

Abstract
Background and Aims:
Intestinal fibrosis is a frequent complication in Crohn’s disease [CD]. The mouse Salmonella typhimurium model, due to its simplicity, reproducibility, manipulability, and penetrance, is an established fibrosis model for drug discovery and preclinical trials. However, the severity of fibrosis and mortality are host- and bacterial strain-dependent, thus limiting the original model. We re-evaluated the S. typhimurium model to optimise fibrosis and survival, using commercially available mouse strains.
Methods:
Fibrotic and inflammatory markers were evaluated across S. typhimurium ΔaroA:C57bl/6 studies performed in our laboratory. A model optimisation study was performed using three commercially available mouse strains [CBA/J, DBA/J, and 129S1/SvImJ] infected with either SL1344 or ΔaroA S. typhimurium. Fibrotic penetrance was determined by histopathology, gene expression, and αSMA protein expression. Fibrosis severity, penetrance, and survival were analysed across subsequent CBA studies.
Results:
Fibrosis severity and survival are both host- and bacterial strain-dependent. Marked tissue fibrosis and 100% survival occurred in the CBA/J strain infected with SL1344. Subsequent experiments demonstrated that CBA/J mice develop extensive intestinal fibrosis, characterised by transmural tissue fibrosis, a Th1/Th17 cytokine response, and induction of pro-fibrotic genes and extracellular matrix proteins. A meta-analysis of subsequent SL1344:CBA/J studies demonstrated that intestinal fibrosis is consistent and highly penetrant across histological, protein, and gene expression markers. As proof-of-concept, we tested the utility of the SL1344:CBA/J fibrosis model to evaluate efficacy of CCG-203971, a novel anti-fibrotic drug.
Conclusion:
The S. typhimurium SL1344:CBA/J model is an optimised model for the study of intestinal fibrosis.
Keywords: IBD, fibrosis, Salmonella typhimurium, mouse strain, model
1. Introduction
Fibrosis, the final common pathway of chronic organ failure, is the leading cause of mortality and morbidity.1–4 In inflammatory bowel disease [IBD], intestinal fibrosis commonly culminates with irreversible organ damage and intestinal blockage, necessitating surgical removal of intestine.5 The incidence and prevalence of IBD worldwide has increased rapidly in the past decade, underscoring its emergence as a global disease.6 Animal models of IBD have been employed for decades, but these are predominantly focused on inflammation, host immune response, and more recently, the gut microbiome.7–9 Though fibrosis occurs in some of these well-characterised models, there is a paucity of intestinal fibrosis-specific animal models.10 Each has limitations with respect to fibrotic penetrance, feasibility, scale-ability, and clinical relevance.7
Although no model fully replicates the complex aetiology of human disease, the murine Salmonella typhimurium model recapitulates much of the pathology of human intestinal fibrosis, including transmural tissue fibrosis, a Th1/Th17 immune response, and induction of pro-fibrotic genes and ECM [extracellular matrix] proteins. As originally described by Grassl et al., the S. typhimurium model has a number of advantages over other colitis and fibrosis models, including reliable disease induction, reduced mortality, and high penetrance in a relatively short induction period.11
In this model, eradication of the normal microflora by a single oral dose of streptomycin creates a niche for S. typhimurium colonisation after a single oral inoculum. Chronic S. typhimurium infection of the gut triggers colitis and ultimately tissue fibrosis. In the original model, the most robust fibrosis was observed in 129SvImJ mice infected with wild-type S. typhimurium strain SL1344. However, intestinal fibrosis was observed in several other mouse strains including DBA and C57bl/6, a critical finding as a majority of genetically modified mice are on a C57bl/6 background.7,12 Overall, C57bl/6 mice are less susceptible to organ fibrosis but highly susceptible to SL1344, due to a mutation in the natural bacterial resistance gene Nramp1.13,14 These limitations were circumvented using an attenuated S. typhimurium ΔaroA strain which induces intestinal fibrosis in the C57bl/6 strain without severe systemic disease. However, fibrosis in the ΔaroA:C57bl/6 model differs both qualitatively and quantitatively from fibrosis in the SL1344:129SvImJ model.
In a number of independent experiments with the ΔaroA:C57bl/6 model in our laboratory, intestinal fibrosis presented with a characteristically profound histological response, consistent with the original model description. Other markers of fibrosis, including αSMA protein expression and fibrotic gene expression [IGF-1, TGFβ, COL1A1], although predictive of fibrosis within an individual study, showed considerable inter-study variation. To determine which markers were most predictive of inflammation and fibrosis, we performed a meta-analysis of our ΔaroA:C57bl/6 studies. As we report here, inflammation is robust and highly penetrant in the ΔaroA:C57bl/6 model. However, fibrosis is attenuated and variably penetrant, thus larger numbers of animals are required to detect experimental outcomes.
Evaluation of preclinical anti-fibrotic therapeutics is our ultimate goal and is dependent upon a robust, highly reproducible model with high fibrotic penetrance and animal survival. As originally reported, intestinal fibrosis differs both in penetrance and magnitude depending on bacterial and host strains. Therefore, to explore the utility of the S. typhimurium model specifically for fibrosis research, we assessed fibrosis using the more virulent SL1344 S. typhimurium strain. As originally described, the 129SvImJ mouse strain which lacks the NrampD169 allele is reportedly less susceptible to severe systemic disease and associated mortality, but develops profound tissue fibrosis.11 However, the inherent genetic heterogeneity of the 129 mouse strains and intra-substrain variability are problematic. As we report here, the utility of commercially available 129SvJ mouse strains is limited due to poor survival. Therefore, we optimised survival, reproducibility, and disease penetrance of the mouse S. typhimurium fibrosis model using commercially available and genetically stable mouse strains CBA/J, DBA/J, and 129S1/SvImJ, infected with either ΔaroA or SL1344.
Survival and fibrosis were both host- and bacterial strain-dependent, with the 100% survival and robust fibrosis observed in the SL1344:CBA model. In a series of follow-up studies, fibrotic disease was consistent across a number of markers including histopathological, gene expression, and protein expression markers. A subsequent meta-analysis of our independent SL1344:CBA studies confirmed that fibrotic disease is robust and highly penetrant. In addition, sample size calculations revealed that substantially smaller group sizes are needed to achieve sufficient experimental power in the SL1344:CBA model.
As these studies indicate, the use of the SL1344:CBA model increases the model efficiency, decreases costs, and reduces the number of experimental animals required, thereby providing an optimised rodent fibrosis model for drug discovery and preclinical applications. As proof-of-concept, we tested the utility of the SL1344:CBA fibrosis model to evaluate efficacy of CCG-203971, a novel preclinical anti-fibrotic therapeutic.15,16 Whereas only modest efficacy was observed, this finding underscores the utility of the more sensitive and robust SL1344:CBA model to evaluate novel anti-fibrotic therapeutics.
2. Materials and Methods
2.1. Bacterial and mouse strains
Salmonella enterica serotype typhimurium strains ΔaroA [a generous gift from G. Grassl, Hannover Medical School, Hannover, Niedersachsen, Germany] and wild-type SL1344 [a kind gift from M. O’Riordan, University of Michigan, USA], which are naturally resistant to streptomycin, were grown in Luria broth [LB] containing 100 μg/ml streptomycin at 37°C. Infectious titres were determined by plating triplicate serial dilutions of the inoculum onto LB/streptomycin plates.
Specific pathogen-free female 8–10-week-old C57BL/6J [Stock No. 000664], 129X1/SvJ [Stock No. 000691], 129S1/SvImJ [Stock No. 002448], DBA/1J [Stock No. 000670], and CBA/J [Stock No. 00656] mice were purchased from Jackson Laboratories [Bar Harbor, ME, USA].
Mice received 20 mg streptomycin in 0.1 M Hank’s buffered salt solution [HBSS] to eradicate the commensal microbiota 24 h before infection with either S. typhimurium strain SL1344 or ΔaroA, in 100 µl 0.1 M HEPES buffer [pH 8.0] by oral gavage as previously described.17–20 Control mice received 100 µl 0.1 M HEPES by oral gavage.
All animal experiments were conducted with the approval and oversight of the University of Michigan Institutional Animal Care and Use Committee [IACUC] and strictly complied with National Institutes of Health Guide for Care and Use of Laboratory Animals.
2.2. S. typhimurium mouse experiments
Study characteristics of the studies described below are summarised in Table 1.
Table 1.
