

Abstract
With the recent development of methods for genome editing of human pluripotent stem cells, study of the genetic basis of human diseases has been rapidly advancing. Genome-edited differentiated stem cells have provided new and more accurate insights into genomic underpinnings of diseases for which there have not been adequate treatments, and moving toward clinical application of genome editing holds great promise for acceleration of therapeutic translation. Here we review recent advances in genome-editing technologies and their application to human biology in disease modeling and beyond.
Keywords: ZFNs, TALENs, CRISPR/Cas9, genome editing, iPSC
Graphical abstract
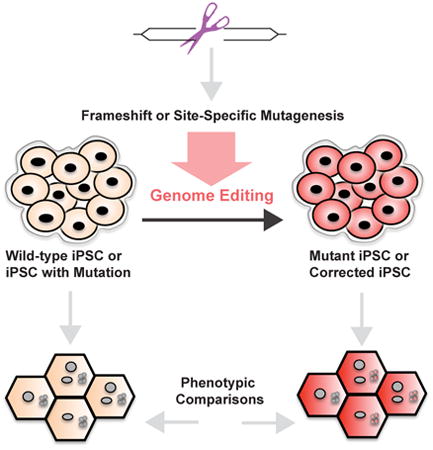
Genome-editing methods involve the formation of a double-stranded DNA break followed by a repair strategy resulting in either a knockout or site-specific mutagenesis. This approach canfacilitate the study of gene function in induced pluripotent stem cells (iPSCs) from healthy individuals or individuals with disease mutations. iPSCs can then be differentiated into the celltype of interest for further study.
Introduction
With increasing availability of human genome-sequencing data from individuals with diseases or disease-related traits, the role of genetics in human disease has become a major focus in translational biomedical research.[1] From genome-wide association studies,[2-4] to functional laboratory follow-up,[1,5-7] and ultimately to development of targeted therapeutics,[2-4,8] the genetic basis of disease has been a hotbed of research geared towards precision medicine—clinical strategies that account for individual variability, e.g., genomics.[9,10] Mechanistic study of potential causal genes for diseases or traits requires genetic manipulation in cellular models, which have advanced in their utility for variant-specific and cell-specific studies with the advent of genome-editing technologies.
Prior to genome editing, viral transgene expression and RNA interference (RNAi) were commonly used for functional studies of specific genes. However, viral vectors and RNAi are falling out of favor due to (1) dysregulated transgene expression from insertion-site mutagenesis and (2) incomplete time-limited ablation of gene function with poor specificity, respectively.[11-13] One approach to circumvent these issues in studying disease mechanisms has been the use of human pluripotent stem cells derived from patients with the disease of interest. With recent advances in technology, induced pluripotent stem cells (iPSCs) can be generated from a skin biopsy or blood sample from any patient, facilitating the derivation, expansion, and differentiation of somatic cells genetically matched to the patient.[14-17] This can offer advantages to studying disease-associated genetic variants in cells that do not contain the severely abnormal karyotypes of immortalized cell lines, cells that are difficult to obtain for primary culture (e.g., neurons), and cells that are difficult to transfect or transduce (e.g., T-cells and macrophages).[15,16,18,19] However, iPSC-based studies also have their own limitations. For example, differences in genetic background between disease cases and healthy controls can confound studies, even those using healthy siblings as controls. Because siblings share only 50% of the genome on average, phenotypic differences observed in studies using disease versus healthy siblings could arise from variants in the unshared portion of the genome rather than the disease-associated mutations being interrogated. In addition, iPSCs from different individuals can vary in terms of genomic methylation patterns where some may retain epigenetic “memory” from the somatic cell of origin from which they were reprogrammed, thus allowing for an iPSC line to differentiate preferentially into some cell types over others.[20-23]
All these limitations of vector-based transgene expression, RNAi, and iPSC studies could potentially be overcome through the use of isogenic (derived from the same parental cell line) genome-edited iPSCs in which wild-type and mutant cell lines differ only in terms of disease mutations. Genome editing allows investigators to introduce a variety of genetic alterations with a high degree of target specificity within a controlled genetic background; these alterations range from single nucleotide modifications to whole-gene addition or deletion. Here we review the different nuclease-based genome-editing technologies based on programmable nucleases, discuss considerations for their use in cellular models, and present their potential and challenges for clinical translation.
Genome Editing Technologies
Zinc Finger Nucleases (ZFNs)
ZFNs comprise a nuclease class used for genome editing through binding of a target DNA sequence by a chimeric enzyme consisting of target site-specific binding domains—adapted from zinc finger transcription factors—fused to the sequence-agnostic nuclease domain of bacterial restriction enzyme FokI. ZFNs are typically designed in pairs to recognize DNA sequences flanking the genomic target site of interest. Each zinc finger domain recognizes a 3-4 basepair (bp) DNA sequence, and multiple domains can be engineered to exist in tandem for each ZFN monomer to bind an extended unique 9-18 bp sequence adjacent to the target site. Upon binding the sequences flanking the excision target, the FokI nuclease domains of the ZFN pair dimerize and generate a double-stranded break (DSB).[24,25] DSBs are then repaired by the cell through one of two methods: non-homologous end joining (NHEJ) or homology-directed repair (HDR) (see Figure). NHEJ repairs the lesion by directly rejoining the DSB ends in an error-prone process that does not require the presence of a repair template. This strategy introduces insertion or deletion mutations (indels) that bridge the break site, and when introduced into a coding portion of the target gene these indels can cause frameshifts that lead to nonsense mediated mRNA decay and/or production of non-functional truncated proteins, thus effectively knocking out the target gene.[24] Unlike NHEJ, HDR involves the use of an exogenous DNA repair template to incorporate desired changes in the DSB repair, i.e., introduction of a specific mutation of interest. The repair template can be a double-stranded DNA vector or a single-stranded DNA oligonucleotide; the single-stranded oligonucleotide can consist of the desired mutation flanked by homology arms of as little as 20 nucleotides in length, offering reasonable repair efficiency that does not require antibiotic selection for screening of correctly targeted clones.[26]
Figure. Overview of genome-editing tools and application to human iPSCs.

