

Abstract
Glioblastoma (GBM) is a primary brain cancer that contains populations of stem-like cancer cells (GSCs) that home to specialized perivascular niches. GSC interactions with their niche influence self-renewal, differentiation and drug resistance, although the pathways underlying these events remain largely unknown. Here, we report that the integrin αvβ8 and its latent transforming growth factor β1 (TGFβ1) protein ligand have central roles in promoting niche co-option and GBM initiation. αvβ8 integrin is highly expressed in GSCs and is essential for self-renewal and lineage commitment in vitro. Fractionation of β8high cells from freshly resected human GBM samples also reveals a requirement for this integrin in tumorigenesis in vivo. Whole-transcriptome sequencing reveals that αvβ8 integrin regulates tumor development, in part, by driving TGFβ1-induced DNA replication and mitotic checkpoint progression. Collectively, these data identify the αvβ8 integrin-TGFβ1 signaling axis as crucial for exploitation of the perivascular niche and identify potential therapeutic targets for inhibiting tumor growth and progression in patients with GBM.
INTRODUCTION
Glioblastoma (GBM) is a heterogeneous and rapidly progressive cancer that arises via genomic alterations in neural stem and progenitor cells of origin.1,2 The initiation, growth and progression of GBM is driven by sub-populations of self-renewing and multipotential tumor-initiating cells, also known as cancer stem cells.3 Stem-like GBM cells (GSCs) have similarities with neural stem and progenitor cells,4 including expression of common molecular markers, for example, Nestin and CD133/Prominin-1.5,6 GSCs also localize to perivascular niches where they exploit stromal-derived growth factors and ECM proteins.7,8 Most cells interact with ECM proteins via integrins, a family of cell surface heterodimeric receptors consisting of α and β subunits.9 Various integrins as well as their ECM protein ligands and intracellular signaling effectors play important roles in brain physiology,10 and alterations in integrin expression and function contribute to the initiation and progression of GBM.11,12
The αv subfamily of integrins is comprised of five members: αvβ1, αvβ3, αvβ5, αvβ6 and αvβ8. αv-containing integrins recognize RGD (arginine-glycine-aspartic acid) peptide motifs in many ECM ligands, including Vitronectin, Fibronectin and Collagen IV, which are abundantly expressed in the brain.13 Various data also link abnormal regulation of αv integrins, particularly αvβ3 and αvβ5, in GBM cell growth and invasiveness.14 However, clinical trials with Cilengitide, an RGD peptide mimetic that inhibits αvβ3 and αvβ5 integrins, have not improved overall survival of patients with GBM,15 revealing that additional factors in tumor cells and/or the microenvironment promote malignant growth and invasion. β8 integrin, encoded by the ITGB8 gene, is a 100 kDa glycoprotein that heterodimerizes exclusively with the 130 kDa αv subunit.16,17 αvβ8 integrin can bind to many ECM proteins, but in vivo data reveal that this integrin is a primary receptor for latent-TGFβ1 and latent-TGFβ3,18 which are produced by cells as inactive complexes. Integrin adhesion to RGD sequences in the ECM-bound latent-TGFβ1 and TGFβ3 complexes mediates ligand activation and receptor signaling.19 In contrast, latent-TGFβ2, which is expressed in the brain microenvironment, lacks the RGD integrin-binding motif and is likely activated via other mechanisms.20 Gene knockout models reveal that glial-expressed αvβ8 integrin regulates angiogenesis in the brain and retina.21–26 Mice lacking αv integrin or β8 integrin in glial cells develop intracerebral hemorrhage and progressive neurological deficits, and these phenotypes are not observed in other integrin mutant models.27 Mutations in the human ITGB8 gene are linked to cerebrovascular pathologies, including brain arteriovenous malformations28,29 and spontaneous forms of intracerebral hemorrhage.30 In the adult brain we have reported that the αvβ8 integrin-TGFβ1 signaling axis is essential for neurogenesis in the subventricular zone, with β8 −/− mice showing reduced neural stem cell self-renewal as well as aberrant neuroglial differentiation and migration.31,32 Functions for αvβ8 integrin in cancer stem cell self-renewal and/or tumor initiation have not been reported.
Here, we have characterized mechanisms by which αvβ8 integrin in primary GBM cells regulates tumor growth and progression. We report the following novel findings: (i) β8 integrin is expressed in perivascular GBM cells in situ; (ii) β8 integrin in freshly sorted GSCs is essential for self-renewal, spheroid formation and lineage commitment in vitro; (iii) β8 integrin expression levels partly correlate with expression of other GSC biomarkers such as CD133 and Sox2; (iv) fractionation of β8 integrin-expressing cells from human GBM samples reveals that β8 integrin is essential for tumor initiation and progression in vivo; and (v) β8 integrin promotes GSC self-renewal and tumorigenesis, in part, by promoting TGFβ receptor signaling and mitotic checkpoint progression.
RESULTS
To characterize roles for ITGB8 in GBM initiation and progression we analyzed integrin protein expression in human GSCs and in resected tumor samples. Cultured primary cells grow in serum-free media as free-floating spheroids (Figure 1a). Cell surface biotinylation and immunoprecipitation experiments revealed high levels of αvβ8 integrin protein in GSCs in comparison with αvβ3 and αvβ5 integrins (Figure 1b). Prior studies have shown that GSCs also show preferential localization to perivascular niches.7 Therefore, we performed immunohistochemistry with fixed human GBM tissue or normal brain tissue using a human-specific anti-β8 integrin antibody. Notably, non-cancerous brain regions adjacent to tumor showed β8 integrin expression in reactive astrocytes (Figures 1c and d and Supplementary Figure 1A). GBM cells that expressed the highest levels of β8 integrin protein showed close proximity to intratumoral blood vessels (Figures 1e–h and Supplementary Figures 1B and D). Immunoblot analyses of detergent-soluble lysates from five different cultured GBM spheroids revealed robust levels of αv and β8 integrin proteins (Figure 1i). Freshly prepared lysates from grade III astrocytomas (n = 3) and grade IV astrocytoma/GBM (n = 7) showed β8 integrin protein expression in most samples analyzed (Figure 1j). In comparison to non-cancerous brain lysates, β8 integrin protein levels were higher in GBM lysates (Supplementary Figure 1E). Next, we queried the open source IVY GBM Atlas Project for spatial expression patterns of integrin mRNA expression in microdisssected and laser-captured tumor regions. ITGAV/αv integrin and ITGB8 mRNAs were detected within cellular regions of GBM (Figure 1k). ITGB8 was absent in intratumoral blood vessels, whereas ITGAV was more abundantly expressed in the vasculature likely due to heterodimerization with other β integrin subunits such as β3 and/or β5. Querying TCGA (The Cancer Genome Atlas) database for human GBM revealed that ITGB8 is a molecular marker for the classical GBM sub-type (Figure 1l). TCGA analyses also revealed that ITGAV and ITGB8 mRNA levels were 1.89-fold and 2.32-fold higher, respectively, in GBM tissue versus non-cancerous brain tissue (data not shown).
Figure 1.
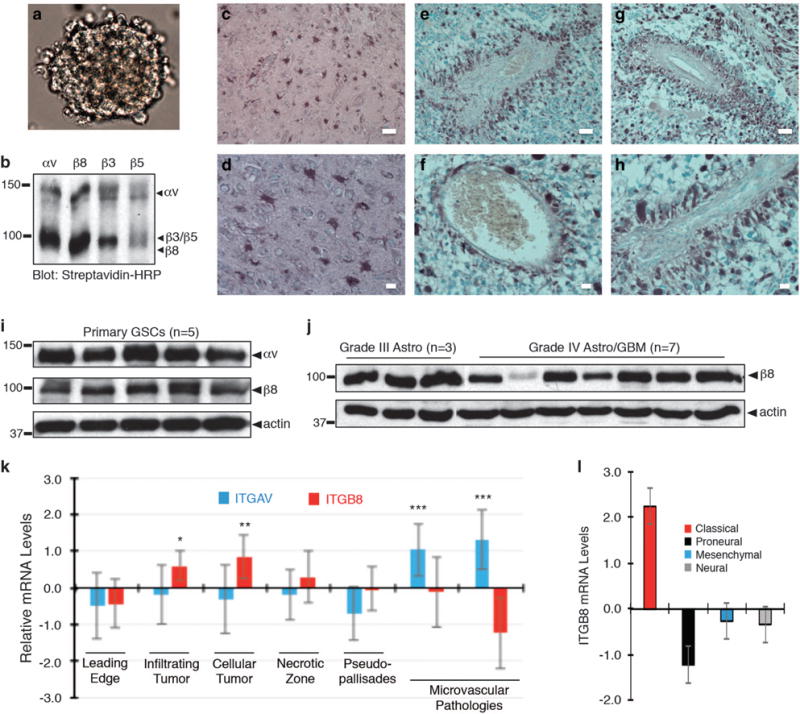
β8 integrin is expressed in cultured GBM spheroids and is enriched in perivascular GBM cells in situ. (a) Primary tumor cells cultured from freshly resected GBM tissue grow as neurosphere-like spheroids in serum-free media containing EGF and bFGF. (b) Cell surface biotinylation and immunoprecipitation experiments identify αvβ8 as a major αv integrin-containing heterodimeric protein expressed in primary human GBM cells. (c–h) Immunohistochemistry staining with an anti-β8 integrin antibody reveals integrin protein expression patterns in non-cancerous brain and human GBM samples. Note that β8 integrin is expressed in reactive astrocytes in the non-cancerous brain (c, d). In contrast, within brain tumors β8 integrin protein is enriched in perivascular GBM cells (e–h). Scale bar, 20 μm. (i); Immunoblot analysis of αv and β8 integrin proteins in human GBM spheroids (n = 5). (j) Immunoblot analysis of β8 integrin protein levels in different tumor lysates from grade III astrocytomas (n = 3) and grade IV GBM lysates (n = 7). (k) Differential expression of ITGAV and ITGB8 mRNAs in various tumor regions based on querying the IVY GBM Atlas Project. (l) Analysis of the TCGA GBM database identifies ITGB8 as a molecular marker for the classical GBM sub-type, *P<0.05, **P<0.01, ***P<0.001.
