

Abstract
The surface receptor FcγRIIIA (CD16a) is encoded by the FCGR3A gene and is acquired by human natural killer (NK) cells during maturation. NK cells bind the Fc portion of IgG via CD16a, and execute antibody-dependent cellular cytotoxicity, which is critical for the effectiveness of several anti-tumor monoclonal antibody therapies. The role of epigenetic regulatory mechanisms controlling transcriptional and post-transcriptional CD16 expression in NK cells is unknown. Here we compared specific patterns of DNA methylation and expression of FCGR3A to FCGR3B, which differ in cell type-specific expression despite displaying nearly identical genomic sequences. We identified a sequence within the FCGR3A promoter that selectively exhibits reduced methylation in CD16a+ NK cells versus CD16a- NK cells and neutrophils. This region contained the transcriptional start site of the most highly expressed CD16a isoform in NK cells. Luciferase assays revealed remarkable cell-type specificity and methylation-dependent activity of FCGR3A- versus FCGR3B-derived sequences. Genomic differences between FCGR3A and FCGR3B are enriched at CpG dinucleotides and mutation of variant CpGs reversed cell-type specificity. We further identified miR-218 as a post-transcriptional negative regulator of CD16a in NK cells. Forced over-expression of miR-218 in NK cells knocked down CD16a mRNA and protein expression. Moreover, miR-218 was highly expressed in CD16a- NK cells compared to CD16a+ NK cells. Taken together, we propose a system of FCGR3A regulation in human NK cells in which CpG dinucleotide sequences and concurrent DNA methylation confer developmental and cell type-specific transcriptional regulation, while miR-218 provides an additional layer of post-transcriptional regulation during the maturation process.
Introduction
The low affinity Fc gamma receptor type IIIA (FcγRIIIA or CD16a) is an activating Fc receptor expressed by natural killer (NK) cells, macrophages, and monocytes. It is coded by the gene, FCGR3A. Surface expression of CD16a is required for antibody-dependent cell-mediated cytotoxicity (ADCC). The efficacy of several clinically effective anti-tumor antibodies involves the engagement of CD16a, which is required for ADCC (1–5). CD16a expression is acquired during NK cell maturation, which can be described in 5 sequential stages involving NK cell developmental intermediates (NKDIs) (6). Stages 1–3 are considered progenitor or immature stages and do not express CD16a. Stage 4 and 5 NK cells are considered mature. Expression of CD16a divides stage 4 (early mature) and stage 5 (late mature) NK cells (7–9). The NK cell population in secondary lymphoid tissues (SLTs) is primarily composed of CD16a- NKDIs including stage 4 NK cells, which are characterized by the CD56bright/CD16a- surface phenotype. SLTs also contain a minor population that is CD56+/CD16a+ and corresponds to a stage 5 NK cell phenotype. In the peripheral blood, this pattern is reversed whereby CD56bright/CD16a- stage 4 NK cells are the minor population while CD56dim/CD16a+ stage 5 NK cells are the predominant population. This suggest that CD56bright/CD16a- NKDIs develop in SLTs before full maturation and traffic into the periphery as stage 5 NK cells (7). Though the function of this receptor has been well characterized (3, 10–18), and it’s post-translational regulation has been described (19), the mechanism(s) of FCGR3A regulation permitting the developmental acquisition of CD16a is not understood.
The lack of knowledge regarding FCGR3A regulation during human NK cell development is due, in part, to inherent difficulties in studying this gene. Cell lines expressing CD16a are notably lacking (20). The closest murine genes, Fcgr3 and Fcgr4, have limited homology (<50%) to human FCGR3A. Furthermore, in humans, a recent genomic segmental duplication event generated two genes, FCGR3A and FCGR3B, that retain a very high degree of sequence homology (>95%) across the gene locus (21). Antibodies that recognize CD16 generally recognize both gene products. Discrimination of these two homologs is highly relevant to immunological studies as FCGR3B cannot mediate ADCC and instead functions as a sink for immune complexes (21, 22).
Despite their nearly identical genomic sequences, FCGR3 homologs are selectively expressed by specific cell types; FCGR3A is expressed by NK cells, monocytes, and macrophages while FCGR3B is expressed by neutrophils (21). Previous work has shown that each FCGR3 homolog uses two distinct alternative promoters within their respective 5′ regions to transcribe at least two unique transcripts (23, 24). In FCGR3A, both promoters confer selective activity in the NK cell line YT, while the FCGR3B promoter alternatively operates myeloid cells, indicating that lineage-specific factors are capable of selectively recognizing sequence differences between FCGR3 homologs (23, 24). However, the mechanism that endows this exquisite specificity and how it selectively develops in separate primary cell lineages it is not understood.
In order to gain insight into mechanisms that might regulate FCGR3A, we focused our studies on NK cells. Epigenetic gene regulation is capable of tightly controlling gene expression in a developmental and tissue-specific manner (25, 26), and may help explain the tissue specificity despite the very high degree of homology between FCGR3A/B. NKDIs do not quickly acquire CD16a surface expression, often requiring a week or more of expansion in vitro before significant CD16a expression is detectable by flow cytometry(27, 28). As the cells acquire CD16a expression, some level of post-transcriptional fine tuning may also be required. To address this possibility, we further sought to identify microRNA (miRNA) regulators of FCGR3A.
Here we identify two types of regulation of CD16a in human NK cells, DNA methylation of the FCGR3A promoter and miR-218 targeting of FCGR3A mRNA. These mechanisms suggest that CD16a expression in repressed in stage 4 NK cells primarily by DNA methylation silencing with concurrent high miR-218 expression. The time required to transition from stage 4 to stage 5 may be necessary to sufficiently modify the FCGR3A promoter methylation patterns and downregulate miR-218, culminating in robust CD16a expression in stage 5 NK cells.