S. typhimurium study characteristics.
| Study | S. typhimurium strain[s] | Mouse strain[s] | Duration [days] | Uninfected [n] | Infected [n] | Comments |
|---|---|---|---|---|---|---|
| ΔaroA:C57bl/6 model | ||||||
| C57-1 | ΔaroA | C57bl/6 | 22 | 5 | 4 | Pilot ΔaroA study |
| C57-2 | ΔaroA | C57bl/6 | 27 | 3 | 8 | |
| C57-5 | ΔaroA | C57bl/6 | 20 | 4 | 4 | Higgins20 |
| C57-6 | ΔaroA | C57bl/6 | 20 | 5 | 5 | |
| C57-7 | ΔaroA | C57bl/6 | 20 | 5 | 5 | |
| C57-8 | ΔaroA | C57bl/6 | 21 | 5 | 5 | Higgins19 |
| C57-10 | ΔaroA | C57bl/6 | 21 | 5 | 5 | Higgins19 |
| C57-11 | ΔaroA | C57bl/6 | 21 | 5 | 5 | Higgins20 |
| C57-12 | ΔaroA | C57bl/6 | 21 | 5 | 5 | Higgins19 |
| SL1344:129X1/SvImJ model | ||||||
| X1-1 | SL1344 | X1 | 13 | 5 | 5 | Pilot SL1344 study |
| X1-2 | SL1344 | X1 | 14 | 5 | 5 | Infectious dose-response |
| SL1344:129S1/SvImJ model | ||||||
| S1-1 | SL1344 | S1 | 19 | 5 | 5 | |
| S1-2 | SL1344 | S1 | 21 | 5 | 5 | |
| Bacterial and host strain comparison | ||||||
| MST-16 | ΔaroA, SL1344 | S1, DBA, CBA | 14 | 5/strain | 0 | Host-response study |
| SL1344:CBA model | ||||||
| MST-29 | SL1344 | CBA | 21 | 5 | 5 | |
| MST-31 | SL1344 | CBA | 21 | 5 | 5 | |
| MST-34 | SL1344 | CBA | 21 | 5 | 5 | |
| MST-36 | SL1344 | CBA | 21 | 5 | 4 | |
ΔaroA, S. typhimurium strain ΔaroA; SL1344, S. typhimurium strain SL1344; X1, 129X1/SvImJ strain; S1, 129S1/SvImJ strain; DBA, DBA/J strain; CBA, CBA/J strain.
2.2.1. ΔaroA:C57bl/6 experiments
For the inaugural ΔaroA:C57bl/6 experiment [C57-1], 10 mice were treated with streptomycin to eradicate the resident microbiota; 24 h after streptomycin treatment, bacterial eradication was confirmed by stool plating onto LB/streptomycin plates. Half the mice [n = 5] were infected with 1 x 106 cfu S. typhimurium ΔaroA by oral gavage, and the other half [n = 5] received vehicle. Mice were sacrificed on Day 22 post infection, when fibrosis is at its zenith. The second ΔaroA:C57bl/6 [C57-2] was performed similarly but with three uninfected and seven ΔaroA-infected mice. Subsequent ΔaroA:C57bl/6 experiments [C57-5, C57-6, C57-7, C57-8, C57-10, C57-11, C57-12] were performed similarly with five mice per group in the both the uninfected and infected groups, with the exception of experiment C57-5 which contained four mice per group.
2.2.2. Wild-type S. typhimurium SL1344 infection in 129X1/SvImJ and 129S1/SvImJ mouse strains
As described in the original model, infection with 3 x 106 or 3 x 108 wild-type S. typhimurium SL1344 induces robust fibrosis in 129SvJ mice.11 However, this 129SvJ strain is an institutional strain and is not available in the USA. Therefore, in our pilot wild-type S. typhimurium SL1344 study [X1-1], we used the 129X1/SvImJ [X1] mouse strain [Jackson Laboratories, Stock No. 000691]. Female 8–10-week-old X1 mice were treated with streptomycin [n = 10]. Half the mice [n = 5] were infected with 3 x 107 cfu SL1344 by oral gavage and uninfected mice [n = 5] received vehicle only. The study was terminated on Day 13 post infection due to high mortality in the SL1344-infected group.
An infectious dose-response study [X1-2] was performed to optimise survival in the SL1344:X1 model using the X1 mouse strain infected with 3 x 102, 1.5 x 103, 3 x 103, 1.5 x 104, 6.5 x 104, or 1.5 x 105 cfu SL1344 [n = 5 per group]. Survival was compared with uninfected mice [n = 5]. Due to high mortality, the study was terminated at Day 15 post infection.
As communicated by Grassl, 129X1/SvImJ mice are more sensitive to wild-type S. typhimurium than another 129 substrain, 129S1/SvImJ [Grassl, personal communication]. Therefore, we tested survival and fibrotic response in the 129S1/SvImJ strain [S1] in response to SL1344 infection. In two separate experiments [S1-1 and S1-2], S1 mice [five per experiment] were infected with SL1344 [3.3 x 105 and 3.7 x 106 cfu, respectively]. Survival and fibrosis were compared with uninfected controls [n = 5 per experiment]. Experiments were terminated at post infection Day 19 and Day 22, respectively.
2.2.3. Bacterial and host strain comparison experiment
In the strain comparison study [MST-16], we compared survival and fibrosis in the S1 strain with two other commercially available mouse strains DBA/1J and CBA/J. Ten animals [8–10-week-old females] from each mouse strain were divided into two groups of five per group and infected with either wild-type SL1344 or ΔaroA. The experiment was terminated on Day 14 post infection.
2.2.4. SL1344: CBA/J studies
In four independent experiments [MST-29, MST-31, MST-34, MST-36], 8–10-week-old female CBA/J mice [n = 5 per experiment] were treated with streptomycin and infected 24 h later with 2.5 to 3.5 x 106 cfu SL1344. In each experiment, a control cohort of uninfected mice [n = 5 per experiment] was treated with streptomycin but then received vehicle. All experiments were terminated at Day 21 post infection.
2.3. Anti-fibrotic therapeutic studies
CCG-203971 [N-[4-chlorophenyl]-1-{[3-[furan-2-yl]phenyl]carbonyl}piperidine-3-carboxamide] was synthesised by the Vahlteich Medicinal Chemistry Core at the University of Michigan.15 Previous studies in C57BL/6 mice demonstrated oral dosing with 100 mg/kg/day of CCG-203971 for 14 days was well tolerated [Neubig, unpublished observations]. Therefore, in our pilot prophylactic treatment study, animals in the drug control and prevention groups received 100 mg/kg/day of CCG-203971 twice daily by oral gavage for 7 days. Animals in the negative control and fibrosis control groups received vehicle [peanut oil] only on the same dosing schedule. After 7 days, all animals received 20 mg of streptomycin by oral gavage to eradicate the gut microbiota, and 24 h later animals in the positive control and prevention groups were infected with 2.4 x 106 cfu SL1344 in 0.1 M HBSS. Negative control and drug control groups received HBSS only. All mice received vehicle or drug twice daily by oral gavage until the end of the study. Due to unexpected deaths in both the prevention and drug control groups, the study was terminated early at Day 11 post infection. Although the toxicity studies in C57BL/6 mice suggested that CCG-203971 was well tolerated for 14 days, longer administration in the CBA/J mice resulted in mortality at Day 16 of administration.
2.4. Gross pathology and tissue collection
Mice were euthanised at the end of each study or when moribund in accordance with Institutional Animal Care and Use Committee [IACUC] humane endpoints. Caecum and distal colon were collected, photographed, measured, and weighed. Tissues were snap-frozen in liquid nitrogen and stored at -80°C before molecular analysis. Caecal contents were collected and serially diluted before plating onto LB/streptomycin plates to determine bacterial titres.19,20
2.5. Histology
Formalin-fixed and paraffin-embedded tissues were stained with haematoxylin and eosin [H&E, inflammatory histology] and Masson’s trichrome [fibrosis] by the University of Michigan CCGC Research Histology and Immunoperoxidase Laboratory [Ann Arbor, MI]. Digital photomicrographs of tissue sections were taken with an Olympus BX microscope [University of Michigan Microscopy and Image Analysis Laboratory]. Histological scoring was performed by a blinded pathologist [DM]. Inflammation was determined using a Wirtz Scale 0 to 4 point scoring system: [0] no inflammation; [1] low level of inflammation with scattered infiltrating mononuclear cells; [2] moderate inflammation with multiple foci; [3] high level of inflammation with increased vascular density and marked wall thickening; and [4] maximal severity of inflammation with transmural leukocyte infiltration and loss of goblet cells.19,21 Fibrosis was scored separately as follows: [0] no fibrosis; [1] mild fibrosis [focal mucosal/submucosal collagen deposition without architectural distortion]; [2] moderate fibrosis [significant mucosal/submucosal collagen deposition with modest distortion of mucosal/submucosal architecture but without obscuring of the mucosal/submucosal border]; and [3] severe fibrosis [extensive mucosal submucosal collagen deposition with marked architectural distortion obscuring the mucosal/submucosal border]. Tissue measurements were quantitated with ImageJ analysis software [NIH, Bethesda, MD].