Next, we utilized fluorescence-activated cell sorting (FACS) methods to fractionate live tumor cells from primary GBM specimens. Recurrent GBM samples were excluded from the study due to possible radiation-induced or chemotherapy-induced genomic alterations. Freshly resected tumor samples were enzymatically dissociated into cell suspensions, and CD31+ vascular endothelial cells, CD45+ lymphocytes and CD11b+ microglial cells/macrophages were removed to enrich suspensions for GBM cells (Figure 2a). We used monoclonal antibodies to sort tumor cells based on expression of β8 integrin. Cells with robust β8 integrin expression were termed ‘β8high’ and those with low/undetectable levels of integrin protein were termed ‘β8low’ cells. Analysis of freshly sorted β8high cells by RT-PCR revealed significant downregulation of TERT and PTEN gene products, supporting that these cell fractions were tumor cells (data not shown). Tumor cells were fractionated from >25 different resected human GBM specimens. Differing amounts of β8 integrin-expressing cells were present, with many tumors (10 of 25 analyzed) containing relatively small percentages (<10%) of β8high cells (Figure 2b and Supplementary Figure 2). When β8high sorted cells were cultured in vitro, they showed robust self-renewal and proliferation capacities as evidenced by spheroid formation in serum-free media (Figures 2c–e). In contrast, β8low cells failed to generate spheroids and instead formed loose aggregates that typically died within a week after isolation. Treatment of β8high GBM cells with an antibody directed against the β8 integrin extracellular region also led to diminished sphere formation (Supplementary Figures 3A and C). Indeed, all low-passage primary GBM spheroid preparation that we have analyzed for integrin expression by FACS contained mostly β8high GBM cells, suggesting a requirement for this integrin in GSC self-renewal in culture.
Figure 2.
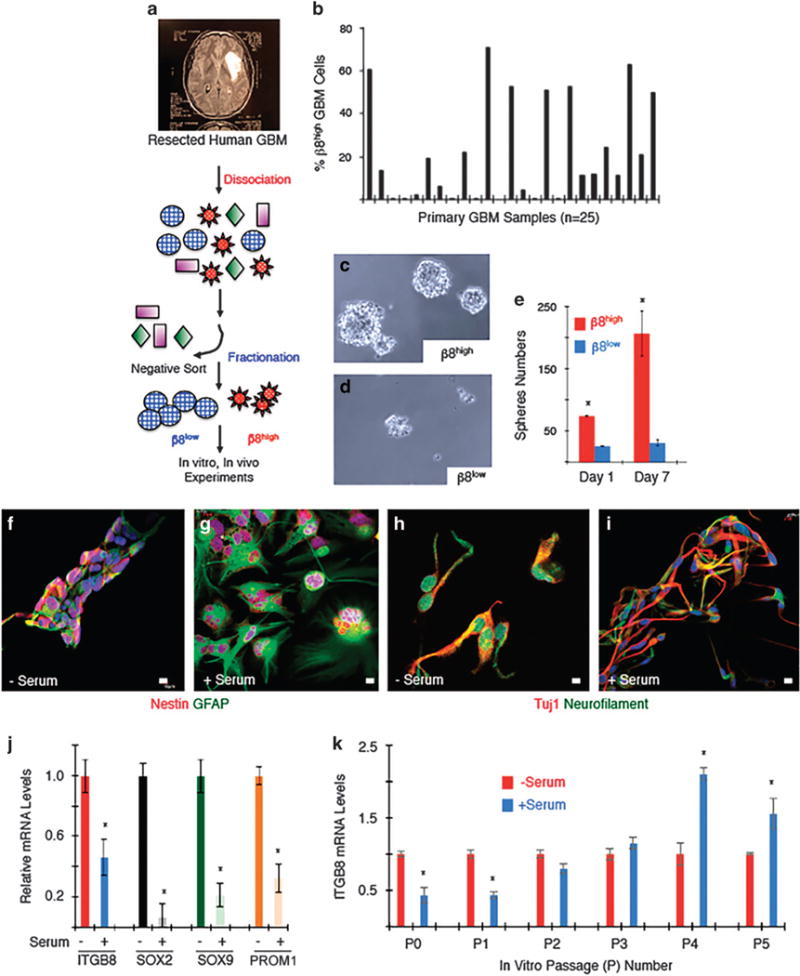
β8 integrin is required for GSC self-renewal and differentiation in vitro. (a) Experimental approach for isolation of β8high and β8low primary GBM cells from human samples followed by in vitro culturing and/or intracranial injection. (b) Summary of β8 integrin protein expression levels as determined by FACS in 25 different freshly resected primary GBM samples. (c, d) β8high GBM cells from sample HBT14 form spheroids and survive in culture (c), whereas β8low cells do not form spheroids and fail to thrive in culture (d). Images shown are of spheroids formed from non-passaged β8high and β8low GBM cells. (e) Quantitation of β8 integrin-dependent sphere formation in vitro. (f–i) Spheroids generated from low-passage β8high GBM cells (HBT41) were grown in the presence or absence of serum and immunofluorescently labeled with anti-Nestin and anti-GFAP to label neural stem cells and astrocytes (f, g) or anti-TUJ1 and anti-neurofilament antibodies to label neurons (h, i). (j) Neural stem cell markers were quantified by RT-PCR using spheroids generated from low-passage β8high GBM cells (HBT28) before and after differentiation via serum exposure. (k) Serum-induced differentiation of β8high GBM cells (HBT32) leads to reduced β8 integrin expression at low passages, but β8 integrin expression increases in more differentiated cells at higher passages.
β8high GSCs expressed the neural stem cell marker Nestin and displayed differentiation capacities toward astroglial (GFAP) and neuronal lineages (Tuj1 and neurofilament) in response to serum treatment (Figure 2f–i). When low-passage GSCs were induced to differentiate, mRNA levels of ITGB8, as well as the neural stem cell markers SOX2 and PROM1, showed downregulation (Figure 2j). These results largely support quantitative transcriptome sequencing data in a separate study,33 showing that ITGB8 is a ‘stem cell classifier’ gene, and that serum-induced differentiation results in diminished ITGB8 expression (Supplementary Figure 3D). Interestingly, although we detected diminished ITGB8 mRNA levels in low-passage GSCs, we found that as GSCs were sequentially passaged, serum-induced differentiation correlated with an upregulation of ITGB8 expression (Figure 2k).
CD133 is a cell surface marker for GSCs, with fractionated CD133+ cells promoting spheroid self-renewal in vitro and generating malignant brain tumors in vivo.6 As shown in Figure 2, serum-induced GSC differentiation leads to a reduction in ITGB8 and CD133 expression. Therefore, we analyzed links between β8 integrin and CD133 by sorting GBM cells from two different freshly resected human GBM samples. As shown in Figures 3a and b, we found 8.9 and 25.9% of GBM cells to be double positive for β8 integrin and CD133, although their co-expression was not completely coincident. A significant percentage of cells showed expression for only one of the two proteins. β8 integrin and CD133 expression was analyzed in four different spheroid cultures generated from unfractionated tumor cells or β8high cells. As shown in Figures 3c–f, all four possible combinations of single positive (β8high or CD133+), double positive (β8high/CD133+) and double negative (β8low/CD133−) populations were detected at varying percentages in the different spheroid cultures. When we used Crispr-Cas9 gene editing strategies to target ITGB8 in cultured spheroids from human brain tumor 32 (HBT32), we found that deletion of ITGB8 correlated with concomitant loss of CD133, with the percentages of CD133+ cells diminishing by nearly 20-fold in spheroids lacking β8 integrin (Figures 3g and h). Similar results were detected when ITGB8 was delete in a second GBM sample (HBT28, data not shown). Fractionation of cells based on β8 integrin and CD133 expression revealed that β8high GSCs showed robust cell growth and viability in vitro, which was not dependent of coincident CD133 expression (Figures 3i and j).
Figure 3.
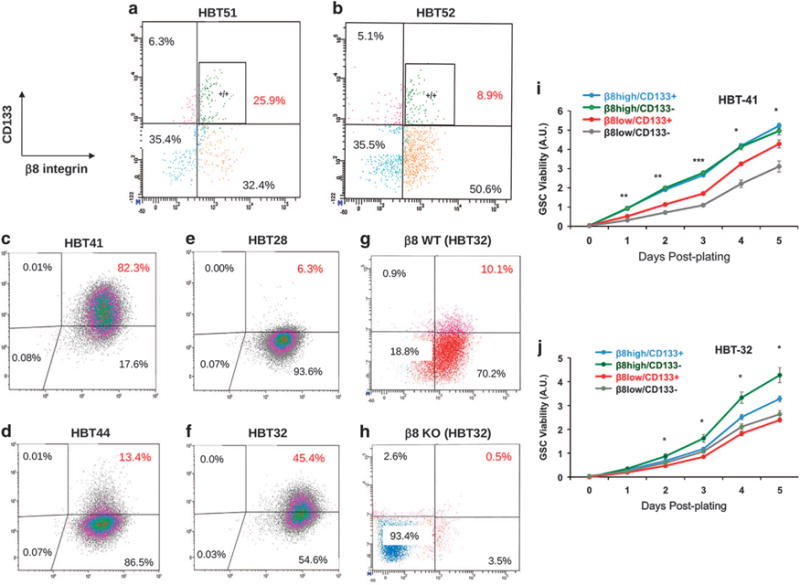
Analysis of β8 integrin and CD133 protein expression in primary GSCs. (a, b) Primary tumor cells from two different freshly resected GBM samples were analyzed for β8 integrin and CD133 expression. Note that the majority of β8high GBM cells do not express CD133. (c–f) Unfractionated primary tumor cells (c, d) from two different freshly resected GBM samples (HBT41 and HBT44) were cultured in serum-free media. Alternatively, β8high GBM cells were fractionated (HBT28 and HBT32) and cultured in vitro (e, f). Note that nearly all GBM cells, whether sorted for β8 integrin or not, express high levels of β8 integrin protein. CD133 protein levels are more variable and do not fully coincide with β8 integrin expression. (g, h) Crispr-Cas9 strategies were used to target ITGB8 in spheroids formed from β8high GBM cells (HBT28) followed by FACS analysis. Note that CD133 is absent following ITGB8 gene targeting. Validation of ITGB8 gene editing via Crispr-Cas9 and absence of integrin protein expression is detailed in Figure 6 and Supplementary Figure 10. (i, j) GBM cells from HBT41 (i) and HBT32 samples (j) were fractionated by FACS based on differential expression of CD133 and β8 integrin. Cell growth and viability were quantified in spheroids every day for 5 days. In comparison with β8high/CD133− cells, note that β8low/CD133+ and β8low/CD133− cell fractions show reduced viability, *P<0.05, **P<0.01, ***P<0.001. β8high/CD133+.
