

Abstract
Introduction
Ewing sarcoma (ES) of the sinonasal tract and associated primitive neuroectodermal tumors are rare. To our knowledge, only 10 case reports of sinonasal ES of the nose or paranasal sinuses have been reported. Furthermore, there has been only 1 case of sinonasal ES arising from the middle turbinate. Recommended management of sinonasal ES stems from the management of its osseous counterpart, ES, but treatment with surgery, radiotherapy, and chemotherapy is varied. Five-year survival rates vary from 21% to 70%, with the lower rates representing patients presenting with metastatic disease.
Case Presentation
A 26-year-old man presented with persistent left-sided nasal obstruction. Endoscopy demonstrated a friable mass in the left nasal cavity originating from the middle turbinate with extension into the nasopharynx, confirmed with computed tomography and magnetic resonance imaging. There was no evidence of metastatic disease on positron emission tomography-computed tomography. Histopathologic results were consistent with sinonasal ES. The result of fluorescent in situ hybridization was positive for the EWS gene translocation. A multidisciplinary tumor board evaluated the patient. The patient then underwent neoadjuvant chemotherapy, followed by definitive endoscopic surgical resection and postoperative radiotherapy.
Discussion
Our literature review found more involvement of the maxillary and ethmoid sinuses compared with the nasal cavity, and that the role of radiation and surgical approach was varied. ES of the sinonasal tract is a rare entity with high mortality, but few standardized treatment protocols exist. Further study and evidence-based treatment protocols are needed. This article outlines the role of relevant imaging, a multidisciplinary team approach, and the optimal timing of surgery, chemotherapy, and radiation.
INTRODUCTION
Ewing sarcoma (ES) is a rare and aggressive tumor that typically involves the long bones of the extremities (skeletal form). The less common extraskeletal form involves soft tissues and rarely manifests in the head and neck region.1 Genetically, the abnormal t(11;22) chromosome translocation is a hallmark of diagnosis.1 ES of the nose or paranasal sinuses is a rare subset of ES, with treatment consisting primarily of chemotherapy followed by surgery and/or radiotherapy.
CASE PRESENTATION
Presenting Concerns
A 26-year-old man presented to the Head and Neck Surgery Clinic with persistent left-sided nasal obstruction. Flexible nasal endoscopy showed a left-sided friable mass originating from the middle turbinate and extending into the nasopharynx. This endoscopic image is not available, but a very similar endoscopic image, performed later in our patient’s course of treatment, is shown in Figure 1. The patient was treated for sinusitis and nasal polyps with oral antibiotics, oral corticosteroids, a steroid nasal spray, and an oral antihistamine. Computed tomography (CT) was scheduled for 1 month after completion of treatment. The CT scan showed a soft-tissue mass in the left posterior nasal cavity.
Figure 1.

Rigid nasal endoscopy image from study performed at halfway point of chemotherapy. A purple friable mass is seen extending from the left posterior lateral nasal wall between the middle and inferior turbinates in the sphenoethmoidal recess. Biopsy specimen showed persistent disease.
Therapeutic Intervention and Treatment
After being lost to follow-up for 3 months, the patient was seen again, and plans were made for a biopsy of the lesion. The biopsy specimen confirmed the diagnosis of ES. Magnetic resonance imaging (MRI) and positron-emission tomography were then performed for staging and treatment planning. Figure 2 shows the extent of the mass as seen on the initial MRI. Pathologic evaluation on hematoxylin-eosin staining revealed cords of tissue infiltrated by cords and nests of neoplasm with small round blue cell features. Scattered mitotic and apoptotic figures were seen (Figures 3 and 4). Immunohistochemistry showed positive staining for CD99 antigen (Figure 5). Findings were supportive of ES/primitive neuroectodermal tumor (PNET). Fluorescent in situ hybridization testing was positive for the t(11;22) translocation associated with the EWS gene in 89% of the cells. Flow cytometry revealed atypical cells that were not further characterized.
Figure 2.

Coronal, T2-weighted magnetic resonance image of the head after contrast enhancement, showing a 4.5-cm enhancing mass centered in the left posterior nasal cavity with extension into the nasopharynx, consistent with Ewing sarcoma/primitive neuroectodermal tumor.
Figure 3.
