

Abstract
The prevention and management of myocardial ischemia/reperfusion (MI/R) injury is an essential part of coronary heart disease surgery and is becoming a major clinical problem in the treatment of ischemic heart disease. Previous studies by our group have demonstrated that pigment epithelium-derived factor (PEDF) improves cardiac function in rats with acute myocardial infarction and reduces hypoxia-induced cell injury. However, the protective function and mechanisms underlying the effect of PEDF in MI/R injury remain to be fully understood. In the present study, the positive effect of PEDF in MI/R injury was confirmed by construction of the adult Sprague-Dawley rat MI/R model. PEDF reduced myocardial infarct size and downregulated cardiomyocyte apoptosis in the I/R myocardium in this model. In addition, PEDF improved cardiac function and increased cardiac functional reserve in rats subjected to MI/R Injury. To further study the protective effect of PEDF and the underlying mechanisms in MI/R injury, a H9c2 cardiomyocyte hypoxia/reoxygenation (H/R) model was constructed. PEDF was confirmed to decrease H/R-induced apoptosis in H9c2 cells, and this anti-apoptotic function was abolished by pigment epithelium-derived factor-receptor (PEDF R) small interfering (si)RNA. Furthermore, administration of PEDF decreased the levels of reactive oxygen species (ROS) and malondialdehyde (MDA) in H/R H9c2 cells. Compared with the H/R group, PEDF decreased mitochondrial ROS, increased the mitochondrial DNA copy number, reduced xanthine oxidase and NADPH oxidase activity, as well as RAC family small GTPase 1 protein expression. However, these effects of PEDF were markedly attenuated by PEDF-R siRNA. To the best of our knowledge, the present study is the first to identify the protective effect of PEDF in MI/R injury, and confirm that the antioxidative effect PEDF occurred via inhibition of ROS generation via PEDF-R under MI/R conditions.
Keywords: pigment epithelium-derived factor, pigment epithelium-derived factor-receptor, reactive oxygen species generation, myocardial ischemia/reperfusion
Introduction
Acute myocardial infarction is a prevalent public health problem which results in severe morbidity and mortality globally (1,2). At present, the standard treatment for acute myocardial infarction is early reperfusion therapy, including thrombolysis, percutaneous coronary intervention and coronary bypass grafting (3). However, the process of reperfusion may cause additional cardiomyocyte dysfunction and death, and this process is generally referred to as myocardial reperfusion injury (MI/R) (4). MI/R injury is a major limitation of certain clinical therapies for coronary artery disease.
The underlying mechanisms of MI/R injury are complex, and at present multiple factors, including excessive generation of reactive oxygen species (ROS), calcium overload and vascular endothelial dysfunction are viewed as being relevant (5). Among these contributors, oxidative stress induced by the burst of ROS that occurs during reperfusion is the critical trigger of MI/R injury (6). An appropriate amount of ROS usually has a cardioprotective effect, while high levels of ROS are deleterious and lead to cardiomyocyte death (7,8). Excessive ROS generated during the MI/R process not only directly and non-specifically oxidize biological macromolecules including DNA, lipids, and proteins, but also damage cells by activating redox-regulated signaling cascades that ultimately lead to cell death (9). The generation of ROS and the clearance of ROS are important factors that affect ROS outbreaks (10). Compared with the ability of ROS clearance, inhibition of ROS generation may be more important in terms of avoiding ROS outbreaks (10). Previous studies have identified major sources of ROS production in heart disorders: The mitochondrial electronic transport chain (ETC) of mitochondria, NADPH oxidases (NOX) and xanthine oxidase (XO) in the cytoplasm (11). Therefore, inhibition of mitochondrial and cytoplasmic ROS formation during the MI/R process, which avoids the outbreak of ROS from their site of origin, may be a key therapeutic approach to effectively protect the myocardium during the MI/R process.
Pigment epithelium-derived factor (PEDF), a 50-kDa secreted glycoprotein, belongs to the superfamily of serine protease inhibitors (12). PEDF is expressed in multiple tissues but is expressed prominently in heart tissue, where it exerts diverse physiological activities (13-15). Previous studies by our group have demonstrated that PEDF protects cardiomyocytes against hypoxia-induced apoptosis and necroptosis via anti-oxidative effects, and PEDF improves cardiac function in rats with acute myocardial infarction via inhibition of vascular permeability and cardiomyocyte apoptosis (14,16). Multiple studies have focused on the effects of PEDF in hypoxic cardiomyocytes. However, the function of PEDF across the whole process of MI/R has rarely been investigated.
PEDF is mainly deposited in the cell membrane, where it interacts with its receptors (17). The PEDF receptor (PEDF-R) and laminin receptor are two crucial receptors among PEDF binding sites (17,18). PEDF-R, an enzyme protein of roughly 55 KDa in size and 504 amino acids in length, exerts potent phospholipase A2 enzymatic activity and lipase activity when it is combined with PEDF (19,20). Previous studies by our group have demonstrated that the effect of PEDF, that may protect cardiomyocytes under hypoxic conditions, is mediated by PEDF-R (21,22). As a result, it is possible to conclude that PEDF-R mediates the protective effect of PEDF in MI/R Injury.
In the present study, the adult Sprague-Dawley rat MI/R model and the H9c2 cardiomyocyte hypoxia/reoxygeneration (H/R) model were established to mimic MI/R injury. The hypothesis that the protective effect of PEDF in MI/R injury and the antioxidative effects of PEDF are based on the inhibition of mitochondrial and cytosolic ROS generation via PEDF-R was investigated. The present study may provide a novel therapeutic target for MI/R injury.