Material and Methods
Isolation of primary human cells from peripheral blood
All human cell work was performed with approval of the Ohio State University Institutional Review Board. Human NK cells were isolated from peripheral blood leukopacks of healthy individuals (American Red Cross) by negative selection with MACSxpress NK Cell Isolation Kit, human (Miltenyi). Enriched cells were collected and labeled for FACS sorting. For DNA isolation of CD16a− and CD16a+ NK cells, we gated on lymphocytes followed by CD3−CD56+ gating and then sorted for either CD56brightCD16a− or CD56dimCD16a+ populations, respectively. NK cells were sorted to >95% purity. Human neutrophils were enriched with CD66abce magnetic beads by positive selection (Miltenyi). Enriched cells were labeled for FACS with CD15 and CD16 antibodies. For DNA isolation, we gated on the CD15+CD16+ population. Cells were sorted to >97% purity.
Antibodies and flow cytometric analysis
The following antibodies were used to stain human peripheral blood cells: CD3 (SK7, BD Biosciences), CD14 (TÜK4, Miltenyi), CD15 (VIMC6, Miltenyi), CD16 (VEP13, Miltenyi), CD16 (3G8, BD Biosciences), and CD56 (N901, Beckman Coulter). Flow cytometry data were analyzed with FlowJo v7.6.1 (Tree Star).
Cell culture
YT (ATCC), K562 (ATCC) and Jurkat (DSMZ, Germany) cells were cultivated in RPMI1640/10% FBS (Gibco) and supplemented with antibiotic-antimycotic (Thermo Fisher Scientific). NKL cells were cultivated in RPMI1640/10% FBS (Gibco) and supplemented with antibiotic-antimycotic (Thermo Fisher Scientific) and 150 IU/mL recombinant human IL-2 (rhIL-2) (La Roche). HEK293T cells were obtained from ATCC. HEK293T cells were cultured in DMEM/10% FBS (Gibco) and supplemented with antibiotics.
Quantitative DNA methylation analysis using MassARRAY
DNA was isolated using the Puregene Core Kit B (Qiagen). 1μL of molecular grade glycogen (Thermo Scientific) was added to each sample and DNA was allowed to precipitate overnight at −20°C followed by resuspension in water. DNA methylation analysis of the CD16 loci was carried out using the MassARRAY EpiTYPER assay (Agena Biosciences) (29). In short, genomic DNA was subjected to bisulfite treatment using the EZ DNA methylation kit (Zymo Research). Regions of interest were amplified with PCR primers specific for the FCGR3A or FCGR3B gene loci (primers listed in Supplementary Table 1) using the hg19 genome assembly from the UCSC genome browser (https://genome.ucsc.edu). PCR products were in vitro transcribed and fragmented with RNase A (Agena) to generate oligonucleotides that were subsequently analyzed via Matrix-Assisted Laser Desorption/Ionization-Time of Flight (MALDI-ToF) mass spectrometry. Ratios of unmethylated versus methylated oligo signals were utilized to calculate the percentage of DNA methylation for individual CpG dinucleotides. To examine the specificity of FCGR3A and FCGR3B assays, amplified sequences included non-CpG sequence variants that produced quantifiable differences in the mass spectra. Primers used for the final analysis were determined to be highly specific.
Luciferase reporter assay
Luciferase reporter assays were carried out as described previously (30). Sequences of the FCGR3A and FCGR3B 5′-untranslated regions were PCR amplified using primers specific for each gene (listed in Supplementary Table 1) and inserted into the CpG-free reporter vector pCpGfree-promoter lucia (Invivogen). Sequences were verified with Sanger Sequencing at the Plant-Microbe Genomics Facility (The Ohio State University). For methylation-specific assays, plasmids were methylated in vitro using M.SssI CpG methyltransferase (Thermo Fisher Scientific). Luciferase assays were carried out in HEK293T cells using TransIT-LT1 transfection reagent (Mirus Bio) according to the manufacturer’s instructions. Jurkat, YT and K562 cells were transfected with a Nucleofector device (Lonza) using Amaxa Cell Line Nucleofector Kit V with programs X-001, O-017 and T-016, respectively. Luciferase activity was assessed 48h after transfection on a DTX880 Multimode Detector (Beckman Coulter). Luciferase signals were normalized to co-transfected pGl4-CMV-firefly luciferase vector (Promega). Luciferase activity values were displayed relative to an unmodified pCpGfree-promoter lucia plasmid.
RNA isolation, miRNA expression assays, and real-time PCR
Total RNA from indicated cell types was extracted using the Total RNA Purification Plus Kit (Norgen Biotek Corp). For miRNA analysis, we used the nCounter ® miRNA Expression Assay from nanoString Technologies interrogating a panel of 800 miRNAs (submitted to Gene Expression Omnibus, https://www.ncbi.nlm.nih.gov/geo, GEO accession number GSE106469). MiRNA expression was also determined using TaqMan MicroRNA Assays (Thermo Fisher Scientific). cDNA from mRNA was generated using the Superscript VILO cDNA Synthesis Kit (Thermo Fisher Scientific) or using SuperScript III reverse transcriptase (cell lines). Real-time PCR was done using Power SYBR Green Master Mix reagent (Thermo Fisher Scientific) and a ViiA 7 Real-Time PCR System (Applied Biosystems). Relative gene expression was calculated as 2^(-Δct) method with Δct=(ct target-ct housekeeping gene). Data were individually normalized to housekeeping gene expression values of β-actin (ACTB), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), TATA box binding protein (TBP) and hypoxanthine phosphoribosyltransferase 1 (HPRT1); the average of the four normalized expression values was used per sample. Quantitative assessment of the ratio of FCGR3A and FCGR3B expression was performed using a MassARRAY iPLEX application (Agena Biosciences) designed for the accurate measurement of allele-specific expression (31). Briefly, 5 primer sets were used to amplify cDNA from purified CD16a+ NK cells and neutrophils that targeted FCGR3A and FCGR3B equally, but flanked at least one discriminatory nucleotide (primers listed in Supplementary Table 1). Oligonucleotides were annealed to the PCR products immediately 5′ to the discriminatory nucleotide followed by single-base extension. Mass spectrometry was used to determine the ratio of FCGR3A to FCGR3B sequences; the reported values represent the average of the 5 assays.