2.6. Quantitative real-time polymerase chain reaction
RNA was extracted from the caecum using the RNeasykit [Qiagen, Valencia, CA]. Reverse transcription of 2 μg of total RNA was performed with the Superscript First Strand RT kit [Invitrogen]. Quantitative real-time polymerase chain reaction [qRT-PCR] of collagen 1 [COL1A1], IGF-1 [insulin-like growth factor 1], TGFβ [transforming growth factor beta], interleukin-1 [IL-1β], tumour necrosis factor alpha [TNF-α], IL-6, IL-17, IL-12p40, and glyceraldehyde 3-phosphate dehydrogenase [GAPDH] was performed with commercial TaqMan gene expression assays [ABI, Foster City, CA]. Q-PCR was performed using a Stratagene Mx3000P real-time PCR system [Stratagene, La Jolla, CA]. Cycling conditions were 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 62°C for 60 s. Gene expression was normalised to GAPDH as the endogenous control and fold-changes [RQ] relative to uninfected controls were calculated using the ΔΔCt-method.22
2.7. Immunoblotting
Immunoblotting was used for the detection of α-smooth muscle actin [αSMA]. Whole tissue was pulverized under liquid nitrogen and lysed in ice-cold radioimmuno precipitation assay buffer [RIPA buffer] with a cocktail of proteinase inhibitors [Roche, Indianapolis, IN].19,20 Protein content was determined using a modified Bradford assay kit [BioRad, Hercules, CA]. Total protein was separated by sodium dodecyl sulfate [SDS] polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride [PVDF] membranes [Amersham Biosciences, Piscataway, NJ].
Membranes were blocked in 5% milk/tris-buffered saline (TBS) and polysorbate 20 [TBST] solution for 1 h at room temperature or overnight at 4°C. αSMA was detected by incubating the membrane overnight at 4°C with mouse antihuman monoclonal antibody [Sigma, St Louis, MO] at 1:5000 dilution in 5% milk/TBST. As a loading control, a mouse antibody for GAPDH [Chemicon, Temecula, CA] was used. Secondary antibody antimouse IgG HRP [Amersham, Piscataway, NJ] was incubated for 1 h at room temperature and the signal was detected by the Pierce detection system [Rockford, IL]. Collagen protein was detected with a rabbit polyclonal antibody for collagen Type I [Rockland, Limerick, PA] at a 1:1000 dilution and HRP-labelled secondary goat-anti-rabbit IgG HRP [Thermo, Waltham, MA] at a 1:10 000 dilution. Autoradiographs were scanned and quantitated using ImageJ analysis software.
2.8. Meta-analysis study selection
Twelve independent S. typhimurium ΔaroA:C57bl/6 studies were performed by our laboratory, five of which have been previously published.19,20 Two studies were excluded due to lack of gene expression data. The other study was excluded since it used a transgenic C57bl/6 strain. Of these, nine were included in the meta-analysis. In a separate meta-analysis, four independent, unpublished S. typhimurium SL1344:CBA/J studies were analysed. Characteristics of the included studies are summarised in Table 1. Detailed study methods for all the ΔaroA:C57bl/6 and SL1344:CBA/J studies are described above in Section 2.2., S. typhimurium mouse experiments.
2.9. Statistical analysis
Primary outcomes were histology, αSMA protein expression, and inflammatory and fibrotic gene expression. Molecular results were compared with histology [‘gold standard’]. Study results are presented as standardised mean differences [SMD] with a 95% confidence interval [CI] based on a random-effects model as described by Mantel–Haenszel.23 Meta-analysis was performed with the metan command in Stata 11.2 [StataCorp, College Station, TX]. The magnitude of the SMDs was interpreted as: small, SMD = 0.2; medium, SMD = 0.5; large, SMD = 0.8; and very large, SMD = 1.3.24 Statistical differences were determined by a two-sided, unpaired Student’s t test. Results with a p-value of < 0.05 were considered statistically significant. Power calculations were determined for the following metrics: αSMA and collagen I protein expression as well as expression of individual fibrotic or inflammatory genes. Sample size was determined with the sampsi command in Stata 11, assuming 80% power and a two-sided alpha of 0.05 [StataCorp, College Station, TX].
3. Results
3.1. Fibrosis in C57bl/6 mice infected with S. typhimurium ΔaroA is variably penetrant.
In a series of pilot experiments [C57-1 and C57-2] to replicate and validate fibrosis in the S. typhimurium ΔaroA:C57bl/6 model, C57bl/6 mice [n = 12] were treated with streptomycin to eradicate the resident microbiota 24 h before infection with 1 x 106 cfu S. typhimurium ΔaroA and were sacrificed on Days 22 and 27 post infection when fibrosis is at its zenith. Control animals [n = 8] received streptomycin and HBSS on the same schedule. In the ΔaroA cohort, chronic infection was characterised by a small, dense caecum with thin, watery contents and heavy caecal colonization [average 4.0 x 108 cfu, p < 0.001]. Caecae from infected mice were smaller [122 versus 163 pixels, p = 0.003], yet ~ 20% heavier [0.67 versus 0.51 g, p < 0.001] indicating possible oedema and/or excessive extracellular collagen deposition. Despite the high bacterial load and grossly pathological caecum, 100% survival was observed in the ΔaroA-infected animals, consistent with the original model. Significant tissue fibrosis was determined by histological staining for tissue collagen and expression of αSMA protein [Figure 1A, C].
Figure 1.

Fibrosis in C57bl/6 mice infected with S. typhimurium ΔaroA is variably penetrant. [A, B] Histological trichrome staining for collagen of uninfected [HBSS] and S. typhimurium ΔaroA-infected C57bl/6 mice in two independent experiments, C57-1 [A] and C57-5 [B], illustrating transmural fibrosis [blue staining] and distortion of tissue architecture by collagen infiltration. [C, D] Expression of αSMA protein in uninfected and infected mice from the same experiments, illustrating the differences in the αSMA response between the C57-1 [C] and C57-5 [D] experiments. Westerns were serially probed with GAPDH [glyceraldehyde 3-phosphate dehydrogenase] to control for protein loading differences. [E, F] Forest plots from a meta-analysis of nine independent S. typhimurium ΔaroA:C57bl/6 studies, illustrating the standardised mean differences [SMD] and 95% confidence intervals [CI] for αSMA protein expression [E] and COL1A1 gene expression [F]. Divergent studies C57-1 [pink] and C57-5 [blue] are highlighted. Random-effects modelling was used.
Concurrent histological inflammation was observed in the ΔaroA-infected mice as evidenced by wall thickening due to a marked increase in the inflammatory cell infiltrate of the mucosa and submucosa, an increase in the density of inflammatory cells surrounding the vasculature, and tissue oedema. No histological inflammation was observed in the uninfected mice. Consistent with the histological findings, induction of pro-inflammatory cytokine genes including Th1 [IL-1β] and Th17 [IL-17] genes was observed. Specifically, IL-1β was induced > 15-fold [p = 0.005] and IL-17 36-fold [p < 0.05] compared with the uninfected controls. In contrast to the originally reported model, we did not observe induction of pro-fibrotic cytokines or extracellular matrix proteins, including TGFβ, IGF-1, and COL1A1.
