto S, Cominelli F, Ley K. Antibody blockade of ICAM-1 and VCAM-1 ameliorates inflammation in the SAMP-1/Yit adoptive transfer model of Crohn’s disease in mice. Gastroenterology 2001;121:1428–36. [DOI] [PubMed] [Google Scholar]
- 27. Su L, Nalle SC, Shen L, et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology 2013;145:407–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tao R, de Zoeten EF, Ozkaynak E, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med 2007;13:1299–307. [DOI] [PubMed] [Google Scholar]
- 29. Zhang HL, Zheng YJ, Pan YD, et al. Regulatory T-cell depletion in the gut caused by integrin β7 deficiency exacerbates DSS colitis by evoking aberrant innate immunity. Mucosal Immunol 2016;9:391–400. [DOI] [PubMed] [Google Scholar]
- 30. Mori K, Yamanishi H, Ikeda Y, et al. Oral administration of carbonic anhydrase I ameliorates murine experimental colitis induced by Foxp3-CD4+CD25- T cells. J Leukoc Biol 2013;93:963–72. [DOI] [PubMed] [Google Scholar]
- 31. Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity 1999;10:387–98. [DOI] [PubMed] [Google Scholar]
- 32. Nguyen GC, Chong CA, Chong RY. National estimates of the burden of inflammatory bowel disease among racial and ethnic groups in the United States. J Crohns Colitis 2014;8:288–95. [DOI] [PubMed] [Google Scholar]
- 33. Loftus EV., Jr Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004;126:1504–17. [DOI] [PubMed] [Google Scholar]
- 34. Fazio VW, Wu JS. Surgical therapy for Crohn’s disease of the colon and rectum. Surg Clin North Am 1997;77:197–210. [DOI] [PubMed] [Google Scholar]
- 35. Achkar JP, Shen B. Medical management of postoperative complications of inflammatory bowel disease: pouchitis and Crohn’s disease recurrence. Curr Gastroenterol Rep 2001;3:484–90. [DOI] [PubMed] [Google Scholar]
- 36. Van Limbergen J, Russell RK, Drummond HE, et al. Definition of phenotypic characteristics of childhood-onset inflammatory bowel disease. Gastroenterology 2008;135:1114–22. [DOI] [PubMed] [Google Scholar]
- 37. Randall J, Singh B, Warren BF, Travis SP, Mortensen NJ, George BD. Delayed surgery for acute severe colitis is associated with increased risk of postoperative complications. Br J Surg 2010;97:404–9. [DOI] [PubMed] [Google Scholar]
- 38. Graham L. AGA reviews the use of corticosteroids, immunomodulators, and infliximab in IBD. Am Fam Physician 2007;75:4106. [Google Scholar]
- 39. Baert FJ, D’Haens GR, Peeters M, et al. Tumor necrosis factor alpha antibody [infliximab] therapy profoundly down-regulates the inflammation in Crohn’s ileocolitis. Gastroenterology 1999;116:22–8. [DOI] [PubMed] [Google Scholar]
- 40. Baert F, Vermeire S, Noman M, Van Assche G, D’Haens G, Rutgeerts P. Management of ulcerative colitis and Crohn’s disease. Acta Clin Belg 2004;59:304–14. [DOI] [PubMed] [Google Scholar]
- 41. Vester-Andersen MK, Prosberg MV, Jess T, et al. Disease course and surgery rates in inflammatory bowel disease: a population-based, 7-year follow-up study in the era of immunomodulating therapy. Am J Gastroenterol 2014;109:705–14. [DOI] [PubMed] [Google Scholar]
- 42. Targan SR, Hanauer SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N Engl J Med 1997;337:1029–35. [DOI] [PubMed] [Google Scholar]
- 43. Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody [adalimumab] in Crohn’s disease: the CLASSIC-I trial. Gastroenterology 2006;130:323–33; quiz 591. [DOI] [PubMed] [Google Scholar]
- 44. Sandborn WJ, Feagan BG, Stoinov S, et al. ; PRECISE 1 Study Investigators. Certolizumab pegol for the treatment of Crohn’s disease. N Engl J Med 2007;357:228–38. [DOI] [PubMed] [Google Scholar]
- 45. Allez M, Karmiris K, Louis E, et al. Report of the ECCO pathogenesis workshop on anti-TNF therapy failures in inflammatory bowel diseases: definitions, frequency and pharmacological aspects. J Crohns Colitis 2010;4:355–66. [DOI] [PubMed] [Google Scholar]