ZFNs are a chimeric enzyme that includes a FokI nuclease (brown oval) attached to DNA-binding zinc finger domains (colored ovals) that each interact with 3-4 bp DNA sequences (each group color-coordinated with corresponding zinc finger domain), allowing for recognition of a 9-18 bp genomic sequence when engineered in tandem. The DNA binding domains flank the target genomic site, where the FokI nucleases make a DSB. TALENs are also a chimeric enzyme but with a FokI nuclease attached to a TALE DNA-binding array, which consists of RVDs (colored rectangles) that each bind to genomic DNA in a 1 RVD to 1 bp ratio. The genomic target site is cleaved by the FokI nucleases, which are flanked by the TALE DNA binding array sequences. Note that the 5′ ends of the TALE arrays begin with a thymine. The CRISPR/Cas9 system consists of a guide RNA (purple) binding to the genomic target (20 bp protospacer) and interacting with the Cas9 nuclease (yellow) that recognizes the PAM site. After a DSB is created by one of these techniques, the DNA can undergo (1) NHEJ in which the blunt ends are rejoined with introduction of additional nucleotides that induce an indel/frameshift mutation (dashes) for gene knockout or (2) HDR in which a repair template is introduced to facilitate the incorporation of a specific point mutation (red letter). This overall genome-editing strategy can then be employed to study gene function in iPSCs. iPSCs derived from humans (beige) can either be wild-type from a healthy individual or with mutation from an individual with the disease of interest. Genome-edited iPSCs (red) can be generated through introduction of a mutation to create a mutant iPSC or corrected iPSC, providing the genetic study complement on an isogenic background. These iPSCs can then be differentiated into the cell type of interest for further study of cell-autonomous phenotypes. iPSCs = induced pluripotent stem cells, ZFNs = zinc finger nucleases, DSB = double-stranded break, TALENs = transcription activator-like effector nucleases, RVD = repeat-variable di-residue, NHEJ = non-homologous end joining, HDR = homology-directed repair.
Although ZFN technology offers advantages over RNAi, its use is still accompanied by challenges worth consideration by investigators new to genome editing. For example, the first steps of engineering ZFNs to target a specific site may prove to be difficult as assembly of desired zinc finger domains to bind an extended nucleotide sequence requires substantial protein engineering expertise.[27] These first steps are still technically challenging and potentially expensive despite publicly available protocols for ZFN screen optimization and alternative ZFN engineering platforms.[28-31] In addition, target-site selection is limited: ZFNs do not facilitate targeting of sequences that are guanine-poor, and ZFN components can only be used for binding sites located every few hundred bp throughout the genome.[11]
Transcription activator-like effector nucleases (TALENs)
Like ZFNs, TALENs are chimeric enzymes with a DNA-binding domain fused to a FokI nuclease domain that generates a DSB within a target region and facilitates repair either through NHEJ or HDR. However, the TALEN's DNA-binding strategy differs in the use of modified TALEs, which consist of DNA binding modules called TAL repeats originally discovered in plant-pathogenic bacteria.[32] Each TAL repeat contains 33-35 amino acids that include a repeat-variable di-residue (RVD), a unit consisting of two adjacent amino acids that specifically recognize and bind to one of four DNA bases.[32,33] With the affinity of specific RVD amino acid combinations for each type of nucleotide, a DNA sequence can be targeted by engineering a tandem array of TAL repeats that contain RVDs in the order of their corresponding target nucleotides, in a 1-RVD to 1-bp ratio (see Figure). TALENs are easier to design than ZFNs through use of the RVD “code” to create de novo extended TAL repeat arrays that bind with high affinity to target genomic sequences, allowing for the construction of hundreds of TALENs at a time[34,35] with robust gene-targeting in human embryonic stem cells and iPSCs. TALENs also do not have the same target-site limitations that ZFNs do, with more potential binding sites as the main requirement is that the 5′ targeted base should be a thymine.[11]
Despite this improvement over ZFNs, widespread use of TALENs has been limited by the suboptimal performance of a significant portion of designed TALEN pairs, thus requiring the screening of a large number of candidate pairs in order to find one with a high level of activity.[35] A potential contributor to variable performance of engineered TALEN pairs is the methylation state of the target region, as the standard TAL repeats cannot bind well to methylated cytosines, often found in CpG islands.[36,37]
CRISPR/Cas9
More recently, the genome editing field has undergone a “revolution” with the rise of clustered regularly interspaced short palindromic repeat (CRISPR)-associated 9 (Cas9) systems, which have been adapted from bacterial immune systems that use CRISPR RNA in conjunction with Cas proteins to direct the cleavage and degradation of invading viral and plasmid DNAs. In response to these invading genomes, bacteria incorporate fragments of the foreign DNA into the CRISPR locus as a nucleic acid “vaccination record”; these DNA sequences are “protospacers” transcribed into “guide RNAs” that can be bound by Cas proteins and that can then recognize other foreign DNAs with the protospacer sequence via complementary binding. Protospacer sequences in DNA must be flanked by protospacer-adjacent motifs (PAMs) that are recognized by Cas proteins directed to target sites by guide RNAs binding to the DNA protospacers; only then will Cas proteins cleave the targeted foreign DNAs.[38,39] For genome editing, this bacterial adaptive immune strategy was re-engineered to use heterologous expression of the Streptococcus pyogenes Cas9 (SpCas9) protein alongside short, investigator-designed guide RNA complexes in mammalian cells. Leveraging the combination of guide RNA-mediated target recognition and Cas9 endonuclease activity, investigators can create a DSB within a genomic target site that has a 20-bp DNA sequence matching the protospacer of the guide RNA and a flanking PAM, which for SpCas9 is a NGG nucleotide sequence (see Figure).[39-41] With this design, sequences can be targeted by changing the 20-bp guide RNA sequence without re-engineering the Cas9 portion, thus allowing for greater ease in engineering CRISPRs. In addition, multiple guide RNAs can be introduced in series within a single vector to allow for multiplexed targeting of multiple sites within the same cell.[39]
Despite its ease of engineering, CRISPR/Cas9 still has its own limitations. For example, SpCas9 genome editing requires the presence of an NGG PAM, which occurs roughly once every 8 bp, and this requirement may lead to less target sequence density than TALENs, which may on average have a dimeric target site every 3 bp.[35,42,43] The PAM requirement may limit the use of CRISPR/Cas9 when target specificity is required for introducing a DSB at a precise location for HDR-mediated repair to introduce a specific mutation. To address this, non-canonical PAM sequences and Cas9-like proteins derived from alternative bacteria have recently been explored to increase the potential for target specificity,[44-46] although their relative efficacy and ease of use in iPSCs compared to established methods remain to be determined. For example, a novel method based on the general concept of CRISPR uses Natronobacterium gregoryi Argonaute, a eukaryotic DNA-guided endonuclease that does not require the presence of a PAM and thus may offer more flexibility in choosing target sequences; more studies are needed to assess precision and efficiency of this recently developed method.[47] In addition, CRISPR/Cas9 efficiency has much room for improvement, as investigators currently need to derive clonal cell lines to study genome-edited cells, a process that can take several weeks to complete. Other issues, including off-target effects, are discussed in more detail below.