Materials and methods
Reagents
Cleaved caspase-3 antibodies (cat. no. 9664) was purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Monoclonal actin (α-sarcomeric; α-sa) antibodies (cat. no. A2172), Dobutamine, Evans Blue dye and 2,3,5-triphenyl-tetrazolium (TTC) were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). The in situ cell death detection kit (cat. no. 11684795910) was purchased from Roche Diagnostics (Indianapolis, IN, USA). β-actin antibodies (cat. no. 66009-1-Ig), PEDF-R rabbit polyclonal antibodies (cat. no. 55190-1-AP) and RAC family small GTPase 1 (rac1) antibodies (cat. no. 24072-1-AP) were purchased from ProteinTech Group, Inc. (Chicago, IL, USA). The MitoSOX™ Red mitochondrial superoxide indicator was purchased from Invitrogen; Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Hoechst 33342 and the Annexin V-allophycocyanin (APC)/propidium iodide (PI) Apoptosis Detection kit were purchased from Nanjing KeyGen Biotech Co., Ltd. (Nanjing, China). Malondialdehyde (MDA; cat. no. S0131), Dihydroethidium (DHE; cat. no. S0063), Dabco 4 and 6-diamidino-2-phenylindole (DAPI; cat. no. C1005) were purchased from Beyotime Institute of Biotechnology (Haimen, China). The TIANamp Genomic DNA kit was purchased from Tiangen Biotech Co., Ltd. (Beijing, China). The NADPH oxidase activity assay kit was purchased from Shanghai Genmed Pharmaceutical Technology Co., Ltd. (Shanghai, China). The xanthine oxidase activity assay kit was purchased from Cayman Chemical Company (Ann Arbor, MI, USA).
Recombinant lentivirus constructs and viral production
Recombinant lentivirus (LV; Shanghai GeneChem Co., Ltd, Shanghai, China) was prepared as described previously (16). PEDF overexpression plasmid (Shanghai GeneChem Co., Ltd.) was successfully constructed and then packaged in 293T cells (American Type Culture Collection, Manassas, VA, USA). PEDF was cloned and ligated into the pGC287 plasmid using AgeI and BamHI sites. The sequences of the PEDF primers used were as follows: Forward, 5′-CGACCGGTGCCACCATGCAGACCCTGG-3′ and reverse, 5′-GGAATTCGGATCCTTAAGTGCTGCTGG-3′. The concentrated titer of virus suspension was 2×1012 TU/l. Transient transfection of H9c2 cells with 20 μM small interfering (si)RNA targeting the PEDF-R genes (5′-GCGGCATTTCAGACAACTTGC-3′) was performed using Lipofectamine® 3000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocols. A free-combination sequence of siPEDF-R was used as negative control (5′-ACGGTTATGCTCGAACTCGCA-3′), all the siRNA were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Preparation of PEDF protein
Recombinant rat PEDF (GenBank accession no. NM_177927) was synthesized by Cusabio Biotech, Co., Ltd. (Wuhan, China). In brief, 293T cells were transfected with the 20 μg recombinant vector pGEX 6P-1 (GE Healthcare, Chicago, IL, USA), containing glutathione S-transferase (GST)-tagged PEDF using Lipofectamine® 3000 according to the manufacturer's protocols. GST-tagged PEDF proteins were purified by high-pressure liquid chromatography purification (>90% purity) and amino-terminal sequence determination (23,24). The resultant proteins were soluble in aqueous solutions.
Animals
Adult male Sprague-Dawley rats (8-10 weeks old, weighing 200-250 g, n=85) were purchased from the Experimental Animal Centre of Xuzhou Medical University (Xuzhou, China) and housed in a controlled environment (humidity, 50-60%). A total of 3 rats were housed per cage and were maintained at room temperature under a 12 h light/dark cycle; rats were provided free access to food and water. The experiments described in this manuscript conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Publication, 8th Edition, 2011, Bethesda, MD, USA) (25). All animal care and experimental protocols were approved by the Animal Care and Use Committee of Xuzhou Medical University (license no. SYXK 2002-0038, Jiangsu, China) and also followed the international guidelines (European Council Directive 2010/63/EU) on the ethical use of animals (26).
Rat MI/R model and intramyocardial gene delivery
The rat MI/R model was produced as described previously (27). Intramyocardial gene delivery was performed one week prior to the MI/R experiment in the rats. PEDF-LV (2×107 TU) in 20 μl enhanced infection solution (pH 7.4, Shanghai GeneChem Co., Ltd.) was delivered with a 20-μl syringe and 25-gauge needle into the myocardium along the left-anterior descending coronary artery (LAD). Sham-operated animals underwent an identical surgical procedure without artery ligation. For the rat MI/R model, after 30 min of ischemia treatment, reperfusion was allowed for 24 h by releasing the ligatures. The animal models were randomly divided into five groups as follows: i) Sham group, surgical procedure without artery ligation (n=18); ii) Sham+P group, surgical procedure without artery ligation, PEDF-LV was transferred prior to surgery (n=18); iii) MI group, 0.5 h ischemia (n=6); iv) MI/R group, 0.5 h ischemia and 24 h reperfusion (n=18); v) MI/R+P group, 0.5 h ischemia and 24 h reperfusion, PEDF-LV was transferred prior to surgery (n=18).
Quantification of myocardial infarct size
Myocardial infarct size was measured by Evans Blue/TTC staining as previously described (28). Briefly, at the end of reperfusion or ischemia, the LAD was again occluded and 1 ml 3% Evans Blue dye was retrogradely injected into the ascending aorta to demarcate the ischemic area at risk (AAR). The heart was rapidly excised and frozen at −20°C prior to being cut into 1 mm thick sections perpendicular to the axis of the LAD. At this point, sections were immediately incubated in 1% TTC in phosphate buffer (pH 7.4) at 37°C for 15 min to discriminate infarcted tissue from viable myocardium. All sections were scanned from both sides using a color CCD camera (FV-10; Fujifilm Holdings Corporation, Tokyo, Japan), and in each slide, infarct area was compared with the AAR using digital planimetry software (Image-Pro Plus 6.0; Media Cybernetics, Inc., Rockville, MD, USA). Following correction with the weight of the sections, the myocardial infarct size was measured and expressed as a percentage of infarct size over total AAR.
Determination of cardiac function and dobutamine stress
Echocardiography was conducted under anesthesia with sodium pentobarbital (30 mg/kg, intraperitoneal; Sigma-Aldrich; Merck KGaA), as described previously (22). Two-dimensional-guided M-mode echocardiography was performed at rest and during dobutamine stress 24 h after reperfusion in order to determine left ventricular (LV) chamber volume at systole and diastole and contractile parameters, including left ventricular end-diastolic dimension (LVEDD), left ventricular end-systolic dimension (LVESD), left ventricular end-diastolic volume (LVEDV) and left ventricular end-systolic volume (LVESV). Left ventricular ejection fraction (EF) and fractional shortening (LVFS) were calculated as follows: EF(%) = (EDV-ESV)/EDV×100. FS(%) = (LVEDD-LVESD)/LVEDD ×100. All measurements represent the mean of at least 3 consecutive cardiac cycles. Dobutamine (1 μg/g body weight; Sigma-Aldrich; Merck KGaA) was given intraperitoneally. Cardiac reserve was investigated 10 min after dobutamine injection.