β8 integrin-dependent tumor initiation and growth in vivo were next quantified using eight different freshly resected patient samples. Live cell sorting methods were used to fractionate β8high and β8low GBM cells (Figure 4a). β8high cells formed neurosphere-like spheroids that expressed of the neural stem cell marker Nestin and the more differentiated biomarker, Vimentin (Figure 4b). Cell surface αvβ8 integrin heterodimeric protein was confirmed in cultured spheroids (Figure 4c). Intracranial implantation of β8high tumor fractions (ranging from 12 500 to 200 000 cells) into non-obese diabetic, severe combined immunodeficient (NOD-SCID) mice led to tumor formation in 24 of 30 (80%) mice (Supplementary Table 1). H&E staining revealed that β8high GBM cells showed diffuse growth patterns, with cells crossing the brain midline and entering the contralateral hemisphere (Figures 4d–f and Supplementary Figure 4). Immunofluorescence analyses of β8high xenograft tumors revealed expression of Nestin and GFAP in human cells identified with a human-specific Vimentin antibody (Figure 4g and Supplementary Figure 5). Tumors were well vascularized as revealed by anti-laminin immunofluorescence, and contained GFAP-expressing reactive astrocytes (Figures 4h and i) and Iba1-expressing microglial cells (Supplementary Figure 5).
Figure 4.
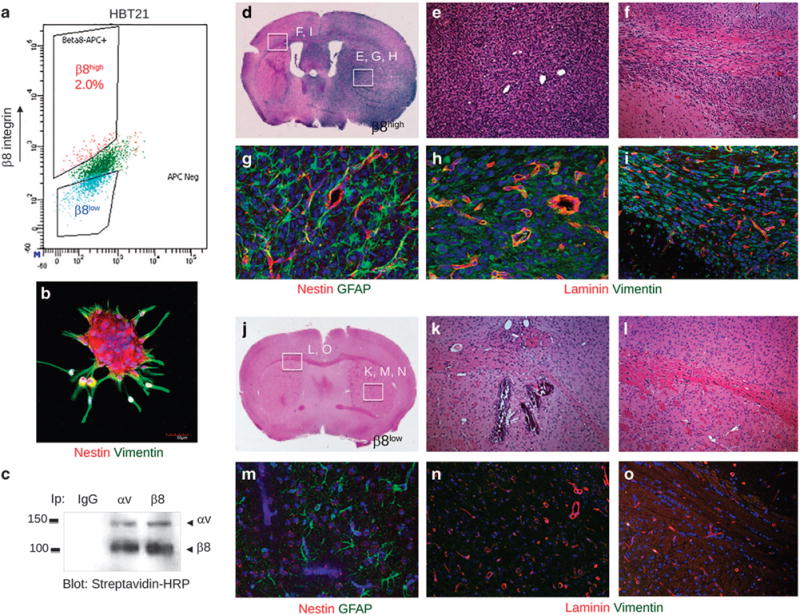
β8 integrin is essential for GBM initiation, growth and invasion in vivo. (a) Representative FACS plot of β8high and β8low primary GBM cell fractions from a freshly resected tumor sample (HBT21). (b) Spheroids formed from sorted β8high cells were labeled with anti-Nestin (red) and anti-Vimentin (green) antibodies. (c) αvβ8 integrin heterodimeric protein is robustly expressed in spheroids formed from sorted β8high cells, as revealed by cell surface biotinylation and co-immunoprecipitation. (d–f) Images of H&E-stained brain sections from mice injected with β8high GBM cells sorted from sample HBT21. Note that β8high cells form diffuse, intracranial tumors that invade along white matter tracts and blood vessels. (g–i) Immunofluorescence analysis of β8 integrin-dependent GBM growth, invasion and angiogenesis in xenograft tumors. β8high GBM cells generated well-vascularized and invasive tumors as revealed by anti-laminin staining to identify vascular basement membranes and anti-vimentin staining to identify human cells. (j–l) Images of H&E-stained tumor sections from mice injected with β8low GBM cells from the HBT21 sample. (m–o) Immunofluorescence analysis of mice injected with β8low GBM cells, revealing absence of vimentin-expressing human tumor cells.
The majority of β8low GBM cell preparations (71%) did not generate brain tumors after injection into NOD-SCID mice (Supplementary Table 1, Figures 4j–o). Analysis of the 29% of β8low GBM cell preparations that generated intracranial tumors in vivo showed re-expression of β8 integrin protein (Figure 5). For example, β8high GBM cells sorted from patient sample HBT32 generated nestin-expressing spheroids in vitro (Figures 5a and b) that were comprised of high percentages of cells that maintained β8 integrin expression (Figure 5c). β8high GBM cells from HBT32 also generated large and invasive intracranial tumors in NOD-SCID mice (Figures 5d and e). Similarly, β8low GBM sorted from HBT32 formed nestin-expressing spheroids in vitro, although they were significantly smaller than those formed from β8high cells (Figures 5a and f). Reanalysis of integrin expression in β8low GBM cells by FACS revealed that 64% of cells expressed β8 integrin (Figure 5g). Immunofluorescence analysis of xenograft tumors formed from β8low cells revealed integrin protein expression in many tumor cells (Figure 5h). The human specificity of the anti-β8 integrin antibody was confirmed by labeling non-cancerous mouse brain regions (Figure 5i). Xenograft brain tumors were also generated with bulk/unfractionated GBM cell preparations from patient sample HBT32 (Supplementary Figure 6A). β8high and β8low cells GBM cells were then sorted from dissected xenografts (Supplementary Figure 6B) and injected into secondary recipient mice. Similar to what we detected with the freshly resected patient samples (Figure 5), brain tumors developed in secondary mice injected with both β8high and β8low cell fractions (Supplementary Figures 6C, and D). Although tumors formed from β8low cells were noticeably smaller and less diffuse, many β8low cancer cells expressed integrin protein (Supplementary Figures 6F, and H). In a third β8low GBM cell fraction that generated tumors in vivo and spheres in vitro (HBT14) we used FACS to show integrin protein expression in β8low spheroids (Supplementary Figures 6I and K). One explanation for these findings is that some β8low cells can adapt after removal from the brain microenvironment by activating β8 integrin expression. Similar findings have been reported for plasticity of CD44 expression in breast cancer stem cells.34 Alternately, small percentages of β8+ GBM cells may contaminate some β8low fractions, leading to sphere formation and tumor initiation.
Figure 5.
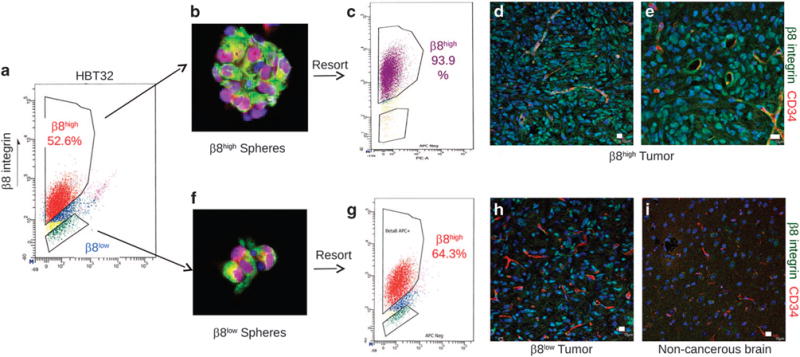
β8 integrin is re-expressed in spheroids and tumors generated from β8low GBM cells. (a) FACS-based fractionation of β8high and β8low tumor cells from a freshly resected primary GBM sample (HBT32). (b, c) β8high GBM cells form spheroids in culture (b) that express the neural stem cell marker Nestin (red) and the more differentiated cell marker Vimentin (green). FACS analysis of spheroids reveals β8 integrin protein expression in nearly all primary GBM cells (c). (d, e) β8high GBM cells fractionated from HBT32 (a) were intracranially implanted into the mouse brain (n = 3 mice injected per cell type). β8high cells formed large and invasive brain tumors in mice and express β8 integrin protein, as revealed with a human-specific anti-β8 integrin protein. Note that perivascular tumor cells express robust levels of β8 integrin protein. (f, g) β8low GBM cells from HBT32 form spheroids in culture (f) that express Nestin (red) and Vimentin (green), although they are smaller than spheroids formed from β8high GBM cells. FACS analysis of β8low spheroids reveals β8 integrin protein expression in 64% of GBM cells (g). (h) Immunofluorescence analysis reveals upregulated integrin protein expression tumors generated from β8low GBM cells. (i) A non-cancerous region of the mouse brain was used to control for specificity of the human-specific β8 integrin antibody. All scale bars are 10 μm.
Crispr-Cas9 gene editing methods were used to inhibit ITGB8 gene expression in low-passage GBM spheroids sorted from sample HBT28. Cells were infected with lentiviruses expressing GFP, Cas9 and gDNAs targeting ITGB8. Immunoblot analyses of GBM cells sorted based on GFP expression confirmed loss of β8 integrin protein in β8KO GSCs, but not in control (β8WT) cells (Figure 6a). FACS confirmed absence of β8 integrin expression on the surface of β8KO cells (Figures 6b and c). H&E staining (Figures 6d and e) and immunofluorescence analyses (Figures 6f and g) revealed that β8WT cells generated malignant tumors in NOD-SCID mice (n = 4). In contrast, β8KO GSCs (Figures 6h–k) formed reduced numbers of spheroids in vitro (data not shown) and were less tumorigenic in vivo (n = 4 injected mice). Similar Crispr-Cas9 gene editing approaches were used to target ITGB8 in GSCs fractionated from two other human GBM samples (HBT32 and HBT36), and diminished spheroid formation in vitro and tumor growth in vivo were also detected (data not shown).