Lentiviral infection of primary human NK cells
Infections were modified from a previously published protocol (32). Briefly, low passage (5–20) HEK293T cells were transfected with pCDH CD511B-1 (System Biosciences), and two packaging plasmids containing VSV-G or gag/pol/tat/rev. pCDH expression vectors contained either no insert or mir-218-2 plus 200bp upstream and downstream. Virus media was concentrated by centrifugation (20,000xg). Human NK cells isolated by FACS sorting were cultured overnight in RPMI/5% human AB serum/antibiotic-antimycotic supplemented with 450IU/mL rhIL-2. NK cells were infected with virus in 96-well round bottom plates. NK cells were infected with a multiplicity of infection of 2–10. Plates were centrifuged for 2 hours at 1800rpm at 32°C and placed in a 37°C incubator overnight. After resting the cells virus-free media, NK cells were subjected to a second round of infection with virus overnight. The next day, virus media was removed again and replaced with RPMI/5% human AB serum/antibiotic-antimycotic supplemented with 450IU/mL rhIL-2 and cultured for 48 hours. NK cells were stained and sorted for live GFP+ cells and used for RNA expression assays.
Results
DNA methylation analysis of FCGR3A
NK cells acquire CD16a as part of their maturation process; less mature NK cells tend to be found in the secondary lymphoid tissue such as the lymph nodes and more mature NK cells in the peripheral blood. To study the acquisition of CD16a by NK cells, we sorted NK subpopulations that precede and succeed this process and obtained CD16a- and CD16a+ subpopulations at >95% purity (Figure 1A and B). We next assessed DNA methylation of the proximal promoter region of FCGR3A in these NK cell subsets. As FCGR3B displays a different tissue-specific expression pattern, yet a high degree of promoter sequence homology compared with FCGR3A, we included both genes in our analysis to identify important regulatory features. FCGR3A and FCGR3B genes are separated by approximately 81 kb on chromosome 1q23.3 and show nearly identical exon/intron architectures and promoter sequences (Figure 2A). Both genes are annotated in the Refseq database as being potentially expressed from at least two alternative transcriptional start sites (TSSs): transcript variants 1 and 3 for FCGR3A, and variants 1 and 2 for FCGR3B (with variants 3 and 2 being homologous between FCGR3A and FCGR3B, respectively). There are two promoters annotated for FCGR3A: the Proximal Promoter (here termed Pprox-A; −198 to −10 bp) and the Medial Promoter-1 (Pmed1-A; −942 to −850 bp) (23, 24). Corresponding regions in FCGR3B were termed Pprox-B and Pmed1-B. To separately interrogate the profile of DNA methylation across both genes, we employed the quantitative MassARRAY system using PCR primers that exploit sequence variations between FCGR3A and FCGR3B genes. Analysis of primary sorted CD16a+ and CD16a- NK cell subsets revealed that CD16a+ NK cells show lower methylation levels within the FCGR3A 5′ region compared to CD16a- NK cells (Figure 2B). CpGs displaying the greatest difference between subsets were focused in Pmed1-A and immediate downstream sequences. The Pprox-A region was partially unmethylated in CD16a- NK cells, whereas the region in the vicinity of Pmed1-A only showed low methylation in CD16a+ NK cells. FCGR3B was highly methylated throughout the entire 5′ region in both CD16a- and CD16a+ NK cells, consistent with selective expression of FCGR3A in the NK lineage (21). We next investigated the NK cell lines YT and NKL, which are CD16a- and CD16a+, respectively. YT cells generally showed high methylation of FCGR3A (and FCGR3B), whereas NKL cells showed lower levels of methylation in FCGR3A, including CpGs immediately proximal and within Pmed1-A (Figure 2C). These data show that pronounced lower levels of DNA methylation are found at the FCGR3A versus FCGR3B promoter in CD16a+ NK cells, and differential surface CD16a expression associates with reduced methylation of CpGs surrounding the Pmed1-A promoter.
Figure 1.
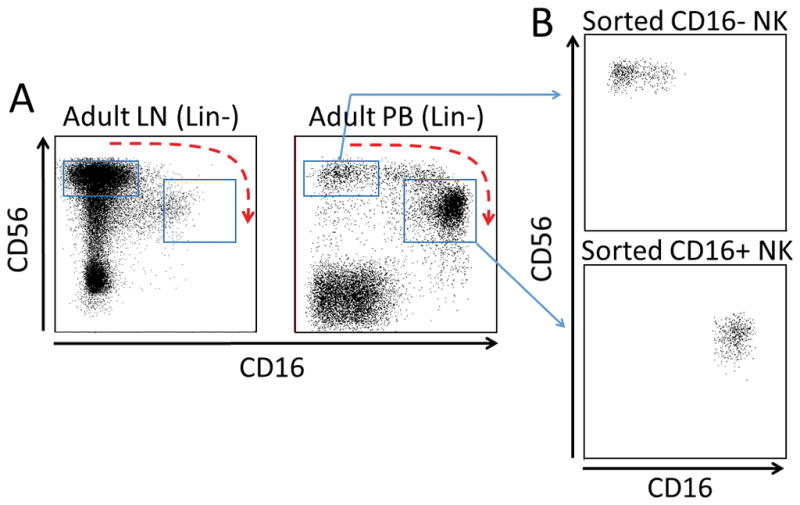
NK cells acquire CD16a during normal maturation. (A) Mononuclear cells (MNCs) from adult axillary lymph node (LN) and adult peripheral blood (PB) were analyzed by flow cytometry by gating on Lin- cells and then NK cell markers CD56 and CD16a. Blue boxes indicate CD56bright/CD16a- NK cells and CD56dim/CD16a+ NK cells. Curved blue arrow indicates the presumed pathway of normal CD16a acquisition by NK cells. (B) Straight blue arrows designate the sorted NK cell populations used in DNA methylation analyses.