- 46. Yanai H, Hanauer SB. Assessing response and loss of response to biological therapies in IBD. Am J Gastroenterol 2011;106:685–98. [DOI] [PubMed] [Google Scholar]
- 47. Reddy JG, Loftus EV., Jr Safety of infliximab and other biologic agents in the inflammatory bowel diseases. Gastroenterol Clin North Am 2006;35:837–55. [DOI] [PubMed] [Google Scholar]
- 48. Velusamy SK, Fine DH, Velliyagounder K. Prophylactic effect of human lactoferrin against Streptococcus mutans bacteremia in lactoferrin knockout mice. Microbes Infect 2014;16:762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kimber I, Cumberbatch M, Dearman RJ, Headon DR, Bhushan M, Griffiths CE. Lactoferrin: influences on Langerhans cells, epidermal cytokines, and cutaneous inflammation. Biochem Cell Biol 2002;80:103–7. [DOI] [PubMed] [Google Scholar]
- 50. Hayashida K, Kaneko T, Takeuchi T, Shimizu H, Ando K, Harada E. Oral administration of lactoferrin inhibits inflammation and nociception in rat adjuvant-induced arthritis. J Vet Med Sci 2004;66:149–54. [DOI] [PubMed] [Google Scholar]
- 51. Machnicki M, Zimecki M, Zagulski T. Lactoferrin regulates the release of tumour necrosis factor alpha and interleukin 6 in vivo. Int J Exp Pathol 1993;74:433–9. [PMC free article] [PubMed] [Google Scholar]
- 52. Pandiyan P, Zhu J. Origin and functions of pro-inflammatory cytokine producing Foxp3+ regulatory T cells. Cytokine 2015;76:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Akin IM, Atasay B, Dogu F, et al. Oral lactoferrin to prevent nosocomial sepsis and necrotizing enterocolitis of premature neonates and effect on T-regulatory cells. Am J Perinatol 2014;31:1111–20. [DOI] [PubMed] [Google Scholar]
- 54. Yen CC, Lin CY, Chong KY, et al. Lactoferrin as a natural regimen for selective decontamination of the digestive tract: recombinant porcine lactoferrin expressed in the milk of transgenic mice protects neonates from pathogenic challenge in the gastrointestinal tract. J Infect Dis 2009;199:590–8. [DOI] [PubMed] [Google Scholar]
- 55. Hadis U, Wahl B, Schulz O, et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 2011;34:237–46. [DOI] [PubMed] [Google Scholar]
- 56. Hu S, Chen M, Wang Y, et al. mTOR inhibition attenuates dextran sulfate sodium-induced colitis by suppressing T cell proliferation and balancing TH1/TH17/Treg profile. PLoS One 2016;11:e0154564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jhunjhunwala S, Chen LC, Nichols EE, Thomson AW, Raimondi G, Little SR. All-trans retinoic acid and rapamycin synergize with transforming growth factor-β1 to induce regulatory T cells but confer different migratory capacities. J Leukoc Biol 2013;94:981–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Collins CB, Aherne CM, Kominsky D, et al. Retinoic acid attenuates ileitis by restoring the balance between T-helper 17 and T regulatory cells. Gastroenterology 2011;141:1821–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Brunstein CG, Miller JS, Cao Q, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood 2011;117:1061–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Di Ianni M, Falzetti F, Carotti A, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 2011;117:3921–8. [DOI] [PubMed] [Google Scholar]
- 61. Bluestone JA, Buckner JH, Fitch M, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med 2015;7:315ra189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Laffan AM, McKenzie R, Forti J, et al. Lactoferrin for the prevention of post-antibiotic diarrhoea. J Health Popul Nutr 2011;29:547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brines RD, Brock JH. The effect of trypsin and chymotrypsin on the in vitro antimicrobial and iron-binding properties of lactoferrin in human milk and bovine colostrum. Unusual resistance of human apolactoferrin to proteolytic digestion. Biochim Biophys Acta 1983;759:229–35. [DOI] [PubMed] [Google Scholar]