Figure 6.
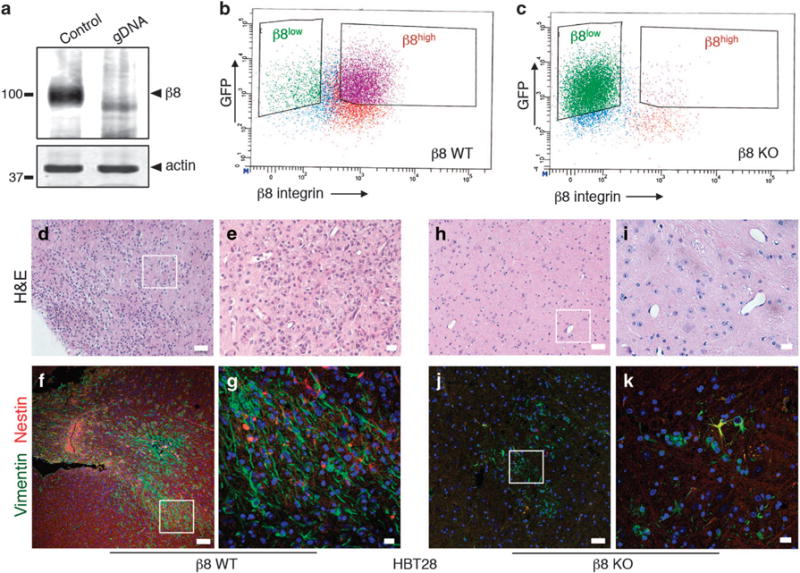
Genetically targeting β8 integrin in low-passage GBM cells leads to defective tumor initiation in vivo. (a) Immunoblot analysis showing absence of β8 integrin protein expression in β8KO cells generated via Crispr-Cas9 gene editing strategies. (b, c) FACS plot from β8WT (b) and β8KO (c) GBM cells, revealing loss of β8 integrin protein expression on the GBM cell surface. (d, e) Images of H&E-stained tumor sections from mice injected with β8WT GBM cells, revealing larger and more invasive tumors derived from β8WT cells. (f, g) Immunofluorescence analysis of xenograft tumors derived from β8WT GBM cells. The anti-vimentin antibody (green) is specific for human cells and anti-nestin (red) labels neural stem cells. (h, i) Brain sections from mice injected with β8KO GBM cells were stained with H&E, revealing small and minimally invasive tumors. (j, k) Immunofluorescence analysis with anti-vimentin and anti-nestin antibodies of mouse brains injected with β8KO GBM cells. Note that there are fewer human cells in β8KO-derived tumors as revealed by anti-vimentin immunofluorescence staining. All scale bars are 10 μm.
To analyze tumor cell interactions with blood vessels in the brain microenvironment, we imaged localization of GFP-expressing β8high GBM cells after intracranial implantation. As shown in Supplementary Figure 7A, GFP+ tumor cells showed close juxtaposition to cerebral blood vessels. Co-culturing β8high GBM cells with vascular endothelial cells in three-dimensional ECM also confirmed close interactions with blood vessel-like networks (data not shown). VEGF-A is a potent angiogenic factor produced by many tumor cells, and agents that therapeutically target VEGF-A and its receptors have been tested in clinical trials.35 Therefore, we quantified β8 integrin-dependent levels of VEGF-A expression. Enzyme-linked immunosorbent assay (ELISA)-based experiments with freshly sorted β8high and β8low GBM cells (Supplementary Figure 7B) revealed similar secreted VEGF-A protein levels. Similarly, we did not detect differences in TGFβ1 protein levels by ELISA using conditioned media taken from β8high and β8low GBM cells (Supplementary Figure 7C).
Although β8 integrin in freshly sorted GBM cells promoted spheroid formation in vitro and robust tumor growth in vivo, it is important to note that sequential passaging of GSCs led to loss of integrin-dependent tumorigenic effects. In primary GBM spheroids isolated from patient samples that have been sequentially passaged and characterized previously for tumor initiation capacities,36,37 we detected αv integrin/CD51 and CD133 expression by FACS (Supplementary Figure 8). Cells expressing αv integrin were more abundant than those expressing β8 integrin, likely because the αv subunit can pair with multiple β subunits. However, when ITGAV or ITGB8 were targeted using lentiviral-delivered shRNAs effects on tumor initiation and growth after intracranial implantation were not detected (Supplementary Figure 9). Furthermore, genetic deletion of ITGB8 in high passage GCSs using Crispr-Cas9 methods did not inhibit sphere formation in vitro (data not shown) or block tumor initiation in vivo (Supplementary Figure 10). In some sequentially passaged GBM cells we detect progressive loss of β8 integrin expression (data now shown), whereas other passaged spheroid cultures maintain integrin expression (Figure 1d). Interestingly, when high passage GSCs were induced to differentiate, ITGB8 mRNA levels increased (Supplementary Figure 9I), which is opposite to what was found in low-passage GSCs, which showed diminished ITGB8 levels (Figure 2k). These results emphasize the importance of studying GSCs in vivo or at low-passage numbers in vitro, and highlight limitations of studying GSCs that have been maintained under long-term culture conditions.
To characterize β8 integrin-dependent pathways that promote GBM cell self-renewal in vitro as well as tumor initiation and progression in vivo, we performed quantitative RNA sequencing using β8high and β8low cells sorted from three different patient samples. Several gene expression signatures showed enrichment in β8high GBM cells, as revealed by the Kyoto Encyclopedia of Genes and Genomes (KEGG) bioinformatics resource. These pathways include lipid and amino acid biosynthesis, ABC transporters, as well as gene signatures linked to DNA synthesis, repair, and recombination (Figures 7a and b and Supplementary Tables 2 and 3). Of particular interest, β8high GBM cells expressed higher levels of several gene products involved in mitotic checkpoint progression, such as BUB1, PKMYT1, CDC20 and CDK1 (Supplementary Figure 11A). We validated higher CDK1 mRNA levels in five of six β8high GBM cells fractionated from a separate set of patient tumors (Figure 7c). In contrast, β8low GBM cells showed enrichment in mitotic checkpoint inhibitors such as CDKN2D, CDKN1B and CDKN1A, which encodes the CDK1 inhibitor p21cip. Core components of the TGFβ1 signaling pathway, including SMAD2, SMAD4 and TGFB1 were also enriched in β8low GBM cells (Supplementary Figure 11B). Additional pathways that were differentially expressed in β8high versus β8low GBM cells based on transcriptome sequencing are summarized in Supplementary Figures 12 and Supplementary Tables 2 and 3.
Figure 7.
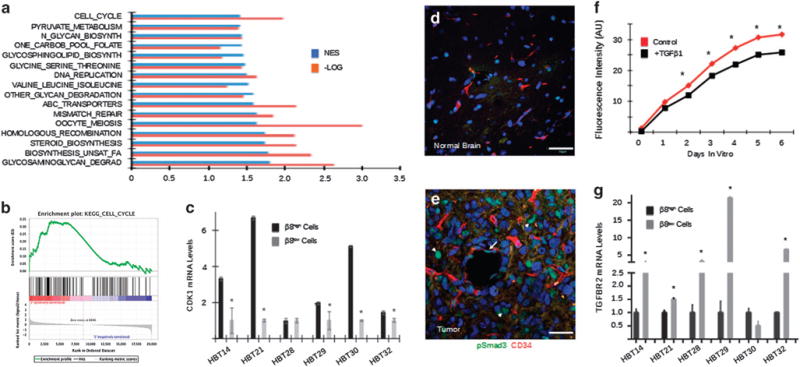
Analysis of β8 integrin-dependent gene expression signatures in GSCs. (a) β8high and β8low GBM cells were fractionated from three different freshly resected primary human tumor samples and analyzed by whole-transcriptome sequencing. Multiple signaling pathways were identified by gene set enrichment analysis based on differential β8 integrin expression. The normalized enrichment score (NES) and the log transformed (−Log) P-values are shown for the top 16 pathways. (b) NES pathway analysis of β8 integrin-dependent cell cycle gene expression signatures in sorted β8high GBM cells. (c) RT-PCR validation showing that CDK1 mRNA expression is significantly downregulated in β8low GBM cells fractionated from five of six human tumor samples. (d, e) Sections from normal mouse brain (d) or from xenograft tumors formed from β8high GBM cells (e) were immunofluorescently labeled with anti-pSmad3 and anti-CD34. Note that TGFβ receptor signaling is active within intratumoral blood vessels (arrow) and in tumor cells (arrowheads). All scale bars are 50 μm. (f) in vitro growth analysis of β8high GBM cells treated with TGFβ1, revealing TGFβ1-dependent growth suppression. (g) RT-PCR data with five different GBM samples showing elevated levels of TGFBR2 mRNA in β8low GBM cells versus β8high GBM cells.
Prior reports have shown that astrocytoma progression from low-grade to grade IV GBM correlates with elevated TGFβ receptor signaling via Smads.38,39 Along these lines, in comparison with normal brain regions, xenograft tumors derived from β8high GBM cells contained elevated levels of phosphorylated Smad3 protein (Figures 7d and e). When β8high GBM cells were treated with exogenous TGFβ1 we detected reduced growth and spheroid formation (Figure 7f). In comparison to β8low cells, β8high GBM cells expressed lower levels of TGFβR2, a major signaling component of the canonical TGFβ pathway (Figure 7g).40 Diminished expression of TGFβR2 in β8high GBM cells suggests that TGFβ1 signaling is more robust in β8low tumor cells and/or stromal cells. Indeed, querying the Brain RNA-Seq database (http://web.stanford.edu/group/barres_lab/brain_rnaseq.html) reveals that cerebral endothelial cells express high levels of TGFβR2 in comparison with other brain cell types (Supplementary Figures 7D, and E). Similarly, analysis of the IVY GBM database (http://glioblastoma.alleninstitute.org/rnaseq/search/index.html) reveals robust TGFβR2 expression in intratumoral blood vessels (Supplementary Figure 7F).