Figure 2.

DNA methylation and isoform-specific expression within the promoter regions of FCGR3A and FCGR3B. A) Representation of the FCGR3 locus spanning approximately 100 kb of chromosome 1q23.3 displaying transcripts annotated in the RefSeq database (adapted from the UCSC Genome Browser). Below, promoter regions of FCGR3A and FCGR3B are enlarged displaying the various transcript variants annotated in the RefSeq database that differ in the 5′ region. MassARRAY amplicons (green bars) used are shown along with the positions of CpG dinucleotides (green markers). TSS, transcriptional start site. B) The MassARRAY EpiTYPER assay was used to interrogate the DNA methylation levels of CpGs across the promoter region of FCGR3A and FCGR3B in a gene-specific manner in sorted NK CD16a+ and CD16a- fractions. Methylation plots are aligned to enlargements of gene promoters (in B) according to the hg19 genome assembly. C, D) DNA methylation levels in NK cell lines and neutrophils, respectively. E) The expression ratio of FCGR3A to FCGR3B in CD16a+ NK cells and neutrophils as determined by the MassARRAY iPLEX assay. F) Quantitative RT-PCR analysis of 5′ transcript variants using primers specific for variant 1 or variants 3 and 2 indicating promoter usage in NK cells, neutrophils, and NK cell lines YT and NKL (both primer pairs do not distinguish FCGR3A vs. FCGR3B). D1-4 represents donor numbers 1–4. Expression values expressed relative to the average of four housekeeping (HK) genes.
To further investigate the role of DNA methylation in the selective expression of FCGR3A, we sorted primary neutrophil samples, which selectively express FCGR3B (21). We found that the CpGs in the FCGR3A promoter that selectively demonstrated low methylation in CD16a+ NK cells were found to be highly methylated in neutrophils; whereas two regions of low methylation in FCGR3B were observed (Figure 2D). These regions corresponded to Prox-B and CpGs adjacent to, but not within, Pmed1-B. Similar to CD16a- NK cells, neutrophils also displayed low methylation of the Pprox-A within FCGR3A, despite both cell types lacking expression of FCGR3A. These data show that in neutrophils the selective expression of FCGR3B is associated with a decrease of FCGR3B methylation mainly at Pprox-B, corresponding to the known promoter usage (33), and, like CD16a- NK cells, low methylation of Pprox-A is not associated with expression.
Promoter-specific DNA methylation associates with FCGR3A/B variant expression
To demonstrate the importance of methylation differences between TSSs, we next evaluated the expression of the transcript variants within FCGR3A and FCGR3B genes in primary cells. As the 5′ exon sequences of FCGR3A and FCGR3B are virtually identical, the sequence identity between genes does not allow the discrimination of variant expression from each gene promoter individually. Thus we first ensured that the relative expression of FCGR3A versus FCGR3B in our samples matched reported cell type-specific expression patterns. For this we measured the expression FCGR3A relative to FCGR3B using a highly quantitative and accurate MassARRAY method for determining transcript expression ratios (31). We confirmed that CD16a+ NK cells are virtually only expressing FCGR3A and neutrophils are expressing only FCGR3B (Figure 2E). Next, using qRT-PCR primers that are specific for variant 1 (FCGR3A and FCGR3B) or variants 3 (FCGR3A) and 2 (FCGR3B), we found that CD16a+ NK cells are strongly expressing variant 3 (and somewhat also variant 1) and, inversely, neutrophils are strongly expressing variant 1 and a small amount of variant 2 (Figure 2F). Taken together, these data show that primary CD16a+ NK cells principally employ the Pmed1-A promoter, whereas neutrophils mainly employ Pprox-B, similar to previous observations in cell lines (23).
Specific activity of Pmed1-A/B is silenced by DNA methylation
To functionally evaluate the role of DNA methylation in the control of the selective activity of FCGR3A and FCGR3B variant promoters, the proximal 5′ region from each gene was split into four separate sections and cloned individually into a promoter-less, luciferase-expressing plasmid devoid of CpG dinucleotides (Figure 3A). Luciferase plasmids were transfected into YT cells (NK lineage), K562 (myeloid lineage) and into Jurkat and HEK293T as negative controls for non-CD16a-expressing lineages. As found previously (23, 24, 34), the Pmed1-A region was selectively activated in YT cells, and not Pmed1-B (Figure 3B). K562 cells selectively activated Pmed1-B, but not Pmed1-A. Other cloned regions did not possess inherent promoter activity in either lineage, including Pprox promoters that are known to require additional flanking enhancer sequences (23, 24). Importantly, in vitro methylation of Pmed1 sequences abolished their activity in both lineages (Figure 3B), corroborating findings in primary cells that DNA methylation of Pmed1-A is likely involved in the regulation of CD16a expression in the NK lineage.
Figure 3.

Analysis of DNA methylation- and lineage-specific activity of FCGR3 promoter sequences. A) Illustration of FCGR3A and FCGR3B sequences cloned into luciferase constructs (black bars). B) Four separate promoter sequence fragments were cloned from either FCGR3A (left) or FCGR3B (right) and transfected in various cell lines. Luciferase assays showing sequence- and gene-specific activity (relative to empty-vector control). Pmed1-A and Pmed1-B were additionally methylated in vitro prior to transfection. Error bars represent SEM of n=3 individual experiments; ND, not done. C) Sequence alignment of Pmed1-A and Pmed1-B with numbered CpGs. All sequence variants are enlarged below; an 8 bp repeat occurring in the vicinity of CpG#6 is highlighted in blue, asterisks below the sequence indicate homology. D) Luciferase activity following site-directed mutagenesis of CpG#s 1&2 of Pmed1-A compared to unaltered FCGR3A and FCGR3B sequences in YT and K562 cells.