- 64. Inubushi T, Kawazoe A, Miyauchi M, et al. Molecular mechanisms of the inhibitory effects of bovine lactoferrin on lipopolysaccharide-mediated osteoclastogenesis. J Biol Chem 2012;287:23527–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Håversen L, Ohlsson BG, Hahn-Zoric M, Hanson LA, Mattsby-Baltzer I. Lactoferrin down-regulates the LPS-induced cytokine production in monocytic cells via NF-kappa B. Cell Immunol 2002;220:83–95. [DOI] [PubMed] [Google Scholar]
- 66. Kim CW, Lee TH, Park KH, Choi SY, Kim J. Human lactoferrin suppresses TNF-α-induced intercellular adhesion molecule-1 expression via competition with NF-κB in endothelial cells. FEBS Lett 2012;586:229–34. [DOI] [PubMed] [Google Scholar]
- 67. Ando K, Hasegawa K, Shindo K, et al. Human lactoferrin activates NF-kappaB through the Toll-like receptor 4 pathway while it interferes with the lipopolysaccharide-stimulated TLR4 signaling. FEBS J 2010;277:2051–66. [DOI] [PubMed] [Google Scholar]
- 68. Zong X, Song D, Wang T, et al. LFP-20, a porcine lactoferrin peptide, ameliorates LPS-induced inflammation via the MyD88/NF-κB and MyD88/MAPK signaling pathways. Dev Comp Immunol 2015;52:123–31. [DOI] [PubMed] [Google Scholar]
- 69. Willnow TE, Goldstein JL, Orth K, Brown MS, Herz J. Low density lipoprotein receptor-related protein and gp330 bind similar ligands, including plasminogen activator-inhibitor complexes and lactoferrin, an inhibitor of chylomicron remnant clearance. J Biol Chem 1992;267:26172–80. [PubMed] [Google Scholar]
- 70. Jiang R, Lönnerdal B. Apo- and holo-lactoferrin stimulate proliferation of mouse crypt cells but through different cellular signaling pathways. Int J Biochem Cell Biol 2012;44:91–100. [DOI] [PubMed] [Google Scholar]
- 71. Takayama Y, Aoki R, Uchida R, Tajima A, Aoki-Yoshida A. Role of CXC chemokine receptor type 4 as a lactoferrin receptor. Biochem Cell Biol 2017;95:57–63. [DOI] [PubMed] [Google Scholar]
- 72. Stragier E, Van Assche G. The use of fecal calprotectin and lactoferrin in patients with IBD. Review. Acta Gastroenterol Belg 2013;76:322–8. [PubMed] [Google Scholar]
- 73. Lamb CA, Mansfield JC. Measurement of faecal calprotectin and lactoferrin in inflammatory bowel disease. Frontline Gastroenterol 2011;2:13–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Collins CB, Aherne CM, Ehrentraut SF, et al. Alpha-1-antitrypsin therapy ameliorates acute colitis and chronic murine ileitis. Inflamm Bowel Dis 2013;19:1964–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Deboer MD. Use of ghrelin as a treatment for inflammatory bowel disease: mechanistic considerations. Int J Pept 2011;2011:189242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. MacManus CF, Campbell EL, Keely S, Burgess A, Kominsky DJ, Colgan SP. Anti-inflammatory actions of adrenomedullin through fine tuning of HIF stabilization. FASEB J 2011;25:1856–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Metelli A, Wu BX, Fugle CW, et al. Surface expression of TGFβ docking receptor GARP promotes oncogenesis and immune tolerance in breast cancer. Cancer Res 2016;76:7106–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Seo YD, Pillarisetty VG. T-cell programming in pancreatic adenocarcinoma: a review. Cancer Gene Ther 2017;24:106–13. [DOI] [PubMed] [Google Scholar]
- 79. Domagala-Kulawik J, Osinska I, Hoser G. Mechanisms of immune response regulation in lung cancer. Transl Lung Cancer Res 2014;3:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ramalingam S, Crawford J, Chang A, et al. ; FORTIS-M Study Investigators. Talactoferrin alfa versus placebo in patients with refractory advanced non-small-cell lung cancer [FORTIS-M trial]. Ann Oncol 2013;24:2875–80. [DOI] [PubMed] [Google Scholar]
- 81. Vincent JL, Marshall JC, Dellinger RP, et al. ; Oral tAlactoferrin in Severe sepsIS Study Investigators. Talactoferrin in severe sepsis: results from the phase II/III Oral tAlactoferrin in severe sepsIS trial. Crit Care Med 2015;43:1832–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.