To further analyze the significance of the αvβ8 integrin-TGFβ1 signaling axis in GBM, we used lentiviruses to forcibly exogenous TGFβR2 in β8high cells fractionated from HBT32 (Figure 8a). Exogenous TGFβR2 expression resulted in reduced growth and sphere formation in vitro (Figure 8b) as well as diminished percentages of cells in the G2-M phase of the cell cycle (Figure 8c). We detected similar growth suppression when TGFβR2 was forcibly expressed in β8high cells fractionated from GBM samples HBT28 and HBT44 (data not shown). TGFβR2 overexpression resulted in reduced levels of endogenous ITGB8 (Figure 8d) and enhanced levels of CDK1 (Figure 8e) and elevated levels of tyrosine phosphorylated Cdk1 protein (Figure 8f). We also detected increased levels of CDKN1A mRNA and p21cip protein in GBM cells displaying elevated canonical TGFβ receptor signaling (Figures 8e and f). Collectively, these data reveal significant TGFβ signaling heterogeneity in GBM cells depending on differential expression of β8 integrin. Tumor cells with high levels of β8 integrin adhere to latent-TGFβ1 and latent-TGFβ3 in the ECM and promote TGFβ activation and signaling in adjacent β8low GBM cells and stromal cells. Robust TGFβ1/3 signaling in β8low GBM cells dampens growth and promotes differentiation. In contrast, β8high GBM cells have low TGFβ receptor signaling due to low endogenous levels of TGFβR2, and as a result can proliferate/self-renew and remain less differentiated. This variable TGFβ responsiveness promotes tumor cell exploitation of the perivascular niche to drive GBM growth, survival and differentiation (Figure 9).
Figure 8.
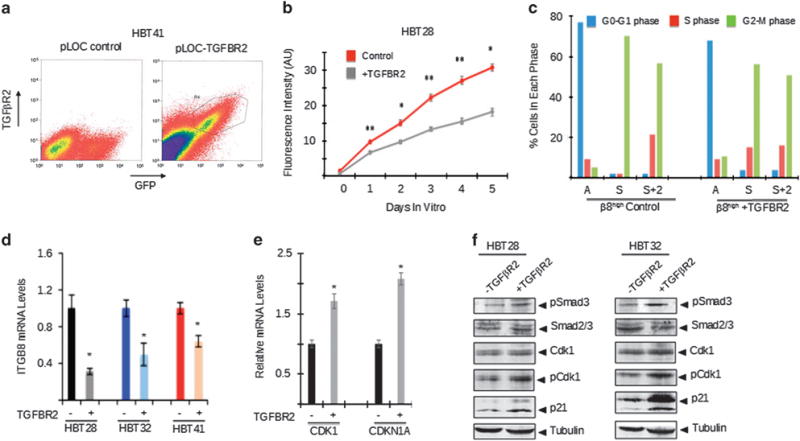
The αvβ8 integrin-TGFβ1 signaling pathway regulates mitotic checkpoint progression in primary GBM cells. (a) GBM cells expressing control pLOC lentivirus (left) or pLOC containing a TGFBR2 cDNA (right) were isolated by FACS based on expression of TGFβR2. (b) β8high GBM cells forcibly expressing TGFβR2 show diminished growth in vitro. (c) Cell cycle phases were analyzed in cultured β8high GBM cells that were asynchronous (A), synchronized (S), or released from synchronization for 2 hours (S+2). Cells were previously infected with control pLOC lentivirus or pLOC expressing TGFβR2. Note that in comparison to controls, TGFβR2 expression results in diminished percentages of cells in the G2-M phase. (d) β8high GBM cells infected with control lentivirus or lentiviruses expressing TGFβR2 were analyzed by RT-PCR, revealing diminished levels of ITGB8 mRNA. (e) β8high GBM cells infected with control lentivirus or lentivirus expressing TGFβR2 were analyzed by RT-PCR, revealing TGFβ1-dependent increases in CDK1 and CDKN1A mRNAs. (f) β8high GBM cells fractionated from two different tumors were infected with pLOC or pLOC-TGFβR2 lentivirus. Detergent-soluble lysates were analyzed by immunoblotting. Note that forced TGFβR2 expression leads to increased levels of phosphorylated Smad3 and Cdk1 proteins as well as elevated expression of p21cip protein.
Figure 9.
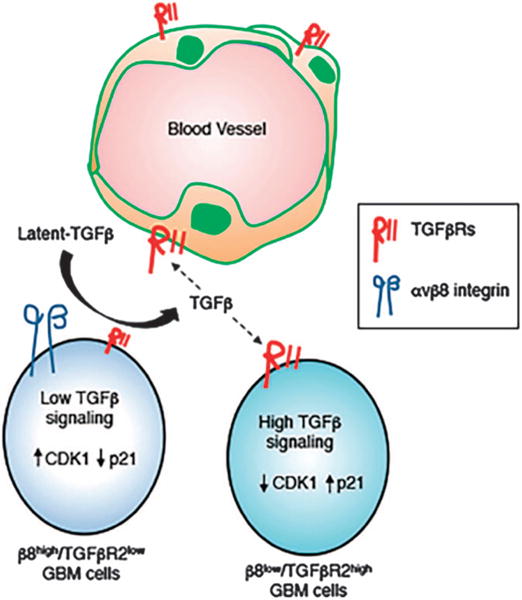
A model for the αvβ8 integrin-TGFβ1 signaling axis in the GBM perivascular niche. β8high GBM cells mediate adhesion to latent-TGFβ1/3 in the ECM, leading to activation of TGFβ receptor signaling in β8low GBM cells and/or in vascular endothelial cells. Varying degrees of TGFβ receptor signaling in β8low versus β8high GBM cells impacts cell cycle progression via CDK1 and CDKN1A/p21cip, thus impacting levels of their proliferation and/or differentiation.
DISCUSSION
In the normal brain αvβ8 integrin-expressing neuroepithelial cells and astrocytes promote robust TGFβ receptor signaling in the vascular endothelium to control sprouting angiogenesis.24,31,32,41 Our data reveal that GSCs hijack this adhesion and signaling axis to enable exploitation of the brain vasculature. The fact that β8high and β8low GBM cells have differential TGFβ1 signaling capacities identifies a likely feedback loop in which more robust TGFβ receptor signaling suppresses integrin expression. This enables β8high GBM cells within the perivascular niche to bind to latent-TGFβ1/3 in the ECM and promote TGFβ receptor signaling in adjacent β8low GBM cells as well as stromal cells. Low levels of TGFβ signaling components in β8high GBM cells enables these cells to avoid the growth-suppressive effects of TGFβ1. It is also possible that αvβ8 integrin-TGFβ1 signaling impacts the differentiation status of GBM cells. Indeed, serum treatment of GSCs leads to diminished expression of ITGB8, PROM1 and SOX2, suggesting that differentiation towards glial and/or neural lineages causes loss of these biomarkers. Furthermore, when we genetically target ITGB8 in GSCs we detect a concomitant loss in CD133 expression. This could be due to GSC differentiation, or alternatively be linked to integrin and adhesion and signaling pathways directly controlling CD133 protein stability at the cell surface and/or regulation of PROM1 gene expression. Changes in differentiation status may also explain why GSC requirements for β8 integrin are diminished following extended passaging. When ITGB8 was genetically targeted in established GBM cell lines, for example, LN229 cells which display more differentiated features, brain tumor initiation is not inhibited.42–44 Along these line, in the U87 GBM cell line inhibition of ITGB8 expression by mir-93 enhances tumor growth and progression.45 Hence, long-term cell passaging and associated differentiation may activate integrin-independent adhesion and signaling pathways or induce expression of other integrins that compensate for loss of β8 integrin in GBM cell lines.
Various studies have shown that TGFβ1 promotes proliferation in established GBM cell lines; however, growth effects on primary cells have been shown to vary significantly.46 For example, TGFβ1 treatment of 10 different primary GSC cultures led to increased proliferation in four GSCs, whereas two GSC preparations showed decreased growth and the other GSCs were non-responsive.38 GSCs that were growth responsive to TGFβ1 had diminished PDGFB gene methylation and enhanced synthesis of PDGFB. These results reveal significant heterogeneity in TGFβ responsiveness related to the differentiation status of primary tumor cell preparations as well as other genetic and/or epigenetic factors. It will be very interesting to analyze β8high GBM cells that did not generate robust xenograft tumors in vivo to determine the methylation status of the PDGFB gene. In addition, we cannot rule out contributions by TGFβ2, which is expressed in the GBM microenvironment but is likely not activated by αvβ8 integrin.47
TGFβ receptor signaling is reported to stimulate p21cip expression via canonical Smad pathways as well as non-canonical pathways involving MAPK1.48 Our transcriptome sequencing data show increased SMAD2 and SMAD4 expression in β8low cells. These cells also express higher levels of TGFβR2, suggesting that increases in p21cip and inhibition of CDK1 are most likely the product of canonical TGFβ receptor signaling. The PKMYT1/Myt1 and Wee1 tyrosine kinases are known to inhibit the CDK1-Cyclin B complex by phosphorylation of CDK1 on Y15, which is crucial for progression through mitosis. As we detect increased CDK1 RNA expression as well as Y15 phosphorylation. In GBM cells it is possible that CDK1 RNA/protein are involved in a TGFβ-regulated signaling loop. RNAi-mediated silencing of Wee1 results in mitotic catastrophe and apoptosis.49 In contrast, a previous report using CRISPR-CAS9 genome editing showed that although these two kinases have a redundant function in normal stem cells, in GSCs PKMYT1 is essential for cell growth and survival.50 Our RNA sequencing data reveal upregulation of PKMYT1 in β8high cells and downregulation of Wee1. Since the cells used for sequencing were not cultured, it could be possible that in vitro culture of GSCs may cause loss of redundancy between PKMYT1 and Wee1. At last, Bub1 is a cell cycle-regulated serine/threonine kinase that is expressed in β8high GBM cells, and has been reported previously to promote GBM growth and progression.51 Interestingly, Bub1 can interact with TGFβR2 and promote Smad phosphorylation and transcriptional activity, suggesting that Bub1 may contribute an additional level of complexity to the αvβ8 integrin-TGFβ1 signaling axis.52
In addition to the cell growth and mitosis gene signatures, the KEGG pathway analyses reveal gene signatures for other interesting pathways. For example multiple ABC transporters, which have roles in multi-drug resistance and are defining features of side-populations of stem cells, are upregulated in β8high GBM cells. Prior reports have shown that GSC self-renewal and resistance to the standard-of-care DNA alkylating agent Temozolomide correlate with high ABCG2 expression.53 GSCs are also resistant to chemotherapy and radiation due to high expression of gene products involved in DNA repair.5 Along these lines, in β8high GBM cells we detect gene expression signatures for DNA replication, mismatch repair and homologous recombination. β8high GBM cells also show upregulated gene signatures related to glycosaminoglycan degradation. It will be interesting to determine how GBM cell degradation of glycosaminoglycans, possible involving hyaluronan and its receptor CD44, impacts the ECM to modulate GSC self-renewal, differentiation and/or invasion. Interestingly, small molecule inhibitors of TGFβ receptors have been shown to target select sub-populations of CD44-expressing cells in GBM and block tumor initiation and progression,54 suggesting links to αvβ8 integrin-expressing cells.