Selective activity of Pmed1A/B requires specific CpG dinucleotides
To identify the basis for lineage-specific expression of Pmed1 we aligned Pmed1-A and -B sequences and found that variants are strongly enriched at CpG dinucleotides. Differences generally disrupt CpGs in FCGR3B versus FCGR3A, including CG>TG transitions at CpG#s 1&2 and an 8 bp indel that disrupts CpG#6 (Figure 3C). To test if these CpGs are involved in selective regulation, we introduced C>T mutations for CpG#s 1&2 in Pmed1-A FCGR3A, which enhances the likeness toward FCGR3B, and then retested these regions in our luciferase-based expression assay (Figure 3D). Mutation of these CpGs abolished selective activity in YT cells. Remarkably, these mutations permitted Pmed1-A to be expressed in K562 cells, as increased likeness toward FCGR3B in the pattern of CpG sequences associated with increased activity in K562 cells. This suggests that in addition to the regulatory function provided by CpG methylation, the CpGs themselves are required to direct selective expression of FCGR3A versus FCGR3B.
Prediction of miRNAs that target FCGR3A
To identify miRNA regulators of FCGR3A we followed the strategy described in Witkos et al (35). We compiled a list of 53 potential miRNA regulators by using TargetScan, DIANA, PITA and miRSVR prediction tools. Separately, we obtained a miRNA expression array of 800 miRNAs in mature human peripheral blood NK cell populations defined as CD56brightCD94high and CD56dimCD94low, which overlap significantly with CD56bright/CD16a- and CD56dim/CD16a+ (Figure S1A–F) (36). We screened for miRNAs that were highly expressed in CD16a- NK cells and poorly expressed in CD16a+ NK cells. We next cross-referenced this list with the list of miRNAs compiled from the prediction tools and identified three miRNAs, miR-92a, miR-133a and miR-218 (Figure 4A). Each of these miRNAs have putative recognition sites in the 3′ untranslated region (UTR) of FCGR3A; miR-92a at UTR positions 255–262, miR-133a at UTR positions 458–471, and miR-218 at UTR positions 843–850 (Figure 4B). We found that this pattern could be validated by real time PCR in freshly sorted CD16a- and CD16a+ primary NK cells for miR-133a and miR-218, but not for miR-92a (Figure 4C).
Figure 4.
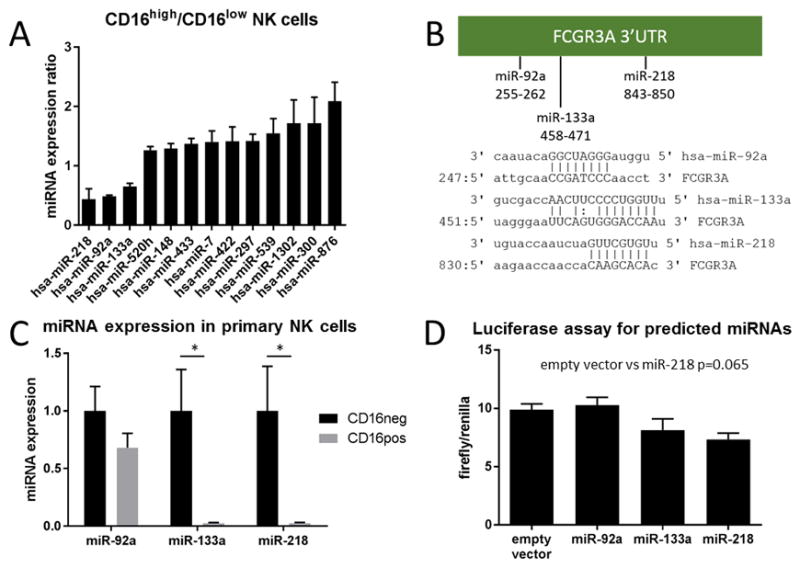
Identification of miR-218 as a potential regulator of FCGR3A. (A) Expression ratio of predicted miRNAs that were present in the miRNA expression array comparing CD16a+ and CD16a- NK cells freshly isolated from adult peripheral blood. A ratio <1 indicates low expression in CD16a+ NK cells while a ratio >1 indicates high expression in CD16a+ NK cells. (B) Predicted miRNA regulators of FCGR3A have putative binding sites in the FCGR3A 3′ UTR. (C) Validation of expression of predicted miRNA regulators of FCGR3A by qPCR (n=3). (D) Luciferase expression as a ratio of firefly/renilla for each expression vector (n=2). Data are presented as mean±SD, * indicates p<0.05.
In order to determine if these putative miRNA regulators could interact with the 3′UTR of FCGR3A mRNA we performed luciferase assays. We cloned the entire 3′UTR of FCGR3A into the luciferase vector pmirGLO downstream of the firefly luc gene, which is driven by a constitutively active promoter. Following co-transfection with an expression vector for one of each of the candidate miRNAs, we found that miR-218 showed trend toward negative regulation of luciferase activity (Figure 4D).
MiR-218 is a negative regulator of CD16a in human NK cells
To validate the activity of miR-218 against FCGR3A, we over-expressed miR-218 in primary human NK cells by lentiviral infection. Transduced cells were sorted based on GFP expression and analyzed by flow cytometry for CD16 expression (Figure S2A). The average transduction efficiency (%GFP+) was 58.0±23.9% for empty vector and 13.8±9.2% for mir-218 (Figure S2B). We found that miR-218 over-expression resulted in decreased CD16a surface expression in NK cells infected with miR-218 virus compared to control virus (Figure 5A and B). We confirmed that miR-218 virus-infected cells did over-express miR-218 (Figure 5C) and also found that this over-expression inversely correlated with FCGR3A mRNA expression (Figure 5D).
Figure 5.