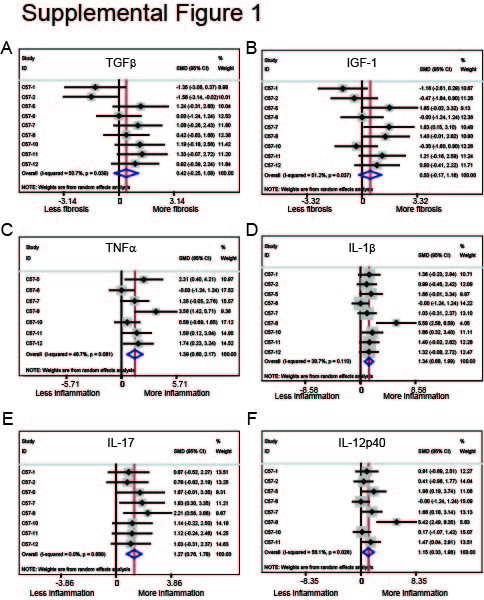
Subsequent ΔaroA:C57bl/6 studies in our laboratory demonstrated induction of intestinal fibrosis by histology and induction αSMA protein expression.19,20 Whereas all studies demonstrated tissue fibrosis as determined by histology, variability in the molecular markers was observed in our ΔaroA:C57bl/6 studies. In approximately a third of the studies, αSMA protein, a marker of activated myofibroblasts and surrogate marker for fibrosis, was not induced despite the presence of histological fibrosis and induction of collagen I [COL1A1] gene expression [Figure 1B, D, F]. In some studies, αSMA and COL1A1 results were discordant. For example, in the pilot C57-1 study, infection with ΔaroA induced αSMA protein without concurrent induction of COL1A1 gene expression. In the C57-5 study, COL1A1 gene expression was induced without concurrent induction of αSMA protein expression [Figure 1A-F]. In addition, we observed in several studies a lack of IGF-1 and TGFβ pro-fibrotic gene induction despite the presence of histological fibrosis. In contrast, inflammatory markers including TNFα, IL-1β, IL-17, and IL12p40, were consistently and significantly induced in all studies in the ΔaroA:C57bl/6 model [Supplementary Figure 1, available as Supplementary data at ECCO-JCC online].
To determine which fibrotic markers are indicative of intestinal fibrosis in the ΔaroA:C57bl/6 model, a meta-analysis of nine independent ΔaroA:C57bl/6 studies from our laboratory was performed [Table 1]. Differences in αSMA protein levels between uninfected [n = 41] and ΔaroA-infected mice [n = 44] were pooled as standardised mean differences [SMDs] using a random-effects model.23 Overall, ΔaroA infection exerted a very large effect on αSMA protein expression [SMD = 2.13, 95% CI: 0.85 - 3.42, p = 0.001, Figure 1E] and a moderate effect on COL1A1 gene expression [SMD = 0.55, 95% CI: 0.001 - 1.10, p = 0.049, Figure 1F]. However, ΔaroA infection did not have a significant effect on TGFβ or IGF-1 gene expression [SMDTGF = 0.42, 95% CI: -0.25 - 1.09, p = 0.22; SMDIGF-1 = 0.50, 95% CI: -0.17 - 1.16, p = 0.14, Table 2, Supplementary Figure 1]. Despite discordance between αSMA protein and COL1A1 gene expression in individual studies, the meta-analysis revealed that both αSMA protein expression and COL1A1 gene expression are positive markers of intestinal fibrosis in this model.
Table 2.
Summary of meta-analysis results and sample size calculations for fibrogenic and inflammatory markers in S. typhimurium ΔaroA:C57bl/6 and S. typhimurium SL1344:CBA/J mouse models.
| S. typhimurium ΔaroA:C57bl/6 | S. typhimurium SL1344:CBA/J | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Metric | SMD | CI [95%] | p-Value | n | Metric | SMD | CI [95%] | p-Value | n |
| αSMA protein | 2.13 | 0.85 -3.42 | 0.001 | 18 | αSMA protein | 3.36 | 2.15 - 4.57 | < 0.001 | 3 |
| collagen protein | 2.99 | 1.77 - 4.21 | < 0.001 | 4 | |||||
| Fibrotic genes | |||||||||
| COL1A1 | 0.55 | 0.001 - 1.10 | 0.049 | 69 | COL1A1 | 0.87 | 0.18 - 1.55 | 0.013 | 17 |
| TGFβ | 0.42 | -0.25 - 1.09 | 0.219 | 90 | TGFβ | 0.94 | 0.25 - 1.63 | 0.007 | 14 |
| IGF-1 | 0.50 | -0.17- 1.16 | 0.142 | 61 | IGF-1 | 1.68 | 0.90 - 2.45 | < 0.001 | 6 |
| Inflammatory genes | |||||||||
| TNFα | 1.39 | 0.60 - 2.17 | 0.001 | 26 | TNFα | 1.85 | 0.95 - 2.76 | < 0.001 | 3 |
| IL-1β | 1.34 | 0.69 - 1.99 | < 0.001 | 16 | IL-1β | 1.41 | 0.57 - 2.24 | 0.001 | 4 |
| IL-17 | 1.27 | 0.76 - 1.78 | < 0.001 | 6 | IL-17 | 1.09 | 0.29 - 1.88 | 0.007 | 12 |
| IL12p40 | 1.15 | 0.33 - 1.96 | 0.006 | 12 | IL12p40 | 1.61 | 0.73 - 2.49 | < 0.001 | 3 |
SMD, standardised mean difference; CI, confidence interval; n, number of mice required per experimental group; αSMA, alpha smooth muscle actin; COL1A1, collagen type I alpha 1; TGFβ, transforming growth factor beta; IGF-1, insulin-like growth factor 1; TNFα, tumour necrosis factor alpha; IL-1β, interleukin-1 beta; IL-17, interleukin 17; IL-12p40, interleukin 12 subunit P40. Effect sizes were determined after Cohen as: small, SMD = 0.2; medium, SMD = 0.5; large, SMD = 0.8; and very large SMD = 1.3.24
To further examine the utility of αSMA protein and COL1A1 gene expression as fibrotic markers, sample size calculations were performed for each fibrotic marker to predict the number of animals required for future ΔaroA:C57bl/6 experiments. Using αSMA protein as a marker would require 18 mice per experimental group, whereas COL1A1 would require a sample size of 69 mice per group to achieve sufficient experimental power [80%] [Table 2]. In contrast, all inflammatory genes [TNFα, IL-1β, IL-17, and IL12p40] were significant predictors of disease. Specifically, infection with ΔaroA had a large to very large effect on TNFα [SMD = 1.39, 95% CI: 0.60 - 2.17, p = 0.001], IL-1β [SMD = 1.34, 95% CI: 0.69 - 1.99, p < 0.001], IL-17 [SMD = 1.27, 95% CI: 0.76 - 1.78, p < 0.001], and IL-12p40 [SMD = 1.15, 95% CI: 0.33 - 1.96, p = 0.006] gene expression [Table 2]. Sample size calculations for each of the inflammatory markers predicted far fewer animals [6 to 26] would be required to detect experimental effects [Table 2].
Though fibrosis occurs in the ΔaroA:C57bl/6 model, the small effect size combined with the requirement for large numbers of experimental animals limits the feasibility of the ΔaroA:C57bl/6 model for fibrotic drug discovery and preclinical studies, particularly where multiple experimental groups are required. Therefore, to explore the utility of the S. typhimurium model for fibrosis research, we assessed fibrosis using the more virulent wild-type SL1344 S. typhimurium strain.
3.2. High mortality is observed in 129SvJ mouse strains infected with S. typhimurium SL1344
As described in the original S. typhimurium model, intestinal fibrosis differs in both penetrance and magnitude, depending on the bacterial strain and the mouse strain. Robust fibrosis was described in 129Sv/ImJ mice infected with wild-type S. typhimurium SL1344. However, this 129SvJ strain is an institutional strain and is unavailable in the USA. Therefore, we attempted to reproduce the fibrotic phenotype in the SL1344:129SvJ model using an equivalent, commercially available 129 strain, 129X1/SvJ [X1]. In our pilot study [X1-1], infection of the X1 mouse strain with SL1344 resulted in a very high death rate with 80% mortality by Day 6 post infection and 100% by Day 13 [Figure 2A]. To reduce mortality, we performed an infectious dose-response experiment in X1 mice, decreasing the infectious dose of SL1344 [3 x 102 to 1.5 x 105 cfu]. Zero mortality was observed only with the lowest infectious dose [3 x 102] group, whereas 5- to 10-fold higher inocula [1.5 x 103 and 3 x 103] produced 33–67% mortality; 100% mortality was observed by Day 7 post infection with inocula greater than 104 cfu [Figure 2B]. These discrepancies between our observed survival rates compared with the reported survival rate in the original SL1344:129SvJ model highlight the problematic nature of 129 strains and the importance of host genetics to susceptibility. Intentional and errant out-crossing of the 129 founder strains has resulted in extensive heterogeneity among the 129 substrains, cumulating in profoundly different experimental responses.25,26 As communicated by Grassl, the X1 strain is more sensitive to S. typhimurium than another 129 substrain, 129S1/SvImJ [Grassl, personal communication]. Therefore, we compared the effects of SL1344 infection in the 129X1/SvImJ [X1] and the 129S1/SvImJ [S1] strains. Though higher mortality was initially observed in the X1 strain by Day 8 post infection, overall survival was comparable between the X1 and S1 strains with > 50% mortality observed by Day 19 post infection [Figure 2C]. Animals that survived past Day 8 developed extensive bowel fibrosis, as evidenced by histological staining for collagen, increased αSMA protein expression, and a significant 5- to 6-fold increase in IGF-1 and TGFβ gene expression [p = 0.04, p = 0.039, respectively, Figure 2D]. These findings, high mortality in the 129 substrains with penetrant and severe fibrosis, suggested that an SL1344 infectious model would be preferred for fibrotic induction, if host mortality was reduced. Therefore, we assessed several commercially available and genetically stable mouse strains for survival and disease penetrance in the SL1344 model.