Other Genome Editing Tools
In addition to ZFNs, TALENs, and CRISPR/Cas9, strategies for genome editing include use of meganucleases,[48] adeno-associated viruses,[49] and adenoviruses.[50,51] These tools have not been as widely used as ZFNs, TALENs, and CRISPR/Cas9 due to lower adaptability. However, given how rapidly all of these genome-editing strategies have been developed, additional modifications to current approaches and the creation of additional novel genome-editing technologies, such as the DNA-guided endonuclease approach described above,[47] are likely to be on the horizon.
Application in Cellular Models
With the feasibility of using genome-editing tools to study gene function, increasing numbers of studies are demonstrating the power of genome editing in disease modeling and potential future therapeutic translation. Here we present examples of recent seminal studies that leverage genome editing for functional studies.
In a study of NOTCH1 nonsense mutations leading to calcific aortic valve disease (CAVD), Theodoris et al. demonstrated that genome-edited iPSCs differentiated into endothelial cells (ECs) can be used to probe the transcriptomic and epigenomic effects of disease-mutation heterozygosity.[52] The investigators used TALENs to correct the disease mutation in iPSCs derived from NOTCH1+/- individuals diagnosed with CAVD to create isogenic control lines. NOTCH1+/- iPSCs, isogenic corrected control iPSCs, and unrelated wild-type control iPSCs were then differentiated into ECs and subjected to shear stress to interrogate whether the disease mutations affected transcriptional responses to this stimulation. They found that shear stress induced aberrant upregulation of pro-osteogenic and inflammatory signaling pathways in NOTCH1+/- iPSC-derived ECs compared to isogenic and unrelated controls. In both static and shear-stress states, NOTCH1+/- iPSC-derived ECs had pro-inflammatory STAT and IRF motifs enriched for H3K27 acetylation, an epigenetic mark of active transcription. Additionally, bisulfite sequencing revealed that NOTCH1+/- iPSC-derived ECs had many differentially methylated regions compared to controls. The investigators showed that genome-edited iPSC-derived cells can reveal important information about epigenetic and transcriptional events in Mendelian diseases.
Genome editing can be used not only to study disease-related intracellular pathways and regulation but also to rescue disease phenotypes in cellular models. This process was first demonstrated in the study of Huntington's disease[53] and Parkinson's disease,[18,54] and more recently the approach was extended to the study of cystic fibrosis (CF). In the CF study, the investigators generated iPSCs from a CF patient homozygous for the disease-associated ΔF508 CFTR mutation, which prevents the proper trafficking of the chloride channel to the cell membrane, leading to aberrant secretions in the lungs that ultimately result in premature respiratory failure and death.[55] They corrected the mutation using CRISPR/Cas9 to create a clonal isogenic control. When stimulated with a cocktail that induces chloride channel currents in wild-type lung epithelial cells, differentiated iPSCs with the ΔF508 CFTR mutation did not exhibit any chloride current response, whereas their CRISPR/Cas9-corrected isogenic controls did respond partially in terms of chloride current and appropriate glycosylation of the CFTR protein. This phenotypic rescue through genome editing holds promise that gene therapy for the ΔF508 CFTR mutation may be a viable approach to improving the health of CF patients.
Current Challenges in Genome-Edited Cellular Models
The potential for achieving a high level of specificity in genetic manipulation is one of the main draws of using genome-editing technologies. Successful design of genome-editing tools can facilitate the precise study of a genetic variant on an isogenic background to limit potential confounders, but genome editing does not guarantee absolute precision given that it involves DSBs, which can induce unanticipated perturbations in the genome if cleavage occurs at off-target sites. Introduction of off-target mutations could have major impact on functional studies in terms of evaluating cellular phenotypes and mechanisms; as genome editing moves toward therapeutic translation, these off-target mutations could be problematic in introducing oncogenic potential and reduced cellular fitness.
Only a limited number of studies have attempted to evaluate and report the targeting specificities of ZFNs, TALENs, and CRISPR/Cas9 systems. Studies addressing specificity of ZFNs and TALENs have demonstrated the challenges of detecting off-target activity for these nuclease-based tools.[56-58] Recently, studies reporting the use of whole-genome sequencing of CRISPR/Cas9-edited stem cell lines revealed a low incidence of off-target mutation, with higher cleavage efficiency for on-target sites.[59-61] Although off-target effects were found to be low, continued improvements are being made to CRISPR/Cas9 technology to improve nuclease specificity. For example, increasing numbers of in silico tools are becoming available to improve target specificity in nuclease engineering.[62] Also, alteration of the Cas9 protein into a single-strand DNA nickase to facilitate the use of two separate guide RNAs to create single-strand breaks on opposite DNA strands have led to reduction of indel formation at computationally predicted off-target sites.[63,64] Furthermore, use of truncated guide RNAs, the development of pairs of RNA-guided FokI nucleases fused to catalytically inactive Cas9, and novel variant Cas9 proteins are new modifications to the CRISPR/Cas9 system that also have been demonstrated to improve target specificity.[65-68]
Even if off-target effects occur with low frequency when using genome-editing tools, sound study design and use of multiple pairs of wild-type and mutated clones will still be needed to address the possibility of off-target effects that may confound an experiment if only one mutated clone with off-target indels were compared to one wild-type clone. In addition, for eventual therapeutic translation, the rapid development of improvements to genome-editing strategies mentioned above will also enhance specificity that will be needed for safe application of these tools for gene therapy.