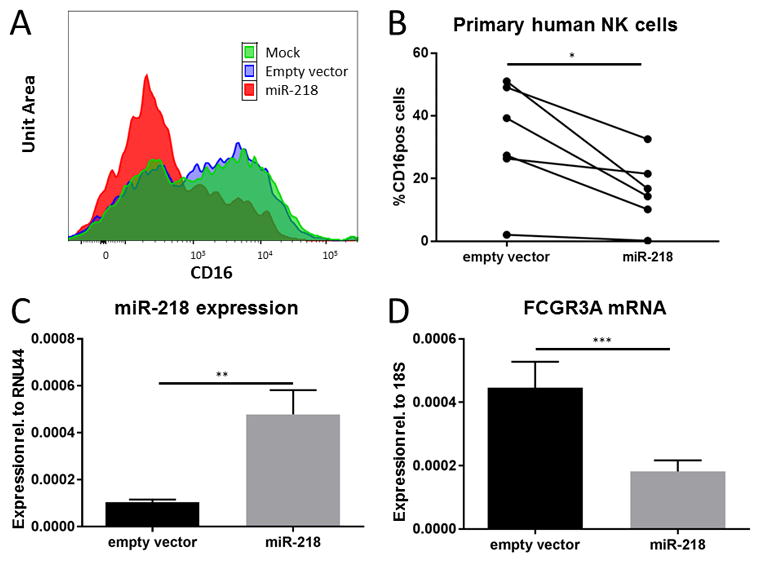
MiR-218 negatively regulates CD16a in primary human NK cells. Primary human NK cells were enriched by magnetic selection to >70% purity and infected with lentivirus containing either miR-218 or empty vector. 48hr after infection, NK cells were sorted as live/CD3-/CD56+/GFP+ lymphocytes. (A) Representative histogram plot (of one of six donors) of CD16a expression in live/CD3-/CD56+/GFP+ primary human NK cells infected with miR-218 or empty vector virus. (B) CD16a expression in primary human NK cells infected with miR-218 or empty vector virus (* indicates p=0.05). (C) Validation of miR-218 over-expression by real-time PCR (** indicates p<0.01). (D) FCGR3A mRNA expression assessed by real-time RT-PCR in sorted NK cells infected with either miR-218 or empty vector (*** indicates p<0.005). (B–D) Data are presented as mean±SD, n=6.
Discussion
Here we describe two important modes of epigenetic control of CD16a expression in primary NK cells that involve regulation at the transcriptional and post-transcriptional levels. Previous work has described that Pmed1-A promotes specific expression of FCGR3A in NK cells (23, 24, 34); however, sequence features alone cannot fully account for the lineage- and stage-specific expression in primary cells. In our study, Pmed1-A and the immediate surrounding sequences were found to be the focal point of reduced methylation in CD16a+ versus CD16a- NK cells, as well as in the CD16a+ NK cell line, NKL. Reduced Pmed1-A methylation was associated with expression of the FCGR3A variant 3 transcript which initiates from Pmed1-A. Interestingly, Pprox-A displayed low methylation regardless of CD16a expression, indicating that the initiation of FCGR3A variant 1 requires additional factor(s) not active in CD16a- NK cells. Indeed, previous work has shown that Pprox-A requires an enhancer element for activity (23, 24, 34). Based on the coordinated expression of FCGR3A variants 1 & 3, we propose that the unmethylated Pmed1-A may provide this function and enhance expression of variant 1. In further support of the role of DNA methylation in the repression of FCGR3 genes, we have made the novel observation that focally reduced methylation of Pprox-B was specifically associated with FCGR3B variant 1 expression in neutrophils. The lack of activity of Pprox-B in K562 luciferase experiments suggests that Pmed1-B may operate as an enhancer for PproxB, however one cannot exclude that K562 are lacking the necessary transcriptional machinery to activate this promoter. Together, these findings indicate that lineage-specific reduction of DNA methylation of key regulatory elements within the FCGR3A and FCGR3B promoter regions permit cell type-specific CD16a versus CD16b expression.
The activity state of a particular promoter or genomic element is dependent upon two primary factors, 1) the epigenetic structure, which controls accessibility, plus 2) the presence of specific interacting trans-activating factors. We observed that the activity of Pmed1-A and Pmed1-B was restricted to YT and K562 cells, respectively, supporting the specific recognition of these sequences in NK and myeloid lineages, as found previously (23, 24, 34). These experiments reveal that YT cells, being of the NK lineage, possess the transcriptional machinery to selectively activate exogenous Pmed1-A; however, YT cells do not express endogenous FCGR3A in concordance with the observation of high endogenous Pmed1-A methylation. Indeed, in vitro methylation of Pmed1-A silenced its activity. As YT cells represent an earlier stage of NK development (37), our findings suggest that the transcriptional machinery required may already be active in earlier stages of NK cell development. We propose that Pmed1-A methylation prevents interaction with the transcriptional machinery and that Pmed1-A demethylation may function as the switch for CD16a expression during the transition from Stage 4 to Stage 5 NK development.
All sequence variations between Pmed1-A and -B involve CpG dinucleotides. Interestingly, a greater number of CpGs are retained in the Pmed1-A region compared to Pmed1-B, concomitant with a greater importance of Pmed1-A for FCGR3A than for FCGR3B in NK cells. Conversely, the Pprox-B promoter that is instead primarily utilized by neutrophils retains two CpGs not present in Pprox-A. Remarkably, mutation of 1 or 2 variant CpGs in Pmed1 was sufficient to disrupt the lineage specificity of wild-type Pmed1-A and Pmed1-B sequences. These findings highlight the dual importance of CpGs in the Pmed1 region, functioning as an off switch when methylated and concurrently endowing selective activity when unmethylated.