Cell culture
The embryonic rat heart-derived H9c2 cell line was obtained from the American Type Culture Collection, and the cells were cultured in DMEM medium supplemented with 10% foetal bovine serum (both from Gibco; Thermo Fisher Scientific, Inc.) and 100 mg/ml penicillin/streptomycin at 37°C in a humidified atmosphere containing 5% CO2. The medium was replaced every 2-3 days, and cells were subcultured or subjected to experimental procedures at 80-90% confluence.
Establishment of H/R model
To establish the H/R model, the culture medium was changed to serum-free low glucose DMEM (Gibco; Thermo Fisher Scientific, Inc.) and placed into a tri-gas incubator (Heal Force Bio-meditech Holdings, Ltd., Shanghai, China) that was purged with 94% N2, 5% CO2, and 1% O2 for 4, 8 and 12 h respectively. Following hypoxia, reoxygenation was initiated by incubating in serum-free high glucose DMEM (Gibco; Thermo Fisher Scientific, Inc.) at 37°C with 5% CO2 for a further 0 or 2 h. H9C2 cells (~80-90% confluence) were treated with or without 10 nM PEDF 1 h prior to H/R. The following experimental groups were included: N group (control, normoxia), N+P group (PEDF, normoxia), H/R group (control, 8 h hypoxia and 2 h reoxygenation), H/R+P group (PEDF, H/R), H/R+PEDF+siPEDF-R group (PEDF+siPEDF-R, H/R), H/R+PEDF+vector group (PEDF+vector, H/R).
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining for apoptosis in vivo and in vitro
Myocardial samples from the rat left ventricle were embedded in optimum cutting temperature compound tissue medium (Sakura Finetek Europe B. V., Flemingweg, The Netherlands), snap-frozen on dry ice and stored at −80°C. Cardiomyocyte apoptosis in vivo was determined by double-labeling TUNEL immunofluorescence staining, which was performed with an In Situ Cell Death Detection kit according to the manufacturer's protocol. Cardiomyocytes were identified using monoclonal actin (α-sa) antibodies. Specimens were blocked with 5% bovine serum albumin (Vicmed Life Sciences, Xuzhou, China) for 30 min at room temperature prior to incubation with primary antibody. Specimens were incubated with anti-α-sa (1:300) overnight at 4°C. Specimens were then washed three times in PBS and incubated with the Goat anti-Mouse immunoglobulin G (IgG; H+L) Cross-Adsorbed secondary antibody (Alexa Fluor®488/green; 1:200; A11001; Thermo Fisher Scientific, Inc.) under light-protected conditions for 1 h at room temperature. DAPI staining conducted at room temperature for 15 min was used to count the total number of nuclei. Cardiomyocyte nuclei with a relatively large diameter are located within myofibers (16). In addition, 2×104 H9c2 cells with Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.) were seeded into each of a 48-well plate. Following the H/R treatments, H9c2 cells were treated with the In Situ Cell Death Detection kit according to the manufacturer's protocol. H9c2 cells were counterstained with DAPI for 30 min at room temperature. The cardiomyocytes and H9c2 cells were observed under a fluorescence microscope (Olympus Corporation, Tokyo, Japan). The percentage of apoptotic cells was calculated as the ratio of the number of TUNEL-positive cells to the total number of cells, which were counted in three different random fields of view.
Western blot analysis
For western blot analysis, the cells were solubilized in lysis buffer (100 mmol/l Tris-HCl, 4% SDS, 20% glycerine, 200 mmol/l dithiothreitol and protease inhibitors; pH 6.8). Total cellular protein was denatured by boiling for 10 min with an equal volume of 2X Tris-glycine SDS buffer. Protein concentration from the supernatant was determined using a BCA protein assay kit (Pierce; Thermo Fisher Scientific, Inc.). A total of 50 ng protein per lane was separated by 12% SDS-PAGE and transferred to nitrocellulose membranes (EMD Millipore, Billerica, MA, USA). Following blocking with 5% non-fat milk/PBS-T for 3 h at room temperature, the membranes were incubated with primary antibodies overnight at 4°C. Cleaved caspase-3, PEDF-R rabbit polyclonal and rac1 antibodies were used at a dilution of 1:1,000; β-actin antibody was used at a dilution of 1:5,000. Then, fluorescence-labeled secondary antibodies was added for 1 h at 37°C: Anti-rabbit IgG H+L DyLight™ 800 4X PEG (1:30,000; cat. no. 5151) and anti-mouse IgG (H+L DyLight™ 680, 1:15,000; cat. no. 5470) (both from Cell Signaling Technology, Inc.) and membranes were scanned by the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA). ImageJ software (v1.50; NIH, Bethesda, MD, USA) was used for quantification.
Determination of apoptosis by flow cytometry
The Annexin V-APC/PI Apoptosis Detection kit was used for the following assay. H9c2 cardiomyocytes (1×106 per group) were collected, washed twice with PBS, resuspended with 500 μl 1X binding buffer, treated with 5 μl Annexin V-APC and 5 μl PI, and placed in the dark at room temperature for 5-15 min. Then, the cells were analyzed using a flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Detection of intracellular ROS generation
DHE was used to stain for intracellular ROS, as previously described (29). DHE, an oxidant-sensitive probe, is widely used for the detection of ROS. Harvested cells (2×104 per well) were incubated with 10 μM DHE for 30 min at 37°C, according to the manufacturer's protocol, then washed with DMEM without FBS three times. Fluorescence was observed under a fluorescence microscope. Fluorescence was calculated by viewing in four randomly selected fields for each group; Image-Pro Plus software (v6.0; Media Cybernetics, Inc.) was used for quantification.