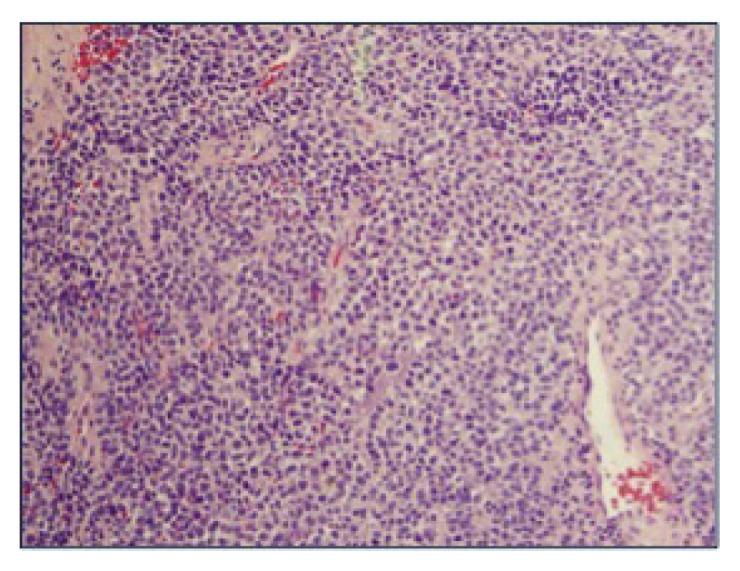
Hematoxylin/eosin-stained tissue section of mass (magnification ×20). Neoplastic cells are small to medium sized and have hyperchromatic nuclei with mild variation in nuclear size. Cytoplasm is fine to granular.
Figure 4.
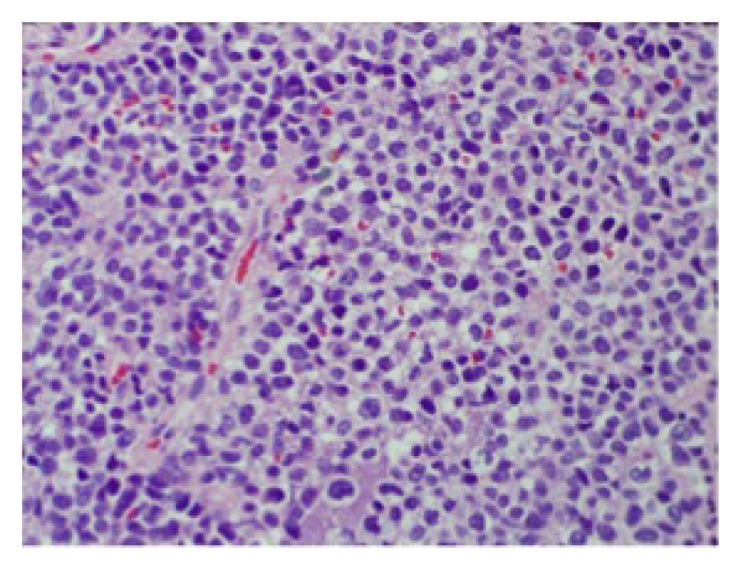
Hematoxylin/eosin-stained tissue section of mass (magnification ×40). Same description as in Figure 3. In addition, cords and sheets of small round blue cells with high nuclear-cytoplasmic ratios are seen.
Figure 5.
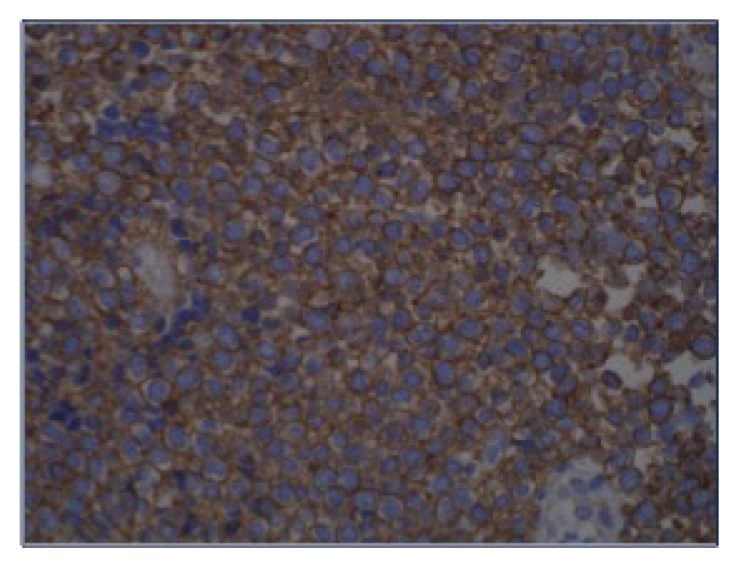
Immunohistochemical CD99 antigen stain (magnification ×40). Tumor cells diffusely express CD99 antigen.