Figure 2.

High mortality is observed in 129SvJ mice infected with S. typhimurium SL1344. Kaplan-Meier survival estimates in 129 substrains 129X1/SvImJ [X1] and 129S1/SvImJ [S1] infected with SL1344. [A] X1 mice infected with SL1344 [dotted line] compared with uninfected mice [solid line]. [B] Infectious dose-dependent survival in X1 mice infected with 3 x 102, 1.5 x 103, 3 x 103, 1.5 x 104, 6.5 x 104, or 1.5 x 105 cfu of S. typhimurium SL1344. None of the mice infected with > 1 x 104 cfu [heavy lines] survived past Day 7 post infection. [C] Survival in X1 [solid line] versus S1 [dotted line] infected with SL1344. Fibrosis in surviving S1 mice as determined by αSMA protein expression [D] and fibrotic gene expression [E]. Protein loading was normalised to GAPDH [glyceraldehyde 3-phosphate dehydrogenase] protein expression. [E] SL1344 induces expression of fibrotic genes IGF-1 and TGFβ [E] in SL1344-infected mice at Day 22 post infection compared with uninfected controls. Relative gene expression was pnormalised to GAPDH gene expression; *p < 0.05.
3.3. Survival is host- and bacterial strain-dependent
A pilot host-response study, comparing 14-day survival and intestinal fibrosis in the S1 strain with two other commercially available mouse strains DBA/1J and CBA/J infected with either wild-type SL1344 or the ΔaroA S. typhimurium strains, was performed. The CBA strain was selected as S. typhimurium-resistant and the DBA and 129S1/SvImJ [S1] strains were selected based on the original model and on Grassl, personal communication.11,27
In all three strains infected with ΔaroA, 100% survival was observed. Notable strain-specific mortality occurred with SL1344 infection, with 0% survival in the DBA mice by Day 7 post infection and 60% survival in the S1 strain at Day 14 post infection. In contrast, 100% survival was observed in the CBA mice infected with SL1344. [Figure 3A].
Figure 3.

Survival and fibrosis are host- and bacterial strain-dependent. [A] Kaplan-Meier survival estimates of CBA/J, 129S1/SvImJ [S1], or DBA mice infected with either wild-type S. typhimurium SL1344 or the ΔaroA mutant; 100% survival at Day 14 was observed in all three mouse strains infected with ΔaroA at Day 14 and CBA/J infected with SL1344 [blue, red, green, and orange lines]; 40% survival was observed in the SL1344-infected S1 mice by Day 14 [pink line]. None of the DBA animals infected with SL1344 survived Dast day 7 post infection [purple line]. [B, C] Trichrome staining of collagen, illustrating transmural tissue fibrosis and architectural distortion with concurrent tissue oedema in mice infected either with ΔaroA [B] or SL1344 [C] at Day 14 post infection. Due to high early mortality, the SL1344-infected DBA histology is from Day 7. [D, E] Western blots of αSMA protein expression in mice infected with ΔaroA [D] compared with SL1344 [E]. Low αSMA expression in the DBA and two of the five S1 mice reflects early-stage disease [before Day 8 post infection and rapid induction of αSMA protein expression]. [F] Quantification of αSMA protein expression normalised to GAPDH [glyceraldehyde 3-phosphate dehydrogenase] expression in ΔaroA- [■] or SL1344 [■]-infected mice. In CBA/J mice, SL1344 infection induced αSMA protein nearly 3-fold compared with ΔaroA infection; *p < 0.05.
3.4. Fibrosis is host- and bacterial strain-dependent
Fibrosis was determined by histology as evaluated by a pathologist [DM] in a blinded manner and by αSMA protein expression. In the ΔaroA cohorts, moderate to severe histological fibrosis was observed in all three mouse strains, with no significant differences between the strains [Figure 3B]. αSMA protein expression was comparable among the three mouse strains. Though αSMA expression trended higher in the S1 strain, this difference was not statistically significant [p = 0.16 versus CBA, p = 0.22 versus DBA] [Figure 3D, F].
Similarly, in the SL1344-infected cohorts, all strains demonstrated similar moderate to severe histological fibrosis [Figure 3C]. Strain-specific variability was observed with higher αSMA protein expression in CBA than in the S1 and DBA strains [Figure 3E]. Undetectable αSMA expression in the DBA and in two of the S1 animals likely reflects early-stage disease, as these animals did not survive past Day 7 post infection. As we have previously published, as well as illustrated in Figure 2D, αSMA protein levels markedly increase after Day 8 post infection and further accumulate over time.17 In the CBA strain, nearly 3-fold higher αSMA protein expression was observed in the SL1344 bacterial strain than in the ΔaroA [2.8-fold, p = 0.027, Figure 3F]. As the CBA mouse strain demonstrated the highest survival and concurrent fibrosis, the SL1344:CBA/J model was further investigated for utility as an intestinal fibrosis model.
3.5. Fibrosis in the SL1344:CBA/J mice infected with SL1344 is highly penetrant and consistent
In a series of four independent experiments, intestinal fibrosis was evaluated in the SL1344:CBA/J model by histopathology, αSMA and collagen protein expression, and fibrotic and inflammatory gene expression. Moderate to severe tissue fibrosis occurred in the CBA strain as determined by histopathology [Figure 4A, B]. Fibrotic proteins αSMA and collagen I were consistently and strongly induced in all experiments [Figure 4C, D, E]. Fibrogenic gene expression was concordant with protein expression as COL1A1, TGFβ, and IGF-1 genes were significantly induced by SL1344 infection in each individual study. In addition, gene expression of inflammatory genes TNFα, IL-1β, IL-17, and IL12p40 were significantly induced [Supplementary Figure 2, available as Supplementary data at ECCO-JCC online].
Figure 4.

Fibrosis in CBA/J mice infected with S. typhimurium SL1344 is highly penetrant and consistent. Histology of uninfected [HBSS] and S. typhimurium SL1344-infected CBA/J mice illustrating profound, transmural tissue fibrosis and collagen infiltration in the infected mice [blue staining, A, right panel]. Across multiple, independent experiments, fibrosis was significantly higher in SL1344-infected mice [n = 19] compared with uninfected mice [n = 20] as indicated by blinded histological fibrosis scoring [B]. [C] Representative western blots of αSMA and collagen I protein expression. Glyceraldehyde 3-phosphate dehydrogenase [GAPDH] expression was used as a loading control for αSMA expression. [D, E] Protein expression from multiple SL1344:CBA/J experiments demonstrates significant induction of both αSMA (as normalided to GAPDH, [D]) and collagen I [E] protein expression. [F, G] Forest plots of fibrotic gene expression from four independent SL1344:CBA/J experiments illustrating the standardised mean differences [SMD] and 95% confidence intervals [CI] for TGFβ [F] and IGF-1 [G] genes. Data are from four independent animal experiments of 20 uninfected and 19 SL1344-infected mice;***pP < 0.001.
To compare the effects of SL1344 on fibrotic and inflammatory markers across the multiple studies, a meta-analysis was performed for the four SL1344:CBA/J studies. Differences in αSMA and collagen I protein expression, fibrotic gene expression, and inflammatory gene expression from uninfected [n = 20] and SL1344 [n = 19] were pooled as SMDs using a random-effects model. SL1344 infection had a very large effect on both αSMA [SMD = 3.36, 95% CI: 2.15 – 4.57, p < 0.001] and collagen protein expression [SMD = 2.99, 95% CI: 1.77 - 4.21, p < 0.001] [Table 2]. In concordance with the fibrotic protein markers, SL1344 infection had a large effect on expression of fibrotic genes COL1A1, TGFβ, and IGF-1 [Table 2 and Supplementary Figure 2].