Detection of mitochondrial (mt)ROS production
To assess the levels of mtROS, MitoSOX™, a fluorochrome specific to anion superoxide produced in the inner mitochondrial compartment (Invitrogen; Thermo Fisher Scientific, Inc.) was used. H9c2 cells (1×104 per well) were seeded into each well of a 48-well plate, underwent their respective treatments as aforementioned, and subsequently loaded with 200 μl MitoSOX™ (5 mM stock in ethanol dissolved in Hank's Balanced Salt Solution to a working solution of 5 μM) for 10 min at 37°C. After washing three times with PBS, nuclei were counter-stained with Hoechst 33342 for 15 min at 37°C. Following three washes with PBS, the sample was observed by a fluorescence microscope (Olympus IX73; Olympus Corporation). Fluorescence was calculated by viewing in five randomly selected fields for each group. Image-Pro Plus software (v6.0; Media Cybernetics, Inc.) was used for quantification.
Quantitative polymerase chain reaction (qPCR)
Total DNA was isolated from cells using the TIANamp Genomic DNA kit, according to the manufacturer's protocol. As mitochondrial DNA (mtDNA) is a primary target of ROS and a reflection of ETC, mtDNA copy number was detected by qPCR using SYBR Green PCR Master Mix (Applied Biosystems; Thermo Fisher Scientific, Inc.). PCR was conducted with a final volume of 20 μl containing 10 μl 2× SYBR-Green PCR Master Mix, 0.1 μM of each primer and 100 μg genomic DNA. The mixture was subjected to qPCR amplification for 95°C for 10 min, 45 cycles (95°C for 10 sec, 60°C for 10 sec, 72°C for 20 sec), 1 cycle (95°C for 1 min, 65°C for 1 min and 97°C with continuous) and then cooled to 40°C for 30 sec using a Roche Light Cycler 480 (Roche Diagnostics). Gene expression was normalized to that of 18s RNA. Gene expression was quantified by using the 2−∆∆Cq method (30). The following primers, synthesized by GenScript (Piscataway, NJ, USA) were used: D-loop forward, 5′-TGGTTCATCGTCCATACGTT-3′ and reverse, 5′-TGACGGCTATGTTGAGGAAG-3′; 18sRNA forward, 5′-CATTCGAACGTCTGCCCTATC-3′ and reverse, 5′-CCTGCTGCCTTCCTTGGA-3′.
Measurement of MDA levels, XO activity and NOX activity
MDA levels, XO activity and NOX activity were measured using the respective detection kits, according to the manufacturers' protocol.
Statistical analysis
The results are expressed as the mean ± standard error of the mean. All statistical analyses were conducted with SPSS 19.0 for Windows (IBM Corp., Armonk, NY, USA). The results were analyzed using unpaired Student's t-test or repeated-measures one-way analysis of variance followed by Duncan's new multiple range method or Fisher's least significant difference test. P<0.05 was considered to indicate a statistically significant difference.
Results
PEDF reduces myocardial infarct size and improves cardiac function under MI/R condition
First, the protective effects of PEDF were verified in a MI/R rat model. As presented in Fig. 1A and B, an irregular small myocardial infarct area was observed in 3 of 6 hearts in the MI group, and infarcted myocar-dium was not observed in the rest of the MI group. However, the MI/R group had a significantly increased myocardial infarct size compared with the MI group. In addition, PEDF treatment resulted in a reduced myocardial infarct size compared with the MI/R group. Next, TUNEL staining was used to analyze the anti-apoptotic effect of PEDF in the I/R myocardium. There was a marked increase in cardiomyocyte apoptosis in MI/R hearts compared with the sham group, and treatment with PEDF reduced this effect (Fig. 1C and D). There was no significant difference between the Sham group and Sham+P group.
Figure 1.

PEDF reduces myocardial infarct size and improves cardiac function under MI/R conditions. (A) Representative figures of the Evans Blue/TTC-stained myocardial tissues in each indicated experimental condition, with (B) quantification of the infarct size (n=6). (C) Representative images of TUNEL staining of cardiomyocyte apoptosis (white arrow), with (D) quantification. Cardiomyocyte apoptosis was measured by TUNEL staining; cardiomyocytes were identified using α-sarcomeric actin antibodies. TUNEL staining for cardiomyocyte apoptosis (red), DAPI for nuclear staining (blue) and α-sarcomeric actin for cardiomyocytes (green) in the border zone of the infarcted left ventricle from all experimental groups (scale bar=20 μm; n=6). (E) Left ventricular ejection fraction prior to dobutamine injection determination by echocardiography (n=6). (F) Left ventricular fractional shortening prior to dobutamine injection determination by echocardiography (n=6). (G) Δ left ventricular ejection fraction following dobutamine injection by echocardiography (n=6). (H) Δ left ventricular fractional shortening following dobutamine injection by echocardiography (n=6). Data are expressed as the mean ± standard error of the mean. *P<0.05, with comparisons indicated by lines. PEDF, pigment epithelium-derived factor; MI/R, myocardial ischemia/reperfusion; TTC, 2,3,5-triphenyltetrazolium; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Cardiac function and cardiac functional reserve were measured using transthoracic M-mode echocardiography prior to and following dobutamine (1 μg/g) injection. The values of ejection fraction and fractional shortening, which are signs of myocardial function, were significantly increased in PEDF-treated rats compared with untreated MI/R hearts (Fig. 1E and F). The Δ ejection fraction and Δ fractional shortening to dobutamine infusions were increased in hearts transfected with PEDF compared with MI/R rats in vivo, which revealed that PEDF may increase cardiac functional reserve (Fig. 1G and H). Together, these results indicated that PEDF has a protective effect against I/R damage in rat hearts.
PEDF suppresses H/R-induced apoptosis in H9c2 cardiomyocytes
To investigate whether PEDF suppresses H9c2 cell apoptosis under H/R conditions, the level of the cleaved caspase-3 following H/R was dected in H9c2 cells at different time points using western blotting. Onset of hypoxia for up to 8 h resulted in an increase in cleaved caspase-3 protein expression, and cleaved caspase-3 expression was also significantly increased in the hypoxia (8 h)/reoxygenation (2 h) group in comparison with the hypoxia (8 h) group (Fig. 2A and B). Treatment with PEDF could significantly reduce the level of the cleaved caspase-3 compared with the H/R group (Fig. 2C and D). To further confirm the observation that PEDF treatment inhibited H9c2 cell apoptosis under H/R condition, flow cytometric detection of early apoptosis was performed. The results demonstrated similar trends (Fig. 2E and F). These observations indicated that PEDF prevented H/R-induced H9c2 cell apoptosis.