The patient’s case was presented to our institution’s multidisciplinary tumor board. A referral was made to a hematologist-oncologist, who recommended chemotherapy. The result of a bone marrow biopsy performed before chemotherapy did not show any evidence of metastatic disease via pathology or fluorescent in situ hybridization analysis.
One month after diagnosis, our patient started chemotherapy with a regimen of vincristine, doxorubicin, and cyclophosphamide alternating with cycles of ifosfamide and etoposide. He underwent 4 cycles of this regimen. Repeated positron-emission tomography/CT performed 2 months after the start of chemotherapy showed resolution of the previous hypermetabolic left-sided posterior nasopharyngeal mass, without fluorodeoxyglucose-avid metastatic disease. However, there was a persistent nonfluorodeoxyglucose-avid, ovoid, soft-tissue density that on repeat biopsy showed persistent disease. The findings during this biopsy are shown in Figure 1. Repeat MRI showed a small (8 mm × 14 mm × 18 mm) enhancing soft-tissue mass in the location of the original lesion. After the patient was evaluated by a radiation oncologist, his case was again presented to our regional tumor board. The decision was made to perform surgical resection with another round of chemotherapy and possible radiation therapy pending surgical margins and final pathology.
The patient was taken to the operating room for endoscopic surgery and removal of the mass centered along the left lateral nasal wall. Surgery was conducted with imaging guidance. A septoplasty was performed for improved exposure. The specimen was resected en bloc, with good exposure. Margins included the posterior antrostomy, inferior turbinate, anterior lateral nasal wall, and uncinate and ethmoid bulla.
Follow-up and Outcomes
All intraoperative margins were negative for tumor. Final pathology confirmed residual disease with negative margins and no evidence of lymphovascular or perineural involvement.
The patient healed well postoperatively and continued his original course of chemotherapy. He did not require radiotherapy because of the absence of high-risk features and negative margin status on final pathology. Twelve-month postoperative follow-up showed no evidence of tumor recurrence.
Figure 6 shows a timeline of the case. Our patient gave written, informed consent for inclusion in this report.
Figure 6.

Case timeline.
CT = computed tomography; FISH = fluorescent in situ hybridization; MRI = magnetic resonance imaging; PET = positron-emission tomography.
DISCUSSION
We describe what we believe is the first reported case of ES originating from the middle turbinate in a young man. By itself, ES is a rare disease, making up only 4% to 6% of all primary bone tumors.2,3 Furthermore, ES involves the head and neck region in only 1% to 4% of cases, and tumors with a sinonasal origin form another rare subset.3 Simons et al4 describes the difficulty in diagnosis of small round blue cell tumors as well as a systemic approach to aid clinicians. ES is described as part of a family of tumors with neuroectodermal differentiation. More specifically, ES is classified as a peripheral PNET.4 Immunohistochemistry often narrows the diagnosis greatly. For example, a lesion that is positive for CD99 antigen and vimentin would be either alveolar rhabdomyosarcoma or ES/PNET. Staining for desmin or myogenin would help direct the clinician toward a diagnosis of ES/PNET in the former scenario and alveolar rhabdomyosarcoma in the latter. Strong membrane-only CD99 positivity would again point toward ES/PNET. Clinical parameters can be of aid as well. Typically ES/PNET presents in patents younger than age 30 years, with men affected more often than women.4 Molecular analysis is particularly useful if the diagnosis is already narrowed to 2 or 3 entities.4 For ES, the t(11;22)(q24;q12) translocation corresponding to EWSR1/FLI-1 fusion is often diagnostic and used for confirmation.4 However, variant translocations are seen in 10% to 15% of ES cases.4
After diagnosis of ES is confirmed, additional necessary studies include CT and MRI to help anatomically define the lesion as a baseline before starting treatment as well as for surgical planning purposes should surgery be pursued. Furthermore, positron-emission tomography should be performed to stage the disease and look for evidence of metastatic disease. Previous studies have found a rate of metastatic disease at diagnosis of around 12.5% for ES of the head and neck, and 20% to 30% for ES of all sites.5 In one of the largest case series of patients who received systemic chemotherapy and in some instances radiotherapy as well, 57% achieved complete remission and 43% achieved partial remission.5 Local remission in this group occurred in 29% of patients, and distant metastasis developed in 46% during the follow-up period.5 Overall survival at 5 years in this group was 53%.5 This highlights the potential aggressiveness of the disease and the need for clinical vigilance and appropriate follow-up care.