Similar to the ΔaroA:C57bl/6 model, large effects were observed in the SL1344 model on COL1A1 gene expression [SMD = 0.87, 95% CI: 0.18 – 1.55, p = 0.013]. In contrast to the ΔaroA:C57bl/6 model where no effects were observed on either TGFβ or IGF-1 gene expression, large effects were observed in the SL1344:CBA/J model for TGFβ [SMDTGF = 0.94, 95% CI: 0.25 - 1.63, p = 0.007, Figure 4F] and IGF-1 gene expression [SMDIGF-1 = 1.68, 95% CI: 0.90 – 2.45, p < 0.001 Figure 4G]. Similar large effects were observed for the inflammatory markers TNFα, IL-1β, IL-17, and IL12p40 [Table 2 and Supplementary Figure 2].
Sample size calculations for each of the fibrotic markers predicted 3 to 17 mice would be required to detect fibrosis in the SL1344:CBA/J model, far fewer animals than the 18 to 90 required in ΔaroA:C57bl/6 model [Table 2]. Additional sample size calculations for the inflammatory markers also revealed a reduction in animal numbers with 3–12 required in the SL1344:CBA/J model versus 6–26 in the C57bl/6 [Table 2]. Whereas fibrosis occurs in the ΔaroA:C57bl/6 model, the SL1344:CBA/J model is an optimal fibrosis model due to a consistent, high degree of intestinal fibrosis with 100% survival. As these studies indicate, the use of the CBA stain increases the model efficiency, decreasing costs, and reducing the number of experimental animals required, thereby providing an optimised rodent fibrosis model for preclinical and drug discovery.
3.6. Evaluation of a novel anti-fibrotic therapeutic in the SL1344:CBA/J mouse model
Developing and evaluating anti-fibrotic therapeutics is our ultimate goal. A number of preclinical and investigational anti-fibrotics are currently in development, but no effective anti-fibrotics exist to treat intestinal fibrosis.28 Our work has demonstrated that pharmacological inhibitors of the MRTF-A/SRF may be an effective approach. As we have demonstrated, novel MRTF-A/SRF inhibitors block fibrogenesis in in vitro intestinal fibrosis models and prevent fibrosis in the mouse bleomycin skin injury model.16,29 Therefore, we used the preclinical investigational compound CCG-203971 to perform a proof-of-concept therapeutic experiment in the SL1344:CBA/J fibrosis model.15,30
To evaluate CCG-203971 as a prophylactic anti-fibrotic, mice in the drug control and therapeutic groups were treated twice daily with 100 mg/kg/day CCG-203971 beginning 1 week before S. typhimurium infection and continuing until sacrifice. Negative and positive control animals received vehicle [100 λ peanut oil] by gavage 2 x/day on the same schedule. As substantial mortality was observed in both the drug control and therapeutic groups [40% and 33%, respectively] [Figure 5A], the experiment was terminated at Day 11 post infection. Though this study did not reach Day 21 post infection as anticipated, we were able to assess effects on early fibrosis, as we have demonstrated fibrosis is initiated around Day 8 post infection.31
Figure 5.

Therapeutic proof-of-concept in the SL1344:CBA/J mouse model. [A–F] In a pilot proof-of-concept experiment, SL1344-infected CBA mice were treated with 100 mg/kg/day CCG-203971 for 18 days, beginning 7 days before SL1344 infection. [A] Kaplan-Meier survival curve illustrating effects of CCG-203971 on animal survival. Anti-fibrotic efficacy of CCG-203971 as determined by expression of collagen I protein expression [B] normalised to glyceraldehyde 3-phosphate dehydrogenase [GAPDH] protein expression and fibrotic (COL1A1 [C] and IGF-1 [D]) and inflammatory (IL-1b [E] and IL-6 [F]) gene expression. *p < 0.05, **p < 0.01 compared with the uninfected control group. ###p < 0.001 compared with the SL1344-infected group.
In this study, no significant effects on gross pathology were observed in mice prophylactically treated with CCG-203971 compared with the SL1344-infected group. Similarly, no differences in histological inflammation and fibrosis, as scored by a blinded pathologist [DM], were observed between the SL1344-infected mice and mice infected with SL1344 and treated with CCG-203971 [data not shown]. However, more sensitive biochemical markers of intestinal fibrosis, including protein and gene expression, suggested anti-fibrotic efficacy.
Biochemical markers of intestinal fibrosis, including collagen I and αSMA protein expression, were quantified. SL1344 infection markedly induced both αSMA and collagen I protein expression. Although CCG-203971 treatment had no effect on αSMA protein expression, collagen I protein expression was significantly repressed 3-fold compared with the SL1344-infected controls [0.030 versus 0.895, p = 0.002] [Figure 5B]
Analysis of transcriptional markers of fibrosis and inflammation demonstrated CCG-203971 efficacy. CCG-203971 treatment significantly repressed the fibrotic genes COL1A1 and IGF-1. Compared with the SL1344-infected group, treatment with CCG-203971 significantly repressed COL1A1 2.5-fold [p = 0.001] and IGF-1 2-fold [p = 0.004] [Figure 5C, D]. Similar effects were observed for inflammatory genes [IL-1β and IL-6]. Specifically, IL-1β expression was repressed 3-fold compared with the SL1344 group [0.36 versus 1.05, p = 0.004] and IL-6 was repressed 4.3-fold compared with SL1344 [0.24 versus 1.04, p < 0.001] [Figure 5E, F]. In addition, CCG-203971 treatment repressed inflammatory gene expression to levels comparable to both the negative [uninfected] control or uninfected drug control groups.
Our analysis of the SL1334:CBA/J model predicted smaller sample sizes would be sufficient to detect anti-fibrotic efficacy. Therefore, we performed a post hoc power analysis of the proof-of-concept therapeutic experiment to determine if the actual sample sizes were congruent with the SL1344:CBA model predictions.
Post hoc sample size calculations were performed to compare the CCG-203971 therapeutic and the SL1344 infected groups for markers significantly repressed by CCG-203971 treatment [i.e. collagen I protein expression, and COL1A1, IGF-1, and IL-1β gene expression] [Table 3]. In the original power analysis of the SL1344:CBA/J model, samples sizes of 4–17 mice were predicted to detect an anti-fibrotic response, depending on the metric. In the therapeutic experiment, sample sizes were consistent with the predicted sample sizes. Original sample size calculations of collagen I protein expression predicted four mice per group. Post hoc calculations determined that three and six mice for the CCG-203971 therapeutic and SL1344 groups, respectively, would be required to achieve an experiment power of 80%, consistent with the SL1344:CBA/J model. Similar sample size congruence was observed for IGF-1 [predicted n = 6, actual n = 2 and 4] and IL-1β [predicted n = 4, actual n = 3 and 6] gene expression. However, for COL1A1 gene expression, the actual sample size was smaller than predicted [n = 2 and 4 versus n = 17]. Whereas CCG-203971 did not definitively ameliorate fibrosis, this inaugural proof-of-concept therapeutic study illustrates the utility of the SL1344:CBA/J model for evaluating preclinical anti-fibrotic therapeutics.
Table 3.