Figure 2.

PEDF protects H9c2 cells against H/R-induced apoptosis. H9c2 cells were exposed to H/R conditions for various durations (0/0, 4/0, 4/2, 8/0, 8/2, 12/0 and 12/2 h). In addition, H9c2 cells treated with or without PEDF (10 nM) were exposed to normoxic or hypoxic/reoxygenation condition for 8/2 h. (A) Western blotting detected the level of the cleaved casp3 protein, with (B) quantification (n=3). (C) Samples were collected and analyzed for the expression of the cleaved casp3 protein by western blotting analysis, with (D) quantification (n=4). (E) Flow cytometric detection of early apoptosis (APC+/PI−), with (F) quantification (n=3). Data are expressed as the mean ± standard error of the mean. *P<0.05, with comparisons indicated by lines. PEDF, pigment epithelium-derived factor; H/R, hypoxia/reoxygenation; casp3, caspase-3; APC, allophycocyanin; PI, propidium iodide; N, negative control.
PEDF protects H9c2 cells against H/R-induced apoptosis via PEDF-R
Next, to investigate whether PEDF-R is involved in the PEDF-mediated repression of cell injury, H9c2 cells were treated with PEDF under normoxic and H/R conditions, and RNA interference assays were used to silence PEDF-R. PEDF-R siRNA significantly reduced PEDF-R expression levels under normoxic conditions in H9c2 cells (Fig. 3). As presented in Fig. 4A and B, PEDF-R siRNA prevented the PEDF-induced reduction of cleaved caspase-3 protein expression under H/R conditions in H9c2 cells. This was not observed in the PEDF+vector group. TUNEL staining was also used to identify apoptotic H9c2 cells, and the results revealed the critical involvement of PEDF-R in the effect of PEDF in H/R injury (Fig. 4C and D). These results suggested that the anti-apoptotic effect of PEDF is dependent on PEDF-R.
Figure 3.
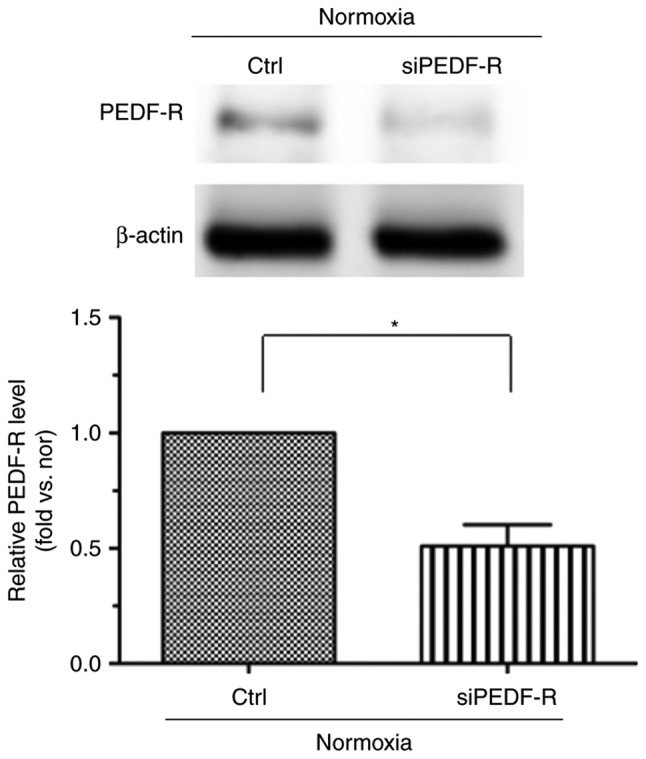
siPEDF-R reduces the levels of PEDF-R under normoxic conditions in H9c2 cells. RNA interference assays were used to silence PEDF-R. Western blot analysis of PEDF-R expression was then performed (n=4). Data are expressed as the mean ± standard error of the mean. *P<0.05, with comparisons indicated by lines. si, small interfering; PEDF-R, pigment epithelium-derived factor receptor.
Figure 4.

PEDF decreases H/R-induced apoptosis via PEDF-R in cultured H9c2 cells. H9c2 cells were maintained in normoxic or H/R conditions for 8/2 h with or without PEDF (10 nM). RNA interference assays were used to silence PEDF-R. (A) Samples were collected for western blotting to analyze the expression of cleaved casp3 protein, with (B) quantification performed using ImageJ software (n=4). (C) Effect of PEDF on H9c2 cells apoptosis, with (D) quantification. TUNEL (red) staining was performed for each group. Nuclei were stained with DAPI (blue). Cells that were TUNEL and DAPI-positive were apoptotic (indicated by the arrows), while DAPI positive were control cells (scale bar=50 μm; n=4). Data are expressed as the mean ± standard error of the mean. *P<0.05, with comparisons indicated by lines. PEDF, pigment epithelium-derived factor; H/R, hypoxia/reoxygenation; PEDF-R, pigment epithelium-derived factor receptor; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; si, small interfering.
PEDF reduces H/R-induced the burst of ROS via PEDF-R
The ROS burst is a key factor of myocardial ischemia/reperfusion injury. Therefore, whether PEDF inhibited the H/R-induced burst of ROS via PEDF-R was investigated. The changes in ROS levels in H9c2 cells are presented in Fig. 5A and B. Compared with the normal group, ROS levels were significantly increased in the H/R groups. Nonetheless, PEDF markedly decreased the level of ROS induced by H/R, while this effect was reversed by PEDF-R siRNA. Intracellular MDA levels were measured to further confirm the antioxidative function of PEDF. In the H/R group, MDA content was increased compared with the normal group. PEDF dampened the MDA increase, but PEDF-R siRNA attenuated this effect (Fig. 5C). These results suggested that PEDF has antioxidant activities in response to H/R injury in H9c2 cells, and these activities are dependent on PEDF-R.
Figure 5.