Based on these data, we propose that αvβ8 integrin will be an effective target to inhibit GBM growth and progression. Developing specific antagonists to αvβ8 integrin should be possible, as its adhesion and signaling functions are distinct from other integrins. For example, the β8 integrin cytoplasmic domain lacks conserved peptide motifs, for example, NPXY, that are present in other integrins and have well-characterized roles in signaling and the extracellular region of β8 integrin lacks a ‘deadbolt’ domain that in other integrins regulates affinity for ECM ligands. Collectively, these data indicate that αvβ8 integrin may exist in a high-affinity adhesion state, and constitutively interact with ECM ligands and activate intracellular signaling pathways. GBM clinical trials with Cilengitide, which target αvβ3 and αvβ5 integrin, have not yielded improvements in overall survival,15 suggesting that additional integrins contribute to tumor malignancy. Antibodies or peptide mimetics that selectively recognize αvβ8 integrin heterodimers may be useful for inhibiting this integrin in GBM cells.
Patients treated with the anti-VEGF inhibitory antibody Bevacizumab55 or other anti-angiogenic drugs show improvements in progression-free survival. However, overall survival rates have not improved owing to acquired resistance and tumor recurrence.56 A significant percentage of patients with recurrent GBM display unusually robust patterns of invasive cell growth.57,58 Molecular studies have shown that Bevacizumab alters interactions between VEGFR-2 and c-Met in GBM cells, thus promoting growth and invasive signaling cascades.59–62 We have reported previously that αvβ8 integrin-mediated signaling via Rho GTPases regulatory factors is essential for tumor cell invasion in the brain.42,63,64 It will be interesting to determine whether αvβ8 integrin or its intracellular signaling effectors are differentially regulated in GSCs following surgery and anti-vascular therapies, thus contributing to tumor recurrence and/or invasion. These studies may extend beyond GBM; for example, ITGB8 expression is upregulated in peripheral nerve sheath tumors,65 and in brain metastases,66 suggesting that targeting this integrin may impact tumor growth in other cancer types. At last, cancer stem cells from many different organs are identified, in part, by expression of integrins.67 For example, tumor-initiating cells isolated from breast adenocarcinomas express αvβ3 integrin68 and cancer stem cells fractionated from esophageal squamous carcinomas express α7β1 integrin.69 Although integrins are used primarily as biomarkers for tumor-initiating cells, they are likely providing critical ECM adhesion functions. Hence, therapeutically targeting integrins may be beneficial for blocking cancer stem cell interactions with ECM ligands in their niche to inhibit tumor growth and progression.
MATERIALS AND METHODS
Experimental mice
All animal procedures performed were according to the Institutional Animal Core and Use Committee-approved protocols of MDACC. NOD-SCID mice were purchased from Jackson Laboratories and used for all experiments involving intracranial injections of mouse astrocytoma cells and human GBM cells. For brain isolation, adult mice were anesthetized and brains were fixed by cardiac perfusion with ice-cold 4% paraformaldehde/phosphate-buffered saline (PBS).
Human GBM tissue samples and primary spheroid cultures
Approval for the use of human specimens was obtained from the Institutional Review Board at the University of Texas MD Anderson Cancer Center. Only freshly resected primary GBM samples were analyzed, with recurrent tumor samples excluded from the study. LN229 GBM cells were purchased from American Tissue Culture Collection. Cells were cultured in Dulbecco’s Modified Eagle’s Medium high-glucose sodium pyruvate supplemented with 1% penicillin/streptomycin and 10% fetal bovine serum, at 37 °C in an atmosphere with 5% CO2. Primary human GBM cells from patient samples were cultured in the following growth media: DMEM-F12 (Mediatech, Manassas, VA, USA), 20 ng/ml EGF and bFGF (Gibco, Hilden, Germany), B27 supplement (Life Technologies, Carlsbad, CA, USA) and one unit per ml penicillin-streptomycin (Gibco). To analyze integrin heterodimers on the surface, cells were biotinylated in PBS containing 0.1 mg/ml normal human serum-biotin (Pierce Chemical Company, Rockford, IL, USA), rinsed with Tris-buffered saline, and lysed in radioimmunoprecipitation assay buffer (10 mMTris, pH 7.4, 1% NP-40, 0.5% deoxycholate, 0.1% sodiu dodecyl sulphate, 150 mM NaCl, 1mM ethylenediaminetetraacetic acid) with protease and phosphatase inhibitors (Roche Holding AG, Basel, Switzerland). Protein concentrations were determined using a bicinchoninic acid assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Membranes were probed with streptavidin-horseradish peroxidase and chemiluminescent reagents (Amersham, Little Chalfont, UK).
Flow cytometry
Resected GBM specimens were manually dissociated using the Brain Tumor Dissociation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Once a single cell suspension was generated, red blood cells were removed by the Lympholyte-H solution (Cedar Lanes Labs, Burlington, NC, USA). Brain stromal cells were negatively sorted using human anti-CD31, ant-CD11b and ant-CD45 mAbs (BD Biosciences, San Jose, CA, USA) for endothelial cells, microglia and lymphocytes, respectively. These antibodies were conjugated to anti-mouse IgG1 microbeads (Miltenyi Biotec) and cells were isolated with a magnet following manufacturer’s specifications. After negative sorting, the remaining cells were labeled with an APC-conjugated anti-β8 integrin mAb (R&D Systems, Minneapolis, MN, USA) and stained for viability with Calcein Violet AM (Thermo Fisher Scientific, Hampton, NH, USA). FACS was performed at the M. D. Anderson Flow Cytometry and Cellular Imaging core facility.
Intracranial tumor cell implantation
All animal procedures were conducted under Institutional Animal Care and Use Committee-approved protocols. NOD-SCID mice were anesthetized and a needle was used to dispense cells in DMEM-F12 media through an intracranially inserted guide screw. Injected cell numbers ranged from 12 500 to 200 000 per animal. Mice were sacrificed upon developing tumor-related neurological deficits.
Whole-transcriptome analysis
β8high and β8low cell pellets were snap frozen and total RNA was isolated using in-house methods at Expression Analysis/Q2 solutions. After RNA quality was validated, six samples with a RIN of ⩾ 7 (n = 3 β8high and n = 3 β8low) were selected. An average of ~ 75 million paired-end reads were generated for each of the six samples. Sequencing quality evaluation, alignment of short sequenced reads, and expression calling using reads per kilobase per million reads values were performed by utilizing the Pipeline for RNA Sequencing Data Analysis (PRADA, PMID: 24695405). The expression values of a total of 20 009 protein-coding genes (Ensembl reference transcriptome version 64) were calculated per the reads per kilobase per million reads value of their longest transcript. The signal-to-noise metric was used to calculate the gene expression differences between β8high and β8low cell samples. KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways were compiled from MsigDB (version 5.2, PMID: 16199517). Unsupervised gene set enrichment analysis for all the KEGG pathways was performed using GSEA (version2.2.3, PMID: 16199517). Normalized enrichment scores and false discovery rate (FDR) values were calculated under a 1000-fold permutation.
Immunoblotting, immunofluorescence and immunohistochemistry
The anti-β8 integrin cytoplasmic domain antibody used for immunoprecipitation and immunoblotting as well as the antibody directed against the β8 integrin extracellular domain used for inhibiting spheroid formation have been detailed previously.44 The rabbit anti-pSMAD3, mouse anti-CDK1, rabbit anti-p21, and rabbit anti-CDK Y15, mouse anti α-Actinin were all purchased from Abcam (Abcam, Cambridge, UK). For immunofluorescence, rabbit anti-GFAP (DAKO, Glostrup, Denmark), chicken anti-Nestin (Neuromics, Minneapolis, MN, USA), rat anti-CD31 (BD Biosciences, San Diego, CA, USA), rat anti-CD34 (GeneTex, Irvine, CA, USA), goat anti-Vimentin (R&D Systems), mouse anti-Neuron-specific class III beta-tubulin (TUJ1) from Sigma-Aldrich (Minneapolis, MN, USA), rabbit anti-Neurofilament (Neuromics), rabbit anti-Laminin (Sigma-Aldrich), mouse anti-NeuN (Chemicon, Billerica, MA, USA), rabbit anti-Myelin Basic Protein (MBP), rabbit anti-Iba1 (Wako, Richmond, VA, USA) were purchased.
For both immunofluorescence and immunohistochemical experiments, formalin-fixed paraffin-embedded sections were incubated at 65 °C for 15 min followed by rehydration in an ethanol series. When required, antigen retrieval was performed at 95 °C for 30 min using DAKO buffer. For immunofluorescence, sections were permeabilized in 0.2% Triton X-100 in PBS for 10 min. Slides were washed with PBS and blocked in 10% serum of the secondary antibody host. Primary antibody incubation was performed overnight and sections were washed three times with 0.2% Triton-PBS for 20 min and incubated with secondary antibody for 1 h. Sections were washed three times and were mounted in Vectashield with DAPI (Vector Labs, Burlingame, CA, USA). For IHC, slides were incubated with primary antibodies overnight and then washed with 0.1% Tween in PBS. Sections were then incubated with 0.3% hydrogen peroxide (Sigma-Aldrich) for 10 min, washed three times with PBS and incubated with secondary antibody for 1 hour. The streptavidin-biotin complex (Thermo Fisher Scientific) was added for 15 min followed by developing with 3,3′-diaminobenzidine Peroxidase Substrate Kit. Sections were then dehydrated and mounted in permount solution (Fisher Scientific, Hampton, NH, USA).