In our survey of miRNA expression in peripheral blood-derived NK cells, we found that the majority of differentially-expressed miRNAs were more highly expressed in the mature CD16a+ NK cell population than in the less mature CD16a- population. This greatly narrowed our search for putative direct negative regulators of FCGR3A mRNA. For three miRNAs, we observed consistent, but modest differences in miRNA expression comparing CD16a- to CD16a+ NK cells. Direct validation of miR-218 expression in CD16a+ and CD16a- primary NK cells demonstrated significant differences in relative expression levels. This difference in scale may be partly due to differences in experimental technique, but also to partly overlapping populations. CD56bright/CD94hi NK cells correspond in many ways to CD16a- stage 4 NK cells, but have some level of CD16a (9, 36). This would explain the modest differences in miRNA expression between NK populations in the expression array and the stark differences in the direct validation by qPCR using CD16a- and CD16a+ NK cells. In addition to the difference in miR-218 expression between CD16a+ and CD16a- NK cells, we further observed direct antagonism of miR-218 against CD16a with over-expression studies in primary NK cells. These results suggest that a layer of microRNA regulation functions in conjunction with FCGR3A methylation-based silencing, potentially for the purpose of controlling spurious transcription that may occur despite promoter repression.
In summary, we propose a system of regulation of FCGR3 homologs which confers profound tissue specificity despite minimal differences in regulatory sequences, and facilitates both strong and fine regulation of FCGR3A expression in NK cells. We have found that well-defined genetic elements with distinct regulatory functions cooperate with epigenetic modifications to ultimately control the cell type-specific expression. This knowledge may provide avenues for improving NK-dependent anti-tumor strategies such as ADCC-based therapeutics and the use of engineered NK cells.
Supplementary Material
Footnotes
References
- 1.Clynes R, Towers T, Presta L, Ravetch J. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 2.Weng W, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21:3940–3947. doi: 10.1200/JCO.2003.05.013. [DOI] [PubMed] [Google Scholar]
- 3.Anegón I, Cuturi M, Trinchieri G, Perussia B. Interaction of Fc receptor (CD16) ligands induces transcription of interleukin 2 receptor (CD25) and lymphokine genes and expression of their products in human natural killer cells. J Exp Med. 1988;167:452–472. doi: 10.1084/jem.167.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carter P, Presta L, Gorman C, Ridgway J, Henner D, Wong W, Rowland A, Kotts C, Carver M, Shepard H. Humanizaiton of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A. 1992;89:4285–4289. doi: 10.1073/pnas.89.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis G, Figari I, Fendly B, Wong W, Carter P, Gorman C, Shepard H. Differential responses of human tumor cell lines to anti-p185HER2 monoclonal antibodies. Cancer Immunol Immunother. 1993;37:255–263. doi: 10.1007/BF01518520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freud A, Yu J, Caligiuri M. Human natural killer cell development in secondary lymphoid tissues. Semin Immunol. 2014;26:132–137. doi: 10.1016/j.smim.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freud A, Caligiuri M. Human natural killer cell development. Immunol Rev. 2006;214:56–72. doi: 10.1111/j.1600-065X.2006.00451.x. [DOI] [PubMed] [Google Scholar]
- 8.Chan A, Hong D, Atzberger A, Kollnberger S, Filer A, Buckley C, McMichael A, Enver T, Bowness P. CD56bright human NK cells differentiate into CD56dim cells: role of contact with peripheral fibroblasts. J Immunol. 2007;179:89–94. doi: 10.4049/jimmunol.179.1.89. [DOI] [PubMed] [Google Scholar]
- 9.Freud A, Yokoyama A, Becknell B, Lee M, Mao H, Ferketich A, Caligiuri M. Evidence for discrete stages of human natural killer cell differentiation in vivo. J Exp Med. 2006;203:1033–1043. doi: 10.1084/jem.20052507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cassatella M, Anegón I, Cuturi M, Griskey P, Trinchieri G, Perussia B. Fc gamma R(CD16) interaction with ligand induces Ca2+ mobilization and phosphoinositide turnover in human natural killer cells. Role of Ca2+ in Fc gamma R(CD16)-induced transcription and expression of lymphokine genes. J Exp Med. 1989;169:549–567. doi: 10.1084/jem.169.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris D, Travis W, Koren H. Induction of activation antigens on human natural killer cells mediated through the Fc-gamma receptor. J Immunol. 1989;143:2401–2406. [PubMed] [Google Scholar]
- 12.Ortaldo J, Mason A, O’Shea J. Receptor-induced death in human natural killer cells: involvement of CD16. J Exp Med. 1995;181:339–344. doi: 10.1084/jem.181.1.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ravetch J, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–290. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 14.Smyth M, Cretney E, Kelly J, Westwood J, Street S, Yagita H, Takeda K, van Dommelen S, Degli-Esposti M, Hayakawa Y. Activation of NK cell cytotoxicity. Mol Immunol. 2005;42:501–510. doi: 10.1016/j.molimm.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 15.Sondel P, Hank J. Antibody-directed, effector cell-mediated tumor destruction. Hematol Oncol Clin North Am. 2001;15:703–721. doi: 10.1016/s0889-8588(05)70243-4. [DOI] [PubMed] [Google Scholar]
- 16.Alderson K, Sondel P. Clinical cancer therapy by NK cells via antibody-dependent cell-mediated cytotoxicity. J Biomed Biotechnol. 2011;2011:379123. doi: 10.1155/2011/379123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trotta R, Puorro K, Paroli M, Azzoni L, Abebe B, Eisenlohr L, Perussia B. Dependence of both spontaneous and antibody-dependent, granule exocytosis-mediated NK cell cytotoxicity on extracellular signal-regulated kinases. J Immunol. 1998;161:6648–6656. [PubMed] [Google Scholar]