PEDF suppresses the levels of ROS and MDA via PEDF-R during H/R. H9c2 cells were maintained in normoxic or H/R conditions for 8/2 h with or without PEDF (10 nM). RNA interference assays were used to silence PEDF-R. (A) ROS level was measured with dihydroethidium fluorescence probes and (B) quantified with Image-Pro Plus software (Scale bar=50 μm; n=4). (C) MDA levels were assessed (n=3). Data are expressed as the mean ± standard error of the mean. *P<0.05, with comparisons indicated by lines. PEDF, pigment epithelium-derived factor; ROS, reactive oxygen species; MDA, malondialdehyde; PEDF-R, pigment epithelium-derived factor receptor; H/R, hypoxia/reoxygenation; si, small interfering.
PEDF decreases H/R-induced mtROS generation via PEDF-R
To study the detailed mechanisms underlying the antioxidative effect of PEDF, which protects cardiomyocytes against H/R-induced apoptosis, our group developed a novel hypothesis: That PEDF attenuates H/R-induced oxidative stress via PEDF-R through inhibition of ROS generation. The main source of ROS is the mitochondria, which produce ROS primarily through single electron transport to molecular oxygen in the ETC (11). In addition, mtDNA is the primary target of the ROS, and a reflection of ETC function (31). Thus, mtROS level and the mtDNA copy number were measured to investigate mitochondrial-derived ROS synthesis. Under H/R conditions, mtROS levels were increased while the mtDNA copy number was decreased. This effect was reversed by PEDF treatment, but PEDF-R siRNA inhibited this effect (Fig. 6). The results demonstrated that PEDF attenuates the H/R-induced burst of ROS by reducing mtROS generation via PEDF-R.
Figure 6.

PEDF reduces H/R-induced mtROS generation via PEDF-R. H9c2 cells were maintained in normoxic or H/R conditions for 8/2 h with or without PEDF (10 nM). RNA interference assays were used to silence PEDF-R. (A) mtROS production was monitored by MitoSOX™ Red in H9c2 cells, with (B) quantification. ROS production was observed by red fluorescence of MitoSOX™ by fluorescence microscopy and analyzed by Image-Pro Plus software (scale bar=20 μm; n=5). (C) mtDNA copy number was quantified by comparing D-loop expression to 18s RNA content using quantitative polymerase chain reaction analysis (n=4). Data are expressed as the mean ± standard error of the mean. *P<0.05, with comparisons indicated by lines. PEDF, pigment epithelium-derived factor; H/R, hypoxia/reoxygenation; mtROS, mitochondrial reactive oxygen species; mtDNA, mitochondrial DNA; si, small interfering; PEDF-R, pigment epithelium-derived factor receptor.
PEDF decreases H/R-induced cytoplasmic ROS generation via PEDF-R
NOX and XO are the main sources of ROS formation in the cytoplasm (11), thus, examination of XO and NOX activity permits the evaluation of cytoplasmic ROS production. XO and NOX activity was increased in H/R and H9c2 cells and, while PEDF reduced thid activity, PEDF-R siRNA reversed this effect (Fig. 7A and B). In addition, the expression of rac1, which has an important effect on NOX activation (32), was detected. Rac1 protein expression was altered in a similar manner to NOX activity (Fig. 7C and D). These observations suggested that PEDF attenuates the H/R-induced ROS burst by reducing cytoplasmic ROS generation via PEDF-R.
Figure 7.

PEDF decreases H/R-induced cytoplasmic ROS generation via PEDF-R. H9c2 cells were maintained in normoxic or H/R conditions for 8/2 h with or without PEDF (10 nM). RNA interference assays were used to silence PEDF-R. (A) XO activity was assessed in all experimental groups using the XO activity assay kit (n=4). (B) NOX activity was assessed in all experimental groups using the NOX activity assay kit (n=4). (C) Western blot analysis of rac1 protein expression, with (D) quantification (n=4). Data are expressed as the mean ± standard error of the mean. *P<0.05, with comparisons indicated by lines. PEDF, pigment epithelium-derived factor; H/R, hypoxia/reoxygenation; ROS, reactive oxygen species; PEDF-R, pigment epithelium-derived factor receptor; XO, xanthine oxidase; NOX, NADPH oxidase; rac1, RAC family small GTPase 1; si, small interfering.
Discussion
In the present study, PEDF was demonstrated to reduce myocardial infarct size and improve cardiac function in a rat MI/R model. In addition, the mechanism underlying effect was verified: PEDF reduced H/R-induced cell injury by attenuating ROS generation, and in particular mtROS generation, via PEDF-R.
A large number of mitochondria are typically present in cardiomyocytes (33). ROS, initially produced by damaged mitochondria, induce the opening of the mitochondrial membrane permeability transition pore, induce the dissipation of mitochondrial membrane potential (ΔΨm) and block the electron transport chain, which leads to the ROS burst (34,35). This process is known as ROS-induced ROS release, and this is one of the factors that induces cell injury (34,35). Mitochondria are not only the main target of ROS damage, but also the primary site of ROS production (6). Therefore, it is important to reduce the generation of mitochondrial (mt)ROS in cardiomyocytes. A previous study by our group demonstrated that PEDF decreases mtROS by inhibiting mitochondrial fission in hypoxic cardiomyocytes (36). However, the explosive generation of mtROS appears during the reperfusion process due to the transient increase of oxygen concentration and the effect of inefficient electron transfer (6). In this case, whether PEDF is able to continue to reduce mtROS generation is worthy of further investigation. In the present study, PEDF was revealed to significantly decreased mtROS levels, increase mitochondrial DNA copy number, reduce xanthine oxidase and NADPH oxidase activity and decrease rac1 protein expression, compared with the H/R group. The results from the present study demonstrated that PEDF may attenuate MI/R-induced mitochondrial and cytosolic ROS formation.
Under the condition of the present study, the results of Evans Blue/TTC staining revealed that small myocardial infarctions occurred in certain individuals from the MI (0.5 h) group. However, the MI (0.5 h)/R (24 h) group had wider range of myocardial infarction compared with the MI (0.5 h) group. In the in vitro experiments, the apoptosis of H9c2 cardiomyocytes was relatively mild 8 h after hypoxia, and was significantly increased in the H (8 h)/R (2 h) group. Based on these results, reperfusion (24 h) following ischemia (0.5 h) were selected in vivo and reoxygenation (2 h) following hypoxia (8 h) were selected in vitro as the optimal time points to study the effect of PEDF in MI/R injury and the underlying mechanisms. The protective effect of PEDF on cardiomyocytes during MI/R process may be best reflected in the protective effect of PEDF on cardiomyocytes during reperfusion.