GBM cell growth, synchronization and ELISA experiments
Primary tumor cells were treated with 2.5 mM Thymidine (Sigma-Aldrich) for 22 h followed by incubation with 150 ng/ml Nocodazole (Sigma-Aldrich) for 17 h. Cells were then fixed in 90% ethanol and incubated with propidium iodide and RNAase (Sigma-Aldrich) prior to cell cycle analysis. In addition, cell pellets were collected for RT-PCR and immunoblotting experiments. The pLOC lentiviral plasmid expressing TGFBR2 was obtained from the M.D. Anderson ORF core facility. For self-renewal assays spheroids were dissociated with Accutase (Thermo Fisher) and single cells were allowed to form new spheroids over 7 days. Alternatively, dissociated cells in complete media were treated with TGFβ1 (32 ng/ml). Cell growth and viability were quantified each day for 4–5 days using the Alamar Blue assay kit (Thermo Fisher). The human TGFβ1 and VEGF-A ELISA kits were purchased from R&D systems (Fisher Scientific).
Reverse transcriptase real-time PCR and shRNAs
Total RNA was extracted using the RNAeasy Mini Kit (Qiagen, Hilden, Germany) and 500 ng of RNA were reversed-transcribed using SuperScript VILO Master Mix (ThermoFisher). For quantitative real-time PCR, cDNA and the following primers were used: ITGB8 5′-ACCAGAAGGAGGTTTTGACG-3′ and 5′-ACCAGAAGGAGGTTTTGACG-3′, CDK1 5′-AAACTACAGGTCAAGTGGTAGCC-3′ and 5′-TCCTGCATAAGCACATCCTGA-3′, CDKN1A 5′-CGATGGAACTTCGACTTTGTCA-3′ and 5′-GCACAAGGGTACAAGACAGTG-3′, GAPDH 5′-CAG AACATCATCCCTGCCTC-3′ and 5′-TGGCAGGTTTTTCTAGACGG-3′, SOX2 5′-AACTTTTGTCGGAGACGGAG-3′ and 5′-CTCGCCATGCTATTGCCG-3, SOX9 5′-ACCAGAACTCCAGCTCCTAC-3′ and 5′-TAGACGGGTTGTTCCCAGT-3′, CD1335′-TGGAGAACATGAACAGCACC-3′ and 5′-GCTGTTGCAGGTTTCACTTG-3′. The sequences for shRNAs used to target ITGAV and ITGB8 in GSCs have been reported elsewhere.44
Crispr/Cas9 gene editing
To target ITGB8 the following gDNA oligos were designed three different gDNAs using open source platforms (http://crispr.mit.edu): gDNA1 5′-GACCTCGCCGGTCGTTTTGC-3′ gDNA2 5′-TGCCTGCAAAACGACCGGCG-3′ gDNA3 5′-TCATATCGGATGGCGAAAAG-3′ gDNA4 5′-CGTGACTTTCGTCTTGGATT-3′ gDNA5 5′-TTCTCCCGTGACTTTCGTCT-3′ gDNA6 5′-ATTAATACCC AGGTGACACC-3′ gDNA7 5′-TTCAGGCTTCTCACGTCGGT-3′ gDNA8 5′-GTATCACCTGTCTAATGATC-3′. gDNAs were inserted in the Lentiviral CRISPR-Cas9 pL-CRISPR.EFS.GFP (Addgene) followed by viral infection of cells. The Surveyor Mutation Detection kit (Integrated DNA Technologies, Coralville, IA, USA) was used to confirm mutation-induced DNA heteroduplexes in ITGB8, but not in potential other genes anlayzed that were predicted to yield possible off-target effects based on gDNA sequences.
Statistical analysis
Student’s t-test was performed to determine statistically significant differences between groups. Excel (Microsoft, Redmond, WA, USA) was used to calculate statistics.
Supplementary Material
Acknowledgments
We would like to thank colleagues in the M.D. Anderson Neurosurgery department for providing freshly resected human samples. This work was supported by grants to JHM from NIH/NINDS (R01NS0 7635 and R01NS078402), the Cancer Prevention and Research Institute of Texas (RP140411), and in part by an NIH/NCI SPORE in Brain Cancer (P50CA127001).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
PAG, JHT, ZC, JEM, QG, EPS and NM performed critical experiments. FFL, GR and GF contributed important experimental reagents. PAG and JHM wrote the manuscript.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer cell. 2009;15:45–56. doi: 10.1016/j.ccr.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129–1133. doi: 10.1038/nature07443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–1217. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanai N, Alvarez-Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med. 2005;353:811–822. doi: 10.1056/NEJMra043666. [DOI] [PubMed] [Google Scholar]
- 5.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 6.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 7.Gilbertson RJ, Rich JN. Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733–736. doi: 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- 8.Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8:610–622. doi: 10.1038/nrn2175. [DOI] [PubMed] [Google Scholar]
- 9.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCarty JH. Cell adhesion and signaling networks in brain neurovascular units. Curr Opin Hematol. 2009;16:209–214. doi: 10.1097/MOH.0b013e32832a07eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delamarre E, Taboubi S, Mathieu S, Berenguer C, Rigot V, Lissitzky JC, et al. Expression of integrin alpha6beta1 enhances tumorigenesis in glioma cells. Am J Pathol. 2009;175:844–855. doi: 10.2353/ajpath.2009.080920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem cell. 2010;6:421–432. doi: 10.1016/j.stem.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weis SM, Cheresh DA. alphaV integrins in angiogenesis and cancer. Cold Spring Harb Perspect Med. 2011;1:a006478. doi: 10.1101/cshperspect.a006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanamori M, Vanden Berg SR, Bergers G, Berger MS, Pieper RO. Integrin beta3 overexpression suppresses tumor growth in a human model of gliomagenesis: implications for the role of beta3 overexpression in glioblastoma multiforme. Cancer Res. 2004;64:2751–2758. doi: 10.1158/0008-5472.can-03-3354. [DOI] [PubMed] [Google Scholar]
- 15.Gilbert MR, Kuhn J, Lamborn KR, Lieberman F, Wen PY, Mehta M, et al. Cilengitide in patients with recurrent glioblastoma: the results of NABTC 03-02, a phase II trial with measures of treatment delivery. J Neurooncol. 2012;106:147–153. doi: 10.1007/s11060-011-0650-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishimura SL, Sheppard D, Pytela R. Integrin alpha v beta 8. Interaction with vitronectin and functional divergence of the beta 8 cytoplasmic domain. J Biol Chem. 1994;269:28708–28715. [PubMed] [Google Scholar]
- 17.Venstrom K, Reichardt L. Beta 8 integrins mediate interactions of chick sensory neurons with laminin-1, collagen IV, and fibronectin. Mol Biol Cell. 1995;6:419–431. doi: 10.1091/mbc.6.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cambier S, Gline S, Mu D, Collins R, Araya J, Dolganov G, et al. Integrin alpha(v) beta8-mediated activation of transforming growth factor-beta by perivascular astrocytes: an angiogenic control switch. Am J Pathol. 2005;166:1883–1894. doi: 10.1016/s0002-9440(10)62497-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Worthington JJ, Klementowicz JE, Travis MA. TGFbeta: a sleeping giant awoken by integrins. Trends Biochem Sci. 2010;36:47–54. doi: 10.1016/j.tibs.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 20.Robertson IB, Rifkin DB. Regulation of the bioavailability of TGF-beta and TGF-beta-related proteins. Cold Spring Harb Perspect Biol. 2016;8:6. doi: 10.1101/cshperspect.a021907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allinson KR, Lee HS, Fruttiger M, McCarty JH, Arthur HM. Endothelial expression of TGFbeta type II receptor is required to maintain vascular integrity during postnatal development of the central nervous system. PLoS One. 2012;7:e39336. doi: 10.1371/journal.pone.0039336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnold TD, Ferrero GM, Qiu H, Phan IT, Akhurst RJ, Huang EJ, et al. Defective retinal vascular endothelial cell development as a consequence of impaired integrin alphaVbeta8-mediated activation of transforming growth factor-beta. J Neurosci. 2012;32:1197–1206. doi: 10.1523/JNEUROSCI.5648-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arnold TD, Niaudet C, Pang MF, Siegenthaler J, Gaengel K, Jung B, et al. Excessive vascular sprouting underlies cerebral hemorrhage in mice lacking alphaVbeta8-TGFbeta signaling in the brain. Development. 2014;141:4489–4499. doi: 10.1242/dev.107193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirota S, Clements TP, Tang LK, Morales JE, Lee HS, Oh SP, et al. Neuropilin 1 balances beta8 integrin-activated TGFbeta signaling to control sprouting angiogenesis in the brain. Development. 2015;142:4363–4373. doi: 10.1242/dev.113746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen HL, Lee YJ, Shin J, Lee E, Park SO, McCarty JH, et al. TGF-beta signaling in endothelial cells, but not neuroepithelial cells, is essential for cerebral vascular development. Lab Invest. 2011;91:1554–1563. doi: 10.1038/labinvest.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Proctor JM, Zang K, Wang D, Wang R, Reichardt LF. Vascular development of the brain requires beta8 integrin expression in the neuroepithelium. J Neuroscience. 2005;25:9940–9948. doi: 10.1523/JNEUROSCI.3467-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCarty JH, Monahan-Earley RA, Brown LF, Keller M, Gerhardt H, Rubin K, et al. Defective associations between blood vessels and brain parenchyma lead to cerebral hemorrhage in mice lacking alphav integrins. Mol Cell Biol. 2002;22:7667–7677. doi: 10.1128/MCB.22.21.7667-7677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma L, Shen F, Jun K, Bao C, Kuo R, Young WL, et al. Integrin beta8 deletion enhances vascular dysplasia and hemorrhage in the brain of adult Alk1 heterozygous mice. Transl Stroke Res. 2016;7:488–496. doi: 10.1007/s12975-016-0478-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su H, Kim H, Pawlikowska L, Kitamura H, Shen F, Cambier S, et al. Reduced expression of integrin alphavbeta8 is associated with brain arteriovenous malformation pathogenesis. Am J Pathol. 2010;176:1018–1027. doi: 10.2353/ajpath.2010.090453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dardiotis E, Siokas V, Zafeiridis T, Paterakis K, Tsivgoulis G, Dardioti M, et al. Integrins AV and B8 gene polymorphisms and risk for intracerebral hemorrhage in Greek and Polish populations. Neuromol Med. 2016;19:69–80. doi: 10.1007/s12017-016-8429-3. [DOI] [PubMed] [Google Scholar]