Therapeutic proof-of-concept post hoc power analysis in the S. typhimurium SL1344:CBA mouse model.
| Metric | Predicted [n] | SL1334 + CCG-203971 | SL1344 |
|---|---|---|---|
| Collagen I protein | 4 | 3 | 6 |
| Gene expression | |||
| COL1A1 | 17 | 2 | 4 |
| IGF-1 | 6 | 2 | 4 |
| IL-1β | 4 | 3 | 6 |
4. Discussion
Fibrosis is the key unsolved problem in IBD, with substantial negative patient outcomes and economic costs. Despite the advent of powerful and effective anti-inflammatory therapeutics, the rate of surgical resection for intestinal fibrosis remains largely unchanged, underscoring the need for effective anti-fibrotic drugs.32 Recently, the number of anti-fibrotic therapeutics in the development pipeline has exponentially risen from two in 2014 to 26 today.33 However, none has been evaluated in intestinal fibrosis models. One limitation is the lack of clinically relevant and robust animal fibrosis models for drug discovery and preclinical testing.7
The development of powerful anti-inflammatory therapeutics has relied heavily on numerous animal models of IBD. However, these models were developed exclusively to measure the inflammatory and immune-system responses characteristic of early-stage human disease. In these models, fibrosis is at best a secondary outcome. Few fibrosis-specific animal models exist; each has limitations with respect to fibrotic penetrance, feasibility, scaleability, and clinical relevance.7
The aetiology of fibrosis in IBD is complex and multifactorial, involving intricate interplay between environmental, microbial [commensal and pathological], immune, and genetic factors.34 Whereas intestinal fibrosis in the mouse S. typhimurium model is triggered by a single infectious agent, both intestinal colonisation and resultant disease is dependent upon disruption of the normal microflora. As our T-RFLP analysis of gut microbiota in the S. typhimurium model has shown, the normal microflora which varies considerably among individual animals and animal facilities is globally eliminated by streptomycin treatment and S. typhimurium colonisation [Higgins, unpublished results]. This process of gut dysbiosis and bacterially induced pathology has similarities to IBD. In human IBD, disruption of gut microbiota by antibiotics or pathological bacteria [e.g. Clostridium difficile] can worsen disease.35,36 In addition, genetic defects in bacterial recognition and killing [NOD2, ATG16L1, IRGM1, and NCF2] are associated with increased risk of IBD and more severe disease.37 Indeed, patients with mutations in the bacteria-sensing gene NOD2 have higher risk of fibrostenotic disease.38
Though the S. typhimurium mouse model, like other animal models, has limitations, this model recapitulates many of the clinical features characteristic of human IBD, including transmural tissue fibrosis, a Th1/Th17 response, myfibroblast activation, and pathological ECM production. As described in the original model, S. typhimurium infection produces fibrosis in several mouse strains, but the phenotype and magnitude of fibrotic response vary across host strains, including C57bl/6.11 Fibrosis in C57bl/6 is a critical finding which enables mechanistic interrogation of fibrosis, as most genetically altered mice, including strains used to interrogate mechanisms of IBD [e.g. NOD2, TGFβ, and MCP-1 transgenic mice], are on a C57bl/6 background.39,40 As our meta-analysis of the ΔaroA:C57bl/6 model illustrates, fibrotic disease presentation and penetrance vary with respect to biochemical markers of fibrosis including αSMA protein expression as well as COL1A1, IGF-1, and TGFβ gene expression. Histological assessment of fibrosis is a key metric, whereas αSMA protein and to lesser extent COL1A1 gene expression are informative, given sufficient sample sizes. With the large inter-experimental variability, caution should be used and biochemical markers alone may be insufficient to gauge response to anti-fibrotic therapies. In addition, larger experimental group sizes are required to detect meaningful differences in fibrosis, increasing both study costs and use of research animals. However, inflammatory markers were highly informative, with far more practicable sample sizes, suggesting that the ΔaroA:C57bl/6 model is appropriate for inflammatory studies but is suboptimal for fibrosis research, particularly for larger drug discovery or preclinical studies.
Other mouse strains, notably the 129Sv/ImJ strain, which lack the Nramp1 mutation, reportedly survive infection with wild-type S. typhimurium SL1344 but develop severe intestinal fibrosis. However, the 129 lineage is plagued by genetic heterogeneity and even intra-strain variation. Though we observed severe fibrosis in 129S1/SvImJ mice, the high mortality in this and another commercially available 129 strain [129X1/SvImJ], limit the utility of 129 strains, particularly for longitudinal drug studies.
Mice are the preferred species to meet the demands of efficient, cost-effective, and sufficiently powered in vivo drug studies. Yet in comparison with other species, mice are relatively resistant to organ fibrosis.41 Complicating matters, fibrotic susceptibility or resistance varies across organs within a specific mouse strain.42 C57bl/6 mice are resistant to renal and cardiac fibrosis but susceptible to pulmonary and hepatic fibrosis, whereas 129S1/SvImJ mice are highly susceptible to renal fibrosis.42 As our pilot S. typhimurium experiment using CBA/J, 129S1/SvImJ, and DBA mouse strains illustrates, both survival and fibrotic disease are host- and bacterial strain-dependent, consistent with the original model.11 Predictably, infection with the metabolic ΔaroA mutant prevents lethality, but attenuates fibrosis. Wild-type S. typhimurium SL1344 is highly lethal in the 129S1/SvImJ and DBA mouse strains. In contrast, the CBA/J mouse strain is resistant to S. typhimurium lethality and develops profound fibrosis similar to human disease with characteristic histological features, a pathological overexpression of ECM [extracellular matrix] proteins, and classical transcriptional markers of fibrosis. These observations are consistent with observations in a surgical model of intestinal fibrosis, demonstrating that CBA/J mice exhibited a greater and more consistent fibrotic response to peptidoglycan-polysaccharide [PG-PS] than either 129S1/SvImJ or DBA mice.43
As we confirmed in a meta-analysis of the SL1344:CBA/J model, protein and transcriptional markers of fibrosis are highly indicative of fibrotic response. In addition, sample size calculations revealed that substantially smaller group sizes are needed to achieve sufficient experimental power. Compared with the ΔaroA:C57bl/6 model, predicted sample sizes in the SL1344:CBA/J model are between 4- and 10-fold lower. The SL1344:CBA/J model is not a pure fibrosis model, as concurrent histological inflammation and inflammatory gene expression are observed. However, as we have previously demonstrated, inflammation can be decoupled from fibrosis.17 Indeed, that may be the most powerful aspect of the S. typhimurium model. Recent work by our group and others, suggests that anti-fibrotic therapy is more effective once inflammation is controlled [T. Pizarro, personal communication, and PDRH., unpublished observations]. In fact, the application of anti-fibrotic therapy in the presence of uncontrolled, active inflammation may have profound adverse effects. As we observed, spironolactone treatment, despite anti-fibrotic efficacy in cardiac fibrosis and in vitro intestinal fibrosis models, produced high mortality in two rodent colitis models and was associated with a 2-fold higher risk of death in patients who have concurrent intestinal inflammation.18
Finally, we demonstrate the utility of the SL1344:CBA/J model in a proof-of-concept study using a CCG-203971, a novel, preclinical anti-fibrotic therapeutic. Although drug toxicity limited full evaluation of anti-fibrotic efficacy, the increased sensitivity of the optimised SL1344:CBA/J model enabled more subtle assessment of anti-fibrotic efficacy, informing future therapeutic study design and modification.
4.1. Conclusions
In conclusion, the S. typhimurium model is a powerful tool for interrogating the mechanisms and therapeutics for intestinal fibrosis. Appropriate selection of host and bacterial strains for the specific scientific application [inflammation versus fibrosis] are required. For mechanistic studies of inflammation, the S. typhimurium ΔaroA:C57bl/6 model is well-suited and can utilize the power of genetically modified mice. However, an a priori power analysis recommended ensuring that experimental groups are adequately powered, particularly for fibrosis studies. Our studies indicate that with low mortality and consistently robust fibrosis, the S. typhimurium SL1344:CBA/J model is an optimal intestinal fibrosis model for preclinical and drug discovery applications. As intestinal fibrosis is a critical unsolved problem in IBD, the S. typhimurium SL1344:CBA/J model may be a crucial tool to identify and evaluate effective anti-fibrotic therapeutics such as CCG-203971, a novel MRTF-A/SRF inhibitor.
Funding
This work was supported by a NIH-NIDDK [grant K08DK080172], and by the Michigan Gastrointestinal Peptide Research Center [MGPRC], [NIDDK 5P30DK034933] to PDRH.
Conflict of Interest
The authors have no conflicts of interest.
Author Contributions
PDRH envisioned and designed the studies, analysed results and edited the manuscript. LJ performed all the experiments, analysed the data, and drafted the manuscript. DM interpreted the histology and edited the manuscript. ER participated in experiments and assisted with the manuscript. SL synthesised CCG-203971. PDRH, RN, SL, ER, and LJ designed the CCG-203971 study. All authors read and approved the final manuscript.
Supplementary Data
Supplementary data are available at ECCO-JCC online.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
The authors appreciate the generosity of Guntram Grassl, University of British Columbia, Vancouver, Canada, for the S. typhimurium ΔaroA strain and insight into mouse strain responses, as well as Mary O’Riordan, University of Michigan, Ann Arbor, MI, for S. typhimurium SL1344. The authors also thank Evan Pouget, Margaret Hart, Kay Sauder, and Kathryn Finley for their assistance with the rodent studies. Also to be thanked are Thersea Pizarro for her unpublished observations in SAMP1/YipFc mouse model, and Guntram Grassl for advice with the 129 mouse strains.