Multiple previous studies have focused on the function of PEDF in hypoxic cardiomyocytes, rather than the functions and mechanisms of PEDF in the MI/R process. A previous study has demonstrated that that PEDF reduces the levels of ROS in hypoxia-reoxygenated human cardiomyocytes (37), consistent with the results of the present study. Furthermore, in addition to detecting the decrease in ROS levels, the present study demonstrated that PEDF has the potential to inhibit the formation of ROS through further studies. To the best of our knowledge, the present study has demonstrated for the first time that PEDF significantly reduces myocardial infarct size in rats undergoing the MI/R process, while protecting cardiac function. In addition, the present study demonstrated that PEDF significantly decreased H9c2 cardiomyocyte apoptosis caused by H/R via PEDF-R in vitro, which is associated with the suppression of ROS generation.
In the present study, a previously unknown association between PEDF and ROS generation in the MI/R process was demonstrated. However, further studies are required to fully explore the detailed mechanisms underlying PEDF-induced regulation of MI/R-induced ROS generation in vivo. In addition, the results derived from H9c2 cells may not represent the same mechanisms as in primary cardiomyocytes. This limitation of the present study means that further research is required. In conclusion, the present study confirmed that PEDF protects cardiomyocytes against MI/R injury by reducing ROS production. The results of the present study suggest that PEDF represents a promising novel therapeutic approach for MI/R injury.
Acknowledgments
We would like to express our thanks to the Research Facility Center for Morphology of Xuzhou Medical University (Xuzhou, China), their research site and facilities provided an effective means to conduct our research.
Footnotes
Funding
The present study was supported by the National Nature Science Foundation of China (grant no. 81570242) and the Natural Science Foundation of Jiangsu Province (grant no. BK20150207).
Availability of data and materials
We declared that materials described in the manuscript, including all relevant raw data, will be freely available to any scientist wishing to use them for non-commercial purposes, without breaching participant confidentiality.
Authors' contributions
QZ, ZL, HD and ZZ conceived and designed the experiments. QZ, ZL, BH, YY, XL, HZ and YZ performed the experiments. QZ, ZL, YL, HD and ZZ analyzed the data. QZ, ZL, FQ, HM and YZ acquired the reagents, materials and tools for analysis. QZ and ZL produced the manuscript.
Ethics approval and consent to participate
The experiments described in this manuscript conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Publication, 8th Edition, 2011, Bethesda, MD, USA) (25). All animal care and experimental protocols were approved by the Animal Care and Use Committee of Xuzhou Medical University (license no. SYXK 2002-0038, Jiangsu, China) and also followed the international guidelines (European Council Directive 2010/63/EU) on the ethical use of animals (26).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Damiani G, Salvatori E, Silvestrini G, Ivanova I, Bojovic L, Iodice L, Ricciardi W. Influence of socioeconomic factors on hospital readmissions for heart failure and acute myocardial infarction in patients 65 years and older: Evidence from a systematic review. Clin Interv Aging. 2015;10:237–245. doi: 10.2147/CIA.S71165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crea F, Battipaglia I, Andreotti F. Sex differences in mechanisms, presentation and management of ischaemic heart disease. Atherosclerosis. 2015;241:157–168. doi: 10.1016/j.atherosclerosis.2015.04.802. [DOI] [PubMed] [Google Scholar]
- 3.Hearse DJ. Myocardial protection during ischemia and reperfusion. Mol Cell Biochem. 1998;186:177–184. doi: 10.1023/A:1006808507605. [DOI] [PubMed] [Google Scholar]
- 4.Hearse DJ, Bolli R. Reperfusion induced injury: Manifestations, mechanisms, and clinical relevance. Cardiovasc Res. 1992;26:101–108. doi: 10.1093/cvr/26.2.101. [DOI] [PubMed] [Google Scholar]
- 5.Turer AT, Hill JA. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am J Cardiol. 2010;106:360–368. doi: 10.1016/j.amjcard.2010.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Minamino T. Cardioprotection from ischemia/reperfusion injury: Basic and translational research. Circ J. 2012;76:1074–1082. doi: 10.1253/circj.CJ-12-0132. [DOI] [PubMed] [Google Scholar]
- 8.Braunersreuther V, Jaquet V. Reactive oxygen species in myocardial reperfusion injury: From physiopathology to therapeutic approaches. Curr Pharm Biotechnol. 2012;13:97–114. doi: 10.2174/138920112798868782. [DOI] [PubMed] [Google Scholar]
- 9.Radak Z, Zhao Z, Goto S, Koltai E. Age-associated neuro-degeneration and oxidative damage to lipids, proteins and DNA. Mol Aspects Med. 2011;32:305–315. doi: 10.1016/j.mam.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 10.Campos JC, Bozi LH, Bechara LR, Lima VM, Ferreira JC. Mitochondrial quality control in cardiac diseases. Front Physiol. 2016;7:479. doi: 10.3389/fphys.2016.00479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Afanas'ev I. ROS and RNS signaling in heart disorders: Could antioxidant treatment be successful? Oxid Med Cell Longev. 2011;2011:293769. doi: 10.1155/2011/293769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Becerra SP, Sagasti A, Spinella P, Notario V. Pigment epithelium-derived factor behaves like a noninhibitory serpin. Neurotrophic activity does not require the serpin reactive loop. J Biol Chem. 1995;270:25992–25999. doi: 10.1074/jbc.270.43.25992. [DOI] [PubMed] [Google Scholar]