Emerging and Future Directions
With the genetic basis of human diseases as a major emphasis in the study of disease pathogenesis as well as in the development of precision medicine strategies, use of genome editing to generate human cell-based disease models has become an increasingly popular approach used in the laboratory setting. Genome-edited cells provide a powerful approach to interrogate cellular mechanism and phenotypes for both Mendelian and complex human diseases in an appropriate genetic background that is not well recapitulated in traditional mouse models. However, cellular models are limited to the study of phenotypes that can be evaluated in the dish, and they do not offer the means to assess complex physiological conditions. To address this issue, investigators have been developing chimeric animal models that house human cells within the in vivo setting of an animal. Although still very inefficient, this has been most successfully accomplished in humanized mouse models of liver disease,[69] in which human iPSC-derived hepatocytes colonize mouse livers via intrasplenic injection and demonstrate successful functional integration into mouse liver parenchyma. Using this approach, investigators can potentially study human genome-edited iPSC-derived differentiated cells in model organisms. Indeed, methods for introducing human pluripotent stem cells into other organ systems are also being developed. For example, one group evaluated the embryonic integration of human iPSC-derived neural crest cells in murine embryonic neural crest development,[70] and another group engrafted human pluripotent stem cell-derived enteric nervous system precursor cells into the post-natal murine colon to treat mice genetically engineered to have megacolon of Hirschsprung's disease.[71] With these emerging methods, in the not-too-distant future improved disease modeling will be able to integrate the disease-appropriate genetic background of iPSCs with the in vivo physiology of animal models. This next step may be able to address the interspecies gap that had previously led to failures of bench-to-bedside translation.
Although chimeric animal models have not become fully established in the study of human diseases, already pre-clinical studies demonstrate that in vivo genome-editing has the potential to be used to correct Mendelian diseases. For example, three groups recently showed that in a mouse model of Duchenne muscular dystrophy, CRISPR/Cas9 delivered through adeno-associated virus (AAV) knocked out exon 23 of the dystrophin gene, restoring the functional form of the dystrophin protein and also improving muscle structure and function.[72-74] Two of these groups used the Staphylococcus aureus ortholog of Cas9 (SaCas9), as SaCas9 is smaller in size than SpCas9 and thus can be more readily packaged into AAV vectors for use in in vivo genome editing.[75] With careful application of AAV for gene therapy already demonstrating an acceptable toxicity profile,[76] clinical translation of genome editing to treat disease mutations in humans is on the horizon.
Conclusion
The rapid advancement of genome-editing technologies in the past decade has opened exciting new avenues in the study of the genetic basis of human disease and in the development of targeted therapeutic strategies that would not have been possible with traditional pharmacological agents. With careful study design, continued improvement in the engineering of genome-editing tools, and appropriate regulatory practices, genome editing will undoubtedly accelerate discoveries in basic science and clinical translation.
Acknowledgments
J.L. is supported by KL2-TR00013910.
Abbreviations
- CF
cystic fibrosis
- CAVD
calcific aortic valve disease
- CRISPR
clustered regularly interspaced short palindromic repeat
- DSB
double-stranded break
- GWAS
genome-wide association studies
- HDR
homology-directed repair
- iPSC
induced pluripotent stem cell
- NHEJ
non-homologous end joining
- RNAi
RNA interference
- RVD
repeat-variable di-residue
- SaCas9
Staphylococcus aureus Cas9
- SpCas9
Streptococcus pyogenes Cas9
- TALEN
transcription activator-like effector nucleases
- ZFN
zinc finger nuclease
Footnotes
Conflicts of Interest: None
Author Contributions: J.L. and K.M. wrote the manuscript. K.M. approved the final version.
References
- 1.Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–197. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- 2.Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, Webb TR, Zeng L, Dehghan A, Alver M, Armasu SM, Auro K, Bjonnes A, Chasman DI, Chen S, Ford I, Franceschini N, Gieger C, Grace C, Gustafsson S, Huang J, Hwang SJ, Kim YK, Kleber ME, Lau KW, Lu X, Lu Y, Lyytikäinen LP, Mihailov E, Morrison AC, Pervjakova N, Qu L, Rose LM, Salfati E, Saxena R, Scholz M, Smith AV, Tikkanen E, Uitterlinden A, Yang X, Zhang W, Zhao W, de Andrade M, de Vries PS, van Zuydam NR, Anand SS, Bertram L, Beutner F, Dedoussis G, Frossard P, Gauguier D, Goodall AH, Gottesman O, Haber M, Han BG, Huang J, Jalilzadeh S, Kessler T, König IR, Lannfelt L, Lieb W, Lind L, Lindgren CM, Lokki ML, Magnusson PK, Mallick NH, Mehra N, Meitinger T, Memon FUR, Morris AP, Nieminen MS, Pedersen NL, Peters A, Rallidis LS, Rasheed A, Samuel M, Shah SH, Sinisalo J, Stirrups KE, Trompet S, Wang L, Zaman KS, Ardissino D, Boerwinkle E, Borecki IB, Bottinger EP, Buring JE, Chambers JC, Collins R, Cupples LA, Danesh J, Demuth I, Elosua R, Epstein SE, Esko T, Feitosa MF, Franco OH, Franzosi MG, Granger CB, Gu D, Gudnason V, Hall AS, Hamsten A, Harris TB, Hazen SL, Hengstenberg C, Hofman A, Ingelsson E, Iribarren C, Jukema JW, Karhunen PJ, Kim BJ, Kooner JS, Kullo IJ, Lehtimäki T, Loos RJF, Melander O, Metspalu A, März W, Palmer CN, Perola M, Quertermous T, Rader DJ, Ridker PM, Ripatti S, Roberts R, Salomaa V, Sanghera DK, Schwartz SM, Seedorf U, Stewart AF, Stott DJ, Thiery J, Zalloua PA, O'Donnell CJ, Reilly MP, Assimes TL, Thompson JR, Erdmann J, Clarke R, Watkins H, Kathiresan S, McPherson R, Deloukas P, Schunkert H, Samani NJ Farrall MCARDIoGRAMplusC4D Consortium. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium, South Asian Type 2 Diabetes (SAT2D) Consortium, Mexican American Type 2 Diabetes (MAT2D) Consortium, Type 2 Diabetes Genetic Exploration by Nex-generation sequencing in muylti-Ethnic Samples (T2D-GENES) Consortium. Mahajan A, Go MJ, Zhang W, Below JE, Gaulton KJ, Ferreira T, Horikoshi M, Johnson AD, Ng MCY, Prokopenko I, Saleheen D, Wang X, Zeggini E, Abecasis GR, Adair LS, Almgren P, Atalay M, Aung T, Baldassarre D, Balkau B, Bao Y, Barnett AH, Barroso I, Basit A, Been LF, Beilby J, Bell GI, Benediktsson R, Bergman RN, Boehm BO, Boerwinkle E, Bonnycastle LL, Burtt N, Cai Q, Campbell H, Carey J, Cauchi S, Caulfield M, Chan JCN, Chang LC, Chang TJ, Chang YC, Charpentier G, Chen CH, Chen H, Chen YT, Chia KS, Chidambaram M, Chines PS, Cho NH, Cho YM, Chuang LM, Collins FS, Cornelis MC, Couper DJ, Crenshaw AT, van Dam RM, Danesh J, Das D, de Faire U, Dedoussis G, Deloukas P, Dimas AS, Dina C, Doney AS, Donnelly PJ, Dorkhan M, van Duijn C, Dupuis J, Edkins S, Elliott P, Emilsson V, Erbel R, Eriksson JG, Escobedo J, Esko T, Eury E, Florez JC, Fontanillas P, Forouhi NG, Forsen T, Fox C, Fraser RM, Frayling TM, Froguel P, Frossard P, Gao Y, Gertow K, Gieger C, Gigante B, Grallert H, Grant GB, Grrop LC, Groves CJ, Grundberg E, Guiducci C, Hamsten A, Han BG, Hara K, Hassanali N, Hattersley AT, Hayward C, Hedman AK, Herder C, Hofman A, Holmen OL, Hovingh K, Hreidarsson AB, Hu C, Hu FB, Hui J, Humphries SE, Hunt SE, Hunter DJ, Hveem K, Hydrie ZI, Ikegami H, Illig T, Ingelsson E, Islam M, Isomaa B, Jackson AU, Jafar T, James A, Jia W, Jöckel KH, Jonsson A, Jowett JBM, Kadowaki T, Kang HM, Kanoni S, Kao WHL, Kathiresan S, Kato N, Katulanda P, Keinanen-Kiukaanniemi KM, Kelly AM, Khan H, Khaw KT, Khor CC, Kim HL, Kim S, Kim YJ, Kinnunen L, Klopp N, Kong A, Korpi-Hyövälti E, Kowlessur S, Kraft P, Kravic J, Kristensen MM, Krithika S, Kumar A, Kumate J, Kuusisto J, Kwak SH, Laakso M, Lagou V, Lakka TA, Langenberg C, Langford C, Lawrence R, Leander K, Lee JM, Lee NR, Li M, Li X, Li Y, Liang J, Liju S, Lim WY, Lind L, Lindgren CM, Lindholm E, Liu CT, Liu JJ, Lobbens S, Long J, Loos RJF, Lu W, Luan J, Lyssenko V, Ma RCW, Maeda S, Mägi R, Männistö S, Matthews DR, Meigs JB, Melander O, Metspalu A, Meyer J, Mirza G, Mihailov E, Moebus S, Mohan V, Mohlke KL, Morris AD, Mühleisen TW, Müller-Nurasyid M, Musk B, Nakamura J, Nakashima E, Navarro P, Ng PK, Nica AC, Nilsson PM, Njølstad I, Nöthen MM, Ohnaka K, Ong TH, Owen KR, Palmer CNA, Pankow JS, Park KS, Parkin M, Pechlivanis S, Pedersen NL, Peltonen L, Perry JRB, Peters A, Pinidiyapathirage JM, Platou CG, Potter S, Price JF, Qi L, Radha V, Rallidis L, Rasheed A, Rathman W, Rauramaa R, Raychaudhuri S, Rayner NW, Rees SD, Rehnberg E, Ripatti S, Robertson N, Roden M, Rossin EJ, Rudan I, Rybin D, Saaristo TE, Salomaa V, Saltevo J, Samuel M, Sanghera DK, Saramies J, Scott J, Scott LJ, Scott RA, Segrè AV, Sehmi J, Sennblad B, Shah N, Shah S, Shera AS, Shu XO, Shuldiner AR, Sigurđsson G, Sijbrands E, Silveira A, Sim X, Sivapalaratnam S, Small KS, So WY, Stancáková A, Stefansson K, Steinbach G, Steinthorsdottir V, Stirrups K, Strawbridge RJ, Stringham HM, Sun Q, Suo C, Syvänen AC, Takayanagi R, Takeuchi F, Tay WT, Teslovich TM, Thorand B, Thorleifsson G, Thorsteinsdottir U, Tikkanen E, Trakalo J, Tremoli E, Trip MD, Tsai FJ, Tuomi T, Tuomilehto J, Uitterlinden AG, Valladares-Salgado A, Vedantam S, Veglia F, Voight BF, Wang C, Wareham NJ, Wennauer R, Wickremasinghe AR, Wilsgaard T, Wilson JF, Wiltshire S, Winckler W, Wong TY, Wood AR, Wu JY, Wu Y, Yamamoto K, Yamauchi T, Yang M, Yengo L, Yokota M, Young R, Zabaneh D, Zhang F, Zhang R, Zheng W, Zimmet PZ, Altshuler D, Bowden DW, Cho YS, Cox NJ, Cruz M, Hanis CL, Kooner J, Lee JY, Seielstad M, Teo YY, Boehnke M, Parra EJ, Chambers JC, Tai ES, McCarthy MI, Morris AP. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46:234–244. doi: 10.1038/ng.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, Bragg-Gresham JL, Chang HY, Demirkan A, Hertog Den HM, Donnelly LA, Ehret GB, Esko T, Feitosa MF, Ferreira T, Fischer K, Fontanillas P, Fraser RM, Freitag DF, Gurdasani D, Heikkilä K, Hyppönen E, Isaacs A, Jackson AU, Johansson A, Johnson T, Kaakinen M, Kettunen J, Kleber ME, Li X, Luan J, Lyytikäinen LP, Magnusson PKE, Mangino M, Mihailov E, Montasser ME, Müller-Nurasyid M, Nolte IM, O'Connell JR, Palmer CD, Perola M, Petersen AK, Sanna S, Saxena R, Service SK, Shah S, Shungin D, Sidore C, Song C, Strawbridge RJ, Surakka I, Tanaka T, Teslovich TM, Thorleifsson G, Van den Herik EG, Voight BF, Volcik KA, Waite LL, Wong A, Wu Y, Zhang W, Absher D, Asiki G, Barroso I, Been LF, Bolton JL, Bonnycastle LL, Brambilla P, Burnett MS, Cesana G, Dimitriou M, Doney ASF, Döring A, Elliott P, Epstein SE, Eyjolfsson GI, Gigante B, Goodarzi MO, Grallert H, Gravito ML, Groves CJ, Hallmans G, Hartikainen AL, Hayward C, Hernandez D, Hicks AA, Holm H, Hung YJ, Illig T, Jones MR, Kaleebu P, Kastelein JJP, Khaw KT, Kim E, Klopp N, Komulainen P, Kumari M, Langenberg C, Lehtimäki T, Lin SY, Lindström J, Loos RJF, Mach F, McArdle WL, Meisinger C, Mitchell BD, Müller G, Nagaraja R, Narisu N, Nieminen TVM, Nsubuga RN, Olafsson I, Ong KK, Palotie A, Papamarkou