- 31.Mobley AK, McCarty JH. beta8 integrin is essential for neuroblast migration in the rostral migratory stream. Glia. 2011;59:1579–1587. doi: 10.1002/glia.21199. [DOI] [PubMed] [Google Scholar]
- 32.Mobley AK, Tchaicha JH, Shin J, Hossain MG, McCarty JH. Beta8 integrin regulates neurogenesis and neurovascular homeostasis in the adult brain. J Cell Sci. 2009;122:1842–1851. doi: 10.1242/jcs.043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci. 2011;108:7950–7955. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arrillaga-Romany I, Reardon DA, Wen PY. Current status of antiangiogenic therapies for glioblastomas. Expert Opin Investig Drugs. 2014;23:199–210. doi: 10.1517/13543784.2014.856880. [DOI] [PubMed] [Google Scholar]
- 36.Sathyan P, Zinn PO, Marisetty AL, Liu B, Kamal MM, Singh SK, et al. Mir-21-Sox2 axis delineates glioblastoma subtypes with prognostic impact. J Neurosci. 2015;35:15097–15112. doi: 10.1523/JNEUROSCI.1265-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, et al. Glioma-associated cancer-initiating cells induce immunosuppression. Clin Cancer Res. 2010;16:461–473. doi: 10.1158/1078-0432.CCR-09-1983. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Bruna A, Darken RS, Rojo F, Ocana A, Penuelas S, Arias A, et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell. 2007;11:147–160. doi: 10.1016/j.ccr.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 39.Penuelas S, Anido J, Prieto-Sanchez RM, Folch G, Barba I, Cuartas I, et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell. 2009;15:315–327. doi: 10.1016/j.ccr.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 40.Massague J. TGFbeta in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirota S, Liu Q, Lee HS, Hossain MG, Lacy-Hulbert A, McCarty JH. The astrocyte-expressed integrin alphavbeta8 governs blood vessel sprouting in the developing retina. Development. 2011;138:5157–5166. doi: 10.1242/dev.069153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reyes SB, Narayanan AS, Lee HS, Tchaicha JH, Aldape KD, Lang FF, et al. alphav-beta8 integrin interacts with RhoGDI1 to regulate Rac1 and Cdc42 activation and drive glioblastoma cell invasion. Mol Biol Cell. 2013;24:474–482. doi: 10.1091/mbc.E12-07-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tchaicha JH, Mobley AK, Hossain MG, Aldape KD, McCarty JH. A mosaic mouse model of astrocytoma identifies alphavbeta8 integrin as a negative regulator of tumor angiogenesis. Oncogene. 2010;29:4460–4472. doi: 10.1038/onc.2010.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tchaicha JH, Reyes SB, Shin J, Hossain MG, Lang FF, McCarty JH. Glioblastoma angiogenesis and tumor cell invasiveness are differentially regulated by beta8 integrin. Cancer Research. 2011;71:6371–6381. doi: 10.1158/0008-5472.CAN-11-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fang L, Deng Z, Shatseva T, Yang J, Peng C, Du WW, et al. MicroRNA miR-93 promotes tumor growth and angiogenesis by targeting integrin-beta8. Oncogene. 2011;30:806–821. doi: 10.1038/onc.2010.465. [DOI] [PubMed] [Google Scholar]
- 46.Huang JJ, Blobe GC. Dichotomous roles of TGF-beta in human cancer. Biochem Society Transac. 2016;44:1441–1454. doi: 10.1042/BST20160065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frei K, Gramatzki D, Tritschler I, Schroeder JJ, Espinoza L, Rushing EJ, et al. Transforming growth factor-beta pathway activity in glioblastoma. Oncotarget. 2015;6:5963–5977. doi: 10.18632/oncotarget.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rojas A, Padidam M, Cress D, Grady WM. TGF-beta receptor levels regulate the specificity of signaling pathway activation and biological effects of TGF-beta. Biochim Biophys Acta. 2009;1793:1165–1173. doi: 10.1016/j.bbamcr.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakajima H, Yonemura S, Murata M, Nakamura N, Piwnica-Worms H, Nishida E. Myt1 protein kinase is essential for Golgi and ER assembly during mitotic exit. J Cell Biol. 2008;181:89–103. doi: 10.1083/jcb.200708176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Toledo CM, Ding Y, Hoellerbauer P, Davis RJ, Basom R, Girard EJ, et al. Genome-wide CRISPR-Cas9 screens reveal Loss of redundancy between PKMYT1 and WEE1 in glioblastoma stem-like cells. Cell Rep. 2015;13:2425–2439. doi: 10.1016/j.celrep.2015.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ding Y, Hubert CG, Herman J, Corrin P, Toledo CM, Skutt-Kakaria K, et al. Cancer-specific requirement for BUB1B/BUBR1 in human brain tumor isolates and genetically transformed cells. Cancer Discov. 2013;3:198–211. doi: 10.1158/2159-8290.CD-12-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nyati S, Schinske-Sebolt K, Pitchiaya S, Chekhovskiy K, Chator A, Chaudhry N, et al. The kinase activity of the Ser/Thr kinase BUB1 promotes TGF-beta signaling. Science Signal. 2015;8:ra1. doi: 10.1126/scisignal.2005379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wee B, Pietras A, Ozawa T, Bazzoli E, Podlaha O, Antczak C, et al. ABCG2 regulates self-renewal and stem cell marker expression but not tumorigenicity or radiation resistance of glioma cells. Sci Rep. 2016;6:25956. doi: 10.1038/srep25956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anido J, Saez-Borderias A, Gonzalez-Junca A, Rodon L, Folch G, Carmona MA, et al. TGF-beta receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell. 2010;18:655–668. doi: 10.1016/j.ccr.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 55.Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 56.Batchelor TT, Reardon DA, de Groot JF, Wick W, Weller M. Antiangiogenic therapy for glioblastoma: current status and future prospects. Clin Cancer Res. 2014;20:5612–5619. doi: 10.1158/1078-0432.CCR-14-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ellis LM, Reardon DA. Cancer: the nuances of therapy. Nature. 2009;458:290–292. doi: 10.1038/458290a. [DOI] [PubMed] [Google Scholar]
- 59.Jahangiri A, De Lay M, Miller LM, Carbonell WS, Hu YL, Lu K, et al. Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of antiangiogenic therapy resistance. Clin Cancer Res. 2013;19:1773–1783. doi: 10.1158/1078-0432.CCR-12-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell. 2012;22:21–35. doi: 10.1016/j.ccr.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCarty JH. Glioblastoma resistance to anti-VEGF therapy: has the challenge been MET? Clin Cancer Res. 2013;19:1631–1633. doi: 10.1158/1078-0432.CCR-13-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Piao Y, Liang J, Holmes L, Henry V, Sulman E, de Groot JF. Acquired resistance to anti-VEGF therapy in glioblastoma is associated with a mesenchymal transition. Clin Cancer Res. 2013;19:4392–4403. doi: 10.1158/1078-0432.CCR-12-1557. [DOI] [PubMed] [Google Scholar]
- 63.Cheerathodi M, Avci NG, Guerrero PA, Tang LK, Popp J, Morales JE, et al. The cytoskeletal adapter protein spinophilin regulates invadopodia dynamics and tumor cell invasion in glioblastoma. Mol Cancer Res. 2016;14:1277–1287. doi: 10.1158/1541-7786.MCR-16-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee HS, Cheerathodi M, Chaki SP, Reyes SB, Zheng Y, Lu Z, et al. Protein tyrosine phosphatase-PEST and beta8 integrin regulate spatiotemporal patterns of RhoGDI1 activation in migrating cells. Mol Cell Biol. 2015;35:1401–1413. doi: 10.1128/MCB.00112-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Upadhyaya M, Spurlock G, Thomas L, Thomas NS, Richards M, Mautner VF, et al. Microarray-based copy number analysis of neurofibromatosis type-1 (NF1)-associated malignant peripheral nerve sheath tumors reveals a role for Rho-GTPase pathway genes in NF1 tumorigenesis. Hum Mutat. 2012;33:763–776. doi: 10.1002/humu.22044. [DOI] [PubMed] [Google Scholar]
- 66.Schittenhelm J, Klein A, Tatagiba MS, Meyermann R, Fend F, Goodman SL, et al. Comparing the expression of integrins alphavbeta3, alphavbeta5, alphavbeta6, alphavbeta8, fibronectin and fibrinogen in human brain metastases and their corresponding primary tumors. Int J Clin Exp Pathol. 2013;6:2719–2732. [PMC free article] [PubMed] [Google Scholar]
- 67.Hamidi H, Pietila M, Ivaska J. The complexity of integrins in cancer and new scopes for therapeutic targeting. Br J Cancer. 2016;115:1017–1023. doi: 10.1038/bjc.2016.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vaillant F, Asselin-Labat ML, Shackleton M, Forrest NC, Lindeman GJ, Visvader JE. The mammary progenitor marker CD61/beta3 integrin identifies cancer stem cells in mouse models of mammary tumorigenesis. Cancer Res. 2008;68:7711–7717. doi: 10.1158/0008-5472.CAN-08-1949. [DOI] [PubMed] [Google Scholar]
- 69.Ming XY, Fu L, Zhang LY, Qin YR, Cao TT, Chan KW, et al. Integrin alpha7 is a functional cancer stem cell surface marker in oesophageal squamous cell carcinoma. Nat Commun. 2016;7:13568. doi: 10.1038/ncomms13568. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.