- 13.Rychli K, Kaun C, Hohensinner PJ, Dorfner AJ, Pfaffenberger S, Niessner A, Bauer M, Dietl W, Podesser BK, Maurer G, et al. The anti-angiogenic factor PEDF is present in the human heart and is regulated by anoxia in cardiac myocytes and fibroblasts. J Cell Mol Med. 2010;14:198–205. doi: 10.1111/j.1582-4934.2009.00731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao X, Zhang H, Zhuang W, Yuan G, Sun T, Jiang X, Zhou Z, Yuan H, Zhang Z, Dong H. PEDF and PEDF-derived peptide 44mer protect cardiomyocytes against hypoxia-induced apoptosis and necroptosis via anti-oxidative effect. Sci Rep. 2014;4:5637. doi: 10.1038/srep05637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H, Sun T, Jiang X, Yu H, Wang M, Wei T, Cui H, Zhuang W, Liu Z, Zhang Z, Dong H. PEDF and PEDF-derived peptide 44mer stimulate cardiac triglyceride degradation via ATGL. J Transl Med. 2015;13:68. doi: 10.1186/s12967-015-0432-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Wang Z, Feng SJ, Xu L, Shi HX, Chen LL, Yuan GD, Yan W, Zhuang W, Zhang YQ, et al. PEDF improves cardiac function in rats with acute myocardial infarction via inhibiting vascular permeability and cardiomyocyte apoptosis. Int J Mol Sci. 2015;16:5618–5634. doi: 10.3390/ijms16035618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Notari L, Baladron V, Aroca-Aguilar JD, Balko N, Heredia R, Meyer C, Notario PM, Saravanamuthu S, Nueda ML, Sanchez-Sanchez F, et al. Identification of a lipase-linked cell membrane receptor for pigment epithelium-derived factor. J Biol Chem. 2006;281:38022–38037. doi: 10.1074/jbc.M600353200. [DOI] [PubMed] [Google Scholar]
- 18.Bernard A, Gao-Li J, Franco CA, Bouceba T, Huet A, Li Z. Laminin receptor involvement in the anti-angiogenic activity of pigment epithelium-derived factor. J Biol Chem. 2009;284:10480–10490. doi: 10.1074/jbc.M809259200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 20.Hirsch J, Johnson CL, Nelius T, Kennedy R, Riese Wd, Filleur S. PEDF inhibits IL8 production in prostate cancer cells through PEDF receptor/phospholipase A2 and regulation of NFκB and PPARγ. Cytokine. 2011;55:202–210. doi: 10.1016/j.cyto.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 21.Wang XY, Zhang YQ, Lu P, Zhang H, Li Y, Dong H, Zhang Z. PEDF attenuates hypoxia-induced apoptosis and necrosis in H9c2 cells by inhibiting p53 mitochondrial translocation via PEDF-R. Biochem Biophys Res Commun. 2015;465:394–401. doi: 10.1016/j.bbrc.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 22.Lu P, Zhang YQ, Zhang H, Li YF, Wang XY, Xu H, Liu ZW, Li L, Dong HY, Zhang ZM. Pigment epithelium-derived factor (PEDF) improves ischemic cardiac functional reserve through decreasing hypoxic cardiomyocyte contractility through PEDF receptor (PEDF-R) J Am Heart Assoc. 2016;5:e003179. doi: 10.1161/JAHA.115.003179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reim DF, Speicher DW. N-terminal sequence analysis of proteins and peptides. Curr Protoc Protein Sci. 2001 doi: 10.1002/0471140864.ps1110s08. Chapter 11: Unit 11.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Zhao X, Liang G, Sun J, Lin Z, Hu R, Chen P, Zhang Z, Zhou L, Li Y. Recombinant SFRP5 protein significantly alleviated intrahepatic inflammation of nonalcoholic steatohepatitis. Nutr Metab (Lond) 2017;14:56. doi: 10.1186/s12986-017-0208-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.National Research Council . Guide for the Care and Use of Laboratory Animals. 8th edition. The National Academies Press; Washington, DC: 2011. [Google Scholar]
- 26.European Commission Directive 2010/63/EU of the European Parliament and of the council of 22 September 2010 on the protection of animals used for scientific purposes. Off J Eur Union. 2010;L276:33–79. [Google Scholar]
- 27.Zhang Y, Li Y, Wang X, Zhao Q, Lu P, Zhang H, Dong H, Zhang Z. A closed-chest rat model of myocardial isch-aemia/reperfusion supports direct intramyocardial gene delivery. Exp Ther Med. In press. [Google Scholar]
- 28.Black SC, Rodger IW. Methods for studying experimental myocardial ischemic and reperfusion injury. J Pharmacol Toxicol Methods. 1996;35:179–190. doi: 10.1016/1056-8719(96)00051-2. [DOI] [PubMed] [Google Scholar]
- 29.Peshavariya HM, Dusting GJ, Selemidis S. Analysis of dihydroethidium fluorescence for the detection of intracellular and extracellular superoxide produced by NADPH oxidase. Free Radic Res. 2007;41:699–712. doi: 10.1080/10715760701297354. [DOI] [PubMed] [Google Scholar]
- 30.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 31.Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and mitochondrial DNA damage in heart failure. Circ J. 2008;72(Suppl A):A31–A37. doi: 10.1253/circj.CJ-08-0014. [DOI] [PubMed] [Google Scholar]
- 32.Li JM, Zhu HQ, Shen E, Wan L, Arnold JMO, Peng TQ. Deficiency of Rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes. 2010;59:2033–2042. doi: 10.2337/db09-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia-Dorado D, Andres-Villarreal M, Ruiz-Meana M, Inserte J, Barba I. Myocardial edema: A translational view. J Mol Cell Cardiol. 2012;52:931–939. doi: 10.1016/j.yjmcc.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 34.Gustafsson AB, Gottlieb RA. Heart mitochondria: Gates of life and death. Cardiovasc Res. 2008;77:334–343. doi: 10.1093/cvr/cvm005. [DOI] [PubMed] [Google Scholar]
- 35.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 1. 2000;92:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou ZX, Wang Z, Guan QH, Qiu F, Li Y, Liu Z, Zhang H, Dong H, Zhang Z. PEDF inhibits the activation of NLRP3 inflammasome in hypoxia cardiomyocytes through PEDF receptor/phospholipase A2. Int J Mol Sci. 2016;17:E2064. doi: 10.3390/ijms17122064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuo HF, Liu PL, Chong IW, Liu YP, Chen YH, Ku PM, Li CY, Chen HH, Chiang HC, Wang CL, et al. Pigment epithelium-derived factor mediates autophagy and apoptosis in myocardial hypoxia/reoxygenation injury. PLoS One. 2016;11:e0156059. doi: 10.1371/journal.pone.0156059. [DOI] [PMC free article] [PubMed] [Google Scholar]