T, Pomilla C, Pouta A, Rader DJ, Reilly MP, Ridker PM, Rivadeneira F, Rudan I, Ruokonen A, Samani N, Scharnagl H, Seeley J, Silander K, Stancáková A, Stirrups K, Swift AJ, Tiret L, Uitterlinden AG, van Pelt LJ, Vedantam S, Wainwright N, Wijmenga C, Wild SH, Willemsen G, Wilsgaard T, Wilson JF, Young EH, Zhao JH, Adair LS, Arveiler D, Assimes TL, Bandinelli S, Bennett F, Bochud M, Boehm BO, Boomsma DI, Borecki IB, Bornstein SR, Bovet P, Burnier M, Campbell H, Chakravarti A, Chambers JC, Chen YDI, Collins FS, Cooper RS, Danesh J, Dedoussis G, de Faire U, Feranil AB, Ferrières J, Ferrucci L, Freimer NB, Gieger C, Groop LC, Gudnason V, Gyllensten U, Hamsten A, Harris TB, Hingorani A, Hirschhorn JN, Hofman A, Hovingh GK, Hsiung CA, Humphries SE, Hunt SC, Hveem K, Iribarren C, Järvelin MR, Jula A, Kähönen M, Kaprio J, Kesäniemi A, Kivimaki M, Kooner JS, Koudstaal PJ, Krauss RM, Kuh D, Kuusisto J, Kyvik KO, Laakso M, Lakka TA, Lind L, Lindgren CM, Martin NG, März W, McCarthy MI, McKenzie CA, Meneton P, Metspalu A, Moilanen L, Morris AD, Munroe PB, Njølstad I, Pedersen NL, Power C, Pramstaller PP, Price JF, Psaty BM, Quertermous T, Rauramaa R, Saleheen D, Salomaa V, Sanghera DK, Saramies J, Schwarz PEH, Sheu WHH, Shuldiner AR, Siegbahn A, Spector TD, Stefansson K, Strachan DP, Tayo BO, Tremoli E, Tuomilehto J, Uusitupa M, van Duijn CM, Vollenweider P, Wallentin L, Wareham NJ, Whitfield JB, Wolffenbuttel BHR, Altshuler D, Ordovas JM, Boerwinkle E, Palmer CNA, Thorsteinsdottir U, Chasman DI, Rotter JI, Franks PW, Ripatti S, Cupples LA, Sandhu MS, Rich SS, Boehnke M, Deloukas P, Mohlke KL, Ingelsson E, Abecasis GR, Daly MJ, Neale BM, Kathiresan S. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–1352. doi: 10.1038/ng.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, Pirruccello JP, Muchmore B, Prokunina-Olsson L, Hall JL, Schadt EE, Morales CR, Lund-Katz S, Phillips MC, Wong J, Cantley W, Racie T, Ejebe KG, Orho-Melander M, Melander O, Koteliansky V, Fitzgerald K, Krauss RM, Cowan CA, Kathiresan S, Rader DJ. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bauer RC, Tohyama J, Cui J, Cheng L, Yang J, Zhang X, Ou K, Paschos GK, Zheng XL, Parmacek MS, Rader DJ, Reilly MP. Knockout of Adamts7, a Novel Coronary Artery Disease Locus in Humans, Reduces Atherosclerosis in MiceCLINICAL PERSPECTIVE. Circulation. 2015;131:1202–1213. doi: 10.1161/CIRCULATIONAHA.114.012669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauer RC, Sasaki M, Cohen DM, Cui J, Smith MA, Yenilmez BO, Steger DJ, Rader DJ. Tribbles-1 regulates hepatic lipogenesis through posttranscriptional regulation of C/EBPα. J Clin Invest. 2015;125:3809–3818. doi: 10.1172/JCI77095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Plenge RM, Scolnick EM, Altshuler D. Validating therapeutic targets through human genetics. Nat Rev Drug Discov. 2013;12:581–594. doi: 10.1038/nrd4051. [DOI] [PubMed] [Google Scholar]
- 9.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–795. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashley EA. The precision medicine initiative: a new national effort. JAMA. 2015;313:2119–2120. doi: 10.1001/jama.2015.3595. [DOI] [PubMed] [Google Scholar]
- 11.Cox DBT, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nature Medicine. 2015;21:121–131. doi: 10.1038/nm.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- 13.Fedorov Y, Anderson EM, Birmingham A, Reynolds A, Karpilow J, Robinson K, Leake D, Marshall WS, Khvorova A. Off-target effects by siRNA can induce toxic phenotype. RNA. 2006;12:1188–1196. doi: 10.1261/rna.28106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seki T, Yuasa S, Oda M, Egashira T, Yae K, Kusumoto D, Nakata H, Tohyama S, Hashimoto H, Kodaira M, Okada Y, Seimiya H, Fusaki N, Hasegawa M, Fukuda K. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell. 2010;7:11–14. doi: 10.1016/j.stem.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Staerk J, Dawlaty MM, Gao Q, Maetzel D, Hanna J, Sommer CA, Mostoslavsky G, Jaenisch R. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell. 2010;7:20–24. doi: 10.1016/j.stem.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- 17.Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reinhardt P, Schmid B, Burbulla LF, Schöndorf DC, Wagner L, Glatza M, Höing S, Hargus G, Heck SA, Dhingra A, Wu G, Müller S, Brockmann K, Kluba T, Maisel M, Krüger R, Berg D, Tsytsyura Y, Thiel CS, Psathaki OE, Klingauf J, Kuhlmann T, Klewin M, Müller H, Gasser T, Schöler HR, Sterneckert J. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell. 2013;12:354–367. doi: 10.1016/j.stem.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 19.Zhang H, Xue C, Shah R, Bermingham K, Hinkle CC, Li W, Rodrigues A, Tabita-Martinez J, Millar JS, Cuchel M, Pashos EE, Liu Y, Yan R, Yang W, Gosai SJ, VanDorn D, Chou ST, Gregory BD, Morrisey EE, Li M, Rader DJ, Reilly MP. Functional analysis and transcriptomic profiling of iPSC-derived macrophages and their application in modeling Mendelian disease. Circulation Research. 2015;117:17–28. doi: 10.1161/CIRCRESAHA.117.305860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Polo JM, Liu S, Figueroa ME, Kulalert W, Eminli S, Tan KY, Apostolou E, Stadtfeld M, Li Y, Shioda T, Natesan S, Wagers AJ, Melnick A, Evans T, Hochedlinger K. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Vol. 28. Nature Publishing Group; 2010. pp. 848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O'Malley R, Castanon R, Klugman S, Downes M, Yu R, Stewart R, Ren B, Thomson JA, Evans RM, Ecker JR. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishino K, Toyoda M, Yamazaki-Inoue M, Fukawatase Y, Chikazawa E, Sakaguchi H, Akutsu H, Umezawa A. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS Genet. 2011;7:e1002085. doi: 10.1371/journal.pgen.1002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nazor KL, Altun G, Lynch C, Tran H, Harness JV, Slavin I, Garitaonandia I, Muller FJ, Wang YC, Boscolo FS, Fakunle E, Dumevska B, Lee S, Park HS, Olee T, D'Lima DD, Semechkin R, Parast MM, Galat V, Laslett AL, Schmidt U, Keirstead HS, Loring JF, Laurent LC. Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell. 2012;10:620–634. doi: 10.1016/j.stem.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11:636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 25.Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA, Ando D, Urnov FD, Galli C, Gregory PD, Holmes MC, Naldini L. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 26.Soldner F, Laganière J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, Khurana V, Golbe LI, Myers RH, Lindquist S, Zhang L, Guschin D, Fong LK, Vu BJ, Meng X, Urnov FD, Rebar EJ, Gregory PD, Zhang HS, Jaenisch R. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146:318–331. doi: 10.1016/j.cell.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramirez CL, Foley JE, Wright DA, Müller-Lerch F, Rahman SH, Cornu TI, Winfrey RJ, Sander JD, Fu F, Townsend JA, Cathomen T, Voytas DF, Joung JK. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Meth. 2008;5:374–375. doi: 10.1038/nmeth0508-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maeder ML, Thibodeau-Beganny S, Sander JD, Voytas DF, Joung JK. Oligomerized pool engineering (OPEN): an “open-source” protocol for making customized zinc-finger arrays. Nat Protoc. 2009;4:1471–1501. doi: 10.1038/nprot.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, Unger-Wallace E, Sander JD, Müller-Lerch F, Fu F, Pearlberg J, Göbel C, Dassie JP, Pruett-Miller SM, Porteus MH, Sgroi DC, Iafrate AJ, Dobbs D, McCray PB, Cathomen T, Voytas DF, Joung JK. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell. 2008;31:294–301. doi: 10.1016/j.molcel.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sander JD, Dahlborg EJ, Goodwin MJ, Cade L, Zhang F, Cifuentes D, Curtin SJ, Blackburn JS, Thibodeau-Beganny S, Qi Y, Pierick CJ, Hoffman E, Maeder ML, Khayter C, Reyon D, Dobbs D, Langenau DM, Stupar RM, Giraldez AJ, Voytas DF, Peterson RT, Yeh JRJ, Joung JK. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA) Nat Meth. 2011;8:67–69. doi: 10.1038/nmeth.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhakta MS, Henry IM, Ousterout DG, Das KT, Lockwood SH, Meckler JF, Wallen MC, Zykovich A, Yu Y, Leo H, Xu L, Gersbach CA, Segal DJ. Highly active zinc-finger nucleases by extended modular assembly. Genome Research. 2013;23:530–538. doi: 10.1101/gr.143693.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 33.Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 34.Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Research. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK. FLASH assembly of TALENs for high-throughput genome editing. Vol. 30. Nature Publishing Group; 2012. pp. 460–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bultmann S, Morbitzer R, Schmidt CS, Thanisch K, Spada F, Elsaesser J, Lahaye T, Leonhardt H. Targeted transcriptional activation of silent oct4 pluripotency gene by combining designer TALEs and inhibition of epigenetic modifiers. Nucleic Acids Research. 2012;40:5368–5377. doi: 10.1093/nar/gks199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim Y, Kweon J, Kim A, Chon JK, Yoo JY, Kim HJ, Kim S, Lee C, Jeong E, Chung E, Kim D, Lee MS, Go EM, Song HJ, Kim H, Cho N, Bang D, Kim S, Kim JS. A library of TAL effector nucleases spanning the human genome. Vol. 31. Nature Publishing Group; 2013. pp. 251–258. [DOI] [PubMed] [Google Scholar]
- 38.Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 39.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 42.Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, Goodwin MJ, Aryee MJ, Joung JK. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Vol. 32. Nature Publishing Group; 2014. pp. 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller JC, Zhang L, Xia DF, Campo JJ, Ankoudinova IV, Guschin DY, Babiarz JE, Meng X, Hinkley SJ, Lam SC, Paschon DE, Vincent AI, Dulay GP, Barlow KA, Shivak DA, Leung E, Kim JD, Amora R, Urnov FD, Gregory PD, Rebar EJ. Improved specificity of TALE-based genome editing using an expanded RVD repertoire. Nat Meth. 2015;12:465–471. doi: 10.1038/nmeth.3330. [DOI] [PubMed] [Google Scholar]
- 44.Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales APW, Li Z, Peterson RT, Yeh JRJ, Aryee MJ, Joung JK. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Ge X, Yang F, Zhang L, Zheng J, Tan X, Jin ZB, Qu J, Gu F. Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci Rep. 2014;4:5405. doi: 10.1038/srep05405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao F, Shen XZ, Jiang F, Wu Y, Han C. DNA-guided genome editing using the Natronobacterium gregoryi Argonaute. Nature Publishing Group; 2016. [DOI] [PubMed] [Google Scholar]