References
- 1. Collins AJ, Foley RN, Herzog C, et al. Excerpts from the US Renal Data System 2009 annual data report. Am J Kidney Dis 2010;55:S1–420, A6–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Neff GW, Duncan CW, Schiff ER. The current economic burden of cirrhosis. Gastroenterol Hepatol 2011;7:661–71. [PMC free article] [PubMed] [Google Scholar]
- 3. Collard HR, Ward AJ, Lanes S, Cortney Hayflinger D, Rosenberg DM, Hunsche E. Burden of illness in idiopathic pulmonary fibrosis. J Med Econ 2012;15:829–35. [DOI] [PubMed] [Google Scholar]
- 4. Wynn TA. Fibrotic disease and the T[H]1/T[H]2 paradigm. Nat Rev Immunol 2004;4:583–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dhillon S, Loftus EV, Tremaine WJ, et al. The natural history of surgery for Crohn’s disease in a population-based cohort from Olmsted County, Minnesota. Am J Gastroenterol 2005;100:S305. [Google Scholar]
- 6. Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012;142:46–54.e42; quiz e30. [DOI] [PubMed] [Google Scholar]
- 7. Rieder F, Kessler S, Sans M, Fiocchi C. Animal models of intestinal fibrosis: new tools for the understanding of pathogenesis and therapy of human disease. Am J Physiol Gastrointest Liver Physiol 2012;303:G786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kiesler P, Fuss IJ, Strober W. Experimental models of inflammatory bowel diseases. Cell Mol Gastroenterol Hepatol 2015;1:154–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mizoguchi A, Takeuchi T, Himuro H, Okada T, Mizoguchi E. Genetically engineered mouse models for studying inflammatory bowel disease. J Pathol 2016;238:205–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Latella G, Rogler G, Bamias G, et al. Results of the 4th scientific workshop of the ECCO [I]: pathophysiology of intestinal fibrosis in IBD. J Crohns Colitis 2014;8:1147–65. [DOI] [PubMed] [Google Scholar]
- 11. Grassl GA, Valdez Y, Bergstrom KS, Vallance BA, Finlay BB. Chronic enteric salmonella infection in mice leads to severe and persistent intestinal fibrosis. Gastroenterology 2008;134:768–80. [DOI] [PubMed] [Google Scholar]
- 12. Skarnes WC, Rosen B, West AP, et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 2011;474:337–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DeVoss J, Diehl L. Murine models of inflammatory bowel disease [IBD]: challenges of modeling human disease. Toxicol Pathol 2014;42:99–110. [DOI] [PubMed] [Google Scholar]
- 14. Collins FS, Rossant J, Wurst W; International Mouse Knockout Consortium A mouse for all reasons. Cell 2007;128:9–13. [DOI] [PubMed] [Google Scholar]
- 15. Evelyn CR, Bell JL, Ryu JG, et al. Design, synthesis and prostate cancer cell-based studies of analogs of the Rho/MKL1 transcriptional pathway inhibitor, CCG-1423. Bioorg Med Chem Lett 2010;20:665–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haak AJ, Tsou PS, Amin MA, et al. Targeting the myofibroblast genetic switch: inhibitors of myocardin-related transcription factor/serum response factor-regulated gene transcription prevent fibrosis in a murine model of skin injury. J Pharmacol Exp Ther 2014;349:480–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnson LA, Luke A, Sauder K, et al. Intestinal fibrosis is reduced by early elimination of inflammation in a mouse model of IBD: Impact of a ‘top-down’ approach to intestinal fibrosis in mice. Inflamm Bowel Diss 2012;18:460–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson LA, Govani SM, Joyce JC, Waljee AK, Gillespie BW, Higgins PD. Spironolactone and colitis: Increased mortality in rodents and in humans. Inflamm Bowel Dis 2012;18:1315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Higgins PD, Johnson LA, Sauder K, et al. Transient or persistent norovirus infection does not alter the pathology of Salmonella typhimurium induced intestinal inflammation and fibrosis in mice. Comp Immunol Microbiol Infect Dis 2011;34:247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Higgins PD, Johnson LA, Luther J, et al. Prior Helicobacter pylori infection ameliorates Salmonella typhimurium-induced colitis: Mucosal crosstalk between stomach and distal intestine. Inflamm Bowel Dis 2011;17:1398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc 2007;2:541–6. [DOI] [PubMed] [Google Scholar]
- 22. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[-Delta Delta C[T]] Method. Methods 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 23. Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 1959;22:719–48. [PubMed] [Google Scholar]
- 24. Cohen J. Statistical Power Analysis for the Behavioral Sciences. Hillsdale, NJ: L. Erlbaum Associates, 1988. [Google Scholar]
- 25. Schauwecker PE. The relevance of individual genetic background and its role in animal models of epilepsy. Epilepsy Res 2011;97:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Simpson EM, Linder CC, Sargent EE, et al. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat Genet 1997;16:19–27. [DOI] [PubMed] [Google Scholar]
- 27. Roy MF, Malo D. Genetic regulation of host responses to Salmonella infection in mice. Genes Immun 2002;3:381–93. [DOI] [PubMed] [Google Scholar]
- 28. Fibrosis: From basic mechanisms to targeted therapies. In: Keystone Conference, Cluj-Napoca Romania, 8–9 September, 2016 Keystone, CO: IKC, 2016. [Google Scholar]
- 29. Johnson LA, Rodansky ES, Haak AJ, et al. Novel RHO/MRTF/SRF inhibitors block matrix-stiffness and TGF-beta-induced fibrogenesis in human colonic myofibroblasts. Inflamm Bowel Diss 2014;20:154–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bell J, Haak A, Wade SM, et al. Optimisation of novel nipecotic bis[amide] inhibitors of the Rho/MKL1/SRF transcriptional pathway as potential anti-metastasis agents. Bioorg Med Chem Lett 2013;23:3826–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson LA, Luke A, Sauder K, et al. Intestinal fibrosis is reduced by early elimination of inflammation in a mouse model of IBD: Impact of a ‘top-down’ approach to intestinal fibrosis in mice. Inflamm Bowel Dis 2012;18:460––71.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vermeire S, van Assche G, Rutgeerts P. Review article: Altering the natural history of Crohn’s disease–evidence for and against current therapies. Aliment Pharmacol Ther 2007;25:3–12. [DOI] [PubMed] [Google Scholar]
- 33. Fibrosis: From basic mechanisms to targeted therapies. In: Keystone Conference, Cluj-Napoca Romania, 8–9 September, 2016 Keystone, CO: IKC, 2016. [Google Scholar]
- 34. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007;448:427–34. [DOI] [PubMed] [Google Scholar]
- 35. Rao K, Higgins PD. Epidemiology, diagnosis, and management of Clostridium difficile infection in patients with inflammatory bowel disease. Inflamm Bowel Dis 2016;22:1744–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Issa M, Ananthakrishnan AN, Binion DG. Clostridium difficile and inflammatory bowel disease. Inflamm Bowel Dis 2008;14:1432–42. [DOI] [PubMed] [Google Scholar]
- 37. Chu H, Khosravi A, Kusumawardhani IP, et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science 2016;352:1116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rieder F, Lawrance IC, Leite A, Sans M. Predictors of fibrostenotic Crohn’s disease. Inflamm Bowel Dis 2011;17:2000–7. [DOI] [PubMed] [Google Scholar]
- 39. Prattis S, Jurjus A. Spontaneous and transgenic rodent models of inflammatory bowel disease. Lab Anim Res 2015;31:47–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Waterston RH, Lindblad-Toh K, Birney E; Mouse Genome Sequencing Consortium Initial sequencing and comparative analysis of the mouse genome. Nature 2002;420:520–62. [DOI] [PubMed] [Google Scholar]
- 41. Pucilowska JB, Williams KL, Lund PK. Fibrogenesis. IV. Fibrosis and inflammatory bowel disease: cellular mediators and animal models. Am J Physiol Gastrointest Liver Physiol 2000;279:G653–9. [DOI] [PubMed] [Google Scholar]
- 42. Walkin L, Herrick SE, Summers A, et al. The role of mouse strain differences in the susceptibility to fibrosis: a systematic review. Fibrogen Tiss Repair 2013;6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reingold L, Rahal K, Schmiedlin-Ren P, et al. Development of a peptidoglycan-polysaccharide murine model of Crohn’s disease: effect of genetic background. Inflamm Bowel Dis 2013;19:1238–44. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.