Although radiation therapy was not used in our case, its use should be considered. Despite substantial associated morbidity, including visual disturbances, pituitary dysfunction, and xerostomia, the report by Lepera et al6 in 2016 advocates for the use of radiation therapy in select cases of sinonasal ES (eg, for cases of suboptimal resection). No study to date has shown clear evidence of increased survival with adjuvant radiotherapy. In our case, the patient underwent evaluation by a multidisciplinary tumor board that believed that the patient would have a good response to chemotherapy and could be treated with salvage surgery before radiation therapy, if radiation therapy was needed. As mentioned in Lepera et al, given the favorable surgical margins in our case, radiation therapy was ultimately avoided, and chemotherapy was given postoperatively only because the patient was in the midst of his chemotherapy protocol when the residual disease was identified.
Previous studies that routinely reported radiotherapy along with chemotherapy as an initial management option primarily included large tumors (> 10 cm).5 This may be because of decreased access to imaging modalities and health care, a hypothesis outlined by the authors of the largest series to date.5 As in our case, surgery is often not recommended as the first-line treatment given the substantial morbidity associated with large head and neck tumor resection. However, with the earlier detection of tumors and the development of advanced endoscopic techniques, treatment patterns may shift. Given the rarity of this disease, no uniform follow-up regimen is currently recommended. Previous authors have suggested regular follow-up every 3 months for the first year, every 6 months for the second year, and then yearly thereafter.5,6 Our patient followed a similar follow-up plan, with imaging as needed on the basis of clinical suspicion of tumor recurrence. Of course, any follow-up plan will depend on the aggressiveness of the tumor and patient or clinician limitations.
CONCLUSION
ES of the sinonasal tract is a rare entity that has a high mortality and for which few standardized treatment protocols exist. This report outlines the role of relevant imaging, pathologic, and cytogenetic studies; the importance of a multidisciplinary team; the timing of surgery; and a proposed chemotherapy regimen. Our patient received chemotherapy up front, followed by surgical salvage therapy for residual disease and completion of chemotherapy without evidence of recurrence at 12-month follow-up. In our literature review, we underscore the gaps in the current research literature, including the role of radiation therapy, various surgical options, and the ideal follow-up period.
Acknowledgments
Daniel Baiyee, MD, provided pathologic and cytogenetic information and described the pathologic slides.
Kathleen Louden, ELS, of Louden Health Communications provided editorial assistance.
Footnotes
Disclosure Statement
The author(s) have no conflicts of interest to disclose.
References
- 1.Negru ME, Sponghini AP, Rondonotti D, et al. Primary Ewing’s sarcoma of the sinonasal tract, eroding the ethmoid and sphenoid sinus with intracranial extension: A rare case report. Mol Clin Oncol. 2015 Jul;3(4):807–10. doi: 10.3892/mco.2015.548. DOI: https://doi.org/10.3892/mco.2015.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hafezi S, Seethala RR, Stelow EB, et al. Ewing’s family of tumors of the sinonasal tract and maxillary bone. Head Neck Pathol. 2011 Mar;5(1):8–16. doi: 10.1007/s12105-010-0227-x. DOI: https://doi.org/10.1007/s12105-010-0227-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol. 2010 Feb;11(2):184–92. doi: 10.1016/S1470-2045(09)70286-4. DOI: https://doi.org/10.1016/S1470-2045(09)70286-4. [DOI] [PubMed] [Google Scholar]
- 4.Simons SA, Bridge JA, Leon ME. Sinonasal small round blue cell tumors: An approach to diagnosis. Semin Diagn Pathol. 2016 Mar;33(2):91–103. doi: 10.1053/j.semdp.2015.09.010. DOI: https://doi.org/10.1053/j.semdp.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 5.Allam A, El-Husseiny G, Khafaga Y, et al. Ewing’s sarcoma of the head and neck: A retrospective analysis of 24 cases. Sarcoma. 1999;3(1):11–5. doi: 10.1080/13577149977811. DOI: https://doi.org/10.1080/13577149977811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lepera D, Volpi L, Facco C, et al. Endoscopic treatment of Ewing sarcoma of the sinonasal tract. J Craniofac Surg. 2016 Jun;27(4):1001–6. doi: 10.1097/SCS.0000000000002701. DOI: https://doi.org/10.1097/SCS.0000000000002701. [DOI] [PubMed] [Google Scholar]