- 48.Grizot S, Smith J, Daboussi F, Prieto J, Redondo P, Merino N, Villate M, Thomas S, Lemaire L, Montoya G, Blanco FJ, Pâques F, Duchateau P. Efficient targeting of a SCID gene by an engineered single-chain homing endonuclease. Nucleic Acids Research. 2009;37:5405–5419. doi: 10.1093/nar/gkp548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khan IF, Hirata RK, Wang PR, Li Y, Kho J, Nelson A, Huo Y, Zavaljevski M, Ware C, Russell DW. Engineering of human pluripotent stem cells by AAV-mediated gene targeting. Mol Ther. 2010;18:1192–1199. doi: 10.1038/mt.2010.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suzuki K, Mitsui K, Aizawa E, Hasegawa K, Kawase E, Yamagishi T, Shimizu Y, Suemori H, Nakatsuji N, Mitani K. Highly efficient transient gene expression and gene targeting in primate embryonic stem cells with helper-dependent adenoviral vectors. Proceedings of the National Academy of Sciences. 2008;105:13781–13786. doi: 10.1073/pnas.0806976105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu GH, Suzuki K, Qu J, Sancho-Martinez I, Yi F, Li M, Kumar S, Nivet E, Kim J, Soligalla RD, Dubova I, Goebl A, Plongthongkum N, Fung HL, Zhang K, Loring JF, Laurent LC, Izpisua Belmonte JC. Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell. 2011;8:688–694. doi: 10.1016/j.stem.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Theodoris CV, Li M, White MP, Liu L, He D, Pollard KS, Bruneau BG, Srivastava D. Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of NOTCH1 haploinsufficiency. Cell. 2015;160:1072–1086. doi: 10.1016/j.cell.2015.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.An MC, Zhang N, Scott G, Montoro D, Wittkop T, Mooney S, Melov S, Ellerby LM. Genetic correction of Huntington's disease phenotypes in induced pluripotent stem cells. Cell Stem Cell. 2012;11:253–263. doi: 10.1016/j.stem.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu GH, Qu J, Suzuki K, Nivet E, Li M, Montserrat N, Yi F, Xu X, Ruiz S, Zhang W, Wagner U, Kim A, Ren B, Li Y, Goebl A, Kim J, Soligalla RD, Dubova I, Thompson J, Yates J, Esteban CR, Sancho-Martinez I, Izpisua Belmonte JC. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature. 2012;491:603–607. doi: 10.1038/nature11557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Firth AL, Menon T, Parker GS, Qualls SJ, Lewis BM, Ke E, Dargitz CT, Wright R, Khanna A, Gage FH, Verma IM. Functional Gene Correction for Cystic Fibrosis in Lung Epithelial Cells Generated from Patient iPSCs. CellReports. 2015;12:1385–1390. doi: 10.1016/j.celrep.2015.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Meth. 2011;8:765–770. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, Nowrouzi A, Bartholomae CC, Wang J, Friedman G, Holmes MC, Gregory PD, Glimm H, Schmidt M, Naldini L, Kalle von C. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol. 2011;29:816–823. doi: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- 58.Guilinger JP, Pattanayak V, Reyon D, Tsai SQ, Sander JD, Joung JK, Liu DR. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat Meth. 2014;11:429–435. doi: 10.1038/nmeth.2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Veres A, Gosis BS, Ding Q, Collins R, Ragavendran A, Brand H, Erdin S, Cowan CA, Talkowski ME, Musunuru K. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 2014;15:27–30. doi: 10.1016/j.stem.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang L, Grishin D, Wang G, Aach J, Zhang CZ, Chari R, Homsy J, Cai X, Zhao Y, Fan JB, Seidman C, Seidman J, Pu W, Church G. Targeted and genome-wide sequencing reveal single nucleotide variations impacting specificity of Cas9 in human stem cells. Nat Comms. 2014;5:5507. doi: 10.1038/ncomms6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tan EP, Li Y, Velasco-Herrera MDC, Yusa K, Bradley A. Off-target assessment of CRISPR-Cas9 guiding RNAs in human iPS and mouse ES cells. Genesis. 2015;53:225–236. doi: 10.1002/dvg.22835. [DOI] [PubMed] [Google Scholar]
- 62.Lee CM, Cradick TJ, Fine EJ, Bao G. Nuclease Target Site Selection for Maximizing On-target Activity and Minimizing Off-target Effects in Genome Editing. Mol Ther. 2016 doi: 10.1038/mt.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, Church GM. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Vol. 31. Nature Publishing Group; 2013. pp. 833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Vol. 32. Nature Publishing Group; 2014. pp. 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Vol. 32. Nature Publishing Group; 2014. pp. 577–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu PQ, Paschon DE, Miranda E, Ordóñez A, Hannan NRF, Rouhani FJ, Darche S, Alexander G, Marciniak SJ, Fusaki N, Hasegawa M, Holmes MC, Di Santo JP, Lomas DA, Bradley A, Vallier L. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478:391–394. doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cohen MA, Wert KJ, Goldmann J, Markoulaki S, Buganim Y, Fu D, Jaenisch R. Human neural crest cells contribute to coat pigmentation in interspecies chimeras after in utero injection into mouse embryos. Proceedings of the National Academy of Sciences. 2016;113:1570–1575. doi: 10.1073/pnas.1525518113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fattahi F, Steinbeck JA, Kriks S, Tchieu J, Zimmer B, Kishinevsky S, Zeltner N, Mica Y, El-Nachef W, Zhao H, de Stanchina E, Gershon MD, Grikscheit TC, Chen S, Studer L. Deriving human ENS lineages for cell therapy and drug discovery in Hirschsprung disease. Nature. 2016 doi: 10.1038/nature16951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM, Madhavan S, Pan X, Ran FA, Yan WX, Asokan A, Zhang F, Duan D, Gersbach CA. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA, Cong L, Zhang F, Vandenberghe LH, Church GM, Wagers AJ. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, Olson EN. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nathwani AC, Reiss UM, Tuddenham EGD, Rosales C, Chowdary P, McIntosh J, Peruta Della M, Lheriteau E, Patel N, Raj D, Riddell A, Pie J, Rangarajan S, Bevan D, Recht M, Shen YM, Halka KG, Basner-Tschakarjan E, Mingozzi F, High KA, Allay J, Kay MA, Ng CYC, Zhou J, Cancio M, Morton CL, Gray JT, Srivastava D, Nienhuis AW, Davidoff AM. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]