- 18.Perussia B. Fc receptors on natural killer cells. Curr Top Microbiol Immunol. 1998;230:63–88. doi: 10.1007/978-3-642-46859-9_6. [DOI] [PubMed] [Google Scholar]
- 19.Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankario D, Luo X, Cooley S, Verneris M, Walcheck B, Miller J. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17) Blood. 2013;121:3599–3608. doi: 10.1182/blood-2012-04-425397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsuo Y, Drexler H. Immunoprofiling of cell lines derived from natural killer-cell and natural killer-like T-cell leukemia-lymphoma. Leuk Res. 2003;27:935–945. doi: 10.1016/s0145-2126(03)00024-9. [DOI] [PubMed] [Google Scholar]
- 21.Li M, Wirthmueller U, Ravetch J. Reconstitution of human Fc gamma RIII cell type specificity in transgenic mice. J Exp Med. 1996;183:1259–1263. doi: 10.1084/jem.183.3.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fossati G, Moots R, Bucknall R, Edwards S. Differential role of neutrophil Fcgamma receptor IIIb (CD16) in phagocytosis, bacterial killing, and responses to immune complexes. Arthritis Rheum. 2002;46:1351–1361. doi: 10.1002/art.10230. [DOI] [PubMed] [Google Scholar]
- 23.Gessner J, Grussenmeyer T, Kolanus W, Schmidt R. The human low affinity immunoglobulin G Fc receptor III-A and III-B genes. Molecular characterization of the promoter regions. J Biol Chem. 1995;270:1350–1361. doi: 10.1074/jbc.270.3.1350. [DOI] [PubMed] [Google Scholar]
- 24.Gessner J, Grussenmeyer T, Dumbsky M, Schmidt R. Separate promoters from proximal and medial control regions contribute to the natural killer cell-specific transcription of the human FcgammaRIII-A (CD16-A) receptor gene. J Biol Chem. 1996;271:30755–30764. doi: 10.1074/jbc.271.48.30755. [DOI] [PubMed] [Google Scholar]
- 25.Bröske A, Vockentanz L, Kharazi S, Huska M, Mancini E, Scheller M, Kuhl C, Enns A, Prinz M, Jaenisch R, Nerlov C, Leutz A, Andrade-Navarro M, Jacobsen S, Rosenbauer F. DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat Genet. 2009;41:1207–1215. doi: 10.1038/ng.463. [DOI] [PubMed] [Google Scholar]
- 26.Trowbridge J, Snow J, Kim J, Orkin S. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell. 2009;5:442–449. doi: 10.1016/j.stem.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferlazzo G, Thomas D, Lin S, Goodman K, Morandi B, Muller W, Moretta A, Münz C. The abundant NK cells in human secondary lymphoid tissues require activation to express killer cell Ig-like receptors and become cytolytic. J Immunol. 2004;172:1455–1462. doi: 10.4049/jimmunol.172.3.1455. [DOI] [PubMed] [Google Scholar]
- 28.Vitale C, Chiossone L, Morreale G, Lanino E, Cottalasso F, Moretti S, Dini G, Moretta L, Mingari M. Analysis of the activating receptors and cytolytic function of human natural killer cells undergoing in vivo differentiation after allogeneic bone marrow transplantation. Eur J Immunol. 2004;34:455–460. doi: 10.1002/eji.200324668. [DOI] [PubMed] [Google Scholar]
- 29.Ehrich M, Nelson MR, Stanssens P, Zabeau M, Liloglou T, Xinarianos G, Cantor CR, Field JK, van den Boom D. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15785–15790. doi: 10.1073/pnas.0507816102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weigel C, Veldwijk MR, Oakes CC, Seibold P, Slynko A, Liesenfeld DB, Rabionet M, Hanke SA, Wenz F, Sperk E, Benner A, Rosli C, Sandhoff R, Assenov Y, Plass C, Herskind C, Chang-Claude J, Schmezer P, Popanda O. Epigenetic regulation of diacylglycerol kinase alpha promotes radiation-induced fibrosis. Nature communications. 2016;7:10893. doi: 10.1038/ncomms10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei Q, Claus R, Hielscher T, Mertens D, Raval A, Oakes C, Tanner S, de la Chappelle A, Byrd J, Stilgenbauer S, Plass C. Germline allele-specific expression of DAPK1 in chronic lymphocytic leukemia. PLoS One. 2013;8:e55261. doi: 10.1371/journal.pone.0055261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Briercheck E, Trotta R, Chen L, Hartlage A, Cole J, Cole T, Mao C, Banerjee P, Hsu H, Mace E, Ciarlariello D, BLM-B, Garcia-cao I, Scoville S, Yu L, Pilarski R, Carson Wr, Leone G, Pandolfi P, Yu J, Orange J, Caligiuri M. PTEN is a negative regulator of NK cell cytolytic function. J Immunol. 2015;194:1832–1840. doi: 10.4049/jimmunol.1401224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gessner JE, Grussenmeyer T, Schmidt RE. Differentially Regulated Expression of Human IgG Fe Receptor Class III Genes. Immunobiology. 1995;193:341–355. doi: 10.1016/s0171-2985(11)80564-4. [DOI] [PubMed] [Google Scholar]
- 34.Heusohn F, Wirries G, Schmidt R, Gessner J. The Pmed1 gene promoter of human Fc gamma RIIIA can function as a NK/T cell-specific restriction element, which involves binding of Sp1 transcription factor. J Immunol. 2002;168:2857–2864. doi: 10.4049/jimmunol.168.6.2857. [DOI] [PubMed] [Google Scholar]
- 35.Witkos T, Koscianska E, Krzyzosiak W. Practical aspectes of microRNA target prediction. Curr Mol Med. 2011;11:93–109. doi: 10.2174/156652411794859250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu J, Mao H, Wei M, Hughes T, Zhang J, Park I, Liu S, McClory S, Marcucci G, Trotta R, Caligiuri M. CD94 surface density identifies a functional intermediary between the CD56bright and CD56dim human NK-cell subsets. Blood. 2010;115:274–281. doi: 10.1182/blood-2009-04-215491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yodoi J, Teshigawara K, Nikaido T, Fukui K, Noma T, Honjo T, Takigawa M, Sasaki M, Minato N, Tsudo M, Uchiyama T, Maeda M. TCGF (IL 2)-receptor inducing factor(s). I. Regulation of IL 2 receptor on a natural killer-like cell line (YT cells) J Immunol. 1985;134:1